

Abstract
Key points
Leptin, is a 16 kDa pleiotropic peptide not only primarily secreted by adipocytes, but also produced by other tissues, including the heart.
Controversy exists regarding the adverse and beneficial effects of leptin on the heart
We analysed the effect of a non‐hypertensive dose of leptin on cardiac function, [Ca2+]i handling and cellular electrophysiology, which participate in the genesis of pump failure and related arrhythmias, both in control mice and in mice subjected to chronic pressure‐overload by transverse aorta constriction.
We find that leptin activates mechanisms that contribute to cardiac dysfunction under physiological conditions. However, after the establishment of pressure overload, an increase in leptin levels has protective cardiac effects with respect to rescuing the cellular heart failure phenotype.
These beneficial effects of leptin involve restoration of action potential duration via normalization of transient outward potassium current and sarcoplasmic reticulum Ca2+ content via rescue of control sarcoplasmic/endoplasmic reticulum Ca2+ ATPase levels and ryanodine receptor function modulation, leading to normalization of Ca2+ handling parameters.
Abstract
Leptin, is a 16 kDa pleiotropic peptide not only primary secreted by adipocytes, but also produced by other tissues, including the heart. Evidence indicates that leptin may have either adverse or beneficial effects on the heart. To obtain further insights, in the present study, we analysed the effect of leptin treatment on cardiac function, [Ca2+]i handling and cellular electrophysiology, which participate in the genesis of pump failure and related arrhythmias, both in control mice and in mice subjected to chronic pressure‐overload by transverse aorta constriction (TAC). Three weeks after surgery, animals received either leptin (0.36 mg kg–1 day–1) or vehicle via osmotic minipumps for 3 weeks. Echocardiographic measurements showed that, although leptin treatment was deleterious on cardiac function in sham, leptin had a cardioprotective effect following TAC. [Ca2+]i transient in cardiomyocytes followed similar pattern. Patch clamp experiments showed prolongation of action potential duration (APD) in TAC and leptin‐treated sham animals, whereas, following TAC, leptin reduced the APD towards control values. APD variations were associated with decreased transient outward potassium current and Kv4.2 and KChIP2 protein expression. TAC myocytes showed a higher incidence of triggered activities and spontaneous Ca2+ waves. These proarrhythmic manifestations, related to Ca2+/calmodulin‐dependent protein kinase II and ryanodine receptor phosphorylation, were reduced by leptin. The results of the present study demonstrate that, although leptin treatment was deleterious on cardiac function in control animals, leptin had a cardioprotective effect following TAC, normalizing cardiac function and reducing arrhythmogeneity at the cellular level.
Keywords: heart failure, intracellular calcium, leptin
Key points
Leptin, is a 16 kDa pleiotropic peptide not only primarily secreted by adipocytes, but also produced by other tissues, including the heart.
Controversy exists regarding the adverse and beneficial effects of leptin on the heart
We analysed the effect of a non‐hypertensive dose of leptin on cardiac function, [Ca2+]i handling and cellular electrophysiology, which participate in the genesis of pump failure and related arrhythmias, both in control mice and in mice subjected to chronic pressure‐overload by transverse aorta constriction.
We find that leptin activates mechanisms that contribute to cardiac dysfunction under physiological conditions. However, after the establishment of pressure overload, an increase in leptin levels has protective cardiac effects with respect to rescuing the cellular heart failure phenotype.
These beneficial effects of leptin involve restoration of action potential duration via normalization of transient outward potassium current and sarcoplasmic reticulum Ca2+ content via rescue of control sarcoplasmic/endoplasmic reticulum Ca2+ ATPase levels and ryanodine receptor function modulation, leading to normalization of Ca2+ handling parameters.
Abbreviations
- APD
action potential duration
- CVD
cardiovascular disease
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- DAD
delayed after depolarization
- EF
ejection fraction
- FS
fractional shortening
- HF
heart failure
- Ito
transient outward potassium current
- ObR
leptin receptor
- RyR2
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
- TAC
transverse aorta constriction
Introduction
Leptin is a 16 kDa adipokine that regulates food intake and energy metabolism at a central level. It is produced and secreted not only by adipose tissue, but also by other tissues, including the heart. Leptin acts via the leptin‐receptor (ObR), which is present on cells of kidney, pancreas, liver, muscle and heart, as well as the vasculature (Abel et al. 2008; Sweeney, 2010).
Obese patients usually present high circulating leptin levels that are generally consequence of a status of leptin resistance and a down‐regulation of ObRs in the hypothalamus. However, there is experimental evidence showing that leptin resistance does not occur in the cardiac tissue of obese individuals. Several studies have reported that ObRs are not down‐regulated in cardiomyocytes and that they preserve full responsiveness to leptin (Abe et al. 2007; Guzmán‐Ruiz et al. 2010; Stucchi et al. 2011; Leifheit‐Nestler et al. 2013).
Obesity is associated with the prevalence of many cardiovascular diseases (CVDs), including hypertension, coronary disease and heart failure (HF) (Schaffer, 2003). However, the link between obesity, hyperleptinaemia and the development of CVD is not completely understood. Several clinical studies (Sierra‐Johnson et al. 2007; Wannamethee et al. 2007; Liu et al. 2010), but not all (Thøgersen et al. 2004; Brennan et al. 2007), have supported a role of hyperleptinaemia as a risk marker for CVDs. By contrast, disruption of the leptin signalling pathway within the mouse heart has been reported to cause left ventricular hypertrophy (Barouch et al. 2003) and it has been proposed that hyperleptinaemia can play a protective role in HF through activation of ObR signalling in cardiomyocytes (McGaffin et al. 2008; McGaffin et al. 2009). Moreover, several studies have reported a better prognosis and lower mortality in overweight and obese patients with HF than in those patients with HF but normal body mass index (Horwich et al. 2001; Oreopoulos et al. 2008; Lavie et al. 2013).
Almost half of HF patients die from sudden cardiac death, most probably as a result of ventricular arrhythmias. Electrophysiological remodelling, with a reduced expression of potassium channels and a prolonged action potential duration (APD), shows typical features of HF that make the heart more prone to early afterdepolarizations and fatal arrhythmias. In addition, failing hearts show enhanced diastolic Ca2+ leak via cardiac ryanodine receptors (RyR2s) that can generate delayed after depolarizations (DADs) and ventricular arrhythmias. Alteration in intracellular Ca2+ handling and reduced expression of potassium channels have been proposed as mechanisms involved in the genesis of HF related arrhythmias (Nattel et al. 2007; Lehnart et al. 2009).
The aim of the present study was to assess the effect of a non‐hypertensive dose of leptin on cardiac electrical remodelling and sarcoplasmic reticulum Ca2+ leak in a mouse model with pre‐existing pathological hypertrophy and cardiac dysfunction induced by transverse aortic constriction (TAC).
The results obtained demonstrate that, in the presence of a cardiac pathology, leptin treatment has a beneficial effect by normalizing cardiac function and reducing arrhythmogenesis. The mechanisms involved included a significant increase of Kv4.2 and KChIP expression that normalized transient outward potassium current (I to) density and APD reduction and a significant reduction in frequency of Ca2+ waves as a consequence of normalization of Ser‐2814 RyR2 phosphorylation levels. Although the effect of leptin on control animals cardiac function was deleterious, it was not accompanied by increased arrhythmogenicity.
Methods
Ethical approval
All experiments on mice were performed with the approval of the Bioethical Committee of the Complutense University of Madrid, in accordance with the recommendations of the Spanish (Proex 035‐15) and European guidelines (2010/63/EU) and also in accordance to the ethical principles laid down by the French (Ministry of Agriculture) Directives for the care of laboratory animals (B9201901).
Animals
Mice were housed in cages of five and fed ad libitum. Ten‐week‐old C57Bl/6 male mice (Jackson Laboratories, Bar Harbor, ME, USA), under anaesthesia (50 mg kg−1 ketamine plus 8 mg kg−1 xylazine i.p) during the whole procedure, underwent transversal aortic constriction (TAC). The depth of anaesthesia was confirmed by toe pinch. After thoracotomy, the aortic arch was constricted with a thread (6‐0 silk suture) together with a 27‐gauge needle (diameter 0.42 mm). The needle was then removed, leaving a region of stenosis that partially reduced the vascular light. The sham group underwent the same procedure without placement of the suture. Three weeks after surgery, before the establishment of cardiovascular dysfunction (ejection fraction sham 76.41% ± 2.53 vs. TAC 77.00% ± 1.84 and fractional shortening sham 39.99% ± 2.24 vs. TAC 40.29% ± 1.74), animals were randomly divided into four groups (sham vehicle, sham leptin, TAC vehicle and TAC leptin). A micro‐osmotic pump containing either leptin (0.36 mg kg−1 day−1) or vehicle (20 mmol L−1 Tris, 150 nmol L−1 NaCl) was s.c. implanted in every mouse (sham and TAC groups). Mice were finally killed for experiments 3 weeks after osmotic pump implantation.
