

Abstract
Thioesterases catalyze hydrolysis of acyl thioesters to release carboxylic acid or macrocyclization to produce the corresponding macrocycle in the biosynthesis of fatty acids, polyketides, or nonribosomal peptides. Recently, we reported that the thioesterase CmiS1 from Streptomyces sp. MJ635-86F5 catalyzes the Michael addition of glycine to an α,β-unsaturated fatty acyl thioester followed by thioester hydrolysis in the biosynthesis of the macrolactam antibiotic cremimycin. However, the molecular mechanisms of CmiS1-catalyzed reactions are unclear. Here, we report on the functional and structural characterization of the CmiS1 homolog SAV606 from Streptomyces avermitilis MA-4680. In vitro analysis indicated that SAV606 catalyzes the Michael addition of glycine to crotonic acid thioester and subsequent hydrolysis yielding (R)-N-carboxymethyl-3-aminobutyric acid. We also determined the crystal structures of SAV606 both in ligand-free form at 2.4 Å resolution and in complex with (R)-N-carboxymethyl-3-aminobutyric acid at 2.0 Å resolution. We found that SAV606 adopts an α/β hotdog fold and has an active site at the dimeric interface. Examining the complexed structure, we noted that the substrate-binding loop comprising Tyr-53–Asn-61 recognizes the glycine moiety of (R)-N-carboxymethyl-3-aminobutyric acid. Moreover, we found that SAV606 does not contain an acidic residue at the active site, which is distinct from canonical hotdog thioesterases. Site-directed mutagenesis experiments revealed that His-59 plays a crucial role in both the Michael addition and hydrolysis via a water molecule. These results allow us to propose the reaction mechanism of the SAV606-catalyzed Michael addition and thioester hydrolysis and provide new insight into the multiple functions of a thioesterase family enzyme.
Keywords: enzyme mechanism, enzyme structure, hydrolase, secondary metabolism, X-ray crystallography, β-amino acid, Michael addition, hotdog fold, thioesterase
Introduction
Thioesterases (TEs)2 hydrolyze the thioester bond involved in acyl-acyl carrier protein (ACP) and acyl-CoA to release a carboxylic acid. Alternatively, TEs catalyze the macrocyclization to produce a macrolide or macrolactam. TEs play important roles in the biosynthesis of fatty acids, polyketides, and nonribosomal peptides and in the metabolism of lipids (1–4). Currently, TEs are classified into 25 families in the ThYme database based on their amino acid sequence, substrate specificity, and tertiary structure (5, 6). Most of them have either an α/β-hydrolase fold or a hotdog fold. The α/β-hydrolase TEs, which belong to family 2 and families 16–22, possess a conserved catalytic triad (Ser-His-Asp) in the active site (2, 5). The reaction is initiated by the nucleophilic attack of the Ser residue on the carbonyl carbon of the acyl thioester substrate followed by hydrolysis or macrocyclization of the resulting ester intermediate. The α/β-hydrolase TEs release final products or remove aberrant non-extendable units from ACP in polyketide and nonribosomal peptide biosynthesis (2, 3).
The hotdog TEs, which belong to families 4–15 and 24, are structurally and evolutionarily related to a family of dehydratases (DHs) (5, 7). They commonly hydrolyze the thioester bond of the acyl-CoA molecule, but some of them act on acyl-ACP. They exist either as a homodimer of a single hotdog fold or as a monomer of a double hotdog fold with an active site located at the interface between the two hotdogs. They normally do not contain the catalytic His residue that is fully conserved in DHs (7). They mostly possess an acidic residue (Glu/Asp) as a nucleophile for generating a covalent anhydride intermediate or general base for activating a water molecule in catalysis in the active site (5, 7). However, some of them (e.g. families 15 and 24) do not contain any acidic residue in the active site. The family 15 TE CalE7, which is involved in the biosynthesis of calicheamicin, is proposed to use an Arg residue to activate the thioester carbonyl and stabilize the oxyanion intermediate in the reaction (8). The family 24 TE Rv0098, which catalyzes the hydrolysis of a long-chain fatty acyl-CoA in Mycobacterium tuberculosis, is hypothesized to use two Tyr residues and a Asn residue for activation of the thioester carbonyl and stabilization of the oxyanion intermediate, although the reaction mechanism is still unclear (9).
β-Amino acids are frequently found as important components of natural products such as macrolactam polyketide antibiotics (10, 11). These β-amino acids are typically derived from proteinogenic α-amino acids. However, some of the β-amino acids are constructed through the polyketide pathway. In the biosynthesis of the macrolactam antibiotic cremimycin, the starter unit 3-aminononanoic acid is synthesized from non-2-enoyl-ACP and glycine (12). Initially, CmiS1, which belongs to the family 24 TEs, catalyzes the Michael addition of glycine to the β-position of the non-2-enoyl-ACP and subsequent hydrolysis of the thioester to give N-carboxymethyl-3-aminononanoic acid (Fig. 1A). N-Carboxymethyl-3-aminononanoic acid is then converted to 3-aminononanoic acid by a FAD-dependent oxidase, CmiS2. Homologous genes to cmiS1 and cmiS2 are conserved in the biosynthetic gene clusters for related macrolactam polyketides such as heronamide, BE-14106, and ML-449, all of which possess a β-amino medium-chain fatty acid unit at the starter position of their polyketide backbone (13–15), suggesting that the amino groups of these β-amino acids are introduced through the same mechanism.
Figure 1.

Reactions of CmiS1 (A) and SAV606 (B).
CmiS1 is an exceptionally rare TE because it performs a dual function consisting of the Michael addition of glycine and the hydrolysis of a thioester. Although CmiS1 belongs to the same family as Rv0098, whose crystal structure is available, the reported function of Rv0098 is only the hydrolysis of a long-chain fatty acyl-CoA (9). Thus, the reported functions of CmiS1 and Rv0098 are quite different. It is unclear how CmiS1 catalyzes the Michael addition of glycine to an α,β-unsaturated thioester. CmiS1 homologs are distributed not only in macrolactam-producing strains but also in other bacteria. For example, the protein SAV606 from Streptomyces avermitilis MA-4680 (16) shows 51% amino acid sequence identity with CmiS1. Here, we describe the functional characterization and structural determination of SAV606. We find that SAV606 catalyzes the Michael addition of glycine to crotonic acid thioester and subsequent hydrolysis (Fig. 1B). The structure of SAV606 in complex with the product molecule allows us to propose the mechanism of the Michael addition of glycine to an α,β-unsaturated thioester.
Results
Functional analysis of SAV606
To investigate the function of SAV606, we expressed recombinant SAV606 protein in Escherichia coli. Because CmiS1 catalyzes the Michael addition of glycine to non-2-enoyl-ACP and the subsequent hydrolysis of the thioester (12), we speculated that SAV606 catalyzes a similar reaction. We first examined the SAV606 reaction with various lengths of α,β-unsaturated fatty acyl N-acetylcysteamine (NAC) in the presence of glycine. We detected the release of NAC with Ellman's reagent (17) in the reaction with crotonoyl-NAC and glycine (specific activity: 3.3 units/mg, relative activity: 100%), whereas we were not able to observe the release of NAC in the absence of glycine (<0.05%). We did not detect the release of NAC in the reaction with butyryl-NAC in the presence of glycine (<0.05%). Thus, the substrate of SAV606 must be α,β-unsaturated fatty acid thioesters. These results suggest that SAV606 requires glycine for its reaction before the hydrolysis of the thioester, as is the case for the CmiS1 reaction. Release of NAC was not detected from the reaction with non-2-enoyl-NAC and glycine (<0.05%), suggesting that SAV606 has a different substrate specificity from CmiS1 and prefers a shorter chain α,β-unsaturated fatty acid thioester as substrate.
We also analyzed the reaction product by HPLC after derivatization with phenyl isothiocyanate. We observed the accumulation of product derivative from the reaction mixture containing both crotonoyl-NAC and glycine (Fig. 2A). We did not observe the accumulation of product derivative when we used crotonic acid as a substrate instead of crotonoyl-NAC, suggesting that the thioester moiety of an α,β-unsaturated fatty acyl substrate is required for the reaction.
