

Abstract
Key points
Chronic fetal hypoxaemia is a common pregnancy complication associated with intrauterine growth restriction that may influence respiratory outcome at birth.
We investigated the effect of maternal chronic hypoxia for a month in late gestation on signalling pathways regulating fetal lung maturation and the transition to air‐breathing at birth using isobaric hypoxic chambers without alterations to maternal food intake.
Maternal chronic hypoxia in late gestation increases fetal lung expression of genes regulating hypoxia signalling, lung liquid reabsorption and surfactant maturation, which may be an adaptive response in preparation for the successful transition to air‐breathing at birth.
In contrast to other models of chronic fetal hypoxaemia, late gestation onset fetal hypoxaemia promotes molecular regulation of fetal lung maturation. This suggests a differential effect of timing and duration of fetal chronic hypoxaemia on fetal lung maturation, which supports the heterogeneity observed in respiratory outcomes in newborns following exposure to chronic hypoxaemia in utero.
Abstract
Chronic fetal hypoxaemia is a common pregnancy complication that may arise from maternal, placental and/or fetal factors. Respiratory outcome of the infant at birth likely depends on the duration, timing and severity of the hypoxaemic insult. We have isolated the effect of maternal chronic hypoxia (MCH) for a month in late gestation on fetal lung development. Pregnant ewes were exposed to normoxia (21% O2) or hypoxia (10% O2) from 105 to 138 days of gestation (term ∼145 days). At 138 days, gene expression in fetal lung tissue was determined by quantitative RT‐PCR. Cortisol concentrations were determined in fetal plasma and lung tissue. Numerical density of surfactant protein positive cells was determined by immunohistochemistry. MCH reduced maternal (106 ± 2.9 vs. 47 ± 2.8 mmHg) and fetal body weight (4.0 ± 0.4 vs. 3.2 ± 0.9 kg). MCH increased fetal lung expression of the anti‐oxidant marker CAT and decreased expression of the pro‐oxidant marker NOX‐4. MCH increased expression of genes regulating hypoxia signalling and feedback (HIF‐3α, KDM3A, SLC2A1, EGLN‐3). There was no effect of MCH on fetal plasma/lung tissue cortisol concentrations, nor genes regulating glucocorticoid signalling (HSD11B‐1, HSD11B‐2, NR3C1, NR3C2). MCH increased expression of genes regulating sodium (SCNN1‐B, ATP1‐A1, ATP1‐B1) and water (AQP‐4) movement in the fetal lung. MCH promoted surfactant maturation (SFTP‐B, SFTP‐D, ABCA3) at the molecular level, but did not alter the numerical density of surfactant positive cells in lung tissue. MCH in late gestation promotes molecular maturation of the fetal lung, which may be an adaptive response in preparation for the successful transition to air‐breathing at birth.
Keywords: chronic hypoxia, fetal lung, growth restriction
Key points
Chronic fetal hypoxaemia is a common pregnancy complication associated with intrauterine growth restriction that may influence respiratory outcome at birth.
We investigated the effect of maternal chronic hypoxia for a month in late gestation on signalling pathways regulating fetal lung maturation and the transition to air‐breathing at birth using isobaric hypoxic chambers without alterations to maternal food intake.
Maternal chronic hypoxia in late gestation increases fetal lung expression of genes regulating hypoxia signalling, lung liquid reabsorption and surfactant maturation, which may be an adaptive response in preparation for the successful transition to air‐breathing at birth.
In contrast to other models of chronic fetal hypoxaemia, late gestation onset fetal hypoxaemia promotes molecular regulation of fetal lung maturation. This suggests a differential effect of timing and duration of fetal chronic hypoxaemia on fetal lung maturation, which supports the heterogeneity observed in respiratory outcomes in newborns following exposure to chronic hypoxaemia in utero.
Abbreviations
- ABCA3
ATP‐binding cassette, sub‐family A (ABC1), member 3
- ACTB
beta‐actin
- ADM
adrenomedullin
- AQP
aquaporin
- ATP1
sodium potassium adenosine triphosphatase
- CAT
catalase
- CLCN2
chloride channel, voltage‐sensitive 2
- CFTR
cystic fibrosis transmembrane conductance regulator
- COL1A1
collagen type 1 alpha 1
- EGLN
egl‐9 family hypoxia inducible factor
- ELN
elastin
- GC
glucocorticoid
- GPX
glutathione peroxidase
- Hb
haemoglobin
- HIF
hypoxia inducible factor
- HMOX1
heme oxygenase 1
- HSD11B
11β‐hydroxysteroid dehydrogenase
- IUGR
intrauterine growth restriction
- JMJD1A
Jumonji domain containing 1A
- KDM3A
lysine (K)‐specific demethylase 3A
- MCH
maternal chronic hypoxia
- NOS
nitric oxide synthase
- NOX
nicotinamide adenine dinucleotide phosphate‐oxidase
- NR3C1
glucocorticoid receptor
- NR3C2
mineralocorticoid receptor
- PCYT1A
phosphate cytidylyl transferase 1, choline, alpha
- PHD
prolyl hydroxylase domain
- PPIA
peptidylprolyl isomerase
- PR
placental restriction/placentally restricted
- qRT‐PCR
quantitative real time PCR
- RDS
respiratory distress syndrome
- SLC2A1
facilitated glucose transporter‐1
- SCNN1
epithelial sodium channel
- SFTP
surfactant protein
- SOD
super oxide dismutase
- VEGF
vascular endothelial growth factor
- YWHAZ
tyrosine 3‐monooxygenase
Introduction
Fetal chronic hypoxaemia is a common pregnancy complication that can predispose to complications at birth and later in life (Hutter & Jaeggi, 2010; Giussani & Davidge, 2013; Giussani, 2016). Exposure to chronic hypoxaemia in utero may arise due to a variety of environmental, maternal, placental and/or fetal factors and is commonly associated with intrauterine growth restriction (IUGR) (Morrison, 2008). IUGR is a diverse complication of pregnancy and the developing fetus undergoes a range of neuroendocrine and cardiovascular adaptations as a result of reduced oxygen and nutrient supply that affects normal growth and development (Economides et al. 1991; McMillen et al. 2001; Dubiel et al. 2002). There is heterogeneity in the risk of IUGR newborns developing respiratory complications at birth and this is likely influenced by the specific nature of the altered intrauterine environment encountered during fetal development, in addition to the increased likelihood of being born prematurely. We have identified chronic fetal hypoxaemia as a molecular regulator of lung development which may alter the risk of IUGR newborns developing respiratory complications at birth (McGillick et al. 2015, 2016a, b; Orgeig et al. 2015). Many animal models have been used to investigate the effects of IUGR on fetal growth and development, including maternal nutrient restriction, maternal hypoxia and placental insufficiency induced by either uterine artery ligation, uterine carunclectomy, maternal hyperthermia or umbilicoplacental embolisation (McMillen & Robinson, 2005; Morrison, 2008). However, there is limited understanding of the effect of chronic hypoxaemia alone on fetal lung development, as many mammalian models of IUGR associated with chronic hypoxaemia also lead to a reduction in maternal food intake (Robinson et al. 1979; Van Geijn et al. 1980). Maternal undernutrition including a low protein diet is an effective model to induce IUGR and is itself associated with disturbances of molecular signalling and structural airway and vascular development in the lungs of offspring (Maritz et al. 2005; Briana & Malamitsi‐Puchner, 2013; Liu et al. 2014; Zana‐Taieb et al. 2015). In this study using sheep, we have isolated the effect of maternal chronic hypoxia (MCH) for a month in late gestation on fetal development using bespoke isobaric hypoxic chambers without alterations to maternal food intake (Allison et al. 2016a; Brain et al. 2015).
Fetal lung development is regulated by a variety of factors that can be influenced by adverse intrauterine conditions. Hypoxia, particularly in prenatal life, is a stimulus for oxidative stress, which occurs due to an imbalance between the generation of reactive oxygen and nitrosative species and the endogenous anti‐oxidant system that protects cells from the deleterious effects of free radicals (Giussani et al. 2012). Regulation of the pro‐oxidant and anti‐oxidant balance is therefore essential during pregnancy, as complications associated with oxidative stress have adverse implications for the developing fetus (Suzin et al. 2002; Herrera et al. 2014). At the cellular level, reduced oxygen tension in utero is associated with activation of the hypoxia signalling cascade, regulated by the expression of hypoxia inducible factor (HIF)‐α subunits (Benizri et al. 2008). Downstream alterations to expression of hypoxia responsive genes have widespread implications on developing organ systems. For instance, in the fetal mouse lung, loss of HIF‐α signalling leads to delayed lung maturation and increases the risk of respiratory distress syndrome (RDS) at birth (Compernolle et al. 2002). Hypoxia signalling is controlled by a decrease in HIF‐α subunit stability under regulation of the prolyl hydroxylase domain (PHD) enzyme family encoded by the EGLN gene (egl‐9 family hypoxia inducible factor), known as EGLN‐1 (PHD‐2), EGLN‐2 (PHD‐1) and EGLN‐3 (PHD‐3) (Bruick & McKnight, 2001). Our studies in a model of early onset chronic hypoxaemia in the placentally restricted (PR) sheep fetus suggest a differential function of the EGLN/PHD enzymes during periods of acute versus chronic hypoxaemia (Ginouvès et al. 2008; Botting et al. 2014; Orgeig et al. 2015). Hence, alterations to hypoxia signalling in utero may underlie altered lung development and downstream risk of IUGR newborns experiencing respiratory complications at birth.
Reabsorption of fetal lung liquid and surfactant maturation are two important processes that enable the lung to function as the primary organ of gas exchange at birth (Avery & Mead, 1959; Hooper et al. 2015). Perinatal hypoxaemia may impair the feto‐neonatal pulmonary transition leading to neonatal RDS and pulmonary hypertension (Storme et al. 2013). However, there is controversy surrounding lung maturation and respiratory outcomes following exposure to fetal chronic hypoxaemia associated with IUGR in both clinical and animal studies (Gagnon et al. 1999; Braems et al. 2000; Cock et al. 2001; Orgeig et al. 2010; McGillick et al. 2016c). These different models support increased or decreased lung maturation, which parallels with either reduced or greater risk of respiratory complications in IUGR newborns in clinical practice (McGillick et al. 2016c). The different outcomes between studies may be due in part to the timing, severity and duration of the fetal hypoxaemic insult experienced in each model (Gagnon et al. 1999; Morrison, 2008; Orgeig et al. 2010). Hence, it is necessary to tease apart the relative contributions of specific factors, including chronic fetal hypoxaemia, on fetal lung development and their role in the incidence of respiratory complications in IUGR newborns. Herein, we comprehensively investigate the effect of chronic fetal hypoxaemia in a model of late gestation onset IUGR on molecular and structural development of the preterm fetal lung.
Methods
Ethical approval
All procedures were performed under the UK Animals (Scientific Procedures) Act 1986 and were approved by the University of Cambridge Ethical Review Committee. Experiments were designed and reported with reference to the ARRIVE guidelines (Kilkenny et al. 2014). The authors have read, and the experiments comply with the policies and regulations of The Journal of Physiology (Grundy, 2015).
Surgery
At 100 ± 1 days gestation (term, ∼145 days), 16 pregnant Welsh mountain ewes carrying singleton pregnancies as determined by ultrasound scan (Toshiba Medical Systems Europe, Zoetermeer, Netherlands) underwent a laparotomy, as previously described (Brain et al. 2015). Antibiotics (30 mg kg−1 i.m. procaine benzylpenicillin; Depocillin; Intervet UK Ltd, Milton Keynes, UK) and an analgesic agent (1.4 mg kg−1 s.c. carprofen; Rimadyl; Pfizer Let, Sandwich, UK) were administered to the ewe immediately before surgery. Briefly, general anaesthesia was induced by Alfaxan (1.5–2.5 mg kg−1, i.v. administration, alfaxalone; Jurox Ltd, Worcestershire, UK) and maintained with 1.5–2.0% isofluorane (Datex‐Ohmeda Ltd, Hatfield, UK). Following midline abdominal incision and uterotomy, the fetal hind limbs were exposed and the fetal sex was determined. To control for possible sex differences only males were included in this study (female fetuses were assigned to a postnatal study). Fetuses were returned to the intrauterine cavity and catheters were placed in the maternal femoral artery and maternal femoral vein as previously described (Brain et al. 2015). Following surgery, ewes were housed in individual floor pens with a 12:12 h light–dark cycle with free access to hay and water. From 100 days, ewes were fed daily with a maintenance diet to facilitate the monitoring of food intake (Brain et al. 2015).