Drugs and devices
Leptin was purchased from Sigma‐Aldrich (St Louis, MO, USA) and the stock solution was prepared in accordance with the manufacturer's instructions. Fluo‐3 AM was purchased from Molecular Probes/Life Technologies (Eugene, OR, USA) and Collagenase type II was obtained from Worthington Biochemical Corp. (Lakewood, NJ, USA). Caffeine and other products were obtained from Sigma‐Aldrich.
Osmotic minipumps were obtained from Alzet (Cupertino, CA, USA). The 5 MHz transducer Vivid 9 for M‐mode echocardiography was obtained from General Electric Healthcare (Piscataway, NJ, USA).
We used an SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany) for Ca2+ imaging and Ca2+ image analyses were performed using IDL software (Research Systems, Inc., Boulder, CO, USA) and bespoke routines. For the whole‐cell, patch clamp method, we used an Axopatch 1D amplifier with pClamp6 (Axon Instruments, Sunnyvale, CA, USA).
Primary antibodies were: sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) (dilution 1:1000) (Thermo Fisher, Waltham, MA, USA), phospho‐phospholamban (dilution 1:5000) (Ser16 and Thr17), phospholamban (dilution 1:1000) and phospho‐RyR (dilution 1:1000) (Ser2814; Badrilla Ltd, Leeds, UK), RyR (dilution 1:1000) (Affinity Bioreagents, Golden, CO, USA), pCaMKII (Thr286) (dilution 1:1000) and CaMKII (dilution 1:1000) (Cell Signaling. Danvers, MA, USA), Kv4.2 (dilution 1:500) (EMD Millipore, Darmstadt, Germany), voltage‐gated K+ channel‐interacting protein 2 (dilution 1:1000) (KChIP2; Affinity Bioreagents), sodium–calcium exchanger (dilution 1:1000) (NCX; Swant Inc., Marly, Switzerland) and GAPDH (dilution 1:5000) (Ambion, Carlsbad, CA, USA). Specific bands were detected using the Amersham ECL protein detection system (General Electric Healthcare, Piscataway, NJ, USA) and were analysed using ImageJ (NIH, Bethesda, MD, USA).
Echocardiography
Transthoracic echocardiography was performed the day before the experiment using a 5 MHz transducer under 3% isoflurane gas anaesthesia. Two‐dimensional‐guided M‐mode echocardiography was used to determine contractile parameters such as fractional shortening (FS) or ejection fraction (EF).
Cardiomyocyte isolation
Mice were heparinized (4 IU g–1, i.p.) and anaesthetized (sodium pentobarbital, 50 mg kg−1, i.p.). The depth of anaesthesia was determined by assessing the loss of the pedal reflex by toe pinch. Immediately after phase III anaesthesia was reached, a central thoracotomy and heart excision was performed. Ventricular myocytes were isolated using a collagenase perfusion method as described previously (Gómez‐Hurtado et al. 2014; Delgado et al. 2015).
Confocal imaging
Ca2+ tolerant rod shaped myocytes were loaded with the fluorescence Ca2+ dye Fluo‐3 AM (6 μmol L−1) for 30 min. Images were obtained after at least an additional 30 min to allow for complete de‐esterification, with confocal microscopy (objective w.i. 63×) fitted with a white light laser tuned to 500 nm. Excitation was collected at > 510 nm. All experiments were performed at room temperature (23°C).
To record Ca2+ transients, Fluo‐3 loaded cells were field stimulated by two Pt electrodes at 2 Hz until steady‐state before recording. Spontaneous Ca2+ sparks and waves were obtained in quiescent cells after [Ca2+]i transients recordings. For sarcoplasmic reticulum (SR) Ca2+ load estimation, cardiac myocytes were rapidly perfused with 10 mmol L−1 caffeine right after field‐stimulation to empty SR. Because [Ca2+]i reached during caffeine experiments may fall in the non‐linear part of the fluorescence‐Ca2+ curve of Fluo‐3, significant changes in [Ca2+]i may provide small differences in fluorescence. Thus, we calculated [Ca2+]i levels according to the formula and diastolic [Ca2+]i reported previously (Gómez et al. 1996), as well as the Fluo‐3 AM Kd values experimentally measured in cardiomyocytes (Loughrey et al. 2003).
Electrophysiological measurements
APs and K+ currents were recorded using a whole‐cell, patch clamp method with solutions and protocols as described previously (Ouvrard‐Pascaud et al. 2005). Trigger activity was defined as the number of spontaneous AP recorded as a result of DADs sufficiently large to reach the threshold potential for activation of a regenerative inward current.
Western blotting
Cardiac tissue was homogenized and centrifuged at 50 g for 1 min at 4°C to eliminate cellular debris. The supernatants were used for immunoblotting. The extracted proteins (15–30 μg) were separated on SDS‐PAGE gels and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% milk and incubated overnight with primary antibodies.
Enzyme‐linked immunosorbent assay
To analyse leptin concentration in heart homogenates, we used a leptin mouse ELISA Kit KMC2281 Novex® (Life Technologies) in accordance with the manufacturer's instructions.
Statistical analysis
Data are presented as the mean ± SEM. Statistical analysis was performed with Prism, version 5.0 (GraphPad, La Jolla, CA, USA). Gaussian distribution of the samples was assessed by a D'Agostino‐Pearson test. Statistical significance was evaluated by one‐way ANOVA followed by a Bonferroni post hoc test in those samples that followed a normal distribution or by a Kruskal–Wallis test followed by Dunn's post hoc test in those samples that did not follow a normal distribution. For statistical comparison of abnormal diastolic Ca2+ release and trigger activity occurrence, the chi‐squared test was used as reported previously (Ruiz‐Hurtado et al. 2012) and the data are presented as a percentage. P < 0.05 was considered statistically significant.
Results
Chronic leptin treatment slightly impairs cardiac function in healthy mice, but prevents cardiac dysfunction in mice subjected to pressure overload
Analysis of cardiac leptin levels did not show significant differences between sham and TAC mice (Table 1). After 3 weeks of treatment, we observed a 25% increase in cardiac leptin levels either in sham or TAC animals. Neither TAC, nor chronic leptin treatment modified mice body weight (BW) or tibia length (TL); however, TAC induced an increase in heart weight normalized by BW or by TL. This cardiac hypertrophy induced by TAC was reduced by 3 weeks of leptin treatment not only at the whole organ level, but also at the cardiomyocyte level, as confirmed by membrane capacitance measurements (Table 1). Although leptin treatment in sham mice impaired cardiac contractility, it improved cardiac function in TAC animals, as measured by M‐mode echocardiography (Fig. 1 A). As shown in Fig. 1 B and C, on average, 3 weeks of treatment with leptin (sham leptin) slightly but significant reduced EF and FS, although by less amplitude than TAC. When treated for 3 weeks with leptin, TAC mice showed improved EF and FS compared to those TAC mice treated only with vehicle.
Table 1.
Macroscopic parameters of mice, cell capacitances and cardiac leptin levels
| Sham vehicle | Sham leptin | TAC vehicle | TAC leptin | |
|---|---|---|---|---|
| Body weight (g) | 28.72 ± 2.75 | 27.65 ± 2.44 | 27.99 ± 0.58 | 26.69 ± 1.70 |
| Tibia length (mm) | 22.19 ± 0.12 | 22.0 ± 0.11 | 21.86 ± 0.04 | 21.63 ± 0.09 |
| Heart weight/body weight (mg g–1) | 7.84 ± 0.45 | 7.91 ± 0.39 | 10.23 ± 0.69* | 8.78 ± 0.60† |
| Heart weight/tibia length (mg mm–1) | 10.10 ± 2.35 | 9.89 ± 2.16 | 13.11 ± 2.85* | 11.06 ± 1.98† |
| Leptin concentration in heart | ||||
| homogenates (pg μg–1 protein) | 376.3 ± 15.39 | 477.8 ± 4.95* | 412.5 ± 37.55 | 463.4 ± 29.53* |
| Membrane capacitance (pF) | 182.4 ± 7.5 | 187.2 ± 12.6 | 220.4 ± 9.9*** | 183.4 ± 9.2††† |
Data are expressed as the mean ± SEM (* P < 0.05 and *** P < 0.001 vs. sham vehicle; † P < 0.05 and ††† P < 0.001 vs. TAC vehicle; N = 15, n = 33 sham vehicle; N = 14, n = 23 sham leptin; N = 7, n = 34 TAC vehicle; N = 8, n = 34 TAC leptin). N, number of animals; n, number of cells.