Figure 2.

HPLC analysis of phenylthiocyanate derivatives of the product of SAV606 reactions with crotonoyl-NAC and glycine as substrates. PEGASIL ODS SP100 (A) and CHIRAL ART Cellulose-SC (B) columns were used for analysis. HPLC traces (detection at 268 nm) are shown. Au, absorbance units.
To confirm the chemical structure of the reaction product formed by SAV606 from crotonoyl-NAC and glycine, we carried out a large-scale enzymatic reaction. After esterification of the enzyme reaction product with ethanol in acidic conditions, the chemical structure of the resulting ethyl ester was analyzed by NMR and MS. High-resolution fast atom bombardment mass spectrometry (HR-FAB-MS) (positive mode) showed a pseudo-molecular ion peak with m/z 218.1381, which corresponds to [M+H]+ of the molecular formula C10H19NO4, indicating the expected diethyl ester of N-carboxymethyl-3-aminobutyric acid (CMABA). 1H NMR and 1H,1H COSY showed a contiguous 1H spin system from H2 to H4 of the 3-aminobutyrate core structure (Fig. 3). One methylene with an AB-type signal at 3.4 ppm in the 1H NMR spectrum is isolated from the spin system, indicating that the resulting ethyl ester is N-ethoxycarbonylmethyl-3-aminobutyric acid ethyl ester, suggesting that the SAV606 reaction product is CMABA. We also synthesized the authentic compound from 3-aminobutyric acid by esterification with ethanol followed by alkylation with ethyl bromoacetate to confirm the structure. To determine the stereochemistry at the C3 position of the product CMABA, we derivatized the SAV606 product with phenyl isothiocyanate and then analyzed the resulting 3-phenylthiohydantoin-butyric acid by HPLC with a chiral column. By comparing the retention time of the derivative with those of the R- enantiomer of 3-phenylthiohydantoin-butyric acid and its racemic form, the stereochemistry at position C3 was determined to be R- (Fig. 2B).
Figure 3.
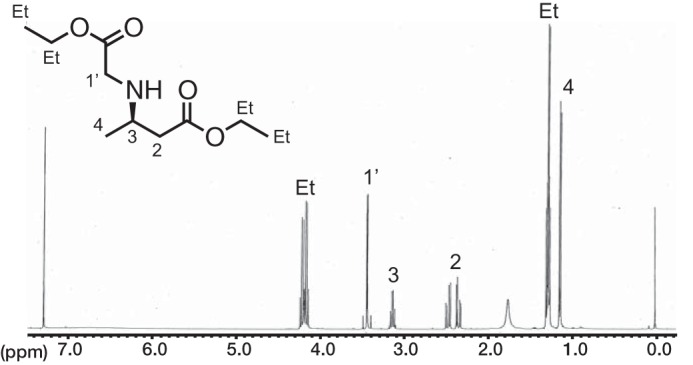
1H NMR spectrum of SAV606 product ethyl ester.
Substrate specificity of SAV606
To analyze the acyl thioester substrate preference of SAV606, we carried out a kinetic analysis. The kcat and Km values of SAV606 for crotonoyl-NAC were found to be 92 min−1 and 0.74 mm (Table 1), respectively, which are comparable with those of CmiS1 for non-2-enoyl-NAC (12). Although SAV606 also shows activity toward hex-2-enoyl-NAC, the Km value was nine times higher than that for crotonoyl-NAC. Next, we analyzed the amino donor preference of SAV606. The Km value of SAV606 for glycine was 26 mm. SAV606 also showed activity toward l-alanine (16% of the activity toward glycine), whereas the enzyme had no activity toward other amino acids such as β-alanine, l-valine, and l-serine.
Table 1.
Steady-state kinetic parameters of SAV606
ND, not detected.
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| mm | min−1 | min−1mm−1 | |
| Crotonoyl-NAC | 0.74 ± 0.14 | 92 ± 6 | 120 |
| Glycine | 26 ± 5 | 91 ± 6 | 3.5 |
| Hex-2-enoyl-NAC | 6.6 ± 0.4 | 17 ± 0.4 | 2.6 |
| Glycine | 120 ± 10 | 16 ± 1 | 0.13 |
| Non-2-enoyl-NAC | ND | ND | ND |
| Glycine | ND | ND | ND |
Analysis of SAV606 reaction with N-carboxymethyl-3-aminobutyryl-NAC
To confirm the reaction pathway, we chemically synthesized racemic N-carboxymethyl-3-aminobutyryl-NAC (CMABA-NAC) as a likely reaction intermediate and examined the reaction of SAV606 with it. We detected the release of NAC with Ellman's reagent, as observed in the reaction with crotonoyl-NAC and glycine. The kcat and Km values of SAV606 for racemic CMABA-NAC were determined to be 23 min−1 and 0.80 mm, respectively. Thus, SAV606 likely catalyzes the Michael addition of glycine to the β-position of the crotonic acid thioester followed by hydrolysis of the thioester to produce (R)-CMABA (Fig. 1B).
Overall structure of SAV606
We determined the crystal structure of apo SAV606 at 2.4 Å resolution (Table 2). There is one SAV606 dimer per crystallographic asymmetric unit. The dimeric interface buries a surface area of 1052 Å2 per monomer (12% of the total monomer surface), indicating that the contact area is large enough for the protein to exist as a dimer. Gel filtration chromatography analysis also showed that SAV606 exists as a homodimer in solution (data not shown). The SAV606 monomer shows a typical α/β hotdog fold that contains a five-stranded antiparallel β-sheet wrapping around one long central helix and two short helices (Fig. 4A). The dimeric structure and α/β hotdog topology are common in TEs and DHs. The structure contains a few disordered loop regions (Asp-56–Gly-58 and Ala-138–Gly-139). The overall structure of SAV606 is similar to that of Rv0098 (PDB code 2PFC; Z-score = 22.0, r.m.s.d. of 1.8 Å, sequence identity of 34%) (9) and that of the family 11 TE YbdB from E. coli (PDB code 4K4C; Z-score = 10.9, r.m.s.d. of 2.5 Å, sequence identity of 13%) (18). SAV606 is also structurally similar to DHs such as FabZ from Pseudomonas aeruginosa (PDB code 1U1Z; Z-score = 13.0, r.m.s.d. of 2.8 Å, sequence identity of 15%) (19) and FabA from P. aeruginosa (PDB code 4B0I; Z-score = 10.7, r.m.s.d. of 3.1 Å, sequence identity of 10%) (20), both of which catalyze the dehydration of (R)-3-hydroxyacyl-ACP.
Table 2.
Structural data collection and refinement statistics
| PDB ID | Datasets |
|
|---|---|---|
| SAV606 |
SAV606-CMABA complex |
|
| 5WSX | 5WSY | |
| Data collection | ||
| Beamline | PF BL-5A | PF BL-5A |
| Wavelength (Å) | 1.00000 | 1.00000 |
| Space group | P212121 | C2221 |
| Unit-cell parameters | ||
| a (Å) | 49.88 | 110.49 |
| b (Å) | 50.10 | 118.31 |
| c (Å) | 125.01 | 71.42 |
| Resolution (Å) | 50.00-2.40 | 50.00-2.00 |
| (outer shell) | (2.52-2.40) | (2.03-2.00) |
| Unique reflections | 12,859 (1,828) | 32,063 (1,578) |
| Redundancy | 6.5 (6.7) | 6.5 (5.7) |
| Completeness (%) | 99.9 (99.9) | 100.0 (99.6) |
| Rmerge (%) | 13.7 (57.8) | 9.1 (55.6) |
| Mean 〈I/σ(I)〉 | 10.8 (2.3) | 21.4 (2.4) |
| Refinement | ||
| Rwork (%) | 20.5 | 17.8 |
| Rfree (%) | 26.1 | 21.5 |
| r.m.s.d. | ||
| Bond lengths (Å) | 0.013 | 0.018 |
| Bond angles (degree) | 1.597 | 1.861 |
| No. of non-hydrogen atoms | ||
| Protein | 2,540 | 2,616 |
| Product | 0 | 22 |
| Solvent | 67 | 136 |
| Average B-factors (Å2) | ||
| Protein | 40.6 | 34.4 |
| Product | 27.4 | |
| Solvent | 39.6 | 38.9 |
| Ramachandran plot | ||
| Favored region (%) | 96.0 | 97.8 |
| Allowed region (%) | 4.0 | 2.2 |
| Outer region (%) | 0.0 | 0.0 |
Figure 4.

