

Abstract
22q11.2 Deletion Syndrome (22qDS) is often complicated by autoimmune diseases. To clarify the causal relationship, we examined the lymphocyte subset distribution and the human leucocyte antigen (HLA) in two female patients (one child and an elderly) with Graves' disease (GD) and 22qDS. Thymus dysgenesis might have contributed to the T-cell imbalance and the lack of negative selection in both cases. Notably, HLA-DR14, a known risk factor for GD in Japanese individuals and the decreased regulatory T-cell numbers that were seen in the pediatric case, may affect the early onset of GD. Central and peripheral tolerance and Th1 cells appeared to be associated with the pathogenesis of GD in 22qDS.
Keywords: Graves' disease, 22q11.2 Deletion Syndrome, HLA, regulatory T-cell
Introduction
22q11.2 deletion syndrome (22qDS) is caused by microdeletions of approximately 0.7-3 million base pairs in size on chromosome 22, resulting in the third and fourth branchial arch dysgenesis (1). 22qDS is also referred to as DiGeorge syndrome. Most children born with 22qDS have de novo mutations, other cases occur via autosomal dominant inheritance. The estimated incidence is 1 in 4,000 people (2). Patients with 22qDS present T-cell immunodeficiency due to thymus dysgenesis, hypoparathyroidism, cardiovascular disease and craniofacial anomalies (1-3). The phenotypes and the onset of 22qDS include psychological disorders, palatal, gastrointestinal and renal abnormalities and autoimmune diseases. Although as many as 33% of 22qDS patients are reported to have coexisting autoimmune diseases, the causal relationship between 22qDS and autoimmunity remains unclear (4). Graves' disease (GD) is a tissue-specific autoimmune disorder (5). In GD, antibodies that stimulate the thyrotropin (TSH) receptor bind to the TSH receptor on thyroid follicular cells, leading to hyperthyroidism. Both genetic and environmental factors contribute to the development of GD. GD is a common cause of thyrotoxicosis in childhood and adolescence. Although a few GD cases with 22qDS have been described (6-9), the pathogenesis of GD in 22qDS has yet to be elucidated. In the present study, we investigated the peripheral lymphocyte subset distribution and the human leukocyte antigen (HLA) in two women with GD and 22qDS. A 19-year-old GD patient and 68-year-old GD patient were examined to investigate the immunological and age-related aspects of the disease.
Case Reports
Case 1
A 19-year-old woman with a 3-month history of occasional tetany visited our hospital. She was diagnosed with GD and was treated with thiamazole (10 mg/day) for 11 years. Her uncle was also treated for GD. No problems were observed at birth. On admission, her blood pressure was 115/73 mmHg, she had a regular pulse rate of 75 beats/min and her body temperature was 36.5℃. Her height was 143 cm and her body weight was 52 kg (BMI 25.6 kg/m2). She had completed her high school education. She had facial abnormalities, including telecanthus, expansion of the nasal apex, and fish mouth. Palatal anomalies, exophthalmos and edema were not seen. Her heart, respiratory sounds, and neurological examination results were completely normal. The clinical activity score (CAS) for thyroid ophthalmopathy and her NOSPECS were both 0. She had a diffusely enlarged soft goiter with a diameter of 15 cm. A laboratory test on admission (Table 1) showed hypocalcemia, which is associated with impaired parathyroid hormone secretion. Elevated serum levels of thyroid hormone in association with suppressed TSH levels were seen. Anti-TSH receptor antibody (Third generation assay: TRAb-3) and thyroid stimulating antibody (TSAb) markedly increased. The titers of anti-thyroglobulin antibody (TgAb), and anti-thyroid peroxidase antibody (TPOAb) were within the normal ranges. No serum gamma globulin abnormalities, including IgA, were observed (data not shown). Her adrenal and pituitary functions were normal (data not shown). HLA-DR14 and DR15 were identified in an HLA-DR typing test. Her chest X-ray, electrocardiogram and echocardiography showed no abnormality (data not shown). Thyroid ultrasonography revealed the markedly diffuse enlargement of the thyroid gland with increased blood flow (Fig. 1). A CT scan of the brain showed no calcification. She had mental retardation with an IQ of 48.8. A Fluorescence in situ hybridization analysis (FISH) confirmed a diagnosis of 22qDS. Laboratory test (Table 2) revealed a total lymphocyte count of 1,710 /μL, which was in the lower normal range. The proportion of CD3+ T-cells decreased with a normal ratio of CD4/CD8. The CD8+ T-cells count was below normal while the Th1/Th2 ratio was mildly increased. The number of regulatory T-cells (Tregs) decreased (the prevalence of CD4+CD25+ was in the lower normal limit in conjunction with a decrease in the total number of T-cells). Her symptoms improved after the administration of alphacalcidol (1 μg/day).