Figure 1. Chronic leptin treatment impairs cardiac function in control but improves cardiac outcomes after TAC mice.
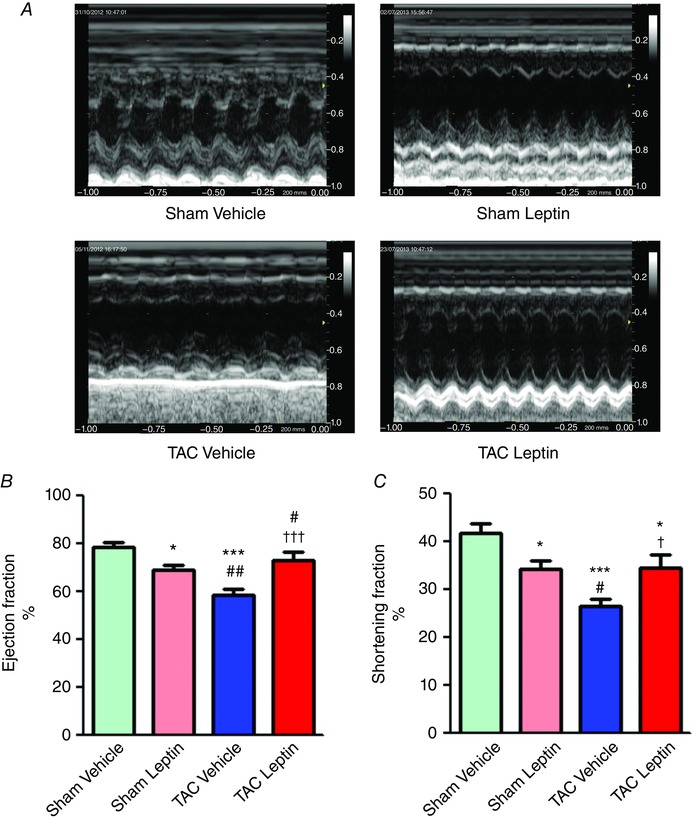
A, representative M‐mode echocardiographic images of sham and TAC mice treated either with vehicle or with leptin. B and C, average ejection fraction (B) and shortening fraction (C), measured by M‐mode echocardiography. Data are the mean ± SEM (n = 6–8 mice). * P < 0.05 and *** P < 0.001 vs. sham vehicle; † P < 0.05 and ††† P < 0.001 vs. TAC vehicle; # P < 0.05 and ## P < 0.01 vs. sham leptin.
Leptin treatment after TAC rescued [Ca2+]i transients and contractile function in cardiomyocytes
Cardiac pump function is dependent on cardiomyocyte contraction, which is activated by Ca2+. To obtain an insight into the cellular mechanism responsible for the modification of cardiac function, we investigated whether leptin modulated [Ca2+]i transients in isolated cardiomyocytes. Figure 2 A shows representative line‐scan images recorded from ventricular myocytes isolated from mice from all four experimental groups. Consistent with the reduced cardiac function observed by echocardiography, 3 weeks of leptin treatment reduced [Ca2+]i transient amplitude (Fig. 2 B) and cell shortening (Fig. 2 C). By contrast, in mice subjected to TAC, where [Ca2+]i transient amplitude and contraction were reduced compared to sham‐operated animals (consistent with failing function), leptin was able to restore [Ca2+]i transient amplitude and cell shortening to levels similar to those observed in control group. The SR Ca2+ content could be responsible for the [Ca2+]i transient amplitude reduction. Indeed, the SR Ca2+ load was reduced in cells isolated from TAC compared to sham animals, as is characteristic of HF. Three weeks of leptin treatment also depressed SR Ca2+ load, although the reduction was smaller than that induced by TAC. The SR Ca2+ load in cells isolated from TAC animals receiving leptin was partially restored (Fig. 2 D and E). As shown in Fig. 2 F, leptin treatment after TAC prevented SERCA2 down‐regulation induced by TAC hearts. This can contribute to restore SR Ca2+ content and [Ca2+]i transient amplitude. Of note, we did not observed changes either in total expression or phosphorylation (in either residue Ser16 or Thr17) of PLB (Fig. 2 G and H).
Figure 2. Leptin treatment depresses cellular [Ca2+]i transients and contraction in sham but restores it in TAC cardiomyocytes.
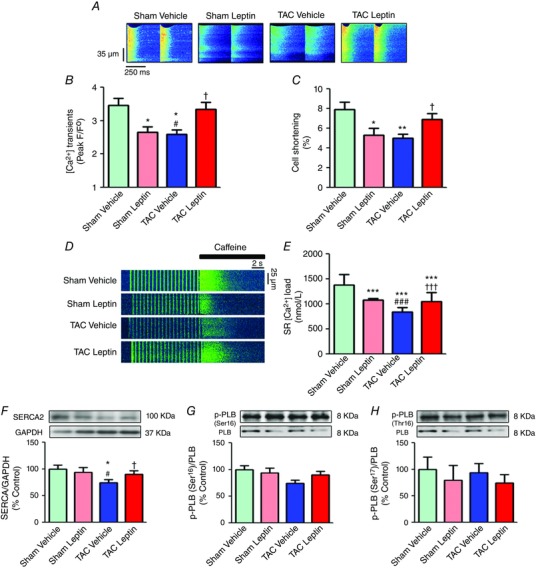
A, representative line‐scan confocal images of [Ca2+]i transients obtained in Fluo‐3 loaded cardiomyocytes from sham and TAC mice treated either with vehicle or with leptin. B, average [Ca2+]i transient amplitude measured as peak F/F 0, where F is the fluorescence trace and F 0 is the fluorescence in the diastolic period in each experimental group. C, cellular shortening expressed in percentage of cell length in each experimental group. D, representative line‐scan images of cardiac myocytes from each experimental group during field stimulation at 2 Hz and rapid 10 mmol L−1 caffeine application. E, average SR content. F, representative immunoblots (top) and average ratio of protein levels (bottom) expressed as a percentage of SERCA normalized by GAPDH. G, representative immunoblots (top) and average ratio of protein levels (bottom) expressed as a percentage of p‐PLB (Ser16) of the total PLB and (H) p‐PLB (Thr17) normalized by the total PLB. Data are the mean ± SEM [n = 4–6 mice; n = 50–70 cells in (B) and (C) and n = 20–40 cells in (D)]. * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. sham vehicle; † P < 0.05 and ††† P < 0.001 vs. TAC vehicle; # P < 0.05 and ### P < 0.001 vs. sham leptin.
Leptin rescued electrophysiological alterations in mice that underwent TAC
Ca2+ release from the SR, which induces contraction, is activated during AP. Cardiac hypertrophy is accompanied by a prolongation of AP duration, mainly as a result of a reduction in the I to (Beuckelmann et al. 1993; Ravens & Cerbai, 2008). To test whether leptin had a modulator effect with respect to these parameters, we carried out patch clamp experiments to measure APs and K+ currents. Figure 3 A shows representative traces of APs recorded in different cardiomyocytes isolated from sham and TAC mice treated either with vehicle or with leptin. Leptin treatment by itself increased APD measured at 20% (APD20) (sham vehicle 1.62 ± 0.13 ms vs. sham leptin 2.28 ± 0.27 ms), 50% (APD50) (sham vehicle 5.12 ± 0.51 ms vs. sham leptin 7.98 ± 1.12 ms) and 90% (APD90) of repolarization (sham vehicle 73.18 ± 8.76 ms vs. sham leptin 107.3 ± 14.58 ms) (Fig. 3 B). Following TAC, cells isolated from vehicle‐treated mice showed the classical hypertrophic induced prolongation of APD measured at 20% (TAC vehicle 3.44 ± 0.85 ms), 50% (TAC vehicle 11.85 ± 1.99 ms) and 90% of repolarization (TAC vehicle 189.12 ± 41.72 ms) (Fig. 3 B). By contrast, leptin treatment shortened the APD in TAC cardiomyocytes (Fig. 3 B) at 20% (TAC leptin 1.69 ± 0.14 ms), 50% (TAC leptin 7.23 ± 0.85 ms) and 90% of repolarization (TAC leptin 148.28 ± 30.03 ms).
Figure 3. Leptin treatment prolonged APD in control mice but reverted it in mice that underwent TAC.