SAV606 structures. A, overall structures of apo SAV606 (orange and magenta) and the complex of SAV606 (cyan and green) with CMABA. The (R)-CMABA molecule is shown as yellow sticks. B, surface representation of the substrate-binding pocket in the complex structure. An Fo − Fc electron density map contoured at 3.0 σ was constructed before incorporation of the (R)-CMABA molecule. The water molecule that interacts with His-59 is shown as a red sphere. C, structural comparison of loop regions between the apo and complex structures. The movements of His-59 and Leu-102 are shown as broken arrows. D, the substrate-binding pocket of the complex structure. Ten residues involved in product recognition are shown as sticks. E, structural comparison of the active site between the apo and complex structures.
Complex structure of SAV606 with (R)-CMABA
To better understand the reaction mechanism of SAV606, we attempted to crystallize SAV606 with glycine, crotonoyl-NAC, or racemic CMABA. We succeeded in the determination of the complex structure with CMABA at 2.0 Å resolution (Table 2 and Fig. 4A). The structure shows clear electron density for (R)-CMABA but not for (S)-CMABA in the substrate-binding pocket, which is located at the dimeric interface (Fig. 4B). The complex structure shows significant ligand-induced conformational changes (r.m.s.d. of 1.30 Å for Cα atoms of chain A). In the complex structure, the substrate-binding loop comprising Tyr-53–Asn-61, which is located before the start of the long central helix (helix 2: Ser-62–Glu-83), shows an ordered structure. In addition, the helix 3 region comprising Leu-94–Pro-103 rotates 20° toward the substrate-binding loop (Tyr-53–Asn-61) from the neighboring subunit in the complex structure. Leu-102 moves ∼6 Å toward the substrate-binding loop from the neighboring subunit and forms hydrophobic contacts with Ile-54 (3.6 Å) and Thr-57 (4.0 Å) (Fig. 4C). Consequently, the substrate-binding loop and the helix 3 region cover the internal substrate-binding pocket in the complex structure, suggesting that they might act as the lid of the substrate-binding pocket.
Substrate-binding pocket
SAV606 has a long tunnel that extends about 14 Å from the surface to the bottom of the substrate-binding pocket (Fig. 4B). The location of the substrate-binding pocket of SAV606 is almost identical to those of other characterized hot dog-type TEs such as Rv0098. The substrate-binding pocket of SAV606 is mainly constituted of 10 residues that include Tyr-53, Ile-54, His-59, Asn-61, and Ser-62 from one subunit and Tyr-20, Asn-70, Met-73, Tyr-74, and Ile-107 from the adjacent subunit (Fig. 4D). In contrast to the canonical hot dog TEs, SAV606 contains no acidic Asp/Glu residue in the active site. The binding of CMABA seemed to induce the movement of some amino acid residues in the substrate-binding pocket. The side chains of His-59, Asn-61, and Tyr-74 rotate to accommodate the CMABA molecule (Fig. 4, C and E). In addition, Tyr-20 and Ile-54 shifts ∼1 Å to be located closer to the glycine moiety of CMABA.
The carboxyl group of the glycine moiety is recognized by Tyr-20 (2.7 Å), Tyr-53 (2.7 Å), and Asn-70 (3.0 Å) (Fig. 4D). The amino group of CMABA is sandwiched between Asn-61 (3.3 Å) and Tyr-74 (3.6 Å). Met-73 forms a hydrophobic contact (3.9 Å) with the terminal methyl group of the butyric acid moiety. Ile-54 and Ile-107 show hydrophobic interactions with the methylene group of the glycine moiety (3.7 Å) and the methylene group of the butyric acid moiety (4.1 Å), respectively. The side-chain hydroxy group and the backbone amide group of Ser-62 and the side-chain amide group of Asn-70 form hydrogen bonds (2.5 Å, 3.2 Å, and 3.2 Å, respectively) with the carboxyl group of the butyric acid moiety. The CMABA molecule is bent through these tight interactions in the substrate-binding tunnel (Fig. 4B). The two carboxyl groups of CMABA are close to each other (3.3 Å). His-59 is positioned near the carboxyl group of the butyric acid moiety, although this residue makes no direct hydrogen bond with the CMABA molecule. His-59 retains a water molecule in the substrate-binding pocket through a hydrogen bond (3.2 Å) (Fig. 4, B and D). This water molecule forms a hydrogen bond (2.9 Å) with the amino group of the glycine moiety and is located close (3.6 Å) to the α-carbon of the butyric acid moiety. Thus, this water molecule seems to be the proton source in the Michael addition step and might also nucleophilically attack the thioester moiety of the reaction intermediate for the hydrolysis.
Mutational study
To identify the catalytically important residues for the Michael addition and hydrolysis reactions, we mutated the seven polar residues in the active site and analyzed the activity of these variants toward crotonoyl-NAC in the presence of glycine by detecting the release of NAC. Y20F, Y53F, N61A, and N70A mutants showed significantly reduced activity compared with the wild type (Table 3). H59A, H59N, and H59Q mutants completely lost activity. These results suggest that these five residues are important for the reaction. In contrast, Ser-62 and Tyr-74 are not critical for catalysis, because S62A and Y74F mutants retained activity (74 and 66% of the wild type, respectively). We further investigated the role of residue His-59 because only the His-59 mutants completely lost activity. To determine which reaction step is affected by the mutation of His-59, we carried out HPLC analysis to investigate whether the crotonoyl-NAC substrate was consumed by the H59A, H59N, and H59Q mutants. We were not able to detect the consumption of crotonoyl-NAC or the formation of the CMABA-NAC intermediate from the reaction of these mutants (Fig. 5), suggesting that the His-59 mutation impaired the first (i.e. the Michael addition) step. In addition, the H59A, H59N, and H59Q mutants were not able to catalyze the hydrolysis of CMABA-NAC (Table 3), suggesting that His-59 mutation also disrupted the hydrolysis of the thioester.
Table 3.
Relative activity of SAV606 mutants
ND, not detected.
| SAV606 | Relative activity with crotonoyl-NAC and glycine | Relative activity with CMABA-NAC |
|---|---|---|
| % | % | |
| Wild type | 100 ± 1 | 100 ± 7 |
| Y20F | 2.6 ± 0.1 | 5.3 ± 0.1 |
| Y53F | 1.3 ± 0.1 | 1.8 ± 0.6 |
| H59A | ND | ND |
| H59N | ND | ND |
| H59Q | ND | ND |
| N61A | 1.3 ± 0.1 | ND |
| S62A | 74 ± 4 | 102 ± 5 |
| N70A | 5.4 ± 0.4 | 15 ± 0.2 |
| Y74F | 67 ± 6 | 38 ± 5 |
Figure 5.