Experimental protocol
At 103 days of gestation, ewes were randomly assigned to either of two experimental groups: normoxic (n = 8) or hypoxic (MCH, n = 8). Ewes allocated to the normoxic group remained housed in individual floor pens for the duration of the experimental protocol. Pregnant ewes assigned to the hypoxic group were housed in bespoke isobaric hypoxic chambers (Telstar Ace, Dewsbury, UK) from 103 days under normoxic conditions and exposed to ∼10% O2 from 105 days by altering the incoming inspirate mixture as described previously (Brain et al. 2015). Maternal feed intake was measured daily and was not different between the two groups (Allison et al. 2016a; Brain et al. 2015).
Arterial blood gas measurements
Maternal arterial blood samples were taken daily for measurement of blood gases (ABL5 blood gas analyser; Radiometer; Copenhagen, Denmark; measurements corrected to 38 °C) and percentage saturation of haemoglobin with oxygen (Sat Hb; measured using a haemoximeter; OSM3, Radiometer) (Brain et al. 2015).
Post‐mortem and tissue collection
Fetuses were evaluated in late gestation (138 days; term = 145 days) when the lung is in the alveolar stage of development similar to human late preterm birth (36–37 weeks of gestation; term = 40 weeks). At 138 days of gestation, ewes in the hypoxic group were transferred from the hypoxic chambers to the post‐mortem laboratory wearing a respiratory hood providing the same hypoxic gas mixture (Brain et al. 2015). All ewes and their fetuses were killed by an overdose of sodium pentobarbitone (0.4 ml kg−1, i.v. administration, Pentoject; Animal Ltd, York, UK) and fetuses were delivered by hysterotomy. A fetal umbilical arterial blood sample was taken for measurement of haemoglobin (Hb; Radiometer; measurements corrected to 39.5 °C) and plasma cortisol concentration. Fetal body and organ weights, bi‐parietal diameter and lower limb length were recorded. A piece of left fetal lung tissue was snap frozen in liquid nitrogen and stored at −80°C for lung tissue cortisol determination and gene expression analysis. A section of right fetal lung tissue was immersion fixed in 4% paraformaldehyde and processed to paraffin for further immunohistochemical analysis. Data from these animals characterising the maternal physiology and cardiovascular outcomes following exposure to MCH have been published previously (Allison et al. 2016a; Brain et al. 2015). As this cohort was not specifically run with lung analysis as a primary outcome, additional samples and analyses such as bronchoalveolar lavage and lung function measures were not available/possible.
Fetal plasma cortisol concentration
Plasma cortisol concentrations in fetal umbilical arterial blood collected at post‐mortem (normoxic, n = 8; hypoxic, n = 8) were measured using a commercially available ELISA kit (RE52061, IBL International, Hamburg, Germany), according to the manufacturer's guidelines as previously described (Brain et al. 2015).
Fetal lung tissue cortisol concentration
Fetal lung tissue (∼50 mg; normoxic, n = 7; hypoxic, n = 6) was sonicated (Kinematica PT‐MR‐3100, Lucerne, Switzerland) in 250 μl of 1× PBS. Cortisol was extracted from the supernatant following addition of 100 μl extraction buffer (Oxford Biomedical Research, Rochester Hills, MI, USA) and 2 ml ethyl ether (VWR, Qld, Australia) and vortexed for 1 min followed by 5 min phase separation. The organic phase (1.2 ml) was dried at 37°C with air for 30 min and re‐suspended with extraction buffer (120 μl). Neat test samples were assayed in addition to the standards on a Cortisol Enzyme Immunoassay according to the manufacturer's guidelines (EA65, Oxford Biomedical Research).
Quantification of mRNA transcripts within the fetal lung
For total RNA extraction, total RNA was extracted from all 16 lung tissue samples (∼50 mg; normoxic, n = 8; hypoxic, n = 8) using QIAzol Lysis Reagent Solution and Qiagen miRNeasy purification columns (Qiagen, Victoria, Australia) and cDNA was synthesised using Superscript III First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) as previously described (McGillick et al. 2015, 2016b).
For quantitative real‐time RT‐PCR (qRT‐PCR) output normalisation three stable reference genes, beta‐actin (ACTB), peptidylprolyl isomerase A (PPIA) (Passmore et al. 2009) and tyrosine 3‐monooxygenase (YWHAZ) (McGillick et al. 2013) were chosen from a panel of candidate reference genes using the geNorm component of qbaseplus 2.0 software (Biogazelle, Zwijnaarde, Belgium) and run in parallel with target genes as described previously (McGillick et al. 2013). Expression of genes regulating oxidative stress, hypoxia signalling, glucocorticoid signalling, lung liquid movement, surfactant maturation and airway remodelling (Table 1) were measured by qRT‐PCR using Fast SYBR® Green Master Mix (Applied Biosystems, Foster City, CA, USA) in a final volume of 6 μl on a ViiA7 Fast Real‐time PCR system (Applied Biosystems) as described previously (McGillick et al. 2013, 2015, 2016b). The abundance of each transcript relative to the abundance of stable reference genes was calculated using DataAssist 3.0 analysis software and is expressed as mRNA mean normalised expression (MNE) ± SD (McGillick et al. 2013).
Table 1.
Evaluation of target genes regulating oxidative stress, hypoxia signalling, glucocorticoid signalling, lung liquid movement (controlled by chloride, sodium and water movement), surfactant maturation and airway remodelling by quantitative real‐time RT‐PCR
| Gene name | Gene symbol | Primer concentration (μm) | Primer reference |
|---|---|---|---|
| Pro‐oxidant markers | |||
| Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase | NOX‐4 | 0.45 | McGillick et al. (2016b) |
| Heme oxygenase‐1 | HMOX‐1 | 0.90 | McGillick et al. (2016b) |
| Inducible nitric oxide synthase | NOS‐2 | 0.45 | McGillick et al. (2016b) |
| Endothelial nitric oxide synthase | NOS‐3 | 0.45 | McGillick et al. (2016b) |
| Anti‐oxidant markers | |||
| Superoxide dismutase enzymes | SOD‐1 | 0.45 | McGillick et al. (2016b) |
| SOD‐2 | 0.45 | ||
| Catalase | CAT | 0.90 | McGillick et al. (2016b) |
| Glutathione peroxidase | GPX | 0.45 | McGillick et al. (2016b) |
| Hypoxia signalling | |||
| Hypoxia inducible factor subunits | HIF‐1α | 0.45 (F), 0.90 (R) | Botting et al. (2014) |
| HIF‐2α | 0.45 | ||
| HIF‐3α | 0.45 | ||
| HIF‐1β | 0.45 | ||
| Vascular endothelial growth factor | VEGF | 0.45 | Botting et al. (2014) |
| Adrenomedullin | ADM | 0.45 | Botting et al. (2014) |
| Lysine (K)‐specific demethylase 3A | KDM3A | 0.45 | Botting et al. (2014) |
| Solute carrier family 2 (facilitated glucose transporter) member 1 | SLC2A1 | 0.45 | Botting et al. (2014) |
| Egl‐9 family hypoxia‐inducible factor enzymes (encoding the prolyl hydroxylase domain (PHD) proteins) | EGLN‐1 (PHD‐2) | 0.45 | Botting et al. (2014) |
| EGLN‐2 (PHD‐1) | 0.45 | ||
| EGLN‐3 (PHD‐3) | 0.45 | ||
| Glucocorticoid signalling | |||
| 11β‐hydroxysteroid dehydrogenase enzyme isoforms | HSD11B‐1 | 0.90 | McGillick et al. (2013) |
| HSD11B‐2 | 0.45 | ||
| Glucocorticoid receptor | NR3C1 | 0.90 | McGillick et al. (2013) |
| Mineralocorticoid receptor | NR3C2 | 0.90 | McGillick et al. (2013) |
| Chloride transport | |||
| Cystic fibrosis transmembrane conductance regulator | CFTR | 0.45 | McGillick et al. (2016b) |
| Chloride channel voltage‐sensitive 2 channel | CLCN2 | 0.45 | McGillick et al. (2016b) |
| Sodium transport | |||
| Epithelial sodium channel subunits | SCNN1‐A | 0.90 | McGillick et al. (2013) |
| SCNN1‐B | 0.45 | ||
| SCNN1‐G | 0.45 | ||
| Sodium potassium adenosine triphosphatase subunits | ATP1‐A1 | 0.45 | McGillick et al. (2013) |
| ATP1‐B1 | 0.45 | ||
| Water transport | |||
| Aquaporins | AQP‐1 | 0.45 | McGillick et al. (2013) |
| AQP‐3 | 0.45 | ||
| AQP‐4 | 0.45 | ||
| AQP‐5 | 0.45 | ||
| Surfactant maturation and lipid transport | |||
| Surfactant proteins | SFTP‐A | 0.30 | Orgeig et al. (2010) |
| SFTP‐B | 0.30 | McGillick et al. (2013) | |
| SFTP‐C | 0.30 | ||
| SFTP‐D | 0.30 | ||
| Phosphate cytidylyltransferase 1, choline, alpha | PCYT1A | 0.45 | McGillick et al. (2015) |
| ATP‐binding cassette, sub‐family A (ABC1), member 3 | ABCA3 | 0.45 | McGillick et al. (2015) |
| Airway remodelling | |||
| Elastin | ELN | 0.90 | McGillick et al. (2015) |
| Collagen type 1 alpha 1 | COL1A1 | 0.45 | McGillick et al. (2015) |
| Reference genes | |||
| Beta‐actin | ACTB | 0.45 | Passmore et al. (2009) |
| Peptidylprolyl isomerase A | PPIA | 0.45 (F), 0.90 (R) | Passmore et al. (2009) |
| Tyrosine 3‐monooxygenase | YWHAZ | 0.45 | McGillick et al. (2013) |
Optimised final concentrations are the same for forward (F) and reverse (R) primers for target and reference genes unless otherwise indicated.
Quantification of SFTP‐B‐positive cells within the fetal lung by immunohistochemistry
Immunohistochemistry was performed (normoxic, n = 6; hypoxic, n = 8), using a rabbit anti‐human mature surfactant protein B (SFTP‐B) antibody (1:500, WRAB‐48604, Seven Hills Bioreagents, Cincinnati, OH, USA) as previously described (Lock et al. 2015; McGillick et al., 2015, 2016b). All sections were counterstained with Mayer's Haematoxylin (Sigma Aldrich, St Louis, MO, USA). Negative control slides were incubated overnight at 4 °C in parallel with test slides under the same experimental conditions as described previously (Lock et al. 2015; McGillick et al. 2015, 2016b). Stained sections were examined using Visiopharm new Computer Assisted Stereological Toolbox (NewCAST) software (Visiopharm, Hoersholm, Denmark) as described previously (McGillick et al. 2013, 2015, 2016b; Lock et al. 2015). Sixty counting frames (600× magnification) of the alveolar epithelium were randomly selected per tissue section. Positive staining was confirmed with the presence of cuboidal shaped cells exhibiting cytoplasmic staining within the alveolar epithelium of lung tissue sections. Point‐counting using an unbiased counting frame with an area of 20,000 μm2 on immersion fixed tissue was used to estimate the numerical density of SFTP‐B‐positive cells within the alveolar epithelium of fetal lung tissue sections as described previously (McGillick et al. 2013; Lock et al. 2015).
Statistical analyses
Our specific research question relates to differences in outcome between the hypoxic (MCH) and normoxic groups in all measures and our data analysis has been carried out in consultation with a biostatistician. In the case of mRNA expression data, all genes were chosen a priori due to their known roles in normal development to prepare the lung for the successful transition to air‐breathing at birth and have previously shown changes in response to altered intrauterine conditions (Walther et al. 1991; Flecknoe et al. 2003; Jesse et al. 2009; McGillick et al. 2013, 2016b). Therefore, we have chosen a statistically significant P value of ≤ 0.05 with no correction for multiple comparisons (Rothman, 1990; Greenland et al. 2016). All data are presented as mean ± SD or MNE ± SD in the case of gene expression data. Maternal blood gas values are presented as an average of the entire sampling period (105–138 days of gestation). Data were analysed using an effect size calculator (Excel) and adjusted P‐values for mean difference (two‐tailed t test) are presented in Table 2 in addition to the effect size and 95% confidence intervals for all measures.
Table 2.