A, representative AP recorded in cardiomyocytes from sham (left) and TAC (right) mice treated either with vehicle or with leptin. B, average APD measured at 20% (left), 50% (middle) and 90% (right) of the repolarization. Data are the mean ± SEM (n = 4–6 mice; n = 18–25 cells). *** P < 0.001 vs. sham vehicle; ††† P < 0.001 vs. TAC vehicle; ### P < 0.001 vs. sham leptin.
To further analyse the mechanisms involved in APD alteration by leptin and/or TAC, we measured the main K+ currents responsible for the AP repolarization phase. In the adult mouse ventricle, outward K+ currents involved in cardiac repolarization consist of three components distinguishable by their specific time and voltage dependency and sensitivity to pharmacological agents: a transient current (I to), a low‐concentration 4‐aminopyridine–sensitive current (I Kur) and a non‐inactivating steady‐state component (I SS) (Brouillette et al. 2004). Three weeks of leptin treatment of sham‐operated mice reduced I to densities (Fig. 4 A, left) without modifying I Kur (Fig. 4 B, left) or I ss (Fig. 4 C, left) densities. In mice subjected to TAC and treated with vehicle (Figures 4 A–C, right), densities of I to and I Kur (but not I ss) were reduced compared to sham cells. Leptin treatment reversed I to density downregulation to levels similar to those observed in control mice (Fig. 4 A, right) but did not modify the reduction of I Kur densities (Fig. 4 B, right). These results are consistent with the AP waveforms (Fig. 3). The decrease in I to density could be the result of a depression in channel expression. We analysed protein expression of the main α subunit that encodes the I to channel in mice (Kv4.2) and also the auxiliary subunit KChIP2 (Patel & Campbell, 2005). Figures 4 D and E show that Kv4.2 and KChIP2 protein expression were reduced in those groups presenting cardiac dysfunction (sham leptin and TAC vehicle) compared to sham vehicle. In TAC hearts, leptin treatment normalized Kv4.2 and KChIP2 protein expression.
Figure 4. TAC‐induced alterations of K+ currents and Kv4.2 and KChIP2 expression are rescued by Leptin.
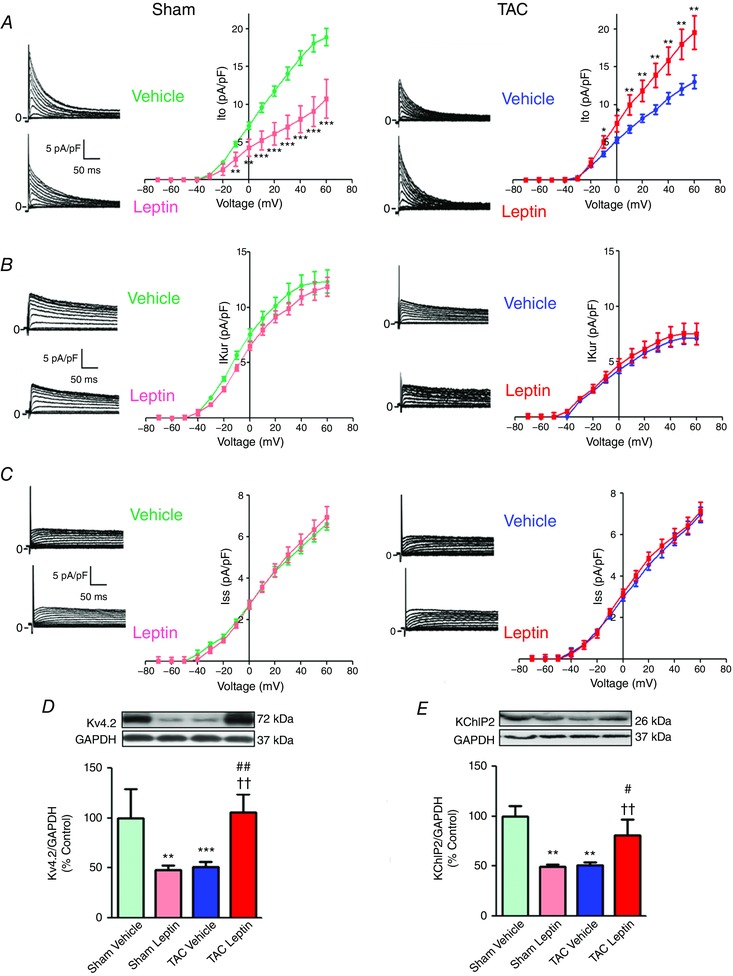
Representative current traces (left) and I–V relationships (right) of I to (A), I Kur (B) and I ss (C), recorded in isolated cardiomyocytes from sham (left) and TAC (right) mice treated either with vehicle or with leptin. The current density and times scales are the same for sham and TAC groups. Data, normalized to cell capacitance, are presented as the mean ± SEM (n = 4–6 mice; n = 10–24 cells). * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. vehicle treated group. For statistical comparisons between sham and TAC, see text. Representative immunoblots (top) and average protein levels normalized by GAPDH and expressed as a percentage of sham vehicle (bottom) for Kv4.2 (D) and KChIP2 (E) in cardiac tissue from sham and TAC mice treated either with vehicle or with leptin. Data are the mean ± SEM (n = 3 or 4 mice). ** P < 0.01 and *** P < 0.001 vs. sham vehicle; †† P < 0.01 vs. TAC vehicle; # P < 0.05 and ## P < 0.01 vs. sham leptin.
Leptin treatment prevents arrhythmic manifestations in cardiac myocytes isolated from TAC mice by reverting CaMKII RyR2 phosphorylation
During AP recordings, we observed oscillations in membrane potential or DADs following completion of the driven AP, which eventually, triggered spontaneous AP (Fig. 5 A). The occurrence of these triggered activities (TA) was greatly enhanced by TAC (Fig. 5 B) and reduced by leptin treatment after TAC. DADs are commonly initiated by non‐electrically driven, propagating spontaneous Ca2+ release from the SR via the RyR2s, termed Ca2+ waves (Pogwizd & Bers, 2004). Consistently, spontaneous Ca2+ wave occurrence (Fig. 5 C and D) was prominent in the TAC group. Although leptin did not promote any effect on Ca2+ wave occurrence in healthy cells, it was able to reduce the occurrence of Ca2+ waves in cardiomyocytes isolated from TAC animals to control levels (Fig. 5 D). In addition, we found that NCX expression was increased in all experimental groups compared to the sham vehicle group (Fig. 5 E). It is widely reported that NCX expression is elevated in hypertrophy and HF, both in humans and animal models (Despa et al. 2002; Pieske et al. 2002; Baartscheer et al. 2003; Louch et al. 2010). The fact that leptin treatment of TAC animals did not prevent this increase tends to rule out a direct NCX involvement in leptin anti‐arrhythmogenic effects.
Figure 5. Leptin treatment reduced the occurrence of trigger activities and spontaneous Ca2+ waves induced by TAC.

A, representative example of DAD and spontaneous AP (TA, triggered activity) recorded in ventricular cardiomyocytes isolated from TAC mouse. B, percentage of cells eliciting TA (n = 4–6 mice; n = 19–28 cells). The number of cells with TA/total analysed cells is noted above each bar. C, representative example of spontaneous Ca2+ wave recorded in TAC cardiomyocyte at rest. D, percentage of cardiomyocytes presenting Ca2+ waves (n = 4–6 mice; n = 46–67 cells). The number of cells with Ca2+ waves/total analysed cells is noted above each bar. E, representative immunoblots (top) and average ratio of protein levels (bottom) expressed as a percentage of NCX normalized by GAPDH. Data are presented as a percentage. ** P < 0.01 and *** P < 0.001 vs. sham vehicle; ††† P < 0.001 vs. TAC vehicle; ### P < 0.001 vs. sham leptin.
