

Abstract
Key points
Layer 2/3 neurons of the prefrontal cortex display higher gain of somatic excitability, responding with a higher number of action potentials for a given stimulus, in fmr1−/y mice.
In fmr1−/y L2/3 neurons, action potentials are taller, faster and narrower.
Outside‐out patch clamp recordings revealed that the maximum Na+ conductance density is higher in fmr1−/y L2/3 neurons.
Measurements of three biophysically distinct K+ currents revealed a depolarizing shift in the activation of a rapidly inactivating (A‐type) K+ conductance.
Realistic neuronal simulations of the biophysical observations recapitulated the elevated action potential and repetitive firing phenotype.
Abstract
Fragile X syndrome is the most common form of inherited mental impairment and autism. The prefrontal cortex is responsible for higher order cognitive processing, and prefrontal dysfunction is believed to underlie many of the cognitive and behavioural phenotypes associated with fragile X syndrome. We recently demonstrated that somatic and dendritic excitability of layer (L) 5 pyramidal neurons in the prefrontal cortex of the fmr1−/y mouse is significantly altered due to changes in several voltage‐gated ion channels. In addition to L5 pyramidal neurons, L2/3 pyramidal neurons play an important role in prefrontal circuitry, integrating inputs from both lower brain regions and the contralateral cortex. Using whole‐cell current clamp recording, we found that L2/3 pyramidal neurons in prefrontal cortex of fmr1−/y mouse fired more action potentials for a given stimulus compared with wild‐type neurons. In addition, action potentials in fmr1−/y neurons were significantly larger, faster and narrower. Voltage clamp of outside‐out patches from L2/3 neurons revealed that the transient Na+ current was significantly larger in fmr1−/y neurons. Furthermore, the activation curve of somatic A‐type K+ current was depolarized. Realistic conductance‐based simulations revealed that these biophysical changes in Na+ and K+ channel function could reliably reproduce the observed increase in action potential firing and altered action potential waveform. These results, in conjunction with our prior findings on L5 neurons, suggest that principal neurons in the circuitry of the medial prefrontal cortex are altered in distinct ways in the fmr1−/y mouse and may contribute to dysfunctional prefrontal cortex processing in fragile X syndrome.
Keywords: action potential, patch clamp, prefrontal cortex, sodium conductance
Key points
Layer 2/3 neurons of the prefrontal cortex display higher gain of somatic excitability, responding with a higher number of action potentials for a given stimulus, in fmr1−/y mice.
In fmr1−/y L2/3 neurons, action potentials are taller, faster and narrower.
Outside‐out patch clamp recordings revealed that the maximum Na+ conductance density is higher in fmr1−/y L2/3 neurons.
Measurements of three biophysically distinct K+ currents revealed a depolarizing shift in the activation of a rapidly inactivating (A‐type) K+ conductance.
Realistic neuronal simulations of the biophysical observations recapitulated the elevated action potential and repetitive firing phenotype.
Abbreviations
- AP
action potential
- FMRP
fragile X mental retardation protein
- mPFC
medial prefrontal cortex
- PFC
prefrontal cortex
- WT
wild‐type
Introduction
The prefrontal cortex contributes to goal‐directed behaviour by exerting top‐down control over numerous cortical and subcortical regions (Miller & Cohen, 2001). Consequently, normal prefrontal function allows for behavioural flexibility and the adaptation of goals and actions to changing contexts and circumstances. Not surprisingly, then, prefrontal cortex impairment is associated with attention deficit, impulsivity and perseverant behaviour (Heilman et al. 1991; Russell, 2002; Euston et al. 2012; Aron et al. 2014). These behavioural phenotypes are hallmarks of fragile X syndrome, the most common form of inherited mental retardation and leading monogenetic cause of autism (Hagerman et al. 2005). Prefrontal dysfunction is thus specifically implicated in many behavioural phenotypes associated with fragile X syndrome.
Like other cortical areas, prefrontal cortex is a laminar structure with excitatory principal neurons residing in layers (L) 2/3, 5, and 6. Pyramidal neurons in these layers receive synaptic inputs arising both within and outside the prefrontal cortex and send projections locally and to other cortical areas (Morishima & Kawaguchi, 2006; Morishima et al. 2011; Ueta et al. 2013; Dembrow & Johnston, 2014). However, the majority of subcortical projections arise from subsets of deep layer pyramidal neurons (reviewed in Dembrow & Johnston, 2014; Harris & Shepherd, 2015). Interlaminar input from L2/3 provides substantial input to these subcortical‐projecting neurons (Otsuka & Kawaguchi, 2011). Thus, L2/3 is capable of shaping downstream cortical activity directly and subcortical activity indirectly via projections to L5/6.
Alterations to connectivity between prefrontal neurons and/or changes to intrinsic neuronal excitability could contribute to prefrontal dysfunction in fragile X syndrome. Indeed, several studies have reported alterations in synaptic function in the prefrontal cortex of the fmr1−/y mouse (Desai et al. 2006; Meredith et al. 2007; Krueger et al. 2011; Testa‐Silva et al. 2012; Martin et al. 2015). In contrast, there are few studies reporting changes to intrinsic neural excitability in the fmr1−/y mouse prefrontal cortex. We recently reported cell type‐specific changes in the functional expression of somatic and dendritic voltage‐gated ion channels in L5 prefrontal cortex (PFC) neurons in the fmr1−/y mouse (Kalmbach et al. 2015). Consequently, dendritic and somatic excitability was altered in pyramidal tract‐projecting layer 5 neurons. A loss of L‐type calcium channel activity in dendritic spines contributes to the deficit in spike timing‐dependent LTP in L2/3 prefrontal neurons in the fmr1−/y mouse (Meredith et al. 2007).
In this study, we addressed whether there are perisomatic changes in voltage‐gated ion channels in L2/3 pyramidal neurons and how those changes affect neuronal function in adult mice. Using whole‐cell current clamp recording, we found that L2/3 neurons in the PFC of fmr1−/y mice fired more action potentials in response to a given current stimulus. Outside‐out patch clamp recordings from the soma of L2/3 neurons revealed significant differences in the functional expression of Na+ and K+ channels between wild‐type and fmr1−/y neurons. Single compartment modelling of our observed changes in Na+ and K+ channels recapitulated the fmr1−/y action potential firing phenotypes. These findings demonstrate that alterations to Na+ and K+ channel function contribute to enhanced firing of L2/3 neurons in the fmr1−/y mouse. These changes may contribute to behavioural phenotypes associated with prefrontal dysfunction in fragile X syndrome.
Methods
Ethical approval
All animal procedures conform to the National Institute of Health guidelines and were approved by University of Texas at Austin Institutional Animal Care and Use Committee.
Slice preparation
Male wild‐type and fmr1−/y mice on C57BL/6 background, 8–16 weeks old, were anaesthetized with an injection of ketamine–xylazine (100/10 mg kg−1 i.p.) cocktail. This age range was chosen for two reasons. First, this was the same age used in our previously published work on the changes in neuronal properties of L5 prefrontal cortical and hippocampal CA1 pyramidal neurons (Kalmbach et al. 2015; Routh et al. 2013; Brager et al. 2012). Second, we are interested in physiological phenotypes associated with fragile X syndrome that persist into adulthood. Mice were perfused through the heart with ice‐cold saline consisting of (in mm): 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 7 dextrose, 205 sucrose, 1.3 ascorbate and 3 sodium pyruvate (bubbled with 95% O2–5% CO2 to maintain pH at ∼7.4) and killed by decapitation. A vibrating tissue slicer (Vibratome 3000, Leica Biosystems Inc., Buffalo Grove, IL, USA) was used to make 300 μm thick coronal sections. Slices were held for 30 min at 35°C in a chamber filled with artificial cerebrospinal fluid (aCSF) consisting of (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, 10 dextrose and 3 sodium pyruvate (bubbled with 95% O2–5% CO2), and then held at room temperature until the time of recording.