Analytical data demonstrating P‐values for mean difference, effect size and 95% confidence intervals for the effect of maternal chronic hypoxia (MCH) compared to Controls on maternal and fetal characteristics, expression of genes regulating lung development, concentrations of cortisol in plasma and lung tissue and evaluation of surfactant producing cells in the fetal lung by immunohistochemistry
| Treatment group (Hypoxic) | Control group (Normoxic) | Confidence interval for difference | Confidence interval for effect size | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Outcome measure | Mean | n | SD | Mean | n | SD | Pooled SD | P‐value for difference in SDs | Mean difference | P‐value for mean diff (2‐tailed t test) | Sediff | Lower | Upper | Effect size | Bias‐corrected (Hedges) | Standard error of E.S. estimate | Lower | Upper | Effect size based on control group SD |
| Maternal Characteristics | |||||||||||||||||||
| (mmHg) | 47 | 8 | 2.8 | 106 | 8 | 2.9 | 2.85 | 0.464 | –59.00 | <0.001 | 1.43 | −61.79 | −56.21 | −20.70 | −19.57 | 3.49 | −44.03 | 4.90 | −20.34 |
| (mmHg) | 29.9 | 8 | 1.5 | 37.3 | 8 | 0.9 | 1.24 | 0.101 | −7.40 | <0.001 | 0.62 | −8.61 | −6.19 | −5.98 | −5.66 | 1.12 | −13.48 | 2.17 | −8.22 |
| pH | 7.5 | 8 | 0.016 | 7.49 | 8 | 0.006 | 0.012 | 0.010 | 0.01 | 0.132 | 0.006 | −0.002 | 0.02 | 0.83 | 0.78 | 0.52 | −2.85 | 4.41 | 1.67 |
| Sat Hb (%) | 80 | 8 | 3.28 | 103.6 | 8 | 1.5 | 2.55 | 0.028 | −23.60 | <0.001 | 1.28 | −26.10 | −21.10 | −9.25 | −8.75 | 1.63 | −20.12 | 2.63 | −15.73 |
| Hb (g dl–1) | 11.7 | 8 | 0.6 | 10 | 8 | 0.4 | 0.51 | 0.153 | 1.70 | <0.001 | 0.26 | 1.20 | 2.20 | 3.33 | 3.15 | 0.75 | −2.09 | 8.39 | 4.25 |
| O2 content | 5.5 | 8 | 0.59 | 6.11 | 8 | 0.71 | 0.65 | 0.319 | −0.61 | 0.083 | 0.33 | −1.25 | 0.03 | −0.93 | −0.88 | 0.52 | −4.55 | 2.78 | −0.86 |
| Fetal characteristics at post‐mortem | |||||||||||||||||||
| Umbilical arterial Hb (g dl–1) | 16.4 | 7 | 1.6 | 13.6 | 7 | 1.4 | 1.51 | 0.33 | 2.81 | 0.004 | 0.81 | 1.24 | 4.39 | 1.87 | 1.75 | 0.63 | −2.65 | 6.15 | 2.07 |
| Body weight (kg) | 3.2 | 7 | 0.9 | 4.0 | 8 | 0.4 | 0.66 | 0.04 | −0.84 | 0.047 | 0.36 | −1.54 | −0.14 | −1.27 | −1.20 | 0.56 | −5.13 | 2.73 | −2.08 |
| Ratio of bi‐parietal diameter to hind limb lower length | 6.5 | 6 | 1.6 | 3.7 | 6 | 0.3 | 1.13 | 0.002 | 2.78 | 0.008 | 0.66 | 1.49 | 4.06 | 2.45 | 2.26 | 0.74 | −2.91 | 7.43 | 8.67 |
| Total brain weight (g) | 42.0 | 7 | 6.5 | 43.4 | 8 | 2.8 | 4.889 | 0.03 | −1.43 | 0.605 | 2.66 | −6.64 | 3.78 | −0.29 | −0.28 | 0.52 | −3.92 | 3.36 | −0.51 |
| Relative brain weight (g kg–1) | 13.9 | 7 | 2.6 | 10.9 | 8 | 0.5 | 1.805 | 0.001 | 2.96 | 0.025 | 0.99 | 0.99 | 4.91 | 1.64 | 1.54 | 0.59 | −2.58 | 5.66 | 5.55 |
| Relative brain/ liver weight ratio | 0.7 | 7 | 0.2 | 0.5 | 8 | 0.0 | 0.114 | 0.002 | 0.25 | 0.005 | 0.06 | 0.13 | 0.38 | 2.21 | 2.08 | 0.64 | −2.41 | 6.58 | 6.35 |
| Total lung weight (g) | 71.3 | 6 | 20.3 | 104.3 | 8 | 21.2 | 20.847 | 0.48 | −33.03 | 0.013 | 11.18 | −54.94 | −11.11 | −1.58 | −1.48 | 0.61 | −5.74 | 2.78 | −1.56 |
| Relative lung weight (g kg–1) | 23.0 | 6 | 3.1 | 26.1 | 8 | 4.2 | 3.799 | 0.26 | −3.06 | 0.145 | 1.96 | −6.90 | 0.79 | −0.80 | −0.75 | 0.56 | −4.66 | 3.16 | −0.72 |
| Pro‐oxidant markers | |||||||||||||||||||
| NOX‐4 | 0.0032 | 7 | 0.0009 | 0.0052 | 7 | 0.0016 | 0.001 | 0.094 | −0.0020 | 0.018 | 0.0007 | −0.003 | −0.001 | −1.54 | −1.44 | 0.60 | −5.64 | 2.76 | −1.25 |
| HMOX‐1 | 0.0265 | 8 | 0.0059 | 0.0287 | 8 | 0.0103 | 0.008 | 0.082 | −0.0022 | 0.611 | 0.0042 | −0.010 | 0.006 | −0.26 | −0.25 | 0.50 | −3.76 | 3.27 | −0.21 |
| NOS‐2 | 0.0184 | 8 | 0.0054 | 0.0185 | 7 | 0.0024 | 0.004 | 0.026 | −0.0001 | 0.963 | 0.0021 | −0.004 | 0.004 | −0.02 | −0.02 | 0.52 | −3.64 | 3.60 | −0.04 |
| NOS‐3 | 0.0064 | 7 | 0.0011 | 0.0074 | 8 | 0.0015 | 0.001 | 0.234 | −0.0010 | 0.162 | 0.0007 | −0.002 | 0.000 | −0.75 | −0.71 | 0.53 | −4.44 | 3.03 | −0.67 |
| Anti‐oxidant markers | |||||||||||||||||||
| SOD‐1 | 0.4009 | 8 | 0.0637 | 0.3874 | 7 | 0.0244 | 0.050 | 0.012 | 0.0135 | 0.593 | 0.0243 | −0.034 | 0.061 | 0.27 | 0.26 | 0.52 | −3.38 | 3.89 | 0.55 |
| SOD‐2 | 0.0420 | 8 | 0.0143 | 0.0364 | 8 | 0.0107 | 0.013 | 0.231 | 0.0056 | 0.391 | 0.0063 | −0.007 | 0.018 | 0.44 | 0.42 | 0.51 | −3.12 | 3.96 | 0.52 |
| CAT | 0.2071 | 8 | 0.0508 | 0.1583 | 8 | 0.0340 | 0.043 | 0.156 | 0.0488 | 0.043 | 0.0216 | 0.006 | 0.091 | 1.13 | 1.07 | 0.53 | −2.67 | 4.81 | 1.44 |
| GPX | 0.0147 | 8 | 0.0059 | 0.0101 | 8 | 0.0045 | 0.005 | 0.246 | 0.0046 | 0.103 | 0.0026 | −0.001 | 0.010 | 0.88 | 0.83 | 0.52 | −2.82 | 4.48 | 1.02 |
| Hypoxia signalling and feedback | |||||||||||||||||||
| HIF‐1α | 0.0366 | 7 | 0.0052 | 0.0376 | 8 | 0.0068 | 0.006 | 0.265 | −0.0010 | 0.753 | 0.0031 | −0.007 | 0.005 | −0.16 | −0.15 | 0.52 | −3.78 | 3.47 | −0.15 |
| HIF‐2α | 0.0720 | 8 | 0.0411 | 0.0578 | 8 | 0.0206 | 0.033 | 0.044 | 0.0142 | 0.403 | 0.0163 | −0.018 | 0.046 | 0.44 | 0.41 | 0.51 | −3.12 | 3.95 | 0.69 |
| HIF‐3α | 0.0417 | 8 | 0.0182 | 0.0219 | 8 | 0.0064 | 0.014 | 0.007 | 0.0198 | 0.018 | 0.0068 | 0.006 | 0.033 | 1.45 | 1.37 | 0.56 | −2.52 | 5.26 | 3.09 |
| HIF‐1β | 0.0385 | 8 | 0.0091 | 0.0355 | 8 | 0.0073 | 0.008 | 0.288 | 0.0030 | 0.480 | 0.0041 | −0.005 | 0.011 | 0.36 | 0.34 | 0.50 | −3.18 | 3.87 | 0.41 |
| VEGF | 0.1614 | 8 | 0.0603 | 0.1312 | 8 | 0.0278 | 0.047 | 0.029 | 0.0302 | 0.227 | 0.0235 | −0.016 | 0.076 | 0.64 | 0.61 | 0.51 | −2.97 | 4.19 | 1.09 |
| ADM | 0.0173 | 7 | 0.0025 | 0.0152 | 8 | 0.0021 | 0.002 | 0.343 | 0.0021 | 0.106 | 0.0012 | 0.000 | 0.004 | 0.92 | 0.86 | 0.54 | −2.92 | 4.65 | 1.00 |
| KDM3A | 0.0610 | 7 | 0.0075 | 0.0532 | 8 | 0.0021 | 0.005 | 0.003 | 0.0078 | 0.032 | 0.0029 | 0.002 | 0.014 | 1.47 | 1.38 | 0.58 | −2.65 | 5.41 | 3.71 |
| SLC2A1 | 0.0220 | 8 | 0.0063 | 0.0114 | 8 | 0.0013 | 0.005 | 0.000 | 0.0106 | 0.002 | 0.0023 | 0.006 | 0.015 | 2.33 | 2.20 | 0.63 | −2.23 | 6.64 | 8.15 |
| EGLN‐1 | 0.0666 | 8 | 0.0170 | 0.0617 | 8 | 0.0072 | 0.013 | 0.019 | 0.0049 | 0.472 | 0.0065 | −0.008 | 0.018 | 0.38 | 0.35 | 0.50 | −3.17 | 3.88 | 0.68 |
| EGLN‐2 | 0.0216 | 8 | 0.0031 | 0.0222 | 8 | 0.0040 | 0.004 | 0.259 | −0.0006 | 0.743 | 0.0018 | −0.004 | 0.003 | −0.17 | −0.16 | 0.50 | −3.66 | 3.35 | −0.15 |
| EGLN‐3 | 0.0253 | 7 | 0.0069 | 0.0090 | 8 | 0.0031 | 0.005 | 0.035 | 0.0163 | 0.0004 | 0.0028 | 0.011 | 0.022 | 3.13 | 2.94 | 0.75 | −2.28 | 8.17 | 5.26 |
| Glucocorticoid signalling | |||||||||||||||||||
| Fetal plasma cortisol (ng ml–1) | 16.6 | 8 | 8.1 | 17.6 | 8 | 8.5 | 8.302 | 0.451 | −1.0000 | 0.813 | 4.1512 | −9.136 | 7.136 | −0.12 | −0.11 | 0.50 | −3.62 | 3.39 | −0.12 |
| Lung tissue cortisol (pg mg–1) | 4.8 | 6 | 2.4 | 4.1 | 7 | 1.6 | 2.010 | 0.174 | 0.7670 | 0.526 | 1.1580 | −1.503 | 3.037 | 0.38 | 0.35 | 0.56 | −3.57 | 4.28 | 0.49 |
| HSD11B‐1 | 0.0064 | 7 | 0.0024 | 0.0064 | 8 | 0.0024 | 0.002 | 0.500 | 0.0000 | 1.000 | 0.0013 | −0.002 | 0.002 | 0.00 | 0.00 | 0.52 | −3.62 | 3.62 | 0.00 |