Enhanced Ca2+ wave occurrence suggests leaky RyR2 channels (Zima et al. 2010). Several studies (Bers, 2002; Ruiz‐Hurtado et al. 2012) previously reported that an increase in CaMKII activation (measurable by CaMKII autophosphorylation at Thr286) and subsequent RyR2 phosphorylation at the CaMKII site (Ser2814) are involved in this effect. To test the involvement of this mechanism in the observed effects, we first analysed the CaMKII autophosphorylation in our four experimental groups. Figure 6 A shows that CaMKII was more activated in TAC mice treated with vehicle than in sham mice, and that leptin treatment was able to reduce this effect on TAC mice, without significantly altering CaMKII activation in sham‐operated animals. Consequently, when we analysed RyR2 phosphorylation, we observed an increase of Ser2814 phosphorylation levels in TAC mice, which was significantly prevented by leptin treatment (Fig. 6 B). We then examined RyR2s activity in situ by visualizing spontaneous Ca2+ sparks. Figure 6 C shows examples of Ca2+ sparks recorded in isolated cardiomyocytes obtained from sham and TAC mice treated either with vehicle or with leptin. Neither TAC, nor leptin treatment significantly modified Ca2+ spark frequency (Fig. 6 D). However, Ca2+ spark amplitude was significantly higher in cells isolated from TAC animals and this effect was blunted by leptin treatment (Fig. 6 E), consistent with RyR S2814 phosphorylation levels (Fig. 6 B). Because RyR2 activity and hence Ca2+ spark occurrence also depends on SR Ca2+ load, we normalized Ca2+ spark frequency by the total amount of Ca2+ stored in the SR. Figure 6 F shows that cardiac myocytes isolated from TAC animals presented significantly more Ca2+ sparks for a given SR Ca2+ load, consistent with RyR2 phosphorylation. Leptin treatment did not alter normalized Ca2+ spark frequency in the sham group but significantly reduced it in the TAC group. Ca2+ waves originate from spontaneous openings of RyR2s as Ca2+ sparks, which then propagate to neighbouring RyR2s clusters. Thus, the spread in time and space of Ca2+ sparks is important with respect to the probability of a Ca2+ spark initiating a Ca2+ wave. Figure 7 shows that leptin treatment diminished Ca2+ spark duration and width in both experimental groups, thus limiting Ca2+ spread and probability to activating an arrhythmogenic Ca2+ wave.
Figure 6. Chronic leptin treatment reduces the TAC‐induced CaMKII activation and RyR2 phosphorylation.

A and B, representative immunoblots (top) and average ratio of protein levels (bottom), expressed as a percentage of p‐CaMKII (Thr286) of the total CaMKII in (A) and p‐RyR2 (Ser2814) normalized by the total RyR2 (B) in hearts (n = 3 or 4 mice) from sham and TAC mice treated either with vehicle or with leptin. C, representative line‐scan images presenting Ca2+ sparks recorded in cardiomyocytes from sham (left two panels) and TAC (right two panels) mice treated either with vehicle or with leptin, as indicated in the labels below each image. Average Ca2+ spark frequency per second and per 100 μm (D), their amplitude as peak F/F 0 (E) and Ca2+ spark frequency per second and per 100 μm normalized to SR load measured as F/F 0 (F) in each group studied (n = 4–6 mice; n = 50–70 cells). Data are the mean ± SEM. * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. sham vehicle; † P < 0.05 and ††† P < 0.001 vs. TAC vehicle; # P < 0.05 and ### P < 0.001 vs. sham leptin.
Figure 7. Ca2+ spark duration and width.
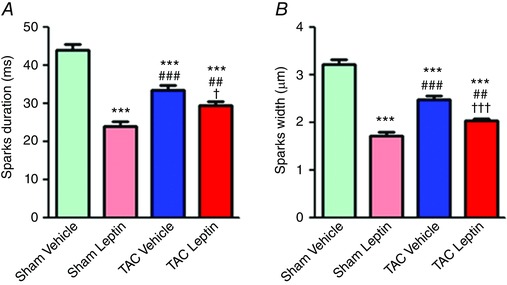
Average Ca2+ spark duration (A) and width (B) in each studied group (N = 4–6 mice; n = 50–70 cells). Data are presented as the mean ± SEM. *** P < 0.001 vs. sham vehicle; † P < 0.05 and ††† P < 0.001 vs. TAC vehicle; ## P < 0.01 and ### P < 0.001 vs. sham leptin.
Discussion
In the last two decades, several studies have determined that plasma leptin levels oscillate in the range 5–15 ng ml−1 in normal weight individuals (Sinha et al. 1996; Ahrén et al. 1997), whereas these levels are significantly increased in obese patients, reaching values in the range 15–500 ng ml−1 (Considine et al. 1996; Soliman et al. 2002).
Moreover, obesity [body mass index (BMI) ≥ 30 kg m–2)] is associated with altered cardiac function. In a follow‐up study conducted over 14 years with 5881 patients from the Framingham study, a gradual increase in HF risk related to an increase in BMI was reported (Kenchaiah et al. 2002). However, it is important to note that, within HF patients, elevated BMI is not a risk factor for worse outcomes (Lavie et al. 2013). This discrepancy between an increased CVD risk associated with obesity and better outcomes of CVD found in obese patients has led to the obesity paradox theory. In the present study,we tested the hypothesis that leptin could have a deleterious or protective effect in cardiac function depending on the previous existence of a cardiac pathology.
Accordingly, we produced TAC in mice, which is characterized by left ventricular hypertrophy development 3 weeks after intervention, whereas, after 6 weeks, systolic dysfunction also occurs (Nakamura et al. 2001). The echocardiographic results confirmed reduced cardiac EF and FS 6 weeks after surgery, validating the model. Leptin treatment in sham‐operated animals induced a smaller but significant depression in cardiac function, consistent with in vitro studies showing impaired cardiomyocyte contractility by leptin (Dong et al. 2006). It is important to note that, after 3 weeks of leptin treatment (0.36 mg kg–1 day–1), we observed neither left ventricular hypertrophy in our mice, nor an increase in the membrane capacitance of the cardiomyocytes isolated from the mice. Although the effects of leptin on cardiac hypertrophy remain controversial, there are previous studies supporting pro‐hypertrophic effects of leptin on isolated neonatal cardiomyocytes (Rajapurohitam et al. 2003; Tajmir et al. 2004; Xu et al. 2004; Madani et al. 2006; Gan et al. 2013). However, our data are consistent with a previous study by Abe et al. (2007) who did not find any differences in left ventricular mass after 4 weeks of chronic leptin infusion with a non‐hypertensive dose (0.32 mg kg–1 day–1). Moreover, it was previously shown that electrical remodelling precedes cellular hypertrophy after myocardial damage (Perrier et al. 2004). Strikingly, contractile function evaluated by M‐mode echocardiography was improved by leptin treatment after TAC, revealing a beneficial effect of leptin in the stressed heart. [Ca2+]i transient analysis of isolated cardiomyocytes obtained from these mice was consistent with the echocardiographic outcomes. Taken together, these results support the suggestion that a rise in leptin levels induces deleterious effects under physiological conditions but may be cardioprotective in pathological situations. Leptin signalling has been shown to have an important role in maintaining ATP formation because knockout of leptin in stressed mice is lethal (Hall et al. 2012). Thus, in the setting of HF, leptin may help, amongst other plausible mechanisms, to refill the SR with Ca2+, providing ATP to the SERCA pump or by increasing SERCA expression levels, which are known to be reduced in HF (Hobai & O'Rourke, 2001). Indeed, HF is characterized by metabolic alterations that contribute to contractile abnormalities. ATP content is decreased and the main energy source is shifted from fatty acid utilization towards glucose in hypertrophy and HF (Sorokina et al. 2007; Ingwall, 2009). There is experimental evidence to support the idea that leptin shifts myocardial metabolism towards fatty acid utilization (Atkinson et al. 2002), which could be beneficial in the setting of HF. Indeed, 3 weeks of leptin treatment after TAC helped preserve the intracellular Ca2+ stores and restored the SERCA levels that were decreased in TAC mice.
Increased arrhythmia susceptibility is a hallmark of HF (Tomaselli & Marbán, 1999). It is well known that alterations in APD as a result of reduced repolarizing K+ currents lead to longer cardiac AP or a long‐QT interval and predispose patients to ventricular arrhythmias (Ravens & Cerbai, 2008). Therefore, patients with impaired K+ channels function are at increased risk for sudden cardiac death. In this regard, it is also well documented that changes in I to function are one of the most important electrophysiological abnormalities related to the left ventricular hypertrophy and HF (Benitah et al. 1993; Beuckelmann et al. 1993; Gómez et al. 1997). Thus, we analysed the electrophysiological remodelling that occurs in our model. We found that leptin under physiological conditions moderately increases APD. Interestingly, in TAC mice, where we observed a marked increase of APDs, leptin treatment completely rescued the APD20 prolongation induced by TAC, and significantly shortened the prolongation at APD50 and APD90. In rodents, the I to current has a major contribution to phase 1 of ventricular repolarization and APD. Therefore, we hypothesize that the effects of leptin in APD could be mediated by the ability of leptin to modulate K+ currents. We found that leptin itself reduced I to densities without modifying I Kur and I ss, which is consistent with APD increases, whereas TAC reduced both I to and I Kur. The AP ‘plateau phase’ is mainly Ca2+‐mediated but, in rodents, as a result of a high prevalence of multiple K+ currents, there is no clear plateau phase and repolarization is rapid (Nerbonne & Kass, 2005). When K+ currents are decreased (not only I to, but also I Kur), this plateau phase become apparent, as observed after TAC. Even if leptin in the TAC model blunted I to reduction, this is not associated with the prevention of I Kur reduction and the plateau phase is still apparent. This is emphasized by the fact that, in sham, leptin treatment induced a reduction of I to without any alteration in I Kur and no obvious appearance of the plateau phase. Nonetheless, we cannot exclude the possibility that another ionic conductance might be involved, notably Ca2+ currents, although leptin did not alter this current in sham‐operated mice (data not shown). However, the most important finding was that I to density was restored by leptin in TAC mice to levels similar to those observed in control mice via a mechanism involving the normalization of protein expression levels of Kv4.2 and KChIP2. Our results are consistent with a previous in vitro study in which leptin induced an increase of I to current and protein expression on 48 h cultured ventricular cardiomyocytes (Gómez‐Hurtado et al. 2014), supporting the effect of leptin with respect to modulating K+ channel expression.