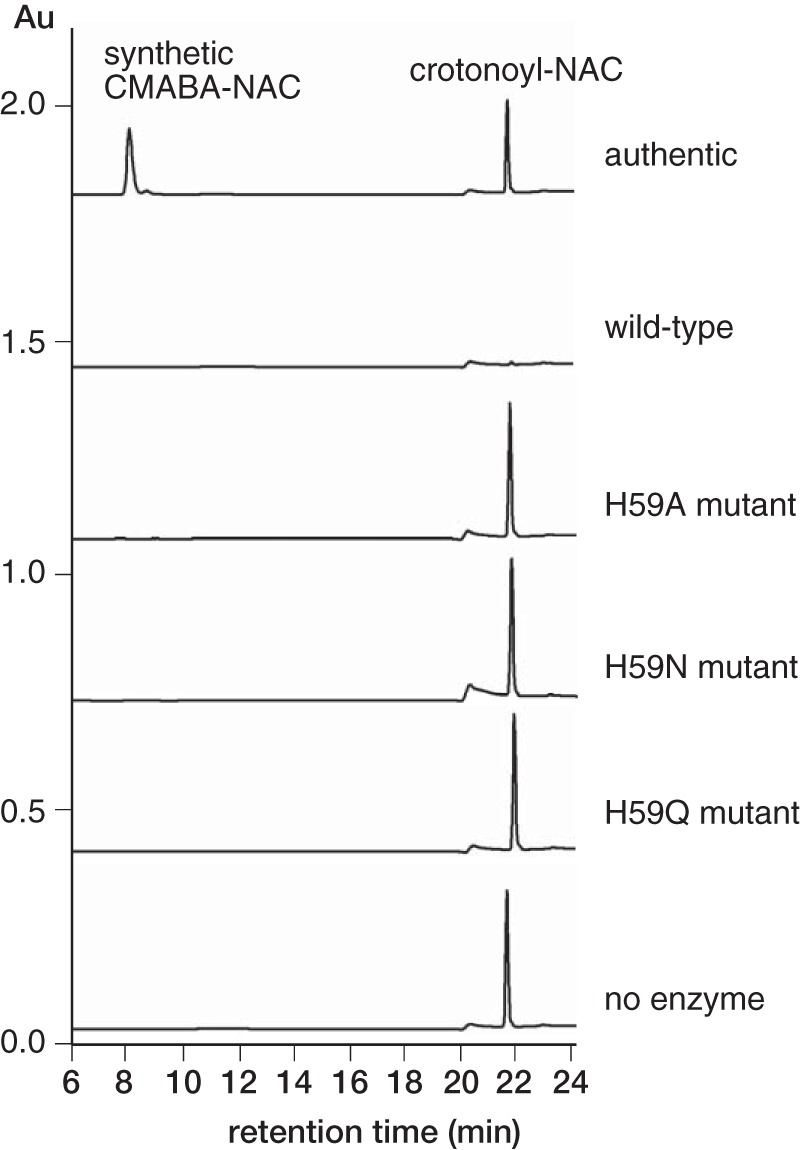
HPLC analysis of the reaction of SAV606 His-59 mutants with crotonoyl-NAC and glycine. HPLC traces (detection at 250 nm) are shown. Au, absorbance units.
The seven polar residues in the active site are fully conserved in CmiS1 (Fig. 6A). In addition, these residues are also conserved in HerU, BecU, and MlaU, which are supposed to be responsible for the biosynthesis of three-amino medium-chain fatty acids in the heronamide, BE-14106, and ML-449 biosynthetic pathways, respectively (13–15). This suggests that these enzymes share the same mechanism as SAV606 to produce N-carboxymethyl-3-amino fatty acids. Interestingly, Met-73 in SAV606 is replaced by Ala in CmiS1 (Fig. 6A). This substitution might be responsible for the difference in the substrate specificity between SAV606 and CmiS1, as judged from the structural observation that SAV606 Met-73 interacts with the terminal methyl group of CMABA.
Figure 6.

Structural comparison of SAV606 with related thioesterases. A, amino acid sequence alignment. The secondary structural elements of SAV606 are indicated above the sequence. Residues in the substrate-binding pocket are shown as cyan and green circles. The cyan dotted line above the sequence indicates the substrate-binding loop of SAV606. B, superimposition of the SAV606 (cyan and green) complex structure with the Rv0098 structure (PDB code 2PFC, gray). The ligand molecules of the SAV606 and Rv0098 structures are shown as yellow and dark gray sticks, respectively.
Discussion
TEs generally catalyze only the hydrolysis of acyl thioesters or macrocyclization (2, 7). However, it was recently reported that some TEs also have protease, peptide ligase, dehydratase, decarboxylase, Claisen cyclase, deacetylase, or transferase activity in addition to the hydrolytic activity (8, 21–26). In this study we found that SAV606 catalyzes both the Michael addition of glycine to an α,β-unsaturated thioester and the hydrolysis of the thioester, as also observed for CmiS1. We determined the crystal structure of SAV606, revealing the structural basis of the Michael addition of glycine to the acyl substrate by this TE. These findings provide new insights into different catalytic functions of TEs.
The complex structure and mutational study allow us to propose the reaction mechanism of SAV606 (Fig. 7). SAV606 first adopts an open conformation so that glycine and crotonic acid thioester would approach the active site. When SAV606 has both glycine and crotonic acid thioester in the active site, the closure of the two hot dogs initiates the Michael addition reaction of glycine to the C3 position of the crotonic acid thioester. Tyr-20, Tyr-53, Asn-61, Asn-70, and Tyr-74 orient the glycine molecule in the proper position for the Michael addition reaction by forming hydrogen bonds. The ammonium ion of the glycine is likely to be deprotonated by His-59 via the water molecule before the Michael addition. Then, the α- position of the butyric acid moiety is protonated by His-59 through the water molecule to produce the CMABA thioester intermediate. His-59 holds the water molecule that is close to both the amino group of the glycine moiety (3.2 Å) and the α-carbon of the butyric acid moiety (3.6 Å) of the CMABA in the complex structure, supporting this proton-shuttling mechanism. The side-chain amide group of Asn-70 and the main-chain amide group of Ser-62 likely stabilize the oxyanion intermediate. In the second step, SAV606 catalyzes the hydrolysis of the CMABA thioester intermediate. His-59 might act as a base and orient the water nucleophile for attack at the carbonyl carbon of the intermediate. The presence of the introduced glycine moiety of the intermediate seems to be necessary for the hydrolysis step because SAV606 did not catalyze the hydrolysis of crotonoyl-NAC in the absence of glycine or butyryl-NAC in the presence of glycine. The substrate-binding loop (Tyr-53–Asn-61) interacts with the glycine moiety but not with the butyryl moiety in the complex structure. This implies the presence of the glycine moiety triggers the closure of the substrate-binding loop, which is crucial for the correct placement of His-59 for hydrolysis. In addition, the carboxyl group of the glycine moiety might assist in the hydrolysis of the thioester by activating the carbonyl group for nucleophilic attack, judging by the short distance (3.3 Å) between the two carboxyl groups of CMABA in the complex structure.
Figure 7.

Proposed reaction mechanism of SAV606.
Structural comparison between SAV606 and Rv0098 revealed that Rv0098 shares all the amino acid residues important for the reaction with SAV606. Tyr-33, Tyr-66, His-72, Asn-74, Asn-83, and Tyr-87 in Rv0098 occupy the same positions as Tyr-20, Tyr-53, His-59, Asn-61, Asn-70, and Tyr-74 in SAV606 (Fig. 6B). Rv0098 has Ala-75 at the position corresponding to Ser-62 in SAV606. We assume that this substitution does not affect the catalysis because the SAV606 S62A mutant retained its activity. Thus, Rv0098 has the essentially same architecture of the active site as SAV606, although the reported function is different. Taken together with the fact that the reported hydrolytic activity of Rv0098 for long-chain acyl-CoA is 102-104-fold less than those of other TEs (9), Rv0098 might require the Michael addition of glycine or other small amino acid(s) to an α,β-unsaturated fatty acid thioester before the hydrolysis of the thioester. Met-73 in SAV606 is substituted by Ala-86 in Rv0098. The small side chain of Ala-86 allows the acyl-binding tunnel to extend over the position of Ala-86 and accommodate a long-chain fatty acyl substrate in the Rv0098 structure (Fig. 6B). This substitution is also observed in CmiS1-type enzymes that utilize a longer acyl substrate than SAV606.