Neurophysiology
Slices were placed in a submerged, heated (32–34°C) recording chamber that was continually perfused (1−2 ml min−1) with bubbled aCSF containing (in mm): 125 NaCl, 3.0 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 10 dextrose, 3 Na+ pyruvate, 0.025 d‐APV, 0.02 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX), 0.005 CGP 55845 and 0.002 gabazine. Slices were viewed either with a Zeiss Axioskop microscope and differential interference optics or a Zeiss AxioExaminer D microscope and Dodt contrast optics. Patch pipettes (4−8 MΩ) were pulled from borosilicate glass and wrapped with parafilm to reduce capacitance. Recordings were made from L2/3 pyramidal neurons in the anterior cingulate and prelimbic areas of the dorsal, medial prefrontal cortex ∼1–2 mm anterior to bregma.
Current clamp cell recordings
The pipette solution contained (in mm): 120 potassium gluconate, 16 KCl, 10 Hepes, 8 NaCl, 7 potassium phosphocreatine, 0.3 Na‐GTP, 4 Mg‐ATP (pH 7.3 with KOH). Neurobiotin (Vector Laboratories, Burlingame, CA, USA; 0.1–0.2%) was also included for histological processing, post hoc cell location determination and morphological reconstruction (see below). In some cases, Alexa 594 (16 μm; Thermo Fisher Scientific, Waltham, MA, USA) was also included in the internal recording solution to determine the recording location. Data were acquired using a Dagan BVC‐700 amplifier (Dagan Corp., Minneapolis, MN, USA) and custom data acquisition software written using Igor Pro (Wavemetrics, Lake Oswego, OR, USA) or AxoGraph X (AxoGraph Scientific, Sydney, Australia) data acquisition software. Data were acquired at 10−50 kHz, filtered at 5−10 kHz and digitized by an ITC‐18 (HEKA Instruments Inc., Holliston, MA, USA) interface. Pipette capacitance was compensated and the bridge was balanced during each recording. Series resistance was monitored and compensated throughout each experiment and was 10−25 MΩ. Voltages are not corrected for the liquid‐junction potential (estimated as ∼8 mV).
Data were analysed using either custom analysis software written in Igor Pro or using AxoGraph X. Input resistance was calculated from the linear portion of the current–voltage relationship in response to a family of 1 s current injections (−150 to +50 pA, 20 pA steps). The functional membrane time constant was defined as the slow component of a double‐exponential fit of the average voltage decay in response to hyperpolarizing current injections (100−300 pA, 2 ms). Single action potentials (APs) were elicited using just‐threshold current injections of various durations. AP threshold was defined as the voltage where the first derivative first exceeded 20 mV ms−1. AP width was measured at half the amplitude between threshold and the peak voltage.
Outside‐out patch clamp recordings
Membrane currents were recorded using an Axopach 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), sampled at 10 kHz, analog filtered at 2 kHz and digitized by an ITC‐18 interface connected to a computer running AxoGraph X. For Na+ channels, the pipette solution contained (in mm): 120 caesium gluconate, 10 CsCl, 10 Hepes, 4 NaCl, 0.1 3,4‐diaminopyradine and 20 tetraethylammonium chloride (pH 7.3 with CsOH). The extracellular saline contained (in mm): 125 NaCl, 3 KCl, 25 NaHCO3, 2 NiCl2, 1 MgCl2, 10 dextrose, 1.3 ascorbate and 3 pyruvate, and TEA and 4AP were added to the extracellular saline. Activation curves were elicited using depolarizing voltage commands (−80 to 30 mV in 10 mV steps) from a holding potential of −90 mV. Steady‐state inactivation curves were elicited using a depolarizing voltage step to 0 mV following a 500 ms prepulse to a family of voltage commands (−70 to −10 mV in 10 mV steps). For K+ channels, the pipette solution was the same as for whole‐cell recording but 1 μm TTX was added to the aCSF. Three separate command protocols were used: (1) depolarizing voltage commands (−70 to 70 mV in 20 mV steps) from a holding potential of −90 mV; (2) the same depolarizing voltage commands following a 200‐ms step to −22 to inactivate A‐type K+ channels; and (3) depolarizing voltage commands (−70 to 70 mV in 20 mV steps) from a holding potential of −22 mV to inactivate all transient K+ channels (Kalmbach et al. 2015). Activation data for both Na+ and K+ currents were fitted to a single Boltzmann function using a least‐squares program. Linear leakage and capacitive currents were digitally subtracted by scaling traces at smaller command voltages in which no voltage‐dependent current was activated.
Neuronal reconstructions
Neurobiotin (Vector Laboratories) was added to the pipette solution (see above, 0.1–0.2%) to facilitate later visualization of individual neurons. Slices were fixed in a 0.1 m phosphate buffer (pH 7.4) with 3% glutaraldehyde at 4°C for at least 48 h before undergoing histological processing using an avidin–horseradish peroxidase system activated by diaminobenzidine (DAB; Vector Laboratories). DAB‐processed slices were mounted in glycerol. Anatomical reconstructions were performed using a compound microscope fitted with a ×40 objective and a computer‐controlled indexing system running Neurolucida 6.0 imaging software (MBF Bioscience, Williston, VT, USA).
Neuronal simulations
To reduce computational cost and to avoid the influences of morphology on spiking patterns (Mainen & Sejnowski, 1996), and motivated by our experimental observations that there are no significant differences in L2/3 neuronal morphology between wild‐type and fmr1−/y mice, we employed a single compartmental model for all simulations with dimensions 60 μm × 60 μm. Passive electrical parameters of the model neuron were set as follows: specific membrane resistance, R m = 17 kΩ cm2, specific membrane capacitance, C m = 1 μF cm−2, and intracellular axial resistivity, R a = 100 Ω cm.
The model neuron expressed six different Hodgkin–Huxley (HH)‐type ion channels; fast Na+ (NaF), delayed rectifier K+ (KDR), A‐type K+ (KA), M‐type K+ (KM), L‐type Ca2+ (CaL) and calcium‐activated small conductance K+ (SK) channels. The kinetic schemes for NaF, KDR and KA channels were taken from Migliore et al. (1999) and the KM channel was modelled as in Rathour & Narayanan (2012a). L‐type Ca2+ (CaL) channel kinetics was adopted form Poirazi et al. (2003) and SK channels were modelled as in Yuen & Durand (1991) and Anirudhan & Narayanan (2015). Reversal potentials for Na+ and K+ channels were set at 55 and –90 mV, respectively. The current through the L‐type Ca2+ channel was modelled using the Goldman–Hodgkin–Katz (GHK) formulation with the default values of external and internal Ca2+ concentrations set at 2 mm and 100 nm, respectively. Calcium decay kinetics was taken from Narayanan & Johnston (2010) and the calcium decay time constant was set to be 30 ms. Default values of these conductances in the base wild‐type model were set as follows (mS cm−2): , , , , and .
All simulations were performed using the NEURON simulation environment (Hines & Carnevale, 1997). For all simulations, membrane potential was fixed at –65 mV and temperature was set at 34°C, and ion channels kinetics were appropriately adjusted based upon their experimentally determined Q 10 factors. The default integration time step was set at 25 μs. All analyses related to action potential properties and firing rate were preformed using custom‐built software written within the Igor Pro programming environment.
Data analysis
Repeated measures analysis of variance (RM‐ANOVA), between‐subjects factors ANOVA, mixed factors ANOVA and post hoc Student's t test were used to test for statistical differences between experimental conditions. The Bonferonni correction was used to correct for multiple comparisons. Error bars represent standard error of the mean (SEM). Statistical analyses were performed using Prism (GraphPad Software, La Jolla, CA, USA). Data are presented in the text as the mean ± SEM.