It is also well established that spontaneous Ca2+ release during diastole can trigger ventricular arrhythmias. When spontaneous SR Ca2+ release occurs and Ca2+ signalling propagates along the myocyte as Ca2+ waves, the Na+/Ca2+ exchanger is activated and is able to generate transient inward currents that result in DADs. If DADs are sufficiently large, they may turn into spontaneous APs and initiate an arrhythmia (Sedej et al. 2010; Galimberti & Knollmann, 2011). In our model, we found a higher occurrence of arrhythmogenic Ca2+ waves only in TAC group, which was related to CaMKII activation and RyR2 hyperphosphorylation by CaMKII. Leptin by itself did not activate CaMKII and decreased Ca2+ spark amplitude without any alteration in frequency. Thus although leptin‐treated sham animals showed cardiac dysfunction, leptin did not enhance arrhythmogenic Ca2+ waves. In TAC animals, however, RyR2 hyperphosphorylation at the CaMKII site, as reported previously (Ruiz‐Hurtado et al. 2012; Fischer et al. 2013; Bers, 2014; Fischer et al. 2014), increased the probability of the opening of RyR2s, as manifested by the enhanced Ca2+ spark frequency relative to the SR Ca2+ load, which contributes to a higher propensity to present Ca2+ waves. This was also normalized by leptin treatment, which prevented CaMKII hyperactivation. CaMKII regulation involves a number of different processes and a number of phosphatases regulated both directly and indirectly by Ca2+. Among these multiple mechanisms, it has been reported that CaMKII autophosphorylation is sensitive to the duration, magnitude and frequency of the calcium transients (De Koninck & Schulman, 1998) and there is also evidence that increased APD enhanced CaMKII activity (Grueter et al. 2007). In this regard, the prevention of increased APD by leptin treatment might contribute to the normalization of CaMKII phosphorylation. On the other hand, CaMKII can also be activated by ROS (Erickson et al. 2008; Curran et al. 2014). In this regard, leptin activation of AMPK (Hardie, 2003) and the subsequent increased expression of the metabolic regulator PGC‐1α (Jäger et al. 2007) might result in increased mitochondrial biogenesis, as well as improved mitochondrial function and oxygen consumption, and therefore reduced ROS production (Lehman et al. 2000).
Altogether, the results of the present study show that leptin activates mechanisms contributing to cardiac dysfunction under physiological conditions. However after the establishment of pressure overload, an increase in leptin levels may have protective cardiac effects, rescuing the cellular HF phenotype. These beneficial effects of leptin involve restoration of APD via I to upregulation and SR Ca2+ content via rescue of control SERCA levels and RyR2 function modulation, leading to normalization of Ca2+ handling parameters. We acknowledge that more in depth studies are needed to fully understand the effects of leptin in the human heart, especially those regarding I to currents regulation, because the main α subunit for I to differs in rodent (Kv4.2) and human (Kv4.3) hearts, although our data indicate leptin being a good candidate involved in the pathophysiological mechanisms related to the obesity paradox (Romero‐Corral et al. 2006; Oreopoulos et al. 2008; Rider et al. 2011; Littnerova et al. 2015; Zamora et al. 2016). In this regard, recent studies (Wannamethee et al. 2014; Mishra et al. 2015) have shown that HF patients with high circulating leptin levels were at a lower risk of mortality from cardiovascular disease, supporting a protective role of leptin once the pathology is established. Although not quite completely understood, this phenomenon has been related to adipose tissue function, and low leptin levels in HF patients have been proposed to reflect a loss of adipose tissue as a result of cachexia, which is associated with reduced survival (Filippatos et al. 2000; Wannamethee et al. 2014). However, our data show that direct effects of leptin on ion channels and Ca2+ handling related proteins could be also involved in the more favourable clinical outcomes observed in overweight and obese HF patients.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
NG‐H, AMG, JPB and CD conceived and designed the work. NG‐H, AD‐R, PM, MF‐V, AV‐B, RA and JS acquired the data. NG‐H, AD‐R, PM, MF‐V, AV‐B, RA, JS, AMG, JPB and CD analysed and interpreted the data. NG‐H, AMG, JPB and CD wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
SAF‐2010‐16377 and SAF2014‐57190R from Ministerio de Economía y Competitividad (MINECO); Agence Nationale de la Recherche (ANR‐13‐BSV1‐0023‐01 and ANR‐15‐CE14‐0005); Fondation pour la recherche médicale (Programme cardiovasculaire 2011); CORDDIM‐Région Ile de France (Équipement jeune équipe 2010; Petit et moyen équipement 2011); ISCIII PI14/01078 and Fondos Feder. The laboratory UMR‐S 1180 is a member of the Laboratory of Excellence LERMIT supported by a grant from the Agence Nationale de la Recherche (ANR‐10‐LABX‐33). NGH was a FPU fellow of the Spanish Ministry of Education.
Translational perspective.
Our data support the idea that high leptin levels contribute to an increased risk of cardiovascular disease under physiological conditions but may be cardioprotective in pathological situations such as heart failure. In the failing transverse aorta constriction (TAC) mouse model, leptin prevents sarcoplasmic reticulum Ca2+ load decline, maintaining physiological protein levels of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2 (SERCA2). Leptin treatment also prevents electrical TAC‐remodelling via potassium current normalization and the prevention of ventricular arrhythmia occurrence as a result of ryanodine receptor activity normalization. These opposite effects of leptin depend on the pathophysiological state of the heart. In conclusion, the present study contributes to a better understanding of the ‘obesity paradox’ phenomenon at the cardiac level.
Acknowledgements
We thank Florence Lefebvre (Inserm, UMR‐S 1180, Châtenay‐Malabry, France) and Manuel Bas (Complutense University, Madrid) for their help with the cell isolation; Françoise Boussac for administrative help; and Valérie Domergue and the AnimEX platform from IPSIT for animal care.
This is an Editor's Choice article from the 1 July 2017 issue.
Contributor Information
Jean‐Pierre Benitah, Email: jean-pierre.benitah@inserm.fr.
Carmen Delgado, Email: cdelgado@iib.uam.es.