Table 1.
Patients Background and Laboratory Data on Admission.
| Case 1 | Case 2 | |||
| Current Age (year) | 19 | 68 | ||
| Age at Onset of GD (year) | 8 | 68 | ||
| Alb | g/dL | 4.3 | 4.1 | |
| Ca | mg/dL | 6.7 | 6.9 | |
| P | mg/dL | 4.8 | 3.5 | |
| Mg | mg/dL | 1.5 | 1.5 | |
| ALP | IU/L | 803 | 360 | |
| Cr | mg/dL | 0.4 | 0.51 | |
| Intact-PTH | pg/mL (10-65) | 34 | 7.0 | |
| TSH | uU/mL (0.35-4.94) | <0.001 | 58.3, | <0.03* |
| FT3 | pg/mL (1.71-3.71) | 5.85 | 1.04 | |
| FT4 | ng/dL (0.70-1.48) | 1.29 | <0.40, | 3.09* |
| TRAb-2 | IU/L (<1.0) | N/A | 1.7** | |
| TRAb-3 | IU/L (<2.0) | 2,080 | N/A | |
| TSAb | % (<120) | 1,072 | 164** | |
| TgAb | IU/mL (<28.0) | 8 | 30.2 | |
| TPOAb | IU/mL (<16.0) | 10.2 | 581.5 | |
Note: underline denotes abnormal values. TSAb: thyroid stimulating antibody TRAb: anti-thyroid-stimulating hormone receptor antibody, TRAb-2: TRAb-second generation assay, TRAb-3: TRAb-third generation assay, TgAb: anti-thyroglobulin antibody, TPOAb: anti-thyroid peroxidase antibody
N/A: not applicable *data measured at onset of GD in another hospital. **measured in euthyroid during L-T4 treatment.
Figure 1.
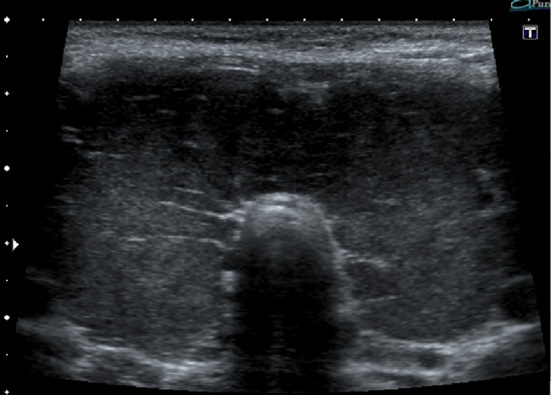
The ultrasound examination of case 1 showed marked diffuse enlargement of the thyroid gland. The size of right lobe was 40.0×35.5×57 mm while the left lobe was 38.8×38.7×67 mm.
Table 2.