The first Michael addition step of SAV606 is analogous to the reverse reaction of dehydration catalyzed by DHs. The water molecule attacks the C3 position of the acyl thioester substrate in the reverse reaction of dehydration, whereas the glycine attacks the C3 position of the crotonic acid thioester in the SAV606 reaction. Because SAV606 shows a moderate structural similarity to DHs such as FabA, we compared the active-site structure of SAV606 with that of FabA. The canonical DH contains a catalytic His-Asp dyad in the active site. In the FabA structure, His-70 from one subunit is placed so as to abstract the proton from the C2 position of the β-hydroxy fatty acid thioester intermediate (20, 27). Asp-84 from the other subunit forms a hydrogen bond with the C3 hydroxy group and is proposed to protonate the C3 hydroxy group for the β-elimination. SAV606 does not have a His-Asp dyad at the corresponding positions and instead has the catalytically important residue His-59 at a different position (Fig. 8). The position of the glycine moiety in the SAV606 structure is also different from that of the C3 hydroxy group of the acyl thioester in the FabA structure. Thus, the position of the molecule (i.e. glycine in the SAV606 reaction and water in the reverse reaction of dehydration) that attacks at the C3 position of the acyl substrate is significantly different between the two structures. We also compared the SAV606 structure with FabA-ACP complex structure (28). The FabA-ACP complex structure showed that Arg-132, Arg-136, Arg-137, and Lys-161 of FabA are important for recognition of ACP. These FabA basic residues are not conserved in SAV606.
Figure 8.
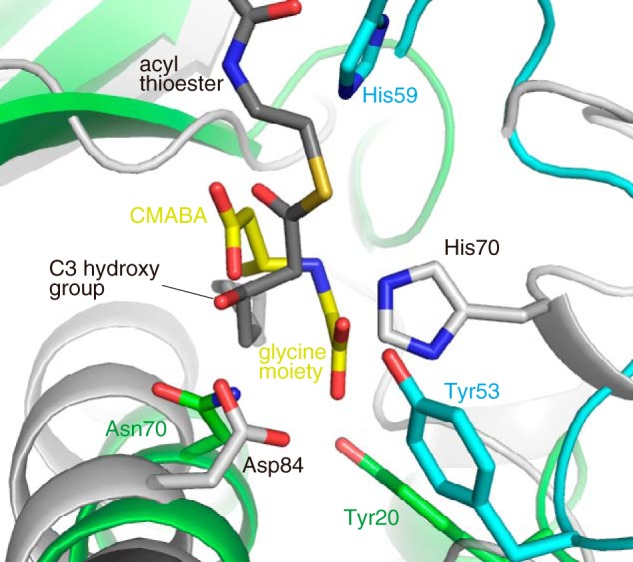
Superimposition of the SAV606 complex structure (cyan and green) on the P. aeruginosa FabA apo structure (PDB code 4FQ9, gray). The (R)-CMABA molecule of the SAV606 structure is shown as yellow sticks. The complex structure of FabA H70N mutant with 3-hydroxydecanoyl-NAC (PDB code 4B0I) is also superimposed, and the 3-hydroxydecanoyl-NAC molecule is shown as dark gray sticks.
The physiological substrate for SAV606 is unclear, although kinetic analysis suggested that crotonic acid thioester is the most likely substrate. SAV606-related metabolites have not been isolated in any culture conditions (29). The gene cluster that includes the sav606 gene also contains a nonribosomal peptide synthetase gene, suggesting that this gene cluster is likely responsible for the biosynthesis of a peptide compound.
In conclusion, we found that SAV606 from S. avermitilis catalyzes the Michael addition of glycine to crotonic acid thioester and the subsequent hydrolysis. We also determined the crystal structure of SAV606 in complex with its product (R)-CMABA. These results develop our understanding of the structure-function relationships of TEs and provide new insight into different catalytic functions of some TEs.
Experimental procedures
Preparation of recombinant SAV606 protein
The artificial sav606 gene that was optimized for overexpression in E. coli was synthesized by Eurofins Genetics (Tokyo, Japan). This sav606 gene was cloned into the expression vector pCold I (Takara Biochemicals, Ohtsu, Japan) by using the NdeI and XhoI restriction sites to form pCold I-sav606. E. coli BL21 (DE3) cells harboring pCold I-sav606 were grown at 37 °C in Luria-Bertani broth containing ampicillin (50 μg/ml). After the optical density at 600 nm reached 0.4, protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (0.2 mm), and the cells were cultured for an additional 20 h at 15 °C. The recombinant SAV606 protein was purified from cell-free extract with a His-60 Ni-Superflow affinity column (Clontech). The protein was then desalted and concentrated using a PD-10 column (GE Healthcare) and an Amicon Ultra 3K centrifugal filter (Merck Millipore, Billerica, MA), respectively. For crystallization, the protein was further treated with Factor Xa protease to remove the His tag and subjected to another round of a His-60 Ni-Superflow affinity chromatography to collect His-tag free protein.
Site-directed mutagenesis
pCold I-sav606 was used for construction of SAV606 mutants. Site-directed mutagenesis was performed with the following oligonucleotides and their complementary oligonucleotides: Y20F, 5′-GTTTTGGTCCCGTTTAAGGACCAC-3′; Y53F, 5′-GTATCGTCGATAAAGCACGATTCTGG-3′; H59A, 5′-GACGATACAGGCGCGCTGAACTCTGTG-3′; H59N, 5′-GACGATACAGGCAATCTGAACTCTGTG-3′; H59Q, 5′-GACGATACAGGCCAGCTGAACTCTGTG-3′; N61A, 5′-ACAGGCCATCTGGCGTCTGTGGAAGTG-3′; S62A, 5′-CATCTGAACGCGGTGGAAGTGAAC-3′; N70A, 5′-AACATCTGCTACGCCCAGATGATGTACTA-3′; Y74F, 5′-AATCAGATGATGTTTTATCTGGTAGCC-3′. The mutations were confirmed by determining the nucleotide sequences. The plasmids were transformed into E. coli BL21 (DE3) cells, and the mutated enzymes were prepared as described above.
Synthesis of acyl-NAC substrates
Crotonic acid (103 mg) was dissolved in CH2Cl2 (5 ml) at 0 °C and stirred for 15 min. A solution of N,N′-dicyclohexylcarbodiimide (274 mg) and 4-(dimethylamino)pyridine (13.7 mg) in CH2Cl2 (5 ml) and NAC (150 mg) were added to the crotonic acid solution simultaneously with two syringes. The mixture was then stirred for 2 days at room temperature. After the slurry was filtered off, the solvent was evaporated. The residue was then purified by silica gel column chromatography (hexane:ethyl acetate = 1:2) to provide crotonoyl-NAC (147 mg). Data are: 1H NMR (400 MHz, CDCl3): δ 6.95 (dq, J = 15.6 Hz, 6.8 Hz, 1H), 6.16 (dq, J = 15.6 Hz, 1.6 Hz, 1H), 3.46 (q, J = 6.0 Hz, 2H), 3.09 (t, J = 6.4 Hz, 2H), 1.96 (s, 3H), 1.91 (dd, J = 6.8 Hz, 1.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 190.4, 170.4, 142.0, 129.9, 39.9, 28.3, 23.3, 18.1; electrospray ionization MS (ESI-MS) (positive mode): m/z 188.1 ([M+H]+).
Non-2-enoyl-NAC was synthesized according to the published method (12). Butyryl-NAC and hex-2-enoyl-NAC were synthesized using a similar method to the crotonoyl-NAC synthesis. From 277 mg of butyric acid, 272 mg of butyryl-NAC was obtained. Data are: 1H NMR (400 MHz, CDCl3): δ 3.44 (q, J = 6.4 Hz, 2H), 3.03 (t, J = 6.2 Hz, 2H), 2.56 (t, J = 7.4 Hz, 2H), 1.97 (s, 3H), 1.70 (sext, J = 7.5 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 200.3, 170.3, 46.0, 39.9, 28.5, 23.3, 19.3, 13.6; ESI-MS (positive mode): m/z 190.1 ([M+H]+). From 107 mg of trans-2-hexenoic acid, 64.5 mg of hex-2-enoyl-NAC was obtained. Data are: 1H NMR (400 MHz, CDCl3): δ 6.94 (dt, J = 15.6 Hz, 6.8 Hz, 1H), 6.14 (dt, J = 15.6 Hz, 1.2 Hz, 1H), 3.46 (q, J = 6.4 Hz, 2H), 3.09 (t, J = 6.4 Hz, 2H), 2.20 (dq, J = 7.2 Hz, 1.6 Hz, 2H), 1.97 (s, 3H), 1.51 (dq, J = 7.6 Hz, 7.2 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 190.6, 170.4, 146.7, 128.5, 39.9, 34.3, 28.3, 23.3, 21.3, 13.8; ESI-MS (positive mode): m/z 216.1 ([M+H]+).