References
- Abe Y, Ono K, Kawamura T, Wada H, Kita T, Shimatsu A & Hasegawa K (2007). Leptin induces elongation of cardiac myocytes and causes eccentric left ventricular dilatation with compensation. Am J Physiol Heart Circ Physiol 292, H2387–H2396. [DOI] [PubMed] [Google Scholar]
- Abel ED, Litwin SE & Sweeney G (2008). Cardiac remodeling in obesity. Physiol Rev 88, 389–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrén B, Larsson H, Wilhelmsson C, Näsman B & Olsson T (1997). Regulation of circulating leptin in humans. Endocrine 7, 1–8. [DOI] [PubMed] [Google Scholar]
- Atkinson LL, Fischer MA & Lopaschuk GD (2002). Leptin activates cardiac fatty acid oxidation independent of changes in the AMP‐activated protein kinase‐acetyl‐CoA carboxylase‐malonyl‐CoA axis. J Biol Chem 277, 29424–29430. [DOI] [PubMed] [Google Scholar]
- Baartscheer A, Schumacher CA, Belterman CN, Coronel R & Fiolet JW (2003). [Na+]i and the driving force of the Na+/Ca2+‐exchanger in heart failure. Cardiovasc Res 57, 986–995. [DOI] [PubMed] [Google Scholar]
- Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP & Hare JM (2003). Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 108, 754–759. [DOI] [PubMed] [Google Scholar]
- Benitah JP, Gomez AM, Bailly P, Da Ponte JP, Berson G, Delgado C & Lorente P (1993). Heterogeneity of the early outward current in ventricular cells isolated from normal and hypertrophied rat hearts. J Physiol 469, 111–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation‐contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bers DM (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 76, 107–127. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Näbauer M & Erdmann E (1993). Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73, 379–385. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Li TY, Kelesidis I, Gavrila A, Hu FB & Mantzoros CS (2007). Circulating leptin levels are not associated with cardiovascular morbidity and mortality in women with diabetes: a prospective cohort study. Diabetologia 50, 1178–1185. [DOI] [PubMed] [Google Scholar]
- Brouillette J, Clark RB, Giles WR & Fiset C (2004). Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol 559, 777–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ & Bauer TL (1996). Serum immunoreactive‐leptin concentrations in normal‐weight and obese humans. N Engl J Med 334, 292–295. [DOI] [PubMed] [Google Scholar]
- Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT & Shannon TR (2014). Nitric oxide‐dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS ONE 9, e87495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koninck P & Schulman H (1998). Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279, 227–230. [DOI] [PubMed] [Google Scholar]
- Delgado C, Ruiz‐Hurtado G, Gómez‐Hurtado N, González‐Ramos S, Rueda A, Benito G, Prieto P, Zaragoza C, Delicado EG, Pérez‐Sen R, Miras‐Portugal MT, Núñez G, Boscá L & Fernández‐Velasco M (2015). NOD1, a new player in cardiac function and calcium handling. Cardiovasc Res 106, 375–386. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Pogwizd SM & Bers DM (2002). Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol 539, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Zhang X & Ren J (2006). Leptin regulates cardiomyocyte contractile function through endothelin‐1 receptor‐NADPH oxidase pathway. Hypertension 47, 222–229. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin‐Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ & Anderson ME (2008). A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedak PW, Verma S, Weisel RD, Skrtic M & Li RK (2005). Cardiac remodeling and failure: from molecules to man (part III). Cardiovasc Pathol 14, 109–119. [DOI] [PubMed] [Google Scholar]
- Filippatos GS, Tsilias K, Venetsanou K, Karambinos E, Manolatos D, Kranidis A, Antonellis J, Kardaras F, Anthopoulos L & Baltopoulos G (2000). Leptin serum levels in cachectic heart failure patients. Relationship with tumor necrosis factor‐alpha system. Int J Cardiol 76, 117–122. [DOI] [PubMed] [Google Scholar]
- Fischer TH, Eiringhaus J, Dybkova N, Förster A, Herting J, Kleinwächter A, Ljubojevic S, Schmitto JD, Streckfuß‐Bömeke K, Renner A, Gummert J, Hasenfuss G, Maier LS & Sossalla S (2014). Ca(2+) /calmodulin‐dependent protein kinase II equally induces sarcoplasmic reticulum Ca(2+) leak in human ischaemic and dilated cardiomyopathy. Eur J Heart Fail 16, 1292–1300. [DOI] [PubMed] [Google Scholar]
- Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, Ellenberger D, Förster A, Schmitto JD, Gummert J, Schöndube FA, Hasenfuss G, Maier LS & Sossalla S (2013). Ca2+/calmodulin‐dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 128, 970–981. [DOI] [PubMed] [Google Scholar]
- Galimberti ES & Knollmann BC (2011). Efficacy and potency of class I antiarrhythmic drugs for suppression of Ca2+ waves in permeabilized myocytes lacking calsequestrin. J Mol Cell Cardiol 51, 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan XT, Zhao G, Huang CX, Rowe AC, Purdham DM & Karmazyn M (2013). Identification of fat mass and obesity associated (FTO) protein expression in cardiomyocytes: regulation by leptin and its contribution to leptin‐induced hypertrophy. PLoS ONE 8, e74235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter CE, Colbran RJ & Anderson ME (2007). CaMKII, an emerging molecular driver for calcium homeostasis, arrhythmias, and cardiac dysfunction. J Mol Med (Berl) 85, 5–14. [DOI] [PubMed] [Google Scholar]
- Guzmán‐Ruiz R, Somoza B, Gil‐Ortega M, Merino B, Cano V, Attané C, Castan‐Laurell I, Valet P, Fernández‐Alfonso MS & Ruiz‐Gayo M (2010). Sensitivity of cardiac carnitine palmitoyltransferase to malonyl‐CoA is regulated by leptin: similarities with a model of endogenous hyperleptinemia. Endocrinology 151, 1010–1018. [DOI] [PubMed] [Google Scholar]
- Gómez AM, Bénitah JP, Henzel D, Vinet A, Lorente P & Delgado C (1997). Modulation of electrical heterogeneity by compensated hypertrophy in rat left ventricle. Am J Physiol Heart Circ Physiol 272, H1078–H1086. [DOI] [PubMed] [Google Scholar]
- Gómez AM, Cheng H, Lederer WJ & Bers DM (1996). Ca2+ diffusion and sarcoplasmic reticulum transport both contribute to [Ca2+]i decline during Ca2+ sparks in rat ventricular myocytes. J Physiol 496 (Pt 2), 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Hurtado N, Fernández‐Velasco M, Fernández‐Alfonso MS, Boscá L & Delgado C (2014). Prolonged leptin treatment increases transient outward K⁺ current via upregulation of Kv4.2 and Kv4.3 channel subunits in adult rat ventricular myocytes. Pflügers Arch 466, 903–914. [DOI] [PubMed] [Google Scholar]
- Hall ME, Smith G, Hall JE & Stec DE (2012). Cardiomyocyte‐specific deletion of leptin receptors causes lethal heart failure in Cre‐recombinase‐mediated cardiotoxicity. Am J Physiol Regul Integr Comp Physiol 303, R1241–R1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2003). Minireview: the AMP‐activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144, 5179–5183. [DOI] [PubMed] [Google Scholar]
- Hobai IA & O'Rourke B (2001). Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation‐contraction coupling in canine heart failure. Circulation 103, 1577–1584. [DOI] [PubMed] [Google Scholar]
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Woo MA & Tillisch JH (2001). The relationship between obesity and mortality in patients with heart failure. J Am Coll Cardiol 38, 789–795. [DOI] [PubMed] [Google Scholar]
- Ingwall JS (2009). Energy metabolism in heart failure and remodelling. Cardiovasc Res 81, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St‐Pierre J & Spiegelman BM (2007). AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1alpha. Proc Natl Acad Sci USA 104, 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB & Vasan RS (2002). Obesity and the risk of heart failure. N Engl J Med 347, 305–313. [DOI] [PubMed] [Google Scholar]
- Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV & Ventura HO (2013). Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail 1, 93–102. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM & Kelly DP (2000). Peroxisome proliferator‐activated receptor gamma coactivator‐1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Maier LS & Hasenfuss G (2009). Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev 14, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifheit‐Nestler M, Wagner NM, Gogiraju R, Didié M, Konstantinides S, Hasenfuss G & Schäfer K (2013). Importance of leptin signaling and signal transducer and activator of transcription‐3 activation in mediating the cardiac hypertrophy associated with obesity. J Transl Med 11, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littnerova S, Parenica J, Spinar J, Vitovec J, Linhart A, Widimsky P, Jarkovsky J, Miklik R, Spinarova L, Zeman K, Belohlavek J, Malek F, Felsoci M, Kettner J, Ostadal P, Cihalik C, Spac J, Al‐Hiti H, Fedorco M, Fojt R, Kruger A, Malek J, Mikusová T, Monhart Z, Bohacova S, Pohludkova L, Rohac F, Vaclavik J, Vondrakova D, Vyskocilova K, Bambuch M & Dusek L (2015). Positive influence of being overweight/obese on long term survival in patients hospitalised due to acute heart failure. PLoS ONE 10, e0117142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Butler KR, Buxbaum SG, Sung JH, Campbell BW & Taylor HA (2010). Leptinemia and its association with stroke and coronary heart disease in the Jackson Heart Study. Clin Endocrinol (Oxf) 72, 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G & Sejersted OM (2010). Sodium accumulation promotes diastolic dysfunction in end‐stage heart failure following Serca2 knockout. J Physiol 588, 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughrey CM, MacEachern KE, Cooper J & Smith GL (2003). Measurement of the dissociation constant of Fluo‐3 for Ca2+ in isolated rabbit cardiomyocytes using Ca2+ wave characteristics. Cell Calcium 34, 1–9. [DOI] [PubMed] [Google Scholar]