Lymphocytes Subsets Distributions in Peripheral Blood Mononucleocytes.
| Case 1 | Case 2 | |
|---|---|---|
| CD3 T-cells % (75-85) | 59.9 | 66.1 |
| CD19 B-cells % (5-15) | 30.8 | 22.1 |
| Th1 % | 26.7 | 16.1 |
| Th2 % | 1.0 | 1.1 |
| Th1/Th2 Ratio | 26.7 | 14.6 |
| CD4/CD8 | 1.39 | 1.92 |
| CD4-CD8+% (22-54) | 28.2 | 23.4 |
| CD4-CD8- % (14-38) | 31.3 | 28.4 |
| CD4+CD8- % (23-52) | 39.5 | 46.6 |
| CD4+CD8+% (<7) | 1.0 | 1.3 |
| CD4+CD25+% (6-21) | 7.0 | 14.3 |
| Lym /μL | 1,710 | 1,760 |
Note: Underline Denotes Abnormal Values. Lym: Peripheral Blood Lymphocytes Counts
Case 2
A 68-year-old woman developed GD four months before admission. Thyrotoxicosis with suppressed TSH was observed at the beginning of treatment for GD (Table 1). She was started on thiamazole (15 mg/day). She had been under treatment for schizophrenia and hypertension for more than 20 years. Her family and medical histories were not remarkable. After she became unconscious, she was sent to our hospital. Her height was 139 cm and her body weight was 43 kg (BMI 22.3 kg/m2). Her level of consciousness, based on Glasgow Coma Scale, was E1V3M4. Her blood pressure was 124/73 mmHg and she had a regular pulse rate of 75 beats/min. Her body temperature was 36.9℃. She had telecanthus, a prominent nose with a large tip and hypoplastic nares and a small mouth. No palatal anomalies, murmur, rales or edema were observed. Neurological examinations revealed completely normal results. Her CAS and NOSPECS were both 0. She had diffusely enlarged mild goiter and mild bilateral hearing loss. The laboratory data on admission showed severe hyponatremia (109 mEq/L) and primary hypothyroidism (Table 1). No serum gamma globulin abnormalities were seen (data not shown). Hypocalcemia, due to a lack of parathyroid hormone (PTH) secretion, was observed. Her titers of TgAb and TPOAb were also increased. Her adrenal function and pituitary function were normal (data not shown). Thyroid ultrasonography revealed an enlarged thyroid gland (Fig. 2).
Figure 2.
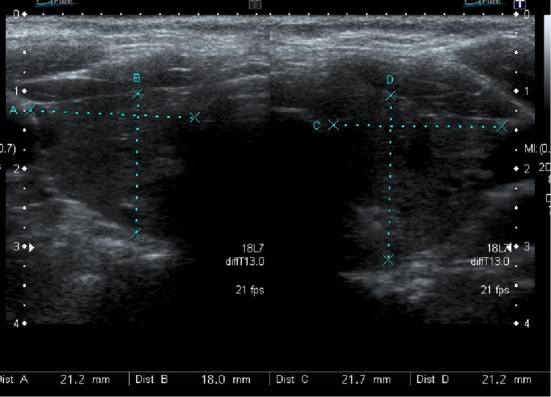
The ultrasound examination of case 2 showed mild diffuse enlargement of the thyroid gland. The right lobe was 21.1×18.0×31.4 mm while the left lobe was 21.7×21.2×38.4 mm.
She was found to have HLA-DR9 and DR15. Chest CT revealed pneumonia in the right upper lobe (Fig. 3A). Brain CT revealed calcification in the bilateral basal ganglia (Fig. 4). FISH testing confirmed a diagnosis of 22qDS. Laboratory tests that were performed after correction of hyponatremia (Table 2) revealed normal lower peripheral lymphocyte counts, decreased CD3+ T-cells, a normal CD4/CD8 ratio, Th1/Th2 ratio and Treg numbers. Her CD8+ T-cell count was decreased. Hyponatremia was thought to have developed due to the use of Angiotensin II receptor blockers (ARB) and antipsychotics as well as hypothyroidism and syndrome of inappropriate secretion of antidiuretic hormone (SIADH) due to pneumonia. Her water intake was restricted to 700 mL/day while sodium and calcium were adequately supplemented. Alphacalcidol (1 μg/day) and L-T4 (25 μg/day) were administered. Even when the patient was euthyroid during L-T4 treatment, her anti-TSH receptor antibody (second-generation kit: TRAb-2) and TSAb levels were elevated. ARB and antipsychotics were replaced with a calcium channel blocker and another antipsychotic. The spontaneous remission of pneumonia was observed (Fig. 3B). Three weeks later, all of her symptoms and her general status improved and the patient was discharged.