| HSD11B‐2 | 0.0037 | 8 | 0.0012 | 0.0044 | 8 | 0.0009 | 0.001 | 0.233 | −0.0007 | 0.210 | 0.0005 | −0.002 | 0.000 | −0.66 | −0.62 | 0.51 | −4.21 | 2.96 | −0.78 |
| NR3C1 | 0.1603 | 8 | 0.0252 | 0.1490 | 8 | 0.0214 | 0.023 | 0.339 | 0.0113 | 0.350 | 0.0117 | −0.012 | 0.034 | 0.48 | 0.46 | 0.51 | −3.09 | 4.00 | 0.53 |
| NR3C2 | 0.0075 | 8 | 0.0022 | 0.0068 | 8 | 0.0020 | 0.002 | 0.404 | 0.0007 | 0.516 | 0.0011 | −0.001 | 0.003 | 0.33 | 0.31 | 0.50 | −3.21 | 3.84 | 0.35 |
| Chloride transport | |||||||||||||||||||
| CFTR | 0.0046 | 8 | 0.001 | 0.004 | 8 | 0.0005 | 0.001 | 0.044 | 0.0006 | 0.160 | 0.0004 | 0.000 | 0.001 | 0.76 | 0.72 | 0.52 | −2.89 | 4.33 | 1.20 |
| CLCN2 | 0.0036 | 8 | 0.0008 | 0.0034 | 8 | 0.0009 | 0.001 | 0.382 | 0.0002 | 0.646 | 0.0004 | −0.001 | 0.001 | 0.23 | 0.22 | 0.50 | −3.29 | 3.73 | 0.22 |
| Sodium transport | |||||||||||||||||||
| SCNN1‐A | 0.0396 | 8 | 0.0111 | 0.0294 | 8 | 0.0113 | 0.011 | 0.482 | 0.0102 | 0.090 | 0.0056 | −0.001 | 0.021 | 0.91 | 0.86 | 0.52 | −2.80 | 4.52 | 0.90 |
| SCNNI‐B | 0.0194 | 8 | 0.0044 | 0.0141 | 7 | 0.0042 | 0.004 | 0.447 | 0.0053 | 0.033 | 0.0022 | 0.001 | 0.010 | 1.23 | 1.16 | 0.56 | −2.76 | 5.07 | 1.26 |
| SCNN1‐G | 0.0085 | 8 | 0.0030 | 0.0078 | 8 | 0.0051 | 0.004 | 0.092 | 0.0007 | 0.744 | 0.0021 | −0.003 | 0.005 | 0.17 | 0.16 | 0.50 | −3.35 | 3.66 | 0.14 |
| ATP1‐A1 | 0.0462 | 8 | 0.0089 | 0.0337 | 8 | 0.0035 | 0.007 | 0.012 | 0.0125 | 0.005 | 0.0034 | 0.006 | 0.019 | 1.85 | 1.75 | 0.59 | −2.37 | 5.86 | 3.57 |
| ATP1‐B1 | 0.0241 | 8 | 0.0058 | 0.0153 | 8 | 0.0039 | 0.005 | 0.158 | 0.0088 | 0.004 | 0.0025 | 0.004 | 0.014 | 1.78 | 1.68 | 0.58 | −2.39 | 5.76 | 2.26 |
| Water movement | |||||||||||||||||||
| AQP‐1 | 0.3166 | 8 | 0.1263 | 0.2146 | 8 | 0.0401 | 0.094 | 0.004 | 0.1020 | 0.061 | 0.0469 | 0.010 | 0.194 | 1.09 | 1.03 | 0.53 | −2.70 | 4.75 | 2.54 |
| AQP‐3 | 0.0021 | 7 | 0.0009 | 0.0013 | 7 | 0.0006 | 0.001 | 0.173 | 0.0008 | 0.079 | 0.0004 | 0.000 | 0.002 | 1.05 | 0.98 | 0.57 | −2.98 | 4.94 | 1.33 |
| AQP‐4 | 0.0166 | 8 | 0.0067 | 0.0091 | 8 | 0.0048 | 0.006 | 0.199 | 0.0075 | 0.023 | 0.0029 | 0.002 | 0.013 | 1.29 | 1.22 | 0.54 | −2.59 | 5.03 | 1.56 |
| AQP‐5 | 0.0359 | 8 | 0.0087 | 0.0311 | 8 | 0.0076 | 0.008 | 0.365 | 0.0048 | 0.259 | 0.0041 | −0.003 | 0.013 | 0.59 | 0.56 | 0.51 | −3.01 | 4.12 | 0.63 |
| Surfactant maturation and lipid transport | |||||||||||||||||||
| SFTP‐A | 0.6379 | 8 | 0.4099 | 0.4339 | 8 | 0.1808 | 0.317 | 0.023 | 0.2040 | 0.227 | 0.1584 | −0.106 | 0.514 | 0.64 | 0.61 | 0.51 | −2.97 | 4.19 | 1.13 |
| SFTP‐B | 1.2957 | 8 | 0.4446 | 0.8471 | 8 | 0.2156 | 0.349 | 0.038 | 0.4486 | 0.028 | 0.1747 | 0.106 | 0.791 | 1.28 | 1.21 | 0.54 | −2.59 | 5.02 | 2.08 |
| SFTP‐C | 5.3256 | 8 | 2.2333 | 3.4354 | 8 | 1.1282 | 1.769 | 0.046 | 1.8902 | 0.058 | 0.8846 | 0.156 | 3.624 | 1.07 | 1.01 | 0.53 | −2.71 | 4.73 | 1.68 |
| SFTP‐D | 0.0446 | 8 | 0.0130 | 0.0294 | 8 | 0.0105 | 0.012 | 0.293 | 0.0152 | 0.023 | 0.0059 | 0.004 | 0.027 | 1.29 | 1.22 | 0.54 | −2.59 | 5.03 | 1.45 |
| PCYT1A | 0.0250 | 8 | 0.0039 | 0.0260 | 8 | 0.0045 | 0.004 | 0.358 | −0.0010 | 0.642 | 0.0021 | −0.005 | 0.003 | −0.24 | −0.22 | 0.50 | −3.74 | 3.29 | −0.22 |
| ABCA3 | 0.0405 | 8 | 0.0121 | 0.0298 | 7 | 0.0065 | 0.010 | 0.064 | 0.0107 | 0.053 | 0.0049 | 0.001 | 0.020 | 1.08 | 1.02 | 0.55 | −2.83 | 4.86 | 1.65 |
| Numerical density of SFTP‐B‐positive cells | 328.0 | 8 | 92.5 | 293.4 | 6 | 33.4 | 73.82 | 0.009 | 34.60 | 0.354 | '35.41 | −34.80 | 104.0 | 0.47 | 0.44 | 0.55 | −3.39 | 4.26 | 1.04 |
| Airway remodelling | |||||||||||||||||||
| ELN | 0.8503 | 7 | 0.1376 | 0.8556 | 7 | 0.2253 | 0.187 | 0.128 | −0.0053 | 0.959 | 0.0998 | −0.201 | 0.190 | −0.03 | −0.03 | 0.53 | −3.77 | 3.72 | −0.02 |
| COL1A1 | 1.8623 | 8 | 0.9092 | 1.4439 | 7 | 0.7295 | 0.831 | 0.288 | 0.4184 | 0.341 | 0.4235 | −0.412 | 1.248 | 0.50 | 0.47 | 0.52 | −3.20 | 4.15 | 0.57 |
Results
Impact of MCH on blood gas status of the ewe and fetus and fetal growth
MCH resulted in decreased partial pressure of oxygen (), partial pressure of carbon dioxide () and the saturation of haemoglobin with oxygen (Sat Hb), but increased haemoglobin content (Hb) in maternal arterial blood throughout the duration of the experimental protocol (Table 2). There was no effect of MCH on maternal pH (Table 2). Fetuses exposed to MCH had increased umbilical arterial Hb content at post‐mortem (Table 2). Fetuses exposed to MCH had lower body weights, increased relative brain weight and elevations in the bi‐parietal diameter to hind lower limb length ratio and brain to liver weight ratio, providing evidence of asymmetric IUGR compared with the normoxic group (Table 2). There was decreased total fetal lung weight, but no effect of MCH on relative fetal lung weight (Table 2).
Expression of genes regulating oxidative and nitrosative stress and anti‐oxidant defence in the fetal lung
There was reduced mRNA expression of the pro‐oxidant marker NOX‐4 (Fig. 1 A) in the fetal lung following exposure to MCH. However, the levels of expression of an oxidative stress marker HMOX‐1 (Fig. 1 B) and nitrosative stress regulators NOS‐2 (Fig. 1 C) and NOS‐3 (Fig. 1 D) were similar between groups. Furthermore, lung mRNA expression of the anti‐oxidant CAT increased (Fig. 1 G) following exposure to MCH, although there was no effect on expression of other anti‐oxidant markers, including SOD‐1 (Fig. 1 E), SOD‐2 (Fig. 1 F) and GPX (Fig. 1 H).
Figure 1. Maternal chronic hypoxia reduces expression of pro‐oxidant marker NOX‐4 and increases expression of anti‐oxidant marker CAT .
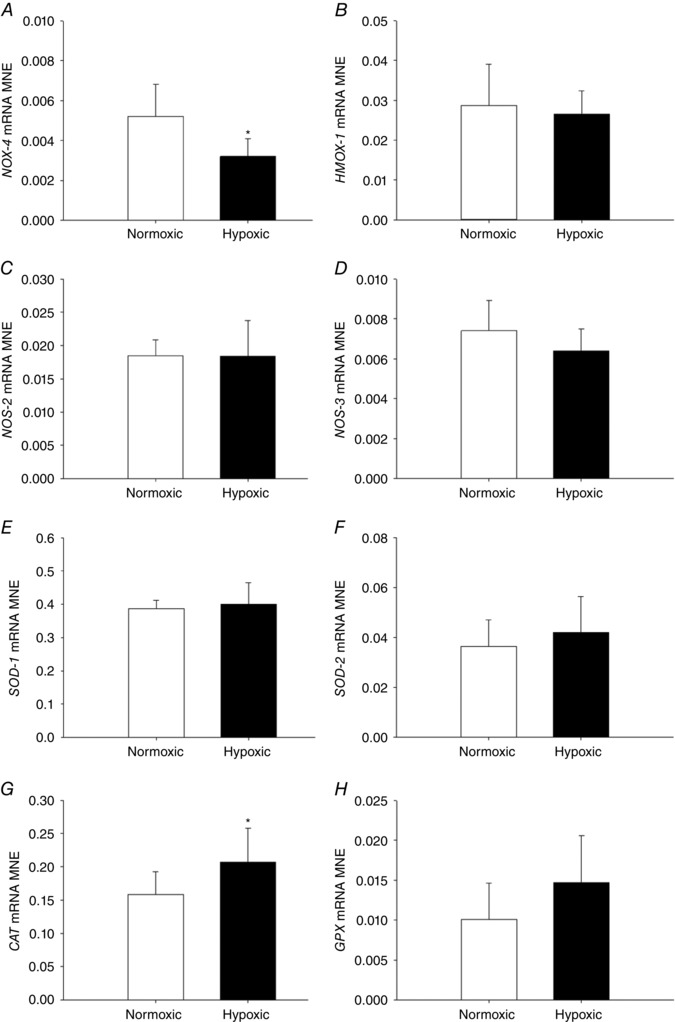
Mean normalised expression (MNE) of genes regulating oxidative and nitrosative stress [NOX‐4 (A), HMOX‐1 (B), NOS‐2 (C) and NOS‐3 (D)] and anti‐oxidant enzymes [SOD‐1 (E), SOD‐2 (F), CAT (G) and GPX (H)] in the lung of the late‐gestation sheep fetus. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Expression of genes regulating hypoxia signalling and feedback in the fetal lung
There was increased fetal lung HIF‐3α mRNA expression (Fig. 2 C) following exposure to MCH, but no effect on gene expression of HIF‐1α (Fig. 2 A), HIF‐2α (Fig. 2 B) or HIF‐1β (Fig. 2 D) subunit expression. There was no effect of MCH on VEGF (Fig. 3 A) or ADM (Fig. 3 B) expression, but there was increased expression of hypoxia responsive genes KDM3A (Fig. 3 C) and SLC2A1 (Fig. 3 D). EGLN‐3 mRNA expression increased (Fig. 4 C), but there was no effect on EGLN‐1 (Fig. 4 A) or EGLN‐2 (Fig. 4 B), in the lung of fetuses following MCH.
Figure 2. Maternal chronic hypoxia increases expression of genes regulating hypoxia signalling.
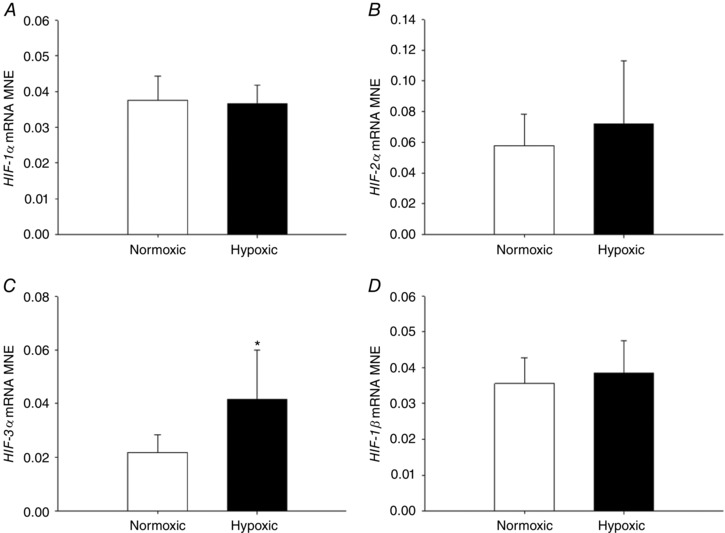
Mean normalised expression (MNE) of genes regulating HIF signalling [HIF‐1α (A), HIF‐2α (B), HIF‐3α (C) and HIF‐1β (D)] in the lung of the late‐gestation sheep fetus. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Figure 3. Maternal chronic hypoxia increases expression of hypoxia responsive genes in the fetal lung.