Results
Recording location
We made whole cell recordings from L2/3 pyramidal neurons from wild‐type and fmr1−/y mice. To confirm the location of our neurons within the medial PFC, all neurons were filled with neurobiotin during whole‐cell recording and the location confirmed post hoc. We found a positive correlation between the input resistance of L2/3 neurons and their location within layer 2/3. Specifically, input resistance was higher for neurons located deeper within L2/3 (Fig. 1 A, wild‐type: 19 cells/8 mice; fmr1−/y: 31 cells/11 mice). In contrast, there was no significant correlation with input resistance and the dorso‐ventral location within the medial prefrontal cortex (mPFC; Fig. 1 B). There was no significant difference in the somatic location of neurons analysed in this study between the wild‐type and fmr1−/y (distance from pia, wild‐type (WT): 160.6 ± 6.09 μm; fmr1−/y: 160.1 ± 6.94 μm; Fig. 1 C). Based on the somatic distance from the dorsal surface, all neurons were located within the anterior cingulate cortex and prelimbic regions of the medial prefrontal cortex (Fig. 1 D).
Figure 1. The location of recorded neurons within the medial prefrontal cortex.
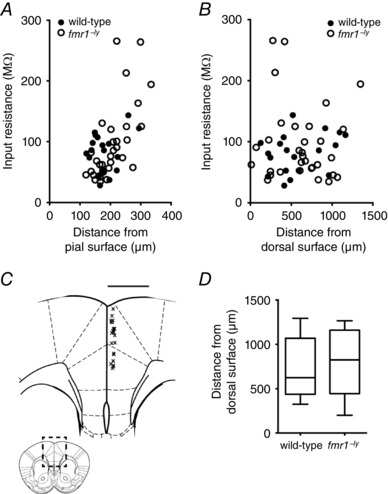
A, input resistance increased as a function of location within layer 2/3. B, there was no correlation between input resistance and the dorso‐ventral location of L2/3 neurons. C, diagram of the area indicated by the dashed box in the inset. Each ‘x’ represents the approximate location of an L2/3 neuron within the mPFC from which whole‐cell recordings were made. Inset, illustration of the rostro‐caudal location within the cortex where electrophysiological recordings were made. D, summary graph showing the dorso‐ventral recording locations for wild‐type and fmr1−/y L2/3 neurons.
Subthreshold properties
We measured the input resistance at multiple membrane potentials (−80 to −60 mV, 5 mV increments) using a series of subthreshold current injections (Fig. 2 A). In both wild‐type and fmr1−/y neurons, input resistance was lower at more hyperpolarized membrane potentials (Fig. 2 B, WT: 14 cells/7 mice; fmr1−/y: 20 cells/9 mice). However, there was no significant difference in input resistance between wild‐type and fmr1−/y neurons at any of the membrane potentials tested. There was also no significant difference in the resting membrane potential between wild‐type and fmr1−/y neurons (WT: −78.2 ± 0.96 mV; fmr1−/y: −77.7 ± 1.2 mV; Fig. 2 C). The membrane time constant, τM, was estimated using the slow component of a double exponential fit to the decay of small changes in membrane potential (∼1–2 mV). Similar to input resistance, τM was shorter at more hyperpolarized membrane potentials for both wild‐type and fmr1−/y neurons with no significant difference in τM between wild‐type and fmr1−/y neurons (Fig. 2 D, WT: 9 cells/4 mice; fmr1−/y: 12 cells/5 mice).
Figure 2. Subthreshold properties are not different between wild‐type and fmr1−/y L2/3 neurons.
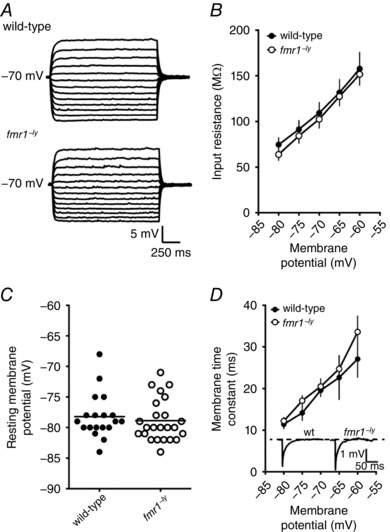
A, representative traces showing the voltage response to a family of current injections used to estimate input resistance. B, summary graph showing that there is no significant difference in input resistance, across a range of voltages, between wild‐type and fmr1−/y neurons (wild‐type: 14 cells/6 mice; fmr1−/y: 20 cells/9 mice). C, there is no significant difference in resting membrane potential between wild‐type and fmr1−/y L2/3 neurons. D, summary graph showing that there is no significant difference in membrane time constant, across a range of voltages, between wild‐type and fmr1−/y neurons (wild‐type: 9 cells/4 mice; fmr1−/y: 12 cells/5 mice). Inset, representative traces showing the voltage response to a small hyperpolarizing injection used to estimate the membrane time constant.
Higher action potential firing in fmr1−/y L2/3 neurons
We used two separate protocols to measure action potential firing (from a holding voltage of −70 mV) in response to a current stimulus. First, we injected a family of 500 ms‐long current steps (50–400 pA, 50 pA steps) and measured the number of action potentials fired during each step (Fig. 3 A). Both wild‐type and fmr1−/y neurons increased the number of action potentials fired with increasing current amplitude (P < 0.001). However, the fmr1−/y neurons fired significantly more action potentials at the higher current injections (300–400 pA) compared to wild‐type neurons (Fig. 3 B; P < 0.05, WT: 14 cells/7 mice; fmr1−/y: 10 cells/6 mice). Consistent with the data in Fig. 2, there was no significant difference in the input resistance between wild‐type and fmr1−/y neurons (107 ± 14.5 vs. 97 ± 7 MΩ). Thus, the gain of action potential output in response to DC current injections was increased in fmr1−/y compared with WT L2/3 neurons.
Figure 3. L2/3 neurons in the fmr1−/y prefrontal cortex fire more action potentials in response to a current stimulus.
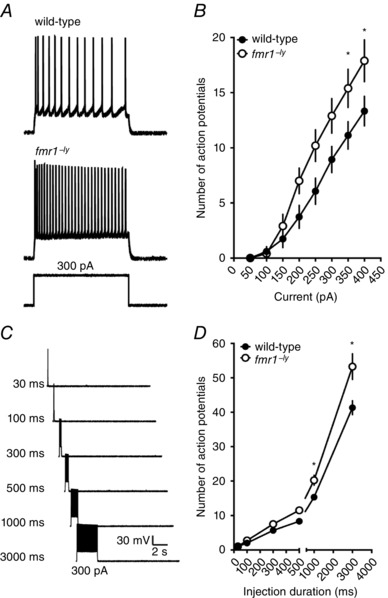
A, representative traces showing the response to a 300 pA current injection in a wild‐type (top) and fmr1−/y (bottom) neuron. B, summary graph showing that fmr1−/y L2/3 neurons fire more action potentials in response to increasing current amplitude compared to wild‐type neurons (wild‐type: 14 cells/7 mice; fmr1−/y: 10 cells/6 mice). C, representative traces showing the response to a 300 pA current injection of increasing duration. D, summary graph showing that fmr1−/y L2/3 neurons fire more action potentials in response to longer duration current injections compared to wild‐type neurons (wild‐type: 6 cells/3 mice; fmr1−/y: 6 cells/2 mice).
In a second set of experiments, we first determined the current necessary to elicit a single action potential during a 30 ms injection. Then the duration was progressively increased, while maintaining the current constant, and the number of action potentials measured (Fig. 3 C). As expected, both wild‐type and fmr1−/y neurons increased the number of action potential fired in response to increased current duration (P < 0.05). In agreement with the results above, fmr1−/y neurons fired significantly more action potentials than wild‐type neurons at the longer durations (500–3000 ms; Fig. 3 D; P < 0.05, WT: 6 cells/3 mice; fmr1−/y: 6 cells/2 mice).