Figure 3.

A: Pulmonary infiltration was seen in the right upper lobe on chest CT. B: The lesions of pulmonary infiltration spontaneously improved after ten days.
Figure 4.
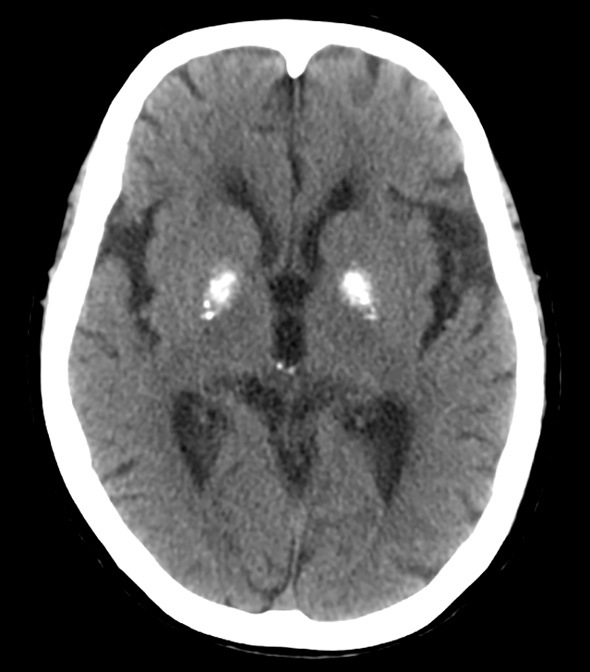
Dense calcification was seen in the bilateral basal ganglia on brain CT.
Discussion
Both cases in this report had diffusely enlarged goiter with hyperthyroidism and elevated anti-TSH receptor antibody levels which supported the diagnosis for GD with genetically-confirmed 22qDS. A Th1 predominance was seen in case 1, which was also reported in pediatric cases with 22qDS (10). Considering that the adult cases with 22qDS are reported to skew towards a Th2 balance in comparison to controls in which the ratio of Th1/Th2 was approximately ten in adults controls and five in adult 22qDS patients (10), case 2, who had a ratio of 14.6, appeared to have a Th1 predominance (Table 1). Th1 plays an important role such as IgG1 type of TSAb formation (11). As described, GD can be seen in 22qDS at any age (1,2). Thymus dysgenesis in 22qDS leads to failure in the negative selection of immunogenic T-cell (12); thus, it may have been associated with the onset of GD in both cases. In case 1, the decreased Treg numbers (13-15) and the inheritance of HLA-DR14, which is a known risk factor for GD in Japanese individuals (16), may have affected the early onset of GD. In addition, DR15, a risk allele among the Han ethnic group in Taiwan, which is geographically close to Japan (17), was observed in both cases-this may have also been related to the development of GD. Thus, impaired central tolerance, which may reflect a dysfunction of negative selection with predisposing HLA, peripheral tolerance (a decreased Tregs count) and Th1 predominance appeared to be associated with the pathogenesis of GD in our 22qDS patients.
The few reports that have described GD in 22qDS patients indicate that it usually occurs in young age (6-9). The prevalence of autoimmune thyroid diseases in 22qDS is 3.3%, which is higher than that in the general population (18). Along with these previously described reports, we further support the notion that GD may be part of the clinical spectrum associated with 22qDS. However, none of the previous studies extensively examined the relationship between the HLA type and the lymphocyte subsets. Piliero et al. investigated the CD3, CD4, CD8 and CD45 populations in patients with 22qDS and found the accelerated conversion of naive T-cells to memory T-cells in patients with 22qDS (19).