- Madani S, De Girolamo S, Muñoz DM, Li RK & Sweeney G (2006). Direct effects of leptin on size and extracellular matrix components of human pediatric ventricular myocytes. Cardiovasc Res 69, 716–725. [DOI] [PubMed] [Google Scholar]
- Mascareno E, Beckles D, Dhar‐Mascareno M & Siddiqui MA (2009). Enhanced hypertrophy in ob/ob mice due to an impairment in expression of atrial natriuretic peptide. Vascul Pharmacol 51, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaffin KR, Sun CK, Rager JJ, Romano LC, Zou B, Mathier MA, O'Doherty RM, McTiernan CF & O'Donnell CP (2008). Leptin signalling reduces the severity of cardiac dysfunction and remodelling after chronic ischaemic injury. Cardiovasc Res 77, 54–63. [DOI] [PubMed] [Google Scholar]
- McGaffin KR, Zou B, McTiernan CF & O'Donnell CP (2009). Leptin attenuates cardiac apoptosis after chronic ischaemic injury. Cardiovasc Res 83, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Harris TB, Hsueh WC, Hue T, Leak TS, Li R, Mehta M, Vaisse C & Sahyoun NR (2015). The association of serum leptin with mortality in older adults. PLoS ONE 10, e0140763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Rokosh DG, Paccanaro M, Yee RR, Simpson PC, Grossman W & Foster E (2001). LV systolic performance improves with development of hypertrophy after transverse aortic constriction in mice. Am J Physiol Heart Circ Physiol 281, H1104–H1112. [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S & Yeh YH (2007). Arrhythmogenic ion‐channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87, 425–456. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM & Kass RS (2005). Molecular physiology of cardiac repolarization. Physiol Rev 85, 1205–1253. [DOI] [PubMed] [Google Scholar]
- Oreopoulos A, Padwal R, Kalantar‐Zadeh K, Fonarow GC, Norris CM & McAlister FA (2008). Body mass index and mortality in heart failure: a meta‐analysis. Am Heart J 156, 13–22. [DOI] [PubMed] [Google Scholar]
- Ouvrard‐Pascaud A, Sainte‐Marie Y, Bénitah JP, Perrier R, Soukaseum C, Nguyen Dinh Cat A, Royer A, Le Quang K, Charpentier F, Demolombe S, Mechta‐Grigoriou F, Beggah AT, Maison‐Blanche P, Oblin ME, Delcayre C, Fishman GI, Farman N, Escoubet B & Jaisser F (2005). Conditional mineralocorticoid receptor expression in the heart leads to life‐threatening arrhythmias. Circulation 111, 3025–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP & Campbell DL (2005). Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol 569, 7–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier E, Kerfant BG, Lalevee N, Bideaux P, Rossier MF, Richard S, Gómez AM & Benitah JP (2004). Mineralocorticoid receptor antagonism prevents the electrical remodeling that precedes cellular hypertrophy after myocardial infarction. Circulation 110, 776–783. [DOI] [PubMed] [Google Scholar]
- Pieske B, Maier LS, Piacentino V, Weisser J, Hasenfuss G & Houser S (2002). Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 106, 447–453. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM & Bers DM (2004). Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 14, 61–66. [DOI] [PubMed] [Google Scholar]
- Rajapurohitam V, Gan XT, Kirshenbaum LA & Karmazyn M (2003). The obesity‐associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res 93, 277–279. [DOI] [PubMed] [Google Scholar]
- Ravens U & Cerbai E (2008). Role of potassium currents in cardiac arrhythmias. Europace 10, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Rider OJ, Petersen SE, Francis JM, Ali MK, Hudsmith LE, Robinson MR, Clarke K & Neubauer S (2011). Ventricular hypertrophy and cavity dilatation in relation to body mass index in women with uncomplicated obesity. Heart 97, 203–208. [DOI] [PubMed] [Google Scholar]
- Romero‐Corral A, Montori VM, Somers VK, Korinek J, Thomas RJ, Allison TG, Mookadam F & Lopez‐Jimenez F (2006). Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet 368, 666–678. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Hurtado G, Gomez‐Hurtado N, Fernandez‐Velasco M, Calderon E, Smani T, Ordonez A, Cachofeiro V, Bosca L, Diez J, Gomez AM & Delgado C (2012). Cardiotrophin‐1 induces sarcoplasmic reticulum Ca‐2 leak and arrhythmogenesis in adult rat ventricular myocytes. Cardiovasc Res 96, 81–89. [DOI] [PubMed] [Google Scholar]
- Schaffer JE (2003). Lipotoxicity: when tissues overeat. Curr Opin Lipidol 14, 281–287. [DOI] [PubMed] [Google Scholar]
- Schram K & Sweeney G (2008). Implications of myocardial matrix remodeling by adipokines in obesity‐related heart failure. Trends Cardiovasc Med 18, 199–205. [DOI] [PubMed] [Google Scholar]
- Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskamper J & Pieske B (2010). Na+‐dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res 87, 50–59. [DOI] [PubMed] [Google Scholar]
- Sierra‐Johnson J, Romero‐Corral A, Lopez‐Jimenez F, Gami AS, Sert Kuniyoshi FH, Wolk R & Somers VK (2007). Relation of increased leptin concentrations to history of myocardial infarction and stroke in the United States population. Am J Cardiol 100, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha MK, Opentanova I, Ohannesian JP, Kolaczynski JW, Heiman ML, Hale J, Becker GW, Bowsher RR, Stephens TW & Caro JF (1996). Evidence of free and bound leptin in human circulation. Studies in lean and obese subjects and during short‐term fasting. J Clin Invest 98, 1277–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman AT, Omar M, Assem HM, Nasr IS, Rizk MM, El Matary W & El Alaily RK (2002). Serum leptin concentrations in children with type 1 diabetes mellitus: relationship to body mass index, insulin dose, and glycemic control. Metabolism 51, 292–296. [DOI] [PubMed] [Google Scholar]
- Sorokina N, O'Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM & Lewandowski ED (2007). Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 115, 2033–2041. [DOI] [PubMed] [Google Scholar]
- Stucchi P, Guzmán‐Ruiz R, Gil‐Ortega M, Merino B, Somoza B, Cano V, de Castro J, Sevillano J, Ramos MP, Fernández‐Alfonso MS & Ruiz‐Gayo M (2011). Leptin resistance develops spontaneously in mice during adult life in a tissue‐specific manner. Consequences for hepatic steatosis. Biochimie 93, 1779–1785. [DOI] [PubMed] [Google Scholar]
- Sweeney G (2010). Cardiovascular effects of leptin. Nat Rev Cardiol 7, 22–29. [DOI] [PubMed] [Google Scholar]
- Tajmir P, Ceddia RB, Li RK, Coe IR & Sweeney G (2004). Leptin increases cardiomyocyte hyperplasia via extracellular signal‐regulated kinase‐ and phosphatidylinositol 3‐kinase‐dependent signaling pathways. Endocrinology 145, 1550–1555. [DOI] [PubMed] [Google Scholar]
- Thøgersen AM, Söderberg S, Jansson JH, Dahlén G, Boman K, Nilsson TK, Lindahl B, Weinehall L, Stenlund H, Lundberg V, Johnson O, Ahrén B & Hallmans G (2004). Interactions between fibrinolysis, lipoproteins and leptin related to a first myocardial infarction. Eur J Cardiovasc Prev Rehabil 11, 33–40. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF & Marbán E (1999). Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res 42, 270–283. [DOI] [PubMed] [Google Scholar]
- Wannamethee SG, Shaper AG, Whincup PH, Lennon L, Papacosta O & Sattar N (2014). The obesity paradox in men with coronary heart disease and heart failure: the role of muscle mass and leptin. Int J Cardiol 171, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannamethee SG, Tchernova J, Whincup P, Lowe GD, Kelly A, Rumley A, Wallace AM & Sattar N (2007). Plasma leptin: associations with metabolic, inflammatory and haemostatic risk factors for cardiovascular disease. Atherosclerosis 191, 418–426. [DOI] [PubMed] [Google Scholar]
- Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF & Luo JD (2004). Leptin induces hypertrophy via endothelin‐1‐reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 110, 1269–1275. [DOI] [PubMed] [Google Scholar]
- Zamora E, Díez‐López C, Lupón J, de Antonio M, Domingo M, Santesmases J, Troya MI, Díez‐Quevedo C, Altimir S & Bayes‐Genis A (2016). Weight loss in obese patients with heart failure. J Am Heart Assoc 4, e002468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM & Blatter LA (2010). Ca²+ spark‐dependent and ‐independent sarcoplasmic reticulum Ca²+ leak in normal and failing rabbit ventricular myocytes. J Physiol 588, 4743–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]