Action potentials are taller and faster in fmr1−/y neurons
We next asked whether the properties of single action potentials were different between L2/3 neurons of fmr1−/y and wild‐type mice. To determine the membrane potential threshold for action potential generation, we used current injections that were just suprathreshold and varied the step duration (Higgs & Spain, 2011; Kalmbach et al. 2015) (Fig. 4 A). Action potential threshold significantly depolarized with increasing current duration for both wild‐type and fmr1−/y neurons (P < 0.0001). However, there was no significant difference in threshold between wild‐type and fmr1−/y neurons at all durations tested (Fig. 4 B, wild‐type: 10 cells/5 mice; fmr1−/y: 8 cells/4 mice). To compare single action potential properties, we elicited action potentials using a 100 ms step with the current adjusted to produce a single action potential with a latency of ∼50 ms (Fig. 4 C). We found that the peak of the action potential was significantly larger in fmr1−/y neurons compared to wild‐type neurons (Fig. 4 D, WT: 13 cells/6 mice; fmr1−/y: 11 cells/5 mice). A closer examination of the kinetics of the action potential waveform revealed significant differences in both the rising and falling phases of the action potential. The maximum rate of depolarization was ∼23% faster (Fig. 4 E) and the time to reach the maximum depolarization rate (relative to threshold) was significantly shorter (WT: 243 ± 4 μs; fmr1−/y: 205 ± 3 μs; P < 0.001) in fmr1−/y neurons. This resulted in a significantly faster rising action potential in fmr1−/y neurons compared to control (WT10–90%: 260 ± 3 μs; fmr1−/y 10–90%: 229 ± 6 μs; P < 0.005). Similarly, we found that maximum repolarization rate of action potential was also faster in fmr1−/y neurons (Fig. 4 F). This resulted in a significantly faster falling phase of the action potential in fmr1−/y neurons compared to control (WT90–10%: 1.17 ± 0.05 ms; fmr1−/y 90–10%: 0.865 ± 0.04 ms; P < 0.001). The net effect of the changes in action potential kinetics produced significantly narrower action potentials in fmr1−/y neurons (Fig. 4 G). Despite the faster action potential repolarization, there was no significant difference between wild‐type and fmr1−/y neurons in the amplitude of the peak afterhyperpolarization (Fig. 4 H).
Figure 4. Action potentials are taller, faster and narrower in fmr1−/y L2/3 neurons.
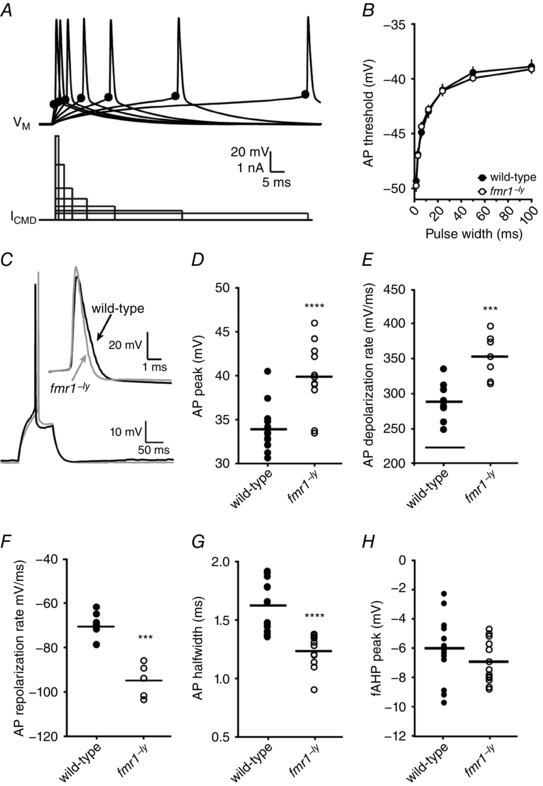
A, representative traces showing a single action potential (top) elicited by variable current injection (bottom). Note the current amplitude was adjusted in each case to elicit the action potential at the end of the current step. The black circle indicates the approximate location of the action potential threshold. B, there was no significant difference in action potential threshold between wild‐type and fmr1−/y neurons at any of the durations tested (wild‐type: 10 cells/5 mice; fmr1−/y: 8 cells/4 mice). C, representative traces showing single action potentials evoked by a 100 ms current injection. Note that the current amplitude was adjusted to elicit the action potential with a latency of ∼50 ms. Inset, representative action potentials from wild‐type (thick) and fmr1−/y (thin) neurons superimposed at threshold. D, summary graph showing that the peak voltage of action potentials in fmr1−/y L2/3 neurons is greater compared to action potentials in wild‐type neurons. E, summary graph showing that the maximum rate of depolarization during action potentials in fmr1−/y L2/3 neurons is greater compared to action potentials in wild‐type neurons. F, summary graph showing that the maximum rate of repolarization during action potentials in fmr1−/y L2/3 neurons is greater compared to action potentials in wild‐type neurons. G, summary graph showing that the half‐width of action potentials in fmr1−/y L2/3 neurons is shorter compared to action potentials in wild‐type neurons (wild‐type: 13 cells/6 mice; fmr1−/y: 11 cells/5 mice). H, summary graph showing that the amplitude of the fast afterhyperpolarization in fmr1−/y neurons is not significantly from wild‐type neurons (wild‐type: 13 cells/6 mice; fmr1−/y: 11 cells/5 mice).
The action potential phenotype persists during repetitive firing
We found that L2/3 neurons in fmr1−/y mice fire more action potentials for larger or prolonged current injections. Therefore, we next asked whether the differences in single action potential properties persisted during repetitive action potential firing. To control for the number of action potentials, trials in which ∼10 action potentials were elicited during the 500 ms current injection were selected for this analysis (mean firing frequency, WT: 20.2 ± 0.2 Hz, n = 14 cells/7 mice; fmr1−/y: 21.7 ± 0.61 Hz, n = 10 cells/6 mice). Both wild‐type and fmr1−/y neurons showed spike frequency adaptation during repetitive action potential firing (Fig. 5 A). However, there was no significant difference in the amount of spike frequency accommodation between wild‐type and fmr1−/y neurons (adaptation index; WT: 1.99 ± 0.41; fmr1−/y: 1.91 ± 0.46). Consistent with our previous result above, there was also no significant difference in the threshold for the first action potential in the train (Fig. 5 B). Although not statistically significant, the threshold for the subsequent action potentials in the train was consistently hyperpolarized by ∼1.7 mV in the fmr1−/y L2/3 neurons compared to wild‐type. Action potential peak voltage, rate of depolarization and repolarization for all action potentials in the train were significantly higher in fmr1−/y neurons (Fig. 5 C–E). As a result, action potentials in fmr1−/y neurons remained significantly narrower compared to wild‐type throughout the train (Fig. 5 F).
Figure 5. The differences between action potentials in wild‐type and fmr1−/y neurons persist during repetitive firing.
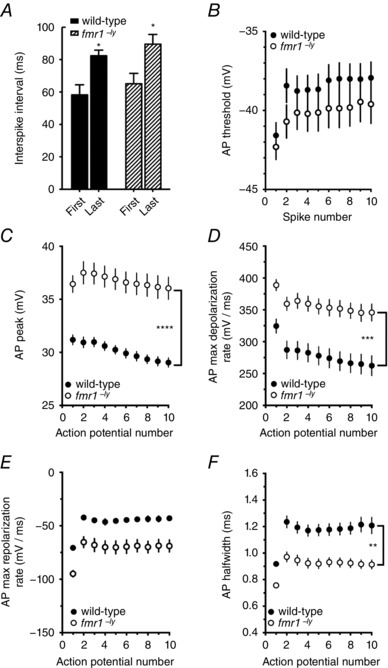
A, summary graph showing the interspike interval (ISI) between the 1st and 2nd action potential (first interval) and 9th and 10th action potential (last interval) in a train of 10 action potentials from wild‐type and fmr1−/y neurons. Note in both wild‐type and fmr1−/y neurons there was a significant increase in ISI between the first and last interval. B, summary graph showing that there was no significant difference in threshold during a train of 10 action potentials from wild‐type and fmr1−/y neurons (wild‐type: 14 cells/7 mice; fmr1−/y: 10 cells/6 mice). C, summary graph showing that action potential peak voltage remained significantly higher throughout the train in fmr1−/y neurons compared to wild‐type neurons. D, summary graph showing that the maximum rate of depolarization during each action potential was significantly faster throughout the train in fmr1−/y neurons compared to wild‐type neurons. E, summary graph showing that the maximum rate of repolarization during each action potential was significantly faster throughout the train in fmr1−/y neurons compared to wild‐type neurons. F, summary graph showing that action potentials remain significantly narrower throughout the train in fmr1−/y neurons compared to wild‐type neurons.