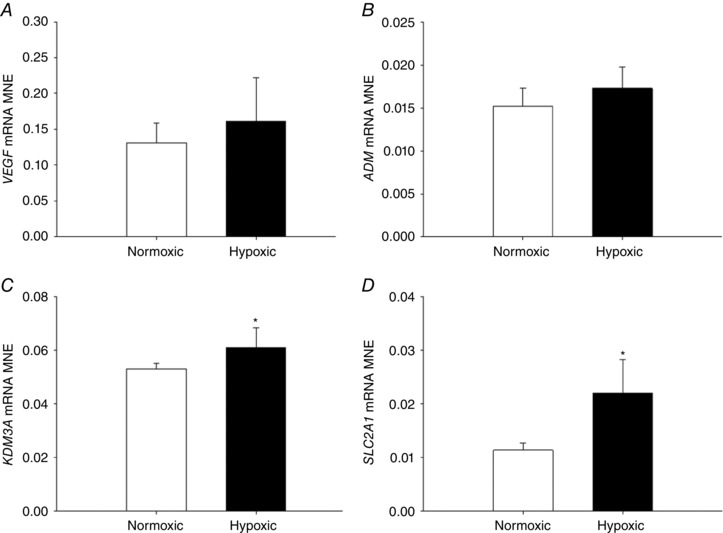
Mean normalised expression (MNE) of genes with hypoxia response elements [VEGF (A), ADM (B), KDM3A (C) and SLC2A1 (D)] in the fetal lung following exposure to maternal chronic hypoxia for a month in late gestation. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Figure 4. Maternal chronic hypoxia increases expression of hypoxia signalling regulatory factor EGLN‐3 .
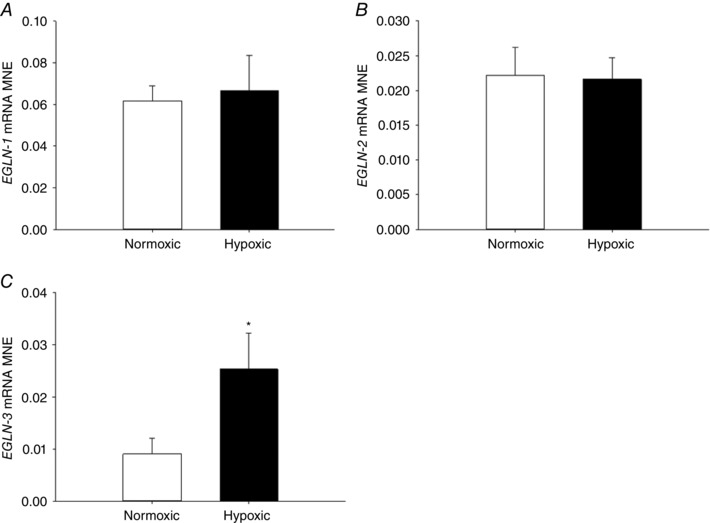
Mean normalised expression (MNE) of genes regulating hypoxia signalling and feedback [EGLN‐1 (A), EGLN‐2 (B) and EGLN‐3 (C)] in the lung of the late‐gestation sheep fetus. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Fetal plasma cortisol, lung tissue cortisol and expression of genes regulating glucocorticoid signalling in the fetal lung
There was no effect of MCH on fetal plasma cortisol concentration (Fig. 5 A) or total cortisol concentration in fetal lung tissue (Fig. 5 B), when measured at 138 days of gestation. There was also no effect of MCH on fetal lung mRNA expression of genes regulating glucocorticoid (GC) availability (HSD11B‐1, Fig. 5 C; HSD11B‐2, Fig. 5 D) or activity (NR3C1, Fig. 5 E; NR3C2, Fig. 5 F) compared with the normoxic group.
Figure 5. No effect of maternal chronic hypoxia on fetal glucocorticoid avaliability or signalling.
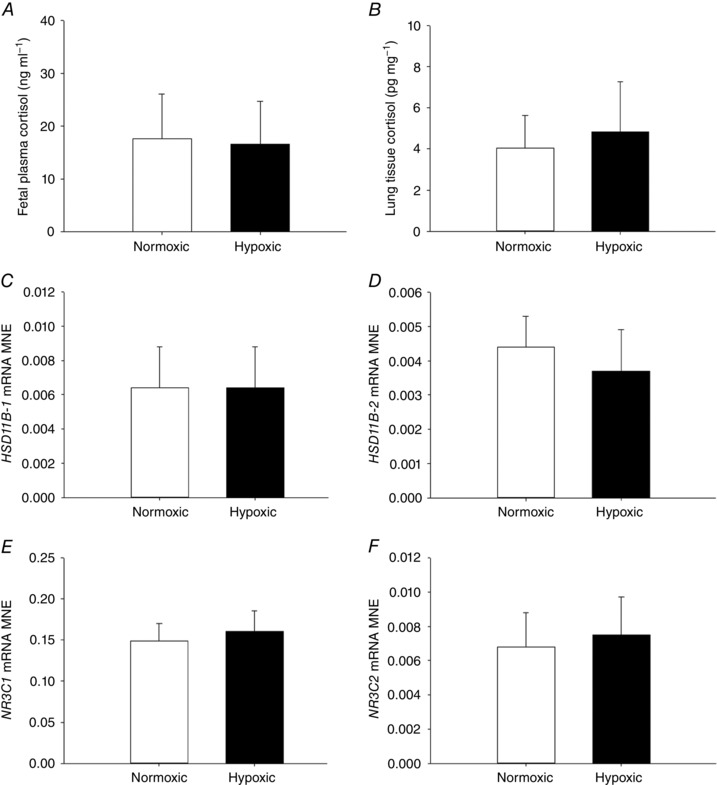
Fetal plasma cortisol (A), total lung tissue cortisol (B) and mean normalised expression (MNE) of genes regulating glucocorticoid availability [HSD11B‐1 (C) and HSD11B‐2 (D)] and activity [NR3C1 (E) and NR3C2 (F)] in the fetal lung. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Expression of genes regulating fetal lung liquid movement
MCH did not alter expression of genes regulating chloride movement (CFTR, Fig. 6 A; CLCN2, Fig. 6 B) in the fetal lung. There was a significant increase in expression of subunits regulating sodium movement, including SCNN1‐B (Fig. 6 D), ATP1‐A1 (Fig. 6 F) and ATP1‐B1 (Fig. 6 G) subunits, but no effect on SCNN1‐A (Fig. 6 C) or SCNN1‐G (Fig. 6 E) subunits, in the fetal lung following exposure to MCH. Exposure to MCH increased mRNA expression of AQP‐4 (Fig. 7 C) in the fetal lung, but had no effect on AQP‐1 (Fig. 7 A), AQP‐3 (Fig. 7 C) or AQP‐5 expression (Fig. 7 D).
Figure 6. Maternal chronic hypoxia increases expression of genes regulating sodium movement in the fetal lung.
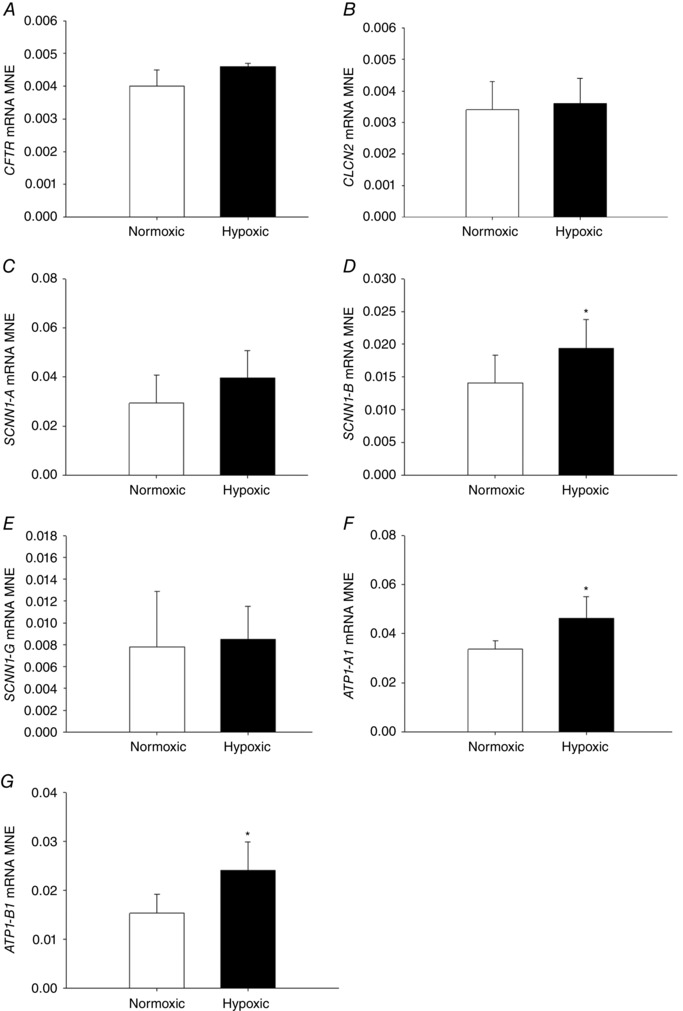
Mean normalised expression (MNE) of genes regulating chloride [CFTR (A) and CLCN2 (B)] and sodium [SCNN1‐A (C), SCNN1‐B (D), SCNN1‐G (E), ATP1‐A1 (F) and ATP1‐B1 (G)] movement in the lung of the late‐gestation sheep fetus. Data expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Figure 7. Maternal chronic hypoxia increases expression of gene regulating water movement in the fetal lung.
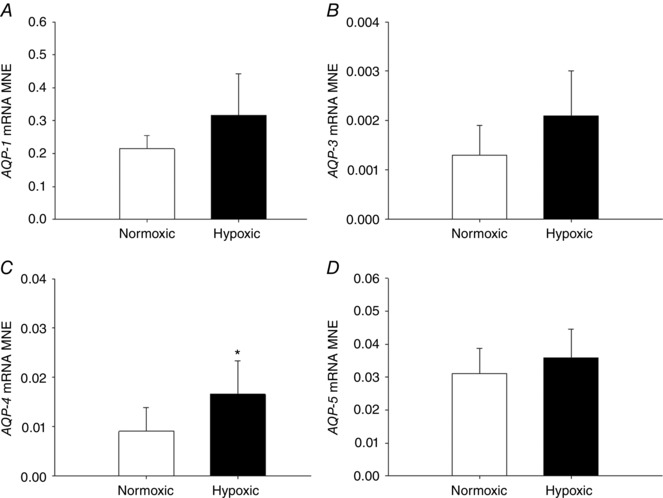
Mean normalised expression (MNE) of genes regulating water movement [AQP‐1 (A), AQP‐3 (B), AQP‐4 (C) and AQP‐5 (D)] in the lung of the late‐gestation sheep fetus. Data expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Molecular and structural regulation of surfactant maturation and airway remodelling in the fetal lung
There was a significant increase in expression of SFTP‐B (Fig. 8 B) and SFTP‐D (Fig. 8 D), but not of SFTP‐A (Fig. 8 A) or SFTP‐C (Fig. 8 C), in the lung of fetuses exposed to MCH compared with the normoxic group. In addition, there was increased expression of the surfactant lipid transporter ABCA3 (Fig. 8 F) following exposure to MCH. There was no effect of MCH on expression of PCYT1A (Fig. 8 E), a rate‐limiting enzyme involved in surfactant phospholipid synthesis or genes regulating airway remodelling (ELN, Fig. 9 A; COL1A1, Fig. 9 B). There was no effect of MCH on the numerical density of SFTP‐B‐positive cells (Fig. 10 E) present in the alveolar epithelium in the fetal lung tissue compared to the normoxic group.
Figure 8. Maternal chronic hypoxia increases expression of genes regulating surfactant maturation and surfactant lipid transport in the fetal lung.