Synthesis of N-ethoxycarbonylmethyl-3-aminobutyric acid ethyl ester
Acetyl chloride (1 ml) was slowly added to ethanol (10 ml) at 0 °C. Then the solution was stirred for 1 h at room temperature. 3-Aminobutyric acid (313 mg) was added to the resulting 10% HCl in ethanol solution. After reflux for 3 h, the solvent was evaporated to provide 3-aminobutyric acid ethyl ester (512 mg).
3-Aminobutyric acid ethyl ester (134 mg) was dissolved in acetonitrile (2.5 ml). K2CO3 (356 mg) was then added to the solution. After stirring the mixture for 15 min at 0 °C, ethyl bromoacetate (121 mg) was added, and the mixture was then stirred for 17 h at room temperature. After evaporation of the solvent, the residue was dissolved in saturated NaHCO3 and extracted with ethyl acetate. After concentration in vacuo, the oil residue was purified with silica gel chromatography (hexane:ethyl acetate = 1:1) to yield N-ethoxycarbonylmethyl-3-aminobutyric acid ethyl ester (69.6 mg). Data are: 1H NMR (400 MHz, CDCl3): δ 4.19 (q, J = 7.7 Hz, 2H), 4.15 (q, J = 7.2 Hz, 2H), 3.44 (d, J = 17.6 Hz, 1H), 3.39 (d, J = 17.2 Hz, 1H), 3.12 (sext, J = 6.4 Hz, 1H), 2.45 (dd, J = 15.2 Hz, 6.4 Hz, 1H), 2.33 (dd, J = 15.2 Hz, 6.0 Hz, 1H), 1.28 (t, J = 7.0 Hz, 3H), 1.26 (t, J = 7.2 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 172.3, 172.0, 60.8, 60.4, 50.0, 48.5, 41.7, 20.4, 14.2; ESI-MS (positive mode): m/z 218.2 ([M+H]+).
Synthesis of 3-phenylthiohydantoin-butyric acid
N-Ethoxycarbonylmethyl-3-aminobutyric acid ethyl ester (69.3 mg) was dissolved in 6 m HCl. After reflux for 4 h, the solvent was evaporated to provide CMABA (45.0 mg), which was used directly for the next step. CMABA (7.7 mg) was dissolved in 2 ml of coupling solution (acetonitrile:pyridine:triethylamine:water = 10:5:2:3). After stirring for 10 min at 0 °C, phenyl isothiocyanate (40 μl) was added. After the mixture was stirred for 4 h at room temperature, it was acidified with 1 m HCl and extracted with ethyl acetate. The organic layer was evaporated to provide crude residue, which was purified by silica gel chromatography (chloroform:methanol = 7:1) to yield 3-phenylthiohydantoin-butyric acid (3.5 mg). Data are: 1H NMR (400 MHz, CDCl3): δ 7.46 (m, 3H), 7.29 (m, 2H), 5.36 (sext, J = 6.8 Hz, 1H), 4.31 (d, 19.2 Hz, 1H), 4.17 (d, J = 19.2 Hz, 1H), 2.83 (dd, J = 15.4 Hz, 6.4 Hz, 1H), 2.78 (dd, J = 15.6 Hz, 6.8 Hz, 1H), 1.46 (d, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 182.5, 173.9, 170.0, 133.0, 129.3, 129.2, 128.4, 48.9, 48.8, 37.8, 29.7, 17.5; ESI-MS (positive mode): m/z 279.1 ([M+H]+).
Synthesis of CMABA-NAC
3-Aminobutyric acid (503 mg), water (2.5 ml), NaOH (206 mg), tert-butanol (2.5 ml), and di-tert-butyl dicarbonate (Boc2O, 1.09 g) were successively added to a flask. The reaction mixture was then stirred at room temperature for 16 h. After acidification with KH2PO4 and H3PO4, the aqueous solution was extracted with ethyl acetate. The combined organic layer was concentrated to obtain Boc-3-aminobutanoic acid (919 mg).
Next, Boc-3-aminobutanoic acid (568 mg) was dissolved in CH2Cl2 (30 ml) and stirred at 0 °C for 15 min. Solutions of N,N′-dicyclohexylcarbodiimide (646 mg) and 4-(dimethylamino)pyridine (38.8 mg) in CH2Cl2 (15 ml) and NAC (402 mg) in CH2Cl2 (15 ml) were added to the solution simultaneously with two syringes. The mixture was then stirred for 2 days at room temperature. After the slurry was filtered off, the solvent was evaporated. The crude residue was then purified by silica column chromatography (hexane:ethyl acetate = 1:2) to provide Boc-3-aminobutyryl-NAC (676 mg).
Boc-3-aminobutyryl-NAC (100 mg) was dissolved in CH2Cl2 (1 ml) at 0 °C and stirred for 5 min. Trifluoroacetic acid (TFA, 2 ml) was slowly added to the solution. After stirring for 2.5 h, the mixture was evaporated to provide crude 3-aminobutyryl-NAC. The crude 3-aminobutyryl-NAC was dissolved in acetonitrile (3 ml), and K2CO3 (253 mg) was added to the solution. After stirring the mixture for 15 min at 0 °C, tert-butyl bromoacetate (84.6 mg) was added, and the mixture was then stirred for 16 h at room temperature. After the mixture was filtered, the solvent was evaporated. The residue was purified by silica gel chromatography (chloroform:methanol = 25:1) to yield N-(tert-butoxycarbonylmethyl)-3-aminobutyryl-NAC (52.1 mg). N-(tert-Butoxycarbonylmethyl)-3-aminobutyryl-NAC (34.4 mg) was dissolved in TFA (1 ml). After stirring for 2.5 h at room temperature, the solution was evaporated. The crude residue was subjected to HPLC chromatography on a PEGASIL ODS SP100 column (5 μm, 250 × 10 mm; Senshu, Tokyo, Japan) with a linear gradient of 5–15% methanol in water containing 0.1% TFA to yield a purified fraction. The fraction was frozen immediately at −80 °C and lyophilized to yield CMABA-NAC (12.2 mg). Data are: 1H NMR (500 MHz, D2O): δ 3.80 (d, J = 16.8 Hz, 1H), 3.75 (d, J = 16.8 Hz, 1H), 3.70 (sext, J = 6.5 Hz, 1H), 3.28 (t, J = 6.3 Hz, 2H), 3.04 (dd, J = 16.6 Hz, 5.8 Hz, 1H), 2.83 (m, 3H), 1.84 (s, 3H), 1.25 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, D2O): δ 199.1, 174.4, 169.5, 51.4, 45.6, 45.2, 38.3, 28.4, 21.8, 15.5; ESI-MS (positive mode): m/z 263.2 ([M+H]+).