Dendritic morphology
Differences in dendritic morphology could influence the onset, shape and kinetics of action potentials by changing the impedance load on the soma/axon (Bekkers & Häusser, 2007; Eyal et al. 2014). We used a Neurolucida system to produce 3D reconstructions and perform morphological analyses on a subset of neurons from the electrophysiological datasets (Fig. 6 A, WT: 7 cells/7 mice; fmr1−/y: 8 cells/8 mice). Sholl analysis revealed that the basal dendritic arbour was significantly more branched than the apical dendritic arbour (two‐way ANOVA, P = 0.03) for both wild‐type and fmr1−/y L2/3 neurons. However, consistent with previous reports (Qin et al. 2011), there was no significant effect of genotype suggesting that the number of dendritic branches for either the basal or apical dendritic arbours between wild‐type and fmr1−/y neurons was not different (Fig. 6 B). Furthermore, there was no significant difference in the total dendritic length, total dendritic surface area, or the number of branch points between wild‐type and fmr1−/y neurons (Fig. 6 C). These results suggest that there are no differences in the dendritic morphology between wild‐type and fmr1−/y L2/3 pyramidal neurons.
Figure 6. Dendritic morphology is not different between WT and fmr1−/y L2/3 neurons.
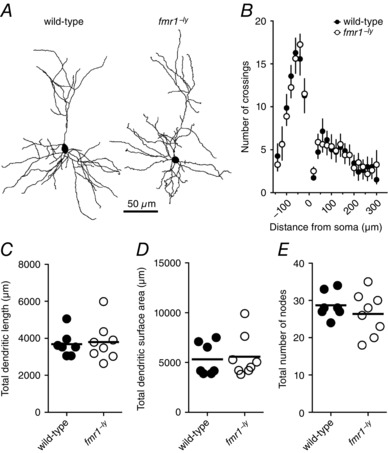
A, representative Neurolucida reconstructions of L2/3 pyramidal neurons from wild‐type and fmr1−/y mice showing dendritic branching patterns (wild‐type: 7 cells/7 mice; fmr1−/y: 8 cells/8 mice). B, Sholl analysis plot showing no significant difference in dendritic branching pattern between wild‐type and fmr1−/y L2/3 neurons. C–E, summary graphs showing no significant difference in dendritic length, surface area, or number of dendritic branch points between wild‐type and fmr1−/y L2/3 neurons.
Na+ conductance
The above results suggested that the amplitude and depolarization rate of action potentials were larger and faster in fmr1−/y neurons. The rising phase of an action potential is largely dictated by the complement and availability of voltage‐gated Na+ channels (Hodgkin & Huxley, 1952a). We made outside‐out patch clamp recordings from the soma of wild‐type and fmr1−/y L2/3 neurons to measure Na+ currents elicited with step voltage commands (−80 to +30 mV, Δ10 mV, 50 ms) from a holding potential of −90 mV (Fig. 7 A). Interestingly, the Na+ current recorded from fmr1−/y neurons was noticeably larger than wild‐type beginning with steps to −20 mV and becoming statistically significant at 0 mV (Fig. 7 B, WT: 12 patches/5 mice; fmr1−/y: 18 patches/5 mice). The maximum Na+ conductance density was significantly higher in fmr1−/y patches compared to wild‐type (Fig. 7 C). There was no significant difference in the area of the outside‐out patches between wild‐type and fmr1−/y neurons (WT: 3.45 ± 0.5 μm2; fmr1−/: 2.87 ± 0.5 μm2). Area was estimated by fitting the decay of the capacitive transient in response to a small voltage step (assuming 1 μF cm−2). The time constant of Na+ channel inactivation (τinact) was estimated from the decay of current evoked during test pulses. Consistent with previous results, τinact significantly decreased with increasing depolarization for both wild‐type and fmr1−/y neurons (Fig. 7 D). However, Na+ channel inactivation was significantly slower in fmr1−/y neurons at the most hyperpolarized potentials tested (Fig. 7 D). (It is worth noting that these differences in Na+ conductance density and kinetics were not observed at room temperature. Nor were there any differences in action potential firing or properties at room temperature, data not shown, see Discussion.) These data suggest that the Na+ current in fmr1−/y L2/3 pyramidal neurons is larger compared to wild‐type neurons. There were no significant differences in the voltage dependence of Na+ channel activation (Fig. 7 E) or steady‐state inactivation (Fig. 7 F). For a summary of the biophysical parameters of somatic Na+ channels recorded in this study, see Table 1.
Figure 7. Somatic Na+ currents were larger and longer lasting in fmr1−/y neurons compared to wild‐type.
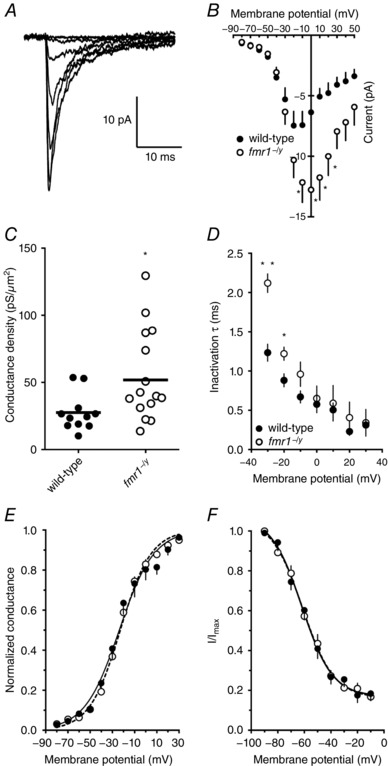
A, representative Na+ current traces recorded from somatic outside‐out patches elicited from a holding potential of –90 mV to the indicated voltage. B, summary graph showing the Na+ currents are larger at more depolarized potentials in patches from fmr1−/y neurons compared to wild‐type (wild‐type: 12 patches/5 mice; fmr1−/y: 18 patches/5 mice). C, summary graph showing the maximum Na+ conductance density is larger in fmr1−/y neurons compared to wild‐type. D, summary graph showing the Na+ current decays more slowly in fmr1−/y neurons compared to wild‐type. E, summary graph showing that there is no significant difference in Na+ channel activation between wild‐type and fmr1−/y patches. F, summary graph showing that there is no significant difference in Na+ channel steady‐state inactivation between wild‐type and fmr1−/y patches.
Table 1.
Biophysical parameters of somatic Na+ and K+ channels recorded in this study
| Parameter | Wild‐type | fmr1−/y |
|---|---|---|
| I Na (room temperature) | ||
| Maximum current (pA) | −12.9 ± 2.5 | −10.8 ± 1.6 |
| τinact @ −30 mV (ms) | 7.4 ± 0.78 | 6.1 ± 0.78 |
| τinact @ +20 mV (ms) | 1.8 ± 0.53 | 1.6 ± 0.2 |
| V 1/2 (mV) | −13.2 ± 1.43 | −12.2 ± 1.51 |
| k | 14.5 ± 1.51 | 15.8 ± 1.47 |
| I Na (32–34°C) | ||
| Maximum current (pA) | −7.5 ± 1.18 | −12.8 ± 1.89 |
| τinact @ −30 mV (ms) | 1.2 ± 0.11 | 2.1 ± 0.12 |
| τinact @ +30 mV (ms) | 0.3 ± 0.09 | 0.3 ± 0.17 |
| V 1/2 (mV) | −27.2 ± 1.71 | −24.4 ± 0.82 |
| k | 12.2 ± 1.67 | 10.9 ± 0.78 |
| I KA (32–34°C) | ||
| Maximum current (pA) | 131 ± 26.2 | 146.1 ± 34.7 |
| τinact @ +50 mV (ms) | 18.7 ± 2.59 | 15.1 ± 4.2 |
| V 1/2 (mV) | −1.6 ± 3.24 | 10.6 ± 3.18 |
| k | 17.4 ± 3 | 16.1 ± 2.6 |
| I K‐SLOW (32–34°C) | ||
| Maximum current (pA) | 131 ± 26.2 | 146.1 ± 34.7 |
| τinact @ +50 mV (ms) | 468.8 ± 75.9 | 479.1 ± 55.8 |
| V 1/2 (mV) | −16.3 ± 3.5 | −13.5 ± 2.8 |
| k | 26.1 ± 4.2 | 24.7 ± 3.2 |
Bold indicates parameters that were significantly different (p < 0.05) between wild‐type and fmr1−/y neurons.