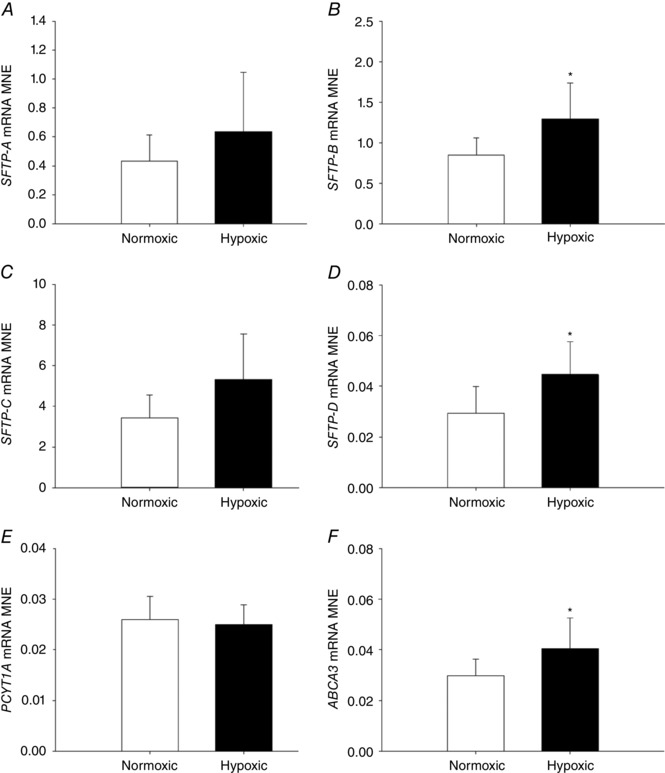
Mean normalised expression (MNE) of genes regulating surfactant maturation [SFTP‐A (A), SFTP‐B (B), SFTP‐C (C), SFTP‐D (D)] and surfactant lipid synthesis and transport [PCYT1A (E) and ABCA3 (F)] in the lung of the late‐gestation sheep fetus. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P≤0.05, P‐value for mean difference between groups.
Figure 9. No effect of maternal chronic hypoxia on expression of genes regulating airway remodelling.
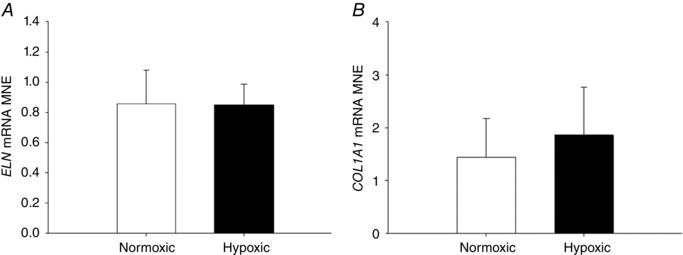
Mean normalised expression (MNE) of ELN (A) and COL1A1 (B) in the lung of the late‐gestation sheep fetus. Data are expressed as mRNA MNE ± SD in normoxic (white bars) and hypoxic (black bars) pregnancies. * P < 0.05, P‐value for mean difference between groups.
Figure 10. No effect of maternal chronic hypoxia on the numerical density of SFTP‐B‐positive cells in the alveolar epithelium of the fetal lung.
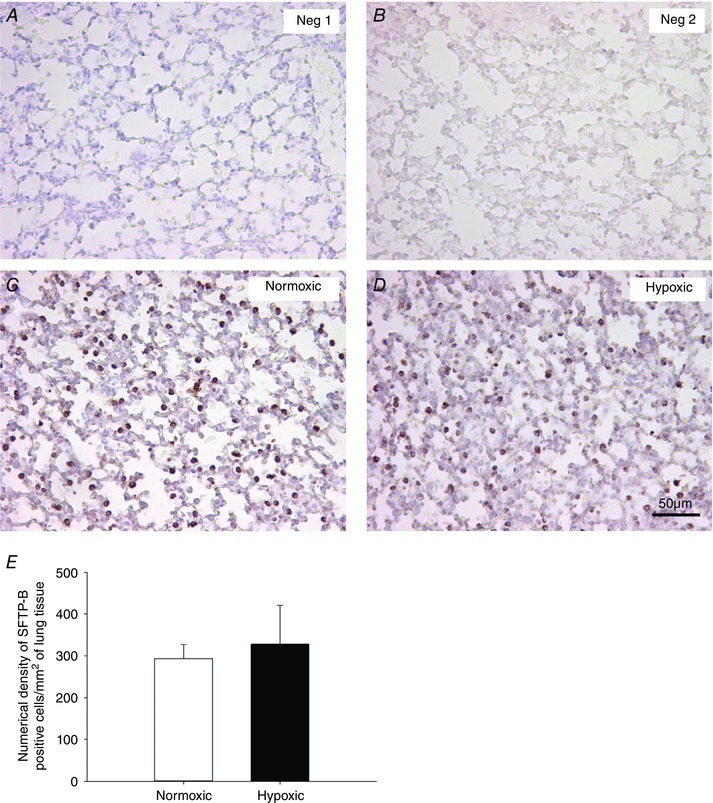
Micrographs demonstrating no primary antibody‐negative control (Neg 1, A), 1:500 rabbit serum negative control (Neg 2, B), SFTP‐B immunoreactivity (brown intracellular precipitate) in the alveolar epithelium of the fetal lung tissue from normoxic (C) and hypoxic (D) pregnancies. There was no significant effect of maternal chronic hypoxia (black bar, E) on the numerical density of SFTP‐B‐positive cells per mm2 of lung tissue in the alveolar epithelium when compared to lung tissue of fetuses from normoxic (white bar, E) pregnancies. Data are expressed as mean ± SD. * P < 0.05, P‐value for mean difference between groups. Micrographs at 200× magnification. Scale bar = 50 μm.
Discussion
We have used isobaric hypoxic chambers that are able to maintain unrestrained pregnant ewes to isolate the effect of exposure to MCH for a month in late gestation on fetal lung maturation (Brain et al. 2015). Exposure to MCH without alteration in maternal food intake promoted increased expression of genes regulating hypoxia signalling, lung liquid reabsorption and surfactant maturation in the fetal lung (Fig. 11). The mechanisms underlying these effects appear to be regulated at the molecular level as there was no difference in the numerical density of SFTP‐B‐positive cells present in the alveolar epithelium. The molecular mechanisms mediating maturational effects of MCH on the fetal lung include alterations to hypoxia but not GC signalling as there was no difference in expression of genes regulating GC signalling or changes to maternal or fetal plasma cortisol concentration, nor elevations in fetal lung tissue cortisol concentration. Combined, the data provide evidence for an adaptive response of the fetal lung to prenatal chronic hypoxaemia to increase the expression of factors essential for lung maturation and the successful transition to the air‐breathing environment at birth.
Figure 11. Overview of study findings on molecular signalling in the fetal lung.
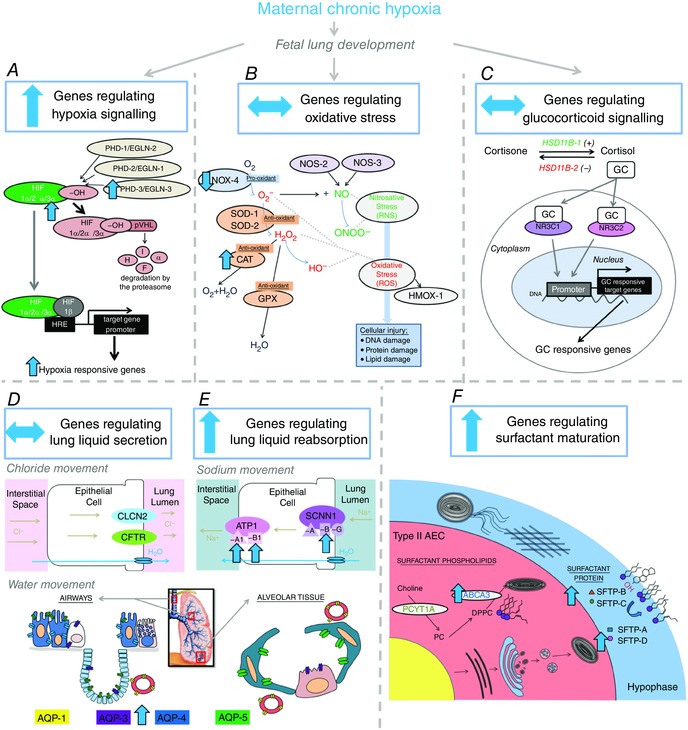
Effect of maternal chronic hypoxia for a month in late gestation on expression of genes regulating hypoxia signalling (A), oxidative stress (B), glucocorticoid signalling (C), lung liquid secretion (chloride movement; D), lung liquid reabsorption (sodium movement; E) and surfactant maturation (F) in the fetal lung. Overall maternal chronic hypoxia increased expression of genes regulating hypoxia signalling (A), lung liquid reabsorption (E) and surfactant maturation (F) in the fetal lung, which may be an adaptive response to aid in the successful transition to the air‐breathing environment at birth.
In most sheep models of IUGR, fetal arterial oxygenation is reduced by ∼5–8 mmHg compared to controls (Morrison, 2008; Herrera et al. 2016). In this sheep model of maternal chronic hypoxia, the change is ∼9–10 mmHg (Brain et al. 2015), akin to those measured in severe IUGR in humans (Hecher et al. 1995). In this model, the level of maternal hypoxia produced decreased mean fetal descending aortic values to 11.5 ± 0.6 mmHg relative to a mean of 20.9 ± 0.5 mmHg in control fetuses of a normoxic pregnancy (Brain et al. 2015). The increased maternal and fetal arterial Hb content represents an established compensatory mechanism to increase the oxygen carrying capacity to tissues in response to MCH. Despite this, exposure to MCH induced significant asymmetric fetal growth restriction, as evidenced by a reduction in fetal body weight, and increases in the ratio of the bi‐parietal diameter to lower limb length and in the brain to liver weight ratio, all accepted indices of persistent fetal brain sparing (Giussani, 2016).
An advantage of this sheep model over rodent models of IUGR in adverse pregnancy (Van Geijn et al. 1980) is that it isolates the contribution of chronic hypoxaemia on fetal growth and development independent of changes in maternal food intake or elevations in circulating maternal stress hormones, despite significant maternal hypoxaemia (Brain et al. 2015). This is important as maternal nutrient restriction and hypoxia can have differential effects on fetal organ development and these effects persist into postnatal life (Williams et al. 2005; Camm et al. 2010). Furthermore, in the present model of MCH, it is unlikely that the observed downstream changes in gene expression in the fetal lung at the time of tissue collection are GC dependent. This is advantageous as alterations in fetal oxygenation and circulating GCs can have differential effects on indices of lung maturation, as evidenced in other sheep models of IUGR resulting in fetal hypoxaemia (Gagnon et al. 1999; Orgeig et al. 2010, 2015; Allison et al. 2016b). Thus, despite the multi‐factorial regulation of organ development by changes in metabolic and endocrine markers in the IUGR pregnancy, a strength of this study is the capacity to isolate the effect of fetal reduced oxygen availability to gain further understanding of the effect of chronic hypoxaemia on the molecular mechanisms regulating fetal lung development in adverse pregnancy.
In this study, only male fetuses were used to control for possible sex differences, which may be viewed as a potential limitation of our experimental design. However, it is important to note that evaluation of data from previous publications in our laboratory has demonstrated no effect of sex on expression of surfactant protein markers in the fetal lung at 133 days of gestation (Orgeig et al. 2015). While there is evidence to support a male respiratory disadvantage at birth, our previous findings in the PR early onset IUGR sheep model suggest that the effects observed between male and female fetuses would be similar in both the Control and the IUGR group in the current study.
Glucocorticoid signalling is essential for normal fetal and lung development and this is exploited clinically with antenatal GC administration to women at risk of preterm delivery to promote fetal lung maturation to ensure the successful transition to newborn life (McGillick et al. 2013; Antenatal Corticosteroid Clinical Practice Guidelines Panel, 2015). In this study, we have observed no effect of MCH on fetal plasma cortisol, lung tissue cortisol concentration, or expression of genes regulating GC availability and activity at the time of tissue collection. The apparent limited effect on GCs in this model is consistent with other sheep models of chronic hypoxia, including exposure to long‐term hypoxaemia induced by living at high altitude from mid‐ to late gestation (Harvey et al. 1993). However, this finding is in contrast to the chronically hypoxaemic PR fetus where there are persistent elevations in circulating GCs and altered expression of factors regulating GC availability and activity throughout gestation (Phillips et al. 1996; Orgeig et al. 2010, 2015). However, it is important to note that investigations in this study were undertaken on plasma and tissue samples collected at post‐mortem. Therefore, it is possible that during the hypoxic insult for a month in late gestation, altered regulation of GC signalling at earlier time points following initiation of the hypoxic insult may have contributed to the overall changes observed in the fetal lung. Importantly, we have demonstrated that there is no difference in maternal catecholamines (Brain et al. 2015) and plasma cortisol concentration between the normoxic and hypoxic pregnancies in this model (104–138 days of gestation; 94.4 ± 16.8 vs. 74.5 ± 13.7 ng ml−1; P = 0.7). Further characterisation of the model in the future will elucidate the specific mechanisms contributing to both maternal and fetal adaptations to MCH.