Isolation of the ethyl ester of the SAV606 product
5 mm crotonoyl-NAC (35 mg) and 200 mm glycine were mixed with 20 μm SAV606 in 50 mm Tris-HCl (pH 7.5) buffer in a total volume of 37.5 ml. The reaction was carried out at 28 °C for 20 h. After the reaction, protein was removed from the reaction mixture by using an Amicon Ultra centrifugal filter. The resulting low-molecular-weight solution was lyophilized. Because it is difficult to isolate the SAV606 product itself, we treated the product with 10% HCl in ethanol solution in reflux for ethyl esterification. The product derivative was extracted with CHCl3 and then purified by silica gel column chromatography (hexane:ethyl acetate = 1:1) to give 20 mg of N-ethoxycarbonylmethyl-3-aminobutyric acid ethyl ester. Data are: 1H NMR (400 MHz, CDCl3): δ 4.18 (q, J = 7.2 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.43 (d, J = 17.2 Hz, 1H), 3.38 (d, J = 17.2 Hz, 1H), 3.10 (sext, J = 6.4 Hz, 1H), 2.44 (dd, J = 15.0 Hz, 7.0 Hz, 1H), 2.32 (dd, J = 15.0 Hz, 5.8 Hz, 1H), 1.26 (t, J = 7.0 Hz, 3H), 1.25 (t, J = 7.2 Hz, 3H), 1.11 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 172.4, 172.1, 60.9, 60.5, 50.0, 48.7, 41.8, 20.5, 14.3; HR-FAB-MS (positive mode): m/z calculated for C10H20NO4: 218.1392 ([M+H]+); found: 218.1381.
Analysis of the stereochemistry of SAV606 product
For determination of the stereochemistry of the SAV606 product, the isolated N-ethoxycarbonylmethyl-3-aminobutyric acid ethyl ester was converted to 3-phenylthiohydantoin-butyric acid as described above. The 3-phenylthiohydantoin-butyric acid obtained was dissolved in methanol. HPLC analysis was carried out using a Hitachi instrument (Chromaster 5110 Pump and UV Detector l-7405; Hitachi, Tokyo, Japan) equipped with a CHIRAL ART Cellulose-SC column (5 μm, 250 × 4.6 mm; YMC, Kyoto, Japan) with an isocratic solvent system (hexane:ethanol:TFA = 70:30:0.1). The flow rate was 1.0 ml/min, and the detection wavelength was 268 nm.
Analysis of SAV606 reaction with acyl-NAC and glycine using Ellman's assay
To investigate the substrate specificity of SAV606 with Ellman's assay (17), 2 mm fatty acyl-NAC (crotonoyl-NAC, butyryl-NAC, hex-2-enoyl-NAC, or non-2-enoyl-NAC) and 200 mm glycine were mixed with 0.5–15 μm SAV606 in 50 mm Tris-HCl (pH 7.5) in a volume of 50 μl. After incubation at 28 °C for 0.5–35 min, the reaction was quenched by the addition of an equal volume of acetonitrile. Ellman's reagent (5 μl of 5,5′-dithiobis(2-nitrobenzoic acid), 20 mm solution in acetonitrile) was then added to each solution. The absorbance at 412 nm was observed with a V-630 UV/Vis spectrometer (Jasco, Tokyo, Japan). One unit was defined as the amount of enzyme that releases 1 μmol of NAC per minute. For determination of kinetic parameters for fatty acyl-NACs (crotonoyl-NAC or hex-2-enoyl-NAC) by Ellman's assay, the concentration of fatty acyl-NAC was varied between 0.25 mm and 15 mm, and the concentration of glycine (200 mm) was fixed. For determination of kinetic parameters for glycine, the concentration of glycine was varied between 1 mm and 200 mm, and the concentration of crotonoyl-NAC (5 mm) was fixed. Steady-state kinetic parameters were determined from the Michaelis-Menten equation.
Analysis of SAV606 reaction with acyl-NAC and glycine by HPLC
For analysis of the SAV606 reaction product by HPLC, 2 mm acyl substrate (crotonoyl-NAC, butyryl-NAC, or crotonic acid) and 5 mm glycine were mixed with 10 μm SAV606 in 50 mm Tris-HCl (pH 7.5) in a volume of 300 μl. After incubation at 28 °C for 3 h, the product was derivatized with phenyl isothiocyanate as described above. The residue was dissolved in methanol. HPLC analysis was carried out using an instrument using a 996 Photodiode Array Detector (Waters, Milford, MA) and an L-6250 Intelligent Pump (Hitachi) equipped with a PEGASIL ODS SP100 column (5 μm, 250 × 4.6 mm; Senshu) with an isocratic solvent system (water:methanol:TFA = 65:35:0.1). The flow rate was 1.0 ml/min, and the detection wavelength was 268 nm.
To investigate whether crotonoyl-NAC was consumed in the presence of SAV606 His-59 mutants, 200 μm crotonoyl-NAC and 100 mm glycine were mixed with 100 μm SAV606 wild type or His-59 mutant in 50 mm Tris-HCl (pH 7.5) in a volume of 50 μl. After incubation at 28 °C for 10 min, the reaction was quenched by the addition of an equal volume of acetonitrile. HPLC analysis was carried out using an instrument using a 996 Photodiode Array Detector and an L-6250 Intelligent Pump equipped with a PEGASIL ODS SP100 column (5 μm, 250 × 4.6 mm) with a gradient of solvents A (0.1% TFA in water) and B (0.1% TFA in methanol): 0–5 min in 5% B, 5–15 min in 10–15% B linear gradient, and 15–25 min in 60% B. The flow rate was 1.0 ml/min and the detection wavelength was 250 nm.
Enzyme assay using CMABA-NAC and Ellman's assay
For the reaction of SAV606 with CMABA-NAC, 2 mm CMABA-NAC was mixed with 1.6–10 μm SAV606 in 100 mm Tris-HCl (pH 7.5) in a volume of 50 μl. After incubation at 28 °C for 5 min, the reaction was quenched by the addition of an equal volume of acetonitrile. 5,5′-Dithiobis(2-nitrobenzoic acid) (5 μl of 20 mm solution in acetonitrile) was then added to each solution. The absorbance at 412 nm was observed with a V-630 UV/Vis spectrometer. For determination of kinetic parameters for CMABA-NAC, the concentration of CMABA-NAC was varied between 0.1 mm and 4 mm. Steady-state kinetic parameters were determined from the Michaelis-Menten equation.
Crystallization, data collection, and structural determination
SAV606 apocrystals were grown using sitting-drop vapor diffusion by mixing protein solution (10 mg/ml in 5 mm Tris-HCl (pH 7.5), 1% glycerol, and 50 mm CoA) with an equal volume of reservoir solution (0.1 m imidazole (pH 9.0) and 1.0 m sodium citrate) at 20 °C. SAV606 cocrystals with CMABA were grown using sitting-drop vapor diffusion by mixing protein solution (10 mg/ml in 5 mm Tris-HCl (pH 7.5), 1% glycerol, and 10 mm synthetic racemic CMABA) with an equal volume of reservoir solution (0.1 m imidazole (pH 9.0) and 1.0 m sodium citrate) at 20 °C. Before X-ray data collection, crystals were soaked in reservoir solution containing 25% (v/v) ethylene glycol or 25% (v/v) glycerol as a cryoprotectant and flash-frozen in the liquid nitrogen stream. All diffraction data were collected at the BL-5A beamline at the Photon Factory (Tsukuba, Japan) and processed with iMosflm (30) or HKL2000 (31). The initial phase was determined by molecular replacement using the Molrep program (32) with the Rv0098 structure (PDB code 2PFC) as a search model. Protein model building of SAV606 was carried out automatically with the ARP/wARP program (33) and subsequently inspected by Coot (34). Refmac (35) was used for refinement of the structures. The non-crystallographic symmetry was not applied as restraints in the refinement. The structural representations were prepared with PyMOL (DeLano Scientific LLC, Palo Alto, CA). The geometries of the final structures were evaluated using the program MolProbity (36).
Author contributions
A. M., F. K., and T. E. designed the research. T. C., A. M., and F. K. performed the experiments. All authors analyzed the data, wrote the manuscript, and approved the final version of the manuscript.
This work was performed with the approval of the Photon Factory Program Advisory Committee (Proposals 2014G530 and 2016G624) and was supported in part by Ministry of Education, Culture, Sports, Science and Technology Grants-in-Aid for Scientific Research in Innovative Areas 16H06451 (to T. E.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5WSX and 5WSY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TE
- thioesterase
- ACP
- acyl carrier protein
- CMABA
- N-carboxymethyl-3-aminobutyric acid
- NAC
- N-acetylcysteamine
- CMABA-NAC
- N-carboxymethyl-3-aminobutyryl-N-acetylcysteamine
- DH
- dehydratase
- ESI-MS
- electrospray ionization mass spectrometry
- HR-FAB-MS
- high-resolution fast atom bombardment mass spectrometry
- r.m.s.d.