K+ conductance
In addition to a significant difference in the rate of action potential depolarization, we observed that the rate of action potential repolarization was significantly faster in fmr1−/y neurons (Fig. 4 F and G). The repolarization phase of action potentials is controlled, in part, by the activation of voltage‐gated K+ channels (Hodgkin& Huxley, 1952b). To better characterize K+ channels, we measured K+ currents from somatic outside‐out patches in a separate set of voltage clamp recordings. There was no significant difference in the area of the outside‐out patches between wild‐type and fmr1−/y neurons (WT: 11.1 ± 0.9 μm2; fmr1−/y: 9.5 ± 1.5 μm2). In agreement with our prior findings in prefrontal layer 5 pyramidal neurons (Kalmbach et al. 2015), the total K+ current measured in L2/3 neurons could be separated by voltage protocols into three distinct currents: a rapidly inactivating, a slowly inactivating, and a non‐inactivating (Fig. 8 A). There was no difference in the maximum amplitude of the total, rapidly inactivating, slowly inactivating, or non‐inactivating K+ currents between wild‐type and fmr1−/y neurons (Fig. 8 B, WT: 9 patches/4 mice; fmr1−/y: 8 patches/3 mice). The τinact values of the rapidly inactivating (WT: 20.1 ± 1.43 ms; fmr1−/y: 19.1 ± 0.9 ms; P = 0.551) and slowly inactivating K+ current (WT: 475.9 ± 62.94 ms; fmr1−/y: 452.5 ± 31.78 ms; P = 0.748) were not significantly different between wild‐type and fmr1−/y neurons and were consistent with previously published values for K+ currents measured in outside patches from cortical neurons (Bekkers, 2000; Kalmbach et al. 2015). Although there was no significant difference in maximum A‐type K+ conductance density (Fig. 8 C), the V 1/2 of the activation curve of rapidly inactivating current was significantly depolarized in the fmr1−/y neurons compared to wild‐type neurons (Fig. 8 D). In contrast, there was no significant difference in either the maximum conductance density (Fig. 8 E) or voltage dependence of activation (Fig. 8 F) of the slowly inactivating K+ current between wild‐type and fmr1−/y neurons. Taken together, our physiological data suggest that differences in somatic Na+ and the A‐type K+ conductance are largely responsible for the faster action potential kinetics and an increase in action potential output observed in fmr1−/y L2/3 neurons. For a summary of the biophysical parameters of somatic K+ channels recorded in this study, see Table 1.
Figure 8. The activation of somatic I KA, but not I K‐SLOW, was depolarized in fmr1−/y neurons compared to wild‐type.
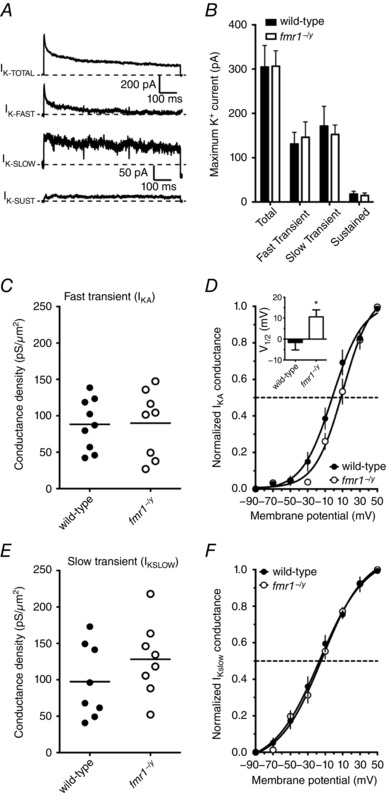
A, representative traces illustrating each of the types of K+ currents recorded from somatic outside‐out patches elicited using the indicated protocol. B, summary graph showing that there was no significant difference in the maximum amplitude of each of the K+ currents measured between wild‐type and fmr1−/y neurons (wild‐type: 9 patches/4 mice; fmr1−/y: 8 patches/3 mice). C, summary graph showing the maximum conductance density for the rapidly inactivating K+ current is not different between fmr1−/y neurons and wild‐type. D, summary graph showing that the voltage dependence of activation of I KA was significantly depolarized in fmr1−/y patches compared to wild‐type patches. E, summary graph showing the maximum conductance density for the slowly inactivating K+ current is not different between fmr1−/y neurons and wild‐type. F, summary graph showing that there is no significant difference in the voltage dependence of activation of I K‐SLOW between wild‐type and fmr1−/y patches.
Simulations
The experimental results described above suggested that firing rate and various properties related to the action potential, including half‐width, max dV/dt and height, were significantly different between prefrontal L2/3 neurons of fmr1−/y and wild‐type mice (Figs 3, 4, 5). These results were accompanied by findings that properties of fast Na+ channels and A‐type K+ channels were also significantly different in PFC L2/3 neurons of fmr1−/y compared to wild‐type mice (Figs 7 and 8). Are these changes in properties of fast Na+ channels and A‐type K+ channels sufficient to bring about differences in phenotypic traits, related to firing rate and action potential properties, between fmr1−/y and wild‐type neurons? To answer this question, we utilized a computational approach. We used a single compartmental model expressing six different ion channels: fast Na+ (NaF), delayed rectifier K+ (KDR), A‐type K+ (KA), M‐type K+ (KM), L‐type Ca++ (CaL) and calcium‐activated small conductance K+ (SK) channels (Fig. 9 A).
Figure 9. Simulations reveal that differences in fast Na+ and A‐type K+ channels between wild‐type and fmr1−/y neurons bring about differences in firing rate and action potential properties.
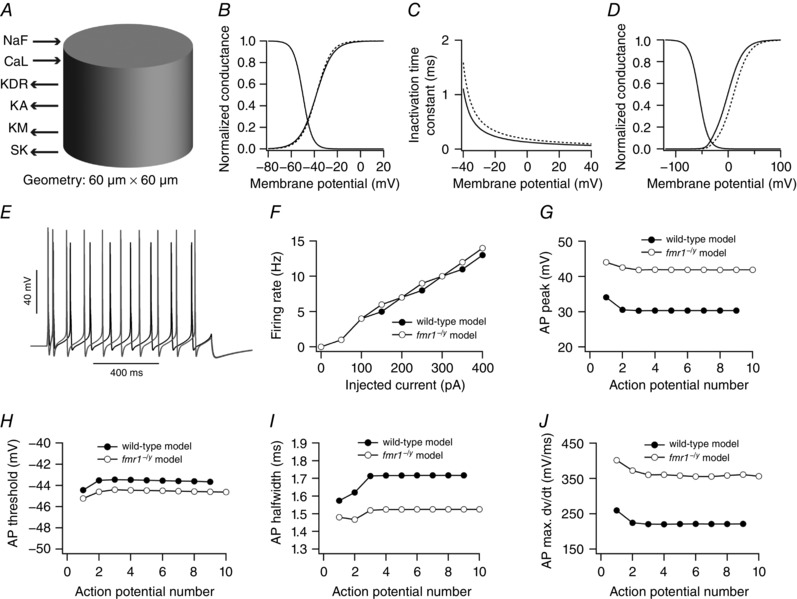
A, single compartmental model, expressing six ion channels, used in this study. B–D, differences in properties of fast Na+ and A‐type K+ channels to define wild‐type and fmr1−/y model neuron: activation curve (B) and inactivation time constant of fast Na+ channels (C), and activation curve of A‐type K+ channels (D). Continuous and dashed lines denote wild‐type and fmr1−/y model neuron, respectively. E, example voltage traces for 300 pA current injection from wild‐type (black) and fmr1−/y (grey) model neuron. F, firing rate profiles of wild‐type and fmr1−/y model neuron. G–J, peak voltage (G), threshold (H), half‐width (I) and maximum dV/dt (J) of action potentials in a spike train obtained from wild‐type and fmr1−/y model neuron.