Although autoimmune diseases frequently develop in 22qDS (1,2,4), the mechanism of susceptibility towards autoimmune diseases, including GD in 22qDS, remains unknown. There are currently no biomarkers to predict the development of autoimmune diseases in 22qDS. Several candidate genes in GD, including the TSH receptor gene, the thyroglobulin gene and the TPO gene are not located in the chromosome 22q11 region. The possible mechanisms by which GD occurs in 22qDS are as follows. 1) Abnormal thymic development resulting in partial immunodeficiency and immunological imbalance can lead to autoimmune diseases, impaired T-cell differentiation or T-cell dysregulation, which may induce autoimmunity (20). The Th1/Th2 imbalance that was seen in our cases, especially in case 1, might have been due to thymus dysgenesis. 2) Since the thymus is incompletely generated due to 22qDS, negative selection in the thymus might be attenuated. It is possible that the abnormal expression of autoimmune regulator (AIRE, located in 21q22.3) contributes to the impairment of negative selection (21). Furthermore, negative selection is largely affected by HLA (12). The higher affinity for protective alleles might prevent the binding and presentation of crucial TSHR epitopes. High-affinity binding during thymic T cell selection might lead to the deletion of cells that are reactive to specific TSHR epitopes. One report described the association of HLA in 22qDS and rheumatoid arthritis (22). Thus, it is logical to consider the genetic susceptibilities of certain HLA types in GD and 22qDS. In this context, a risk allele of DR14 (seen in case 1) and an associated allele of DR15 (seen in both cases) may predispose a patient to GD. 3) Tregs, which are generated in the thymus (23), are reported to be associated with GD when functional impairment occurs (24). Two reports noted that patients with 22qDS had lower numbers of Treg cells in comparison to the control group (13,14). A separate report showed decreased numbers of Tregs in 22qDS patients, especially pediatric patients, thus the decrease in the number of Tregs may have been related to the earlier onset of GD in case 1 (15). 4) The functional characteristics, such as a Th1/Th2 balance of the CD4+ T-cells that respond to TSHR epitopes may vary between each patient. Other helper T-cells, such as Th9, Th17 or cytotoxic T cells may be dysregulated in 22qDS. 5) In both cases, a reduction in the number of CD8+ T cells was observed. This is a common observation in 22qDS and may lead to chronic viral infections, which may affect the peripheral immunity. 6) B-cell abnormalities are not clear in 22qDS, but it is hypothesized that B-cells will be dysregulated to a certain degree in the presence of T cell abnormalities (20).
From an endocrinological perspective, a large study in Norway reported that 22qDS was observed in 15% of hypoparathyroidism cases (3). AIRE gene mutations are seen in autoimmune polyendocrine syndrome type 1 (APS-1: was found in 17% of hypoparathyroidism cases). Endocrine organs, such as the adrenal cortex, ovaries and parathyroid glands are typically affected. Moreover, NALP5, a parathyroid autoantigen was identified in APS-1 (25). However, the relationship of the genotypes and phenotypes between APS-1 and 22qDS is not clear (3).
The prognosis for 22qDS is mainly determined by the degree of cardiac abnormalities and immunodeficiency. An accurate diagnosis and appropriate treatment in more severe cases can improve the prognosis and provide opportunities for a better quality of life.
In conclusion, two cases of GD in patients with 22qDS provided insights into the immunological significance of the pathological mechanisms and the onset of GD. Further investigations in an increased number of cases are warranted to establish the appropriate diagnostic process and treatment interventions.
The authors state that they have no Conflict of Interest (COI).