HIF‐3α is regarded as a sensitive regulator of the tissue response to systemic hypoxia (Heidbreder et al. 2003). Increased HIF‐3α expression has been observed following exposure to a 50% reduction in oxygen tension for 0.5–2 h, with no impact on HIF‐1α, HIF‐2α or HIF‐1β transcription (Heidbreder et al. 2003). The present study confirms increased expression of HIF‐3α in the fetal lung following exposure to MCH for a month in late gestation. Increased HIF‐α signalling is further evidenced by increased expression of downstream genes with hypoxia response elements, including SLC2A1 and KDM3A. An advantage of measuring expression of genes with hypoxia response elements in their promoter region is that they are under direct control of HIF‐α subunit stability and hence act as a more functional outcome than HIF‐α protein expression itself. The increased expression of KDM3A, which encodes the histone demethylase, jumonji domain containing 1A (JMJD1A), suggests an interesting potential role of MCH by epigenetic modifications to the regulation of fetal lung maturation at the transcriptional level (Wellmann et al. 2008). In this model, MCH also increased EGLN‐3 expression, which encodes PHD‐3 in the fetal lung. This finding is consistent with altered pulmonary regulation of hypoxia signalling by increased EGLN/PHD expression, as has been reported for the lung (Orgeig et al. 2015) and heart (Botting et al. 2014) of the chronically hypoxaemic fetus in late gestation as a result of placental restriction.
Increased hypoxia signalling following exposure to MCH occurred together with positive downstream effects on molecular regulation of lung liquid movement and surfactant maturation. Positive effects of hypoxia on both biochemical and physiological determinants of lung liquid movement have been reported previously (Vivona et al. 2001; Dada et al. 2003; Abreu‐Rodríguez et al. 2011). However, there is limited in vivo evidence for the effect of chronic hypoxaemia on molecular mechanisms regulating fetal lung liquid movement. In addition, the timing, duration and severity of the hypoxic insults may have a differential impact on the magnitude of change in these factors (Vivona et al. 2001; Dada et al. 2003; Morrison, 2008; Abreu‐Rodríguez et al. 2011). For instance, we have previously reported that in the chronically hypoxaemic PR fetus, where the insult occurs prior to conception and growth restriction begins early in gestation, there are few effects of chronic hypoxaemia and IUGR on the molecular mechanisms regulating lung liquid movement (McGillick et al. 2016b). In contrast, here, following exposure to MCH for a month in late gestation we show a significant increase in the expression of genes regulating sodium (SCNN1‐B, ATP1‐A1 and ATP1‐B1) and water (AQP‐4) movement in fetal lung tissue. These molecular changes may contribute in part to the control of lung liquid reabsorption in preparation for air‐breathing at birth and play a functional role in regulation of lung liquid movement at the air–liquid interface in the neonatal lung (Hooper et al. 2015).
We also provide mechanistic molecular evidence for upregulation of the surfactant system with increased expression of SFTP‐B and SFTP‐D and the surfactant lipid transporter ABCA3 in the fetal lung following MCH. Interestingly, our data suggest that exposure to MCH regulates surfactant maturation at the molecular level by increasing the functional capacity of surfactant‐producing cells rather than by an overall change to the numerical density of SFTP‐B‐positive cells in fetal lung tissue as determined by immunohistochemistry. Evaluation of fetal lung tissue at the cellular level by immunohistochemistry is advantageous as it provides specific detail regarding functional capability of cells able to produce surfactant in contrast to ultrastructural detail of alveolar epithelial cell differentiation achieved with electron microscopy (Lock et al. 2015). The finding of increased molecular regulation of surfactant maturation in this model of MCH is consistent with other sheep models of fetal hypoxaemia, including utero‐placental embolisation (Gagnon et al. 1999) or short‐term 48 h of maternal inhalational hypoxia in late gestation (Braems et al. 2000). However, these present and past data contrast with findings in the chronically hypoxaemic PR fetus, in which there is reduced SFTP expression in the lung (Orgeig et al. 2010; McGillick 2015 and 2016b). Therefore, it is clear that surfactant maturation in the fetal lung can be differentially regulated by chronic hypoxaemia of varying duration, severity and/or time of onset. Moreover, these findings provide molecular evidence for the impact of chronic hypoxaemia on the heterogeneity observed in respiratory distress syndrome in IUGR neonates at birth (McGillick et al. 2016c).
While we were unable to measure the incidence of fetal breathing movements as the fetuses in this study were not catheterised, in our previous work in chronically hypoxaemic PR fetuses there was no effect on the incidence, amplitude or frequency of breathing movements in late gestation (Poudel et al. 2015; McGillick et al. 2016b). This study supports the notion of chronic hypoxaemia as a molecular regulator of fetal lung development. In contrast, a previous study showed reduced breathing movements measured by diaphragmatic electromyogram across late gestation in a group of hypoxaemic and acidotic growth‐restricted fetuses that did not have brain sparing (Maloney et al. 1982). This parallels with human clinical literature with acidotic and hypoxic fetuses exhibiting reduced breathing movements (Yoneyama et al. 1994). As the fetuses in this model of MCH are not acidotic (Allison et al. 2016a) and exhibit characteristic growth restriction including brain sparing, it is likely that there would be no effect of this model on fetal breathing movements.
Finally, despite no change in the expression of NOS‐2 and NOS‐3 (key regulators of free radical generation contributing to nitrosative stress) or of HMOX‐1 (a molecular marker of oxidative stress), there was reduced expression of the pro‐oxidant marker NOX‐4 and a significant increase in the anti‐oxidant CAT in the fetal lung following exposure to MCH. These data suggest a compensatory shift in the upregulation of anti‐oxidant pathways to offset oxidative stress and maintain homeostasis in the fetal lung. This may be an additional adaptation to protect the fetal lung from the relative hyperoxia following birth and one that appears to be regulated by hypoxia rather than GC signalling in this model of MCH.
The findings from this study highlight the complex regulation of lung development by chronic hypoxaemia associated with IUGR that supports the spectrum of respiratory distress syndrome outcomes that these infants may experience at birth (McGillick et al. 2016c). Despite providing mechanistic evidence for chronic hypoxaemia as a direct molecular regulator of key pathways contributing to fetal lung development, it is important to consider the possibility that overall the effects observed in this model of MCH may also be regulated synergistically or indirectly by a secondary mediator. With regards to alternative pathways that are potential regulators, we have previously investigated markers of cellular proliferation, beta‐adrenergic receptors, interleukin 1‐beta and transforming growth factor‐beta following chronic fetal hypoxaemia in a PR model induced by uterine carunclectomy and found that these were not affected by chronic hypoxaemia (McGillick et al. 2015; Orgeig et al. 2015). Importantly, in this study we have investigated markers of oxidative stress, a process that represents an additional factor that may be potentially altered as a result of the relationship between hypoxia signalling and the generation of reactive oxygen species. Further characterisation of this MCH model in the future will elucidate additional mechanisms influencing fetal organ growth and development in response to the chronically hypoxaemic conditions experienced for a month in late gestation. In late gestation, studies in the PR sheep fetus showed a similar effect of chronic hypoxaemia on molecular regulators of lung maturation at both 133 and 140 days of gestation (Orgeig et al. 2010). These findings suggest that there is a similar response of the lung to chronic hypoxaemia throughout late gestation and, thus, it is reasonable to postulate that if the markers of lung maturation were evaluated closer to term (e.g. 145 days of gestation) there would be similar effects observed in the lung of the fetus following exposure to MCH in this model.
Importantly, this model of chronic hypoxaemia results in cardiovascular deficits similar to the human IUGR fetus (Allison et al. 2016a; Brain et al. 2015). These include fetal cardiovascular dysfunction, characterised by impaired myocardial contractility and diastolic function as well as impaired peripheral vascular reactivity and loss of nitric oxide‐dependent endothelial function (Brain et al. 2015). Interestingly, despite the negative cardiovascular effects of exposure to MCH, the fetus still has the capacity to enhance protective effects such as lung maturation to ensure successful transition to extrauterine life. These findings are consistent with studies showing decreased risk of respiratory distress syndrome in IUGR subpopulations (McGillick et al. 2016c), although this is in contrast to findings in the PR sheep fetus, which support increased risk of respiratory distress syndrome in a subset of IUGR newborns. Combined with our previous findings in a model of early‐onset chronic hypoxaemia and IUGR (Orgeig et al. 2010, 2015; McGillick et al. 2015, 2016b), our work in this area highlights a differential effect of IUGR complicated by chronic hypoxaemia on fetal lung development that may underlie the heterogeneity in clinical newborn respiratory outcomes (McGillick et al. 2016a).
In summary, the data in the present study provide evidence for upregulation of the molecular mechanisms involved in processes vital for lung liquid movement and surfactant production in response to MCH (Fig. 11). This may be an adaptive response anticipating preterm birth triggered by adverse conditions experienced late in gestation. Overall, the study data suggest an increased functional capacity of the lung to ensure the adequate transition to air‐breathing following exposure to late‐gestation onset IUGR induced by MCH for a month in late gestation, thus reducing the risk of respiratory distress at birth and pulmonary dysfunction in later life.
Additional information
Competing interests
The authors have no conflicts of interest.
Author contributions
EVM, SO, DAG and JLM were responsible for the conception and design of the experiments. EVM, SO, BJA, KLB, YN, NI, KLS, ADK, EAH, DAG and JLM were each involved in data acquisition. EVM, SO, DAG and JLM were involved in analysis and interpretation of the data. EVM, SO, DAG and JLM drafted the article. EVM, SO, BJA, KLB, YN, NI, KLS, ADK, EAH, DAG and JLM contributed to and have approved the final version to be published.
Funding
The animal component of the work was funded by the British Heart Foundation (DAG) and the molecular component of the work was funded by a National Health and Medical Research Council (NHMRC) Project Grant (APP1030853, JLM and SO). JLM was funded by an NHMRC Career Development Fellowship (APP1066916).
Translational perspective.
In this study we investigated the effect of maternal chronic hypoxia for a month in late gestation on molecular and structural regulation of fetal lung development. We isolated the effect of chronic hypoxaemia on fetal development using isobaric hypoxic chambers without alterations to maternal food intake. We have provided evidence that maternal chronic hypoxia in late gestation increases the expression of genes regulating hypoxia signalling, lung liquid reabsorption and surfactant maturation in the fetal lung. There was no effect of maternal chronic hypoxia on airway remodelling or structural fetal lung development. Therefore, we provide evidence for chronic fetal hypoxaemia as a molecular regulator of lung maturation. In contrast to other models of intrauterine growth restriction including early‐onset chronic fetal hypoxaemia leading to reduced fetal lung maturation, late‐onset fetal chronic hypoxaemia promotes molecular regulation of fetal lung development which may be an adaptive response in preparation for the successful transition to air‐breathing at birth. Overall the findings from this study provide evidence for a differential effect of timing and duration of fetal chronic hypoxaemia on expression of factors regulating fetal lung maturation. This provides evidence for the heterogeneity observed in respiratory outcomes in newborns at birth following exposure to chronic hypoxaemia in utero. Greater understanding of the intrauterine environment encountered in growth‐restricted pregnancies may lead to identification of newborns at altered risk of respiratory complications at birth in clinical practice.