Values of all six conductances were hand‐tuned such that the base model neuron was able to reproduce various experimental observations obtained from wild‐type neurons including firing rate, and action potential peaks, thresholds and maximum dV/dt in spike train. One measurement, action potential half‐widths in a spike train, did not match quantitatively to their experimental counterparts, although qualitatively, the trend of these measurements within the spike train were similar to that of experimental observations (Fig. 9 I vs. 5 F). This discrepancy could be attributed to the differences between channel kinetics of native PFC L2/3 neuronal ion channels and channel kinetics present in the model neuron. Furthermore, apart from and in conjunction with differences in ion channels kinetics, the model neuron expressed only six types of conductances whereas native neurons express other ion channels not present in our model. Nonetheless, values of action potential half‐widths in our model were within the experimentally observed ranges when single action potentials were used to quantify these properties (Fig. 4).
The properties of fast Na+ channels and A‐type K+ channels were changed in accordance with the experimental observations to convert the wild‐type model neuron into an fmr1−/y model neuron (Figs 7 and 8). Specifically, maximum Na+ conductance was increased by 1.5‐fold, slope of the activation curve of fast Na+ channels was increased (Fig. 9 B), inactivation time constant of fast Na+ channels was increased (Fig. 9 C) and activation curve of A‐type K+ conductance was shifted to depolarized potentials by 8 mV (Fig. 9 D). Strikingly, we found that our model neuron was able to reproduce various experimental observations. Specifically, in the fmr1−/y model neuron the action potential peak value was increased (Fig. 9 G), threshold and half‐width were decreased (Fig. 9 H and I), and maximum dV/dt was increased (Fig. 9 J), as compared to the wild‐type neuron model. Although the firing rate in the fmr1−/y model neuron was increased compared to wild‐type neuron model (Fig. 9 F), this increase was less compared to the experimental counterpart (Fig. 3 B). One possible reason for this is that the fast afterhyperpolarization was larger in the fmr1−/y model neuron compared to the wild‐type due to greater activation of delayed rectifier type K+ current (data not shown). Taken together, these results suggest that changes in properties of fast Na+ channels and A‐type K+ channels were sufficient to explain some of the differences in phenotypic traits related to firing rate and action potential properties between fmr1−/y and wild‐type neurons of PFC L2/3.
To what extent do these modelling results hold? Are these results influenced by the choice of conductance values of six ion channels in wild‐type neurons? To answer these questions, we employed powerful global sensitivity analysis (Prinz et al. 2003, 2004; Rathour & Narayanan, 2012b, 2014; Anirudhan & Narayanan, 2015). Randomizing the conductance values of six ion channels generated a population of wild‐type neuronal models to perform a global sensitivity analysis. Specifically, conductances of six ion channels were assigned a range such that they uniformly and independently span a neighbourhood of their default values present in our base wild‐type model (mS cm−2): , , , , and . With all conductances randomized, we generated 5000 model neurons, each comprising a unique set of values for six conductances. The validity of these models was tested against the experimental measurements. Each model neuron had to satisfy 13 constraints related to firing rate and action potential properties, in order to become a valid model neuron (Table 2). As a test of this validation procedure, we constrained only the properties of the first action potential in a spike train while the properties of rest of the action potentials were unconstrained. Employing this validation procedure on each model yielded 25 valid wild‐type neuronal models (0.5% of total models).
Table 2.
Bounds on measurements to define a wild‐type valid model population
| Constraints imposed on model's firing rate | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Injected current (pA) | 0 | 50 | 100 | 150 | 200 | 250 | 300 | 350 | 400 |
| Experimentally observed Firing rate range (Hz) | 0 | 0 | 0–6 | 0–10 | 1–13 | 2–16 | 4–19 | 6–21 | 8–23 |
| Firing rate range used for model neurons (Hz) | 0 | 0–1 | 0–5 | 2–7 | 3–9 | 5–12 | 8–15 | 11–18 | 13–21 |
| Constraints imposed on the properties of first action potential in a spike train | ||
|---|---|---|
| Measurements | Experimentally observed range | Range used for model neurons |
| Threshold (mV) | –45 to –39 | −45 to −43 |
| Peak (mV) | 29 to 32 | 30 to 35 |
| Half‐width (ms) | 0.8 to 1.1 | 1.5 to 2 |
| Maximum dV/dt (mV ms−1) | 253 to 390 | 250 to 300 |
Looking at the distribution of conductance values of six ion channels across the 25 valid wild‐type model neurons, we found that there was a very narrow range for fast Na+ conductance, while other conductances were relatively distributed across their assigned ranges (Fig. 10 A). To generate a population of fmr1−/y models we transformed these 25 valid wild‐type neuronal models into fmr1−/y neuronal models by incorporating the aforementioned changes in properties of fast Na+ and A‐type K+ channels. As at the single neuron level (Fig. 9), we found that the firing rate of the fmr1−/y model population was higher compared to the wild‐type model population (Fig. 10 B). We also found that in the fmr1−/y model population action potential height and maximum dV/dt were increased (Fig. 10 C and F), while threshold and half‐width were decreased (Fig. 10 D and E), compared to the wild‐type model population as measured experimentally. It should be noted that we only constrained the properties of the first action potential in the spike train while properties of the rest of the action potentials in train were automatically optimized. Similar to our observation at the single neuron level, the increase in firing rate of the fmr1−/y model population was less compare to the experimental counterpart (Fig. 3 B vs. 10 B). Nonetheless, these results suggest that irrespective of the choice of the conductance values of six ion channels, changes in properties of fast Na+ and A‐type K+ channels could explain experimentally observed phenotypic traits related to firing rate and action potential properties of PFC L2/3 fmr1−/y and wild‐type neurons.
Figure 10. Differences in firing rate and action potential properties of wild‐type and fmr1−/y model neurons are independent of the choice of the conductance values in wild‐type neuronal models.
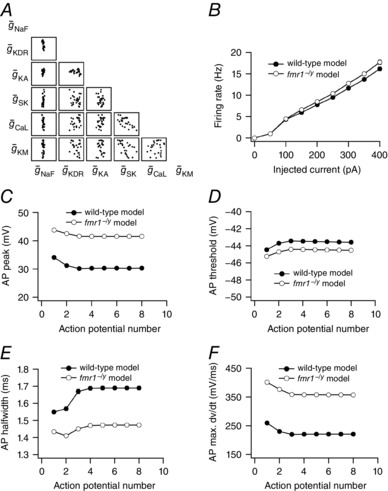
A, scatter plot matrix depicting variability in conductance values across 25 wild‐type neuronal models. B, population averaged firing rate profiles obtained from wild‐type and fmr1−/y model population. C–F, population averaged peak voltage (C), threshold (D), half‐width (E) and maximum dV/dt (F) of action potentials in a spike train obtained from wild‐type and fmr1−/y model population. For B–F, data is presented as mean ± SEM (n = 25).