- root mean square deviation
- Boc
- t-butoxycarbonyl.
References
- 1. Finzel K., Lee D. J., and Burkart M. D. (2015) Using modern tools to probe the structure-function relationship of fatty acid synthases. Chembiochem 16, 528–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horsman M. E., Hari T. P., and Boddy C. N. (2016) Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: logic gate or a victim of fate? Nat. Prod. Rep. 33, 183–202 [DOI] [PubMed] [Google Scholar]
- 3. Du L., and Lou L. (2010) PKS and NRPS release mechanisms. Nat. Prod. Rep. 27, 255–278 [DOI] [PubMed] [Google Scholar]
- 4. Hunt M. C., Siponen M. I., and Alexson S. E. (2012) The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. Biochim. Biophys. Acta 1822, 1397–1410 [DOI] [PubMed] [Google Scholar]
- 5. Cantu D. C., Chen Y., and Reilly P. J. (2010) Thioesterases: a new perspective based on their primary and tertiary structures. Protein Sci. 19, 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cantu D. C., Chen Y., Lemons M. L., and Reilly P. J. (2011) ThYme: a database for thioester-active enzymes. Nucleic Acids Res. 39, D342–D346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Labonte J. W., and Townsend C. A. (2013) Active site comparisons and catalytic mechanisms of the hot dog superfamily. Chem. Rev. 113, 2182–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kotaka M., Kong R., Qureshi I., Ho Q. S., Sun H., Liew C. W., Goh L. P., Cheung P., Mu Y., Lescar J., and Liang Z. X. (2009) Structure and catalytic mechanism of the thioesterase CalE7 in enediyne biosynthesis. J. Biol. Chem. 284, 15739–15749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang F., Langley R., Gulten G., Wang L., and Sacchettini J. C. (2007) Identification of a type III thioesterase reveals the function of an operon crucial for Mtb virulence. Chem. Biol. 14, 543–551 [DOI] [PubMed] [Google Scholar]
- 10. Kudo F., Miyanaga A., and Eguchi T. (2014) Biosynthesis of natural products containing β-amino acids. Nat. Prod. Rep. 31, 1056–1073 [DOI] [PubMed] [Google Scholar]
- 11. Miyanaga A., Kudo F., and Eguchi T. (2016) Mechanisms of β-amino acid incorporation in polyketide macrolactam biosynthesis. Curr. Opin. Chem. Biol. 35, 58–64 [DOI] [PubMed] [Google Scholar]
- 12. Amagai K., Takaku R., Kudo F., and Eguchi T. (2013) A unique amino transfer mechanism for constructing the β-amino fatty acid starter unit in the biosynthesis of the macrolactam antibiotic cremimycin. Chembiochem 14, 1998–2006 [DOI] [PubMed] [Google Scholar]
- 13. Zhu Y., Zhang W., Chen Y., Yuan C., Zhang H., Zhang G., Ma L., Zhang Q., Tian X., Zhang S., and Zhang C. (2015) Characterization of heronamide biosynthesis reveals a tailoring hydroxylase and indicates migrated double bonds. Chembiochem 16, 2086–2093 [DOI] [PubMed] [Google Scholar]
- 14. Jørgensen H., Degnes K. F., Sletta H., Fjaervik E., Dikiy A., Herfindal L., Bruheim P., Klinkenberg G., Bredholt H., Nygård G., Døskeland S. O., Ellingsen T. E., and Zotchev S. B. (2009) Biosynthesis of macrolactam BE-14106 involves two distinct PKS systems and amino acid processing enzymes for generation of the aminoacyl starter unit. Chem. Biol. 16, 1109–1121 [DOI] [PubMed] [Google Scholar]
- 15. Jørgensen H., Degnes K. F., Dikiy A., Fjaervik E., Klinkenberg G., and Zotchev S. B. (2010) Insight into the evolution of macrolactam biosynthesis through cloning and comparative analysis of the biosynthetic gene cluster for a novel macrocyclic lactam, ML-449. Appl. Environ. Microbiol. 76, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ikeda H., Ishikawa J., Hanamoto A., Shinose M., Kikuchi H., Shiba T., Sakaki Y., Hattori M., and Omura S. (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 21, 526–531 [DOI] [PubMed] [Google Scholar]
- 17. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 [DOI] [PubMed] [Google Scholar]
- 18. Wu R., Latham J. A., Chen D., Farelli J., Zhao H., Matthews K., Allen K. N., and Dunaway-Mariano D. (2014) Structure and catalysis in the Escherichia coli hotdog-fold thioesterase paralogs YdiI and YbdB. Biochemistry 53, 4788–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kimber M. S., Martin F., Lu Y., Houston S., Vedadi M., Dharamsi A., Fiebig K. M., Schmid M., and Rock C. O. (2004) The structure of (3R)-hydroxyacyl-acyl carrier protein dehydratase (FabZ) from Pseudomonas aeruginosa. J. Biol. Chem. 279, 52593–52602 [DOI] [PubMed] [Google Scholar]
- 20. Moynié L., Leckie S. M., McMahon S. A., Duthie F. G., Koehnke A., Taylor J. W., Alphey M. S., Brenk R., Smith A. D., and Naismith J. H. (2013) Structural insights into the mechanism and inhibition of the β-hydroxydecanoyl-acyl carrier protein dehydratase from Pseudomonas aeruginosa. J. Mol. Biol. 425, 365–377 [DOI] [PubMed] [Google Scholar]
- 21. He H. Y., Tang M. C., Zhang F., and Tang G. L. (2014) cis-Double bond formation by thioesterase and transfer by ketosynthase in FR901464 biosynthesis. J. Am. Chem. Soc. 136, 4488–4491 [DOI] [PubMed] [Google Scholar]
- 22. Lou L., Chen H., Cerny R. L., Li Y., Shen Y., and Du L. (2012) Unusual activities of the thioesterase domain for the biosynthesis of the polycyclic tetramate macrolactam HSAF in Lysobacter enzymogenes C3. Biochemistry 51, 4–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gehret J. J., Gu L., Gerwick W. H., Wipf P., Sherman D. H., and Smith J. L. (2011) Terminal alkene formation by the thioesterase of curacin A biosynthesis: structure of a decarboxylating thioesterase. J. Biol. Chem. 286, 14445–14454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Korman T. P., Crawford J. M., Labonte J. W., Newman A. G., Wong J., Townsend C. A., and Tsai S. C. (2010) Structure and function of an iterative polyketide synthase thioesterase domain catalyzing Claisen cyclization in aflatoxin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 6246–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vagstad A. L., Hill E. A., Labonte J. W., and Townsend C. A. (2012) Characterization of a fungal thioesterase having Claisen cyclase and deacetylase activities in melanin biosynthesis. Chem. Biol. 19, 1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lackner G., Bohnert M., Wick J., and Hoffmeister D. (2013) Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chem. Biol. 20, 1101–1106 [DOI] [PubMed] [Google Scholar]
- 27. Leesong M., Henderson B. S., Gillig J. R., Schwab J. M., and Smith J. L. (1996) Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure 4, 253–264 [DOI] [PubMed] [Google Scholar]
- 28. Nguyen C., Haushalter R. W., Lee D. J., Markwick P. R., Bruegger J., Caldara-Festin G., Finzel K., Jackson D. R., Ishikawa F., O'Dowd B., McCammon J. A., Opella S. J., Tsai S. C., and Burkart M. D. (2014) Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 505, 427–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikeda H., Kazuo S. Y., Omura S. (2014) Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J. Ind. Microbiol. Biotechnol. 41, 233–250 [DOI] [PubMed] [Google Scholar]
- 30. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 32. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Morris R. J., Perrakis A., and Lamzin V. S. (2002) ARP/wARP and automatic interpretation of protein electron density maps. Acta Crystallogr. D Biol. Crystallogr. 58, 968–97512037299 [Google Scholar]
- 35. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 36. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]