References
- 1.McDonald-McGinn DM, Sullivan KE, Marino B, et al. . 22q11.2 deletion syndrome. Nat Rev Dis Primers 1: 15071, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnside RD. 22q11.21 Deletion Syndromes: a review of proximal, central, and distal deletions and their associated features. Cytogenet Genome Res 146: 89-99, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Astor MC, Løvås K, Debowska A, et al. . Epidemiology and health related quality of life in hypoparathyroidism in Norway. J Clin Endocrinol Metab 101: 3045-3053, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gennery AR, Barge D, O'Sullivan JJ, Flood TJ, Abinun M, Cant AJ. Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch Dis Child 86: 422-425, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akamizu T, Mori T, Nakao K. Pathogenesis of Graves' disease: molecular analysis of anti-thyrotropin receptor antibodies. Endocr J 44: 633-646, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Brown JJ, Datta V, Browning MJ, Swift PG. Graves' disease in DiGeorge syndrome: patient report with a review of endocrine autoimmunity associated with 22q11.2 deletion. J Pediatr Endocrinol Metab 17: 1575-1579, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Ham Pong AJ, Cavallo A, Holman GH, Goldman AS. DiGeorge syndrome: long-term survival complicated by Graves disease. J Pediatr 106: 619-620, 1985. [DOI] [PubMed] [Google Scholar]
- 8.Kawame H, Adachi M, Tachibana K, et al. . Graves' disease in patients with 22q11.2 deletion. J Pediatr 139: 892-895, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Kawamura T, Nimura I, Hanafusa M, et al. . DiGeorge syndrome with Graves' disease: A case report. Endocr J 47: 91-95, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Zemble R, Luning Prak E, McDonald K, McDonald-McGinn D, Zackai E, Sullivan K. Secondary immunologic consequences in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Immunol 136: 409-418, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapoport B, McLachlan SM. Graves' hyperthyroidism is antibody-mediated but is predominantly a Th1-type cytokine disease. J Clin Endocrinol Metab 99: 4060-4061, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kappler JW, Roehm N, Marrack P. T-cell tolerance by clonal elimination in the thymus. Cell 49: 273-280, 1987. [DOI] [PubMed] [Google Scholar]
- 13.McLean-Tooke A, Barge D, Spickett GP, Gennery AR. Immunologic defects in 22q11.2 deletion syndrome. J Allergy Clin Immunol 122: 362-367, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan KE, McDonald-McGinn D, Zackai EH. CD4(+) CD25(+) T-cell production in healthy humans and in patients with thymic hypoplasia. Clin Diagn Lab Immunol 9: 1129-1131, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klocperk A, Grecová J, Šišmová K, Kayserová J, Froñková E, Šedivá A. Helios expression in T-regulatory cells in patients with di George Syndrome. J Clin Immunol 34: 864-870, 2014. [DOI] [PubMed] [Google Scholar]
- 16.Ueda S, Oryoji D, Yamamoto K, et al. . Identification of independent susceptible and protective HLA alleles in Japanese autoimmune thyroid disease and their epistasis. J Clin Endocrinol Metab 99: E379-E383, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Chen PL, Fann CS, Chu CC, et al. . Comprehensive genotyping in two homogeneous Graves' disease samples reveals major and novel HLA association alleles. PLoS One 6: e16635, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi JH, Shin YL, Kim GH, et al. . Endocrine manifestations of chromosome 22q11.2 microdeletion syndrome. Horm Res 63: 294-299, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Piliero LM, Sanford AN, McDonald-McGinn DM, Zackai EH, Sullivan KE. T-cell homeostasis in humans with thymic hypoplasia due to chromosome 22q11.2 deletion syndrome. Blood 103: 1020-1025, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Gennery AR. Immunological aspects of 22q11.2 deletion syndrome. Cell Mol Life Sci 69: 17-27, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol 4: 350-354, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan KE, McDonald-McGinn DM, Driscoll DA, et al. . Juvenile rheumatoid arthritis-like polyarthritis in chromosome 22q11.2 deletion syndrome (DiGeorge anomalad/velocardiofacial syndrome/conotruncal anomaly face syndrome). Arthritis Rheum 40: 430-436, 1997. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe N, Wang YH, Lee HK, et al. . Hassall's corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 436: 1181-1185, 2005. [DOI] [PubMed] [Google Scholar]
- 24.González-Amaro R, Marazuela M. T regulatory (Treg) and T helper 17 (Th17) lymphocytes in thyroid autoimmunity. Endocrine 52: 30-38, 2016. [DOI] [PubMed] [Google Scholar]
- 25.Alimohammadi M, Björklund P, Hallgren A, et al. . Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med 358: 1018-1028, 2008. [DOI] [PubMed] [Google Scholar]