References
- Abreu‐Rodríguez I, Silva RS, Martins AP, Soveral G, Toledo‐Aral JJ, López‐Barneo J & Echevarría M (2011). Functional and transcriptional induction of aquaporin‐1 gene by hypoxia; analysis of promoter and role of Hif‐1α. PloS One 6, e28385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison B, Brain K, Niu Y, Kane A, Herrera E, Thakor A, Botting K, Cross C, Itani N & Skeffington K (2016a). Fetal in vivo continuous cardiovascular function during chronic hypoxia. J Physiol 594, 1247–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison BJ, Hooper SB, Coia E, Zahra VA, Jenkin G, Malhotra A, Sehgal A, Kluckow M, Gill AW & Sozo F (2016b). Ventilation induced lung injury is not exacerbated by growth restriction in preterm lambs. Am J Physiol Lung Cell Mol Physiol 310, L213–223. [DOI] [PubMed] [Google Scholar]
- Antenatal Corticosteroid Clinical Practice Guidelines Panel . (2015). Antenatal corticosteroids given to women prior to birth to improve fetal, infant, child and adult health: Clinical Practice Guidelines. Liggins Institute, The University of Auckland, Auckland, New Zealand. [Google Scholar]
- Avery ME & Mead J (1959). Surface properties in relation to atelectasis and hyaline membrane disease. AMA J Dis Child 97, 517–523. [DOI] [PubMed] [Google Scholar]
- Benizri E, Ginouves A & Berra E (2008). The magic of the hypoxia‐signaling cascade. Cell Mol Life Sci 65, 1133–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botting KJ, McMillen IC, Forbes H, Nyengaard JR & Morrison JL (2014). Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia‐responsive genes. J Am Heart Assoc 3, e000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braems GA, Yao LJ, Inchley K, Brickenden A, Han VKM, Grolla A, Challis JRG & Possmayer F (2000). Ovine surfactant protein cDNAs: use in studies on fetal lung growth and maturation after prolonged hypoxemia. Am J Physiol Lung Cell Mol Physiol 278, L754–L764. [DOI] [PubMed] [Google Scholar]
- Brain KL, Allison BJ, Youguo N, Cross C, Itani N, Kane A, Herrera EA & Giussani D (2015). Induction of controlled hypoxic pregnancy in large mammalian species. Physiol Rep 3, e12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briana DD & Malamitsi‐Puchner A (2013). Small for gestational age birth weight: impact on lung structure and function. Paediatr Respir Rev 14, 256–262. [DOI] [PubMed] [Google Scholar]
- Bruick RK & McKnight SL (2001). A conserved family of prolyl‐4‐hydroxylases that modify HIF. Science 294, 1337–1340. [DOI] [PubMed] [Google Scholar]
- Camm EJ, Hansell JA, Kane AD, Herrera EA, Lewis C, Wong S, Morrell NW & Giussani DA (2010). Partial contributions of developmental hypoxia and undernutrition to prenatal alterations in somatic growth and cardiovascular structure and function. Am J Obstet Gynecol 203, e424–495. [DOI] [PubMed] [Google Scholar]
- Cock ML, Albuquerque CA, Joyce BJ, Hooper SB & Harding R (2001). Effects of intrauterine growth restriction on lung liquid dynamics and lung development in fetal sheep. Am J Obstet Gynecol 184, 209–216. [DOI] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E & Lupu F (2002). Loss of HIF‐2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8, 702–710. [DOI] [PubMed] [Google Scholar]
- Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM & Sznajder JI (2003). Hypoxia‐induced endocytosis of Na, K‐ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC‐ζ. J Clin Invest 111, 1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel M, Gunnarsson GO & Gudmundsson S (2002). Blood redistribution in the fetal brain during chronic hypoxia. Ultrasound Obstet Gynecol 20, 117–121. [DOI] [PubMed] [Google Scholar]
- Economides DL, Nicolaides KH & Campbell S (1991). Metabolic and endocrine findings in appropriate and small for gestational age fetuses. J Perinatal Med 19, 97–105. [DOI] [PubMed] [Google Scholar]
- Flecknoe S, Wallace M, Cock M, Harding R & Hooper S (2003). Changes in alveolar epithelial cell proportions during fetal and postnatal development in sheep. Am J Physiol Lung Cell Mol Physiol 285, L664–L670. [DOI] [PubMed] [Google Scholar]
- Gagnon R, Langridge J, Inchley K, Murotsuki J & Possmayer F (1999). Changes in surfactant‐associated protein mRNA profile in growth‐restricted fetal sheep. Am J Physiol Lung Cell Mol Physiol 276, L459–L465. [DOI] [PubMed] [Google Scholar]
- Ginouvès A, Ilc K, Macías N, Pouysségur J & Berra E (2008). PHDs overactivation during chronic hypoxia “desensitizes” HIF‐α and protects cells from necrosis. Proc Natl Acad Sci USA 105, 4745–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani D & Davidge S (2013). Developmental programming of cardiovascular disease by prenatal hypoxia. J Dev Orig Health Dis 4, 328–337. [DOI] [PubMed] [Google Scholar]
- Giussani DA ( 2016). The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol 594, 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, Blake EZ, Horder KA, Thakor AS & Hansell JA (2012). Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One 7, e31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenland S, Senn S, Rothman K, Carlin J, Poole C, Goodman S & Altman D (2016). Statistical tests, P values, confidence intervals, and power: a guide to misinterpretations. Eur J Epidemiol 31, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey LM, Gilbert RD, Longo LD & Ducsay CA (1993). Changes in ovine fetal adrenocortical responsiveness after long‐term hypoxemia. Am J Physiol Endocrinol Metab 264, E741–E747. [DOI] [PubMed] [Google Scholar]
- Hecher K, Snijders R, Campbell S & Nicolaides K (1995). Fetal venous, intracardiac, and arterial blood flow measurements in intrauterine growth retardation: relationship with fetal blood gases. Am J Obstet Gynecol 173, 10–15. [DOI] [PubMed] [Google Scholar]
- Heidbreder M, Fröhlich F, Jöhren O, Dendorfer A, Qadri F & Dominiak P (2003). Hypoxia rapidly activates HIF‐3α mRNA expression. FASEB J 17, 1541–1543. [DOI] [PubMed] [Google Scholar]
- Herrera EA, Krause B, Ebensperger G, Reyes RV, Casanello P, Parra‐Cordero M & Llanos AJ (2014). The placental pursuit for an adequate oxidant balance between the mother and the fetus. Front Pharmacol 5, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera EA, Rojas RT, Krause BJ, Ebensperger G, Reyes RV, Giussani DA, Parer JT & Llanos AJ (2016). Cardiovascular function in term fetal sheep conceived, gestated and studied in the hypobaric hypoxia of the Andean altiplano. J Physiol 594, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SB, Polglase GR & Roehr CC (2015). Cardiopulmonary changes with aeration of the newborn lung. Paediatr Respir Rev 16, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter D & Jaeggi E (2010). Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int J Pediatr 2010, 401323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesse NM, McCartney J, Feng X, Richards EM, Wood CE & Keller‐Wood M (2009). Expression of ENaC subunits, chloride channels, and aquaporins in ovine fetal lung: ontogeny of expression and effects of altered fetal cortisol concentrations. Am J Physiol Regul Integr Comp Physiol 297, R453–R461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M & Altman DG (2014). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Animals 4, 35–44. [DOI] [PubMed] [Google Scholar]
- Liu X, Lin Y, Tian B, Miao J, Xi C & Liu C (2014). Maternal protein restriction alters VEGF signaling and decreases pulmonary alveolar in fetal rats. Int J Clin Exp Pathol 7, 3101. [PMC free article] [PubMed] [Google Scholar]
- Lock MC, McGillick EV, Orgeig S, Zhang S, McMillen IC & Morrison JL (2015). Mature surfactant protein‐B expression by immunohistochemistry as a marker for surfactant system development in the fetal sheep lung. J Histochem Cytochem 63, 866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney J, Bowes G, Brodecky V, Dennett X, Wilkinson M & Walker A (1982). Function of the future respiratory system in the growth retarded fetal sheep. J Dev Physiol 4, 279–297. [PubMed] [Google Scholar]
- Maritz GS, Morley CJ & Harding R (2005). Early developmental origins of impaired lung structure and function. Early Hum Dev 81, 763–771. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S, Giussani DA & Morrison JL (2016a). Chronic hypoxaemia as a molecular regulator of fetal lung development: implications for risk of respiratory complications at birth. Paediatr Respir Rev 21, 3–10. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S, McMillen IC & Morrison JL (2013). The fetal sheep lung does not respond to cortisol infusion during the late canalicular phase of development. Physiol Rep 1, e00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S & Morrison JL (2015). Structural and molecular regulation of lung maturation by intratracheal VEGF administration in the normally grown and placentally restricted fetus. J Physiol 594, 1399–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S & Morrison JL (2016b). Regulation of lung maturation by prolyl hydroxylase domain (PHD) inhibition in the lung of the normally grown and placentally restricted fetus in late gestation. Am J Physiol Regul Integr Comp Physiol 310, R1226–R1243. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S, Williams MT & Morrison JL (2016c). Risk of respiratory distress syndrome and efficacy of glucocorticoids: are they the same in the normally grown and growth restricted infant? Reprod Sci 23, 1459–1472. [DOI] [PubMed] [Google Scholar]
- McMillen IC, Adams MB, Ross JT, Coulter CL, Simonetta G, Owens JA, Robinson JS & Edwards LJ (2001). Fetal growth restriction: adaptations and consequences. Reproduction 122, 195–204. [DOI] [PubMed] [Google Scholar]
- McMillen IC & Robinson JS (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85, 571–633. [DOI] [PubMed] [Google Scholar]
- Morrison JL (2008). Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35, 730–743. [DOI] [PubMed] [Google Scholar]
- Orgeig S, Crittenden TA, Marchant C, McMillen IC & Morrison JL (2010). Intrauterine growth restriction delays surfactant protein maturation in the sheep fetus. Am J Physiol Lung Cell Mol Physiol 298, L575–583. [DOI] [PubMed] [Google Scholar]
- Orgeig S, McGillick EV, Botting KJ, Zhang S, McMillen IC & Morrison JL (2015). Increased lung prolyl hydroxylase and decreased glucocorticoid receptor are related to decreased surfactant protein in the growth restricted sheep fetus. Am J Physiol Lung Cell Mol Physiol 309, L84–97. [DOI] [PubMed] [Google Scholar]
- Passmore M, Nataatmadja M & Fraser JF (2009). Selection of reference genes for normalisation of real‐time RT‐PCR in brain‐stem death injury in Ovis aries . BMC Mol Biol 10, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ID, Simonetta G, Owens JA, Robinson JS, Clarke IJ & McMillen IC (1996). Placental restriction alters the functional development of the pituitary‐adrenal axis in the sheep fetus during late gestation. Pediatr Res 40, 861–866. [DOI] [PubMed] [Google Scholar]
- Poudel R, McMillen IC, Dunn SL, Zhang S & Morrison JL (2015). Impact of chronic hypoxemia on blood flow to the brain, heart, and adrenal gland in the late‐gestation IUGR sheep fetus. Am J Physiol Regul Integr Comp Physiol 308, R151–R162. [DOI] [PubMed] [Google Scholar]
- Robinson J, Kingston E, Jones C & Thorburn G (1979). Studies on experimental growth retardation in sheep. The effect of removal of a endometrial caruncles on fetal size and metabolism. J Dev Physiol 1, 379–398. [PubMed] [Google Scholar]
- Rothman KJ (1990). No adjustments are needed for multiple comparisons. Epidemiology 1, 43–46. [PubMed] [Google Scholar]
- Storme L, Aubry E, Rakza T, Houeijeh A, Debarge V, Tourneux P, Deruelle P, Pennaforte T & Group FCDHS (2013). Pathophysiology of persistent pulmonary hypertension of the newborn: impact of the perinatal environment. Arch Cardiovasc Dis 106, 169–177. [DOI] [PubMed] [Google Scholar]
- Suzin J, Karowicz‐Bilińska A & Sieroszewski P (2002). Evaluation of oxidative stress indices during treatment in pregnant women with intrauterine growth retardation. Med Sci Monit 8, CR211–CR216. [PubMed] [Google Scholar]
- Van Geijn HP, Jr Kaylor W, Nicola KR & Zuspan FP (1980). Induction of severe intrauterine growth retardation in the Sprague‐Dawley rat. Am J Obstet Gynecol 137, 43–47. [DOI] [PubMed] [Google Scholar]
- Vivona ML, Matthay M, Chabaud MB, Friedlander G & Clerici C (2001). Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: reversal by β‐adrenergic agonist treatment. Am J Respir Cell Mol Biol 25, 554–561. [DOI] [PubMed] [Google Scholar]
- Walther FJ, Wade AB, Warburton D & Forman HJ (1991). Ontogeny of antioxidant enzymes in the fetal lamb lung. Exp Lung Res 17, 39–45. [DOI] [PubMed] [Google Scholar]
- Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK & Bührer C (2008). Hypoxia upregulates the histone demethylase JMJD1A via HIF‐1. Biochem Biophys Res Commun 372, 892–897. [DOI] [PubMed] [Google Scholar]
- Williams SJ, Campbell ME, McMillen IC & Davidge ST (2005). Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol Regul Integr Comp Physiol 288, R360–R367. [DOI] [PubMed] [Google Scholar]
- Yoneyama Y, Shin S, Iwasaki T, Power GG & Araki T (1994). Relationship between plasma adenosine concentration and breathing movements in growth‐retarded fetuses. Am J Obstet Gynecol 171, 701–706. [DOI] [PubMed] [Google Scholar]
- Zana‐Taieb E, Pham H, Franco‐Montoya M, Jacques S, Letourneur F, Baud O, Jarreau P & Vaiman D (2015). Impaired alveolarization and intra‐uterine growth restriction in rats: a postnatal genome‐wide analysis. J Pathol 235, 420–430. [DOI] [PubMed] [Google Scholar]