Discussion
Higher order processing in the prefrontal cortex underlies executive control over other brain regions. Aberrant function of the prefrontal cortex is believed to contribute to many neurological phenotypes of fragile X syndrome and autistic behaviours including impulsivity, attention deficit, obsessive compulsivity and cognitive inflexibility (Hagerman et al. 2005). We previously reported that altered functional expression of voltage‐gated K+ and h‐channels leads to altered input–output properties of layer 5 pyramidal neurons in the prefrontal cortex of the fmr1−/y mouse (Kalmbach et al. 2015). In this study, we demonstrate that layer 2/3 pyramidal neurons in the prefrontal cortex of the fmr1−/y mouse also have altered input–output properties. However, unlike L5 neurons, which have a hyperpolarized action potential threshold due in part to a downregulation of a KV1‐like current (Kalmbach et al. 2015), the action potential threshold in L2/3 fmr1−/y neurons is not significantly different from wild‐type. Instead, we found that action potentials were significantly taller and faster in L2/3 fmr1−/y neurons compared to wild‐type.
A larger dendritic impedance load, due to a more elaborate dendritic arbour, would increase the onset and kinetics of action potentials (Eyal et al. 2014). We found no significant difference in either the basal or the apical dendritic arbours between wild‐type and fmr1−/y L2/3 neurons. A greater density of dendritic spines would also increase the impedance load and result in faster kinetics. There are conflicting reports as to whether there is an increase in dendritic spine density in fmr1−/y neurons (Comery et al. 1997; Nimchinsky et al. 2001; Grossman et al. 2006). However, a study looking at L2/3 neurons in the prefrontal cortex found no significant difference in dendritic spine density between wild‐type and fmr1−/y neurons (Meredith et al. 2007). Taken together these results suggest that the differences in action potential kinetics we observed were not due to a change in dendritic load on the soma or axon initial segment. We found that somatic Na+ currents were larger and longer lasting in fmr1−/y L2/3 neurons compared to wild‐type. We also found that activation of a rapidly inactivating A‐type K+ current was depolarized by ∼10 mV compared to wild‐type. Realistic conductance‐based simulations showed that these changes in Na+ and K+ channel function reproduced the action potential kinetic and firing rate phenotypes.
Consistent with previously published data, we found no difference in resting membrane potential, input resistance, or time constant in L2/3 neurons of fmr1−/y prefrontal cortex (Meredith et al. 2007). It should be noted that Meredith et al. reported no differences in action potential properties. One potential source for this discrepancy is age of the mice used in the studies; we used 8‐ to 16‐week‐old mice while Meredith and Mansvelder used 3‐ to 4‐week‐old mice.
Potential mechanisms
Our data suggest that an increase in Na+ conductance (Fig. 7) and a depolarizing shift in the voltage dependence of a rapidly inactivating K+ conductance (Fig. 8) contribute to the elevated firing rate and higher rate of action potential depolarization and repolarization in L2/3 neurons in the prefrontal cortex of the fmr1−/y mouse. In central nervous system neurons, Na+ channels are composed of a pore‐forming α subunit (NaV1.1, NaV1.2, NaV1.3, or NaV1.6) and auxiliary β subunits (NaVβ1–4) (Catterall, 2012). The mRNA for two Na+ channel α subunits, NaV1.2 (SCN2A) and NaV1.6 (SCN8A), are known targets of fragile X mental retardation protein (FMRP; Darnell et al. 2011). If FMRP represses translation of SCN2A or SCN8A, then its loss would increase the expression of NaV1.2 and NaV1.6 in L2/3 neurons respectively. Alternatively, the total expression of α subunits may be unaltered but the surface expression could be higher in fmr1−/y neurons. The simplest explanation for the higher somatic Na+ conductance density would be an increase in somatic Na+ channel expression in fmr1−/y L2/3 pyramidal neurons. However, we found that there was no difference in the conductance density when measured at room temperature. This suggests that the loss of FMRP does not result in a constitutive elevation in somatic Na+ channel expression.
The biophysical properties and surface expression of Na+ channel α subunits is regulated in part by β subunits (Patino & Isom, 2010). Accordingly a change in the expression or association of the Na+ channel pore‐forming subunit with β subunits could alter the kinetics and/or surface expression of Na+ channels. One potential mechanism underlying the greater conductance density in fmr1−/y neurons would be an increase in the activation to inactivation balance. It is possible that the activation rate for Na+ channels in fmr1−/y neurons is faster compared to wild‐type neurons. We could not directly test this hypothesis due to very rapid activation rate of Na+ channels. We also found that the rate of Na+ current inactivation was slower in fmr1−/y neurons (at more hyperpolarized potentials, Fig. 7 D). The β subunits of Na+ channels modulate the rate of fast inactivation (Goldin, 2003). The presence of β1 and β3 speed the rate of inactivation of NaV1.2, 1.3 and 1.4 (McCormick et al. 1998; Stevens et al. 2001). A loss of expression of β1 or β3 or reduced association with α subunits could result in slower inactivation in fmr1/y neurons.
In cortical neurons ankyrin G is responsible for the clustering of Na+ channels at the axon initial segment (Zhou et al. 1998). The mRNA for ankyrin G is also a target of FMRP (Darnell et al. 2011) suggesting that loss of FMRP would increase the expression of ankyrin G and thereby increase in the density of Na+ channels. Interestingly, both the β1 and β2 subunits of Na+ channels play a role in recruitment of ankyrin and the clustering of Na+ channels (Malhotra et al. 2000, 2002). How the loss of FMRP affects the expression of α and β Na+ channel subunits as well as ankyrin proteins remains to be explored.
A recent study found that layer III pyramidal neurons in the entorhinal cortex of the fmr1−/y mouse have more persistent Na+ current compared to wild‐type (Deng & Klyachko, 2016). Although they did not directly test for changes in the transient Na+ current, they did report that the maximal rise speed of action potentials was significantly faster in fmr1−/y neurons. The increase in persistent Na+ current contributed to a hyperpolarized action potential threshold in fmr1−/y neurons (Deng & Klyachko, 2016). Although we did not measure persistent Na+ currents, we did not find a difference in action potential threshold between wild‐type and fmr1−/y L2/3 prefrontal neurons. There are several potential explanations for the difference in these observations. First, we and others have shown that identified channelopathies in fmr1−/y mice are cell‐type specific (Brager & Johnston, 2014; Contractor et al. 2015). One possibility is that Na+ channels are altered in a cell type‐specific manner between the prefrontal and entorhinal cortex. Second, there is a developmental increase in a slowly inactivating component of the Na+ current in neocortical neurons (Huguenard et al. 1988). As we did not observe a double exponential decay of the Na+ current, it is possible that there is minimal contribution of this slowly inactivating component in L2/3 pyramidal neurons of the PFC. Lastly, the drugs used to confirm the action of the persistent Na+ current in entorhinal neurons are not completely specific and can have some effect on the transient Na+ current (Park et al. 2013).
We found that the activation of A‐type K+ channels was depolarized by 10 mV in the fmr1−/y L2/3 neurons. These results are in contrast to our previous findings where we found that the activation of A‐type K+ channels in CA1 neurons was hyperpolarized in fmr1−/y mice (Routh et al. 2013), or unaffected in L5 pyramidal neurons of the fmr1−/y PFC (Kalmbach et al. 2015). One potential mechanism for the shift in activation is activity of protein kinases. Stimulation of protein kinase A or protein kinase C will produce a 10–15 mV shift in the activation of A‐type K+ channels in CA1 pyramidal neurons (Hoffman & Johnston, 1998). One caveat to this is that the molecular identity of the A‐type K+ current in PFC neurons in not known. The ability of protein kinases to shift the activation of A‐type K+ channels may be restricted to the KV4.2 subunit, the primary pore forming subunit in CA1 neurons. We previously demonstrated that in L5 neurons the I KA is mediated by KV4 channels (Kalmbach et al. 2015). However, we cannot say whether these channels are composed of KV4.2 or KV4.3 subunits. Interestingly, in ventricular myocytes the β1 subunit of Na+ channels can associate with the KV4 channels that underlie the rapidly inactivating K+ current (Deschênes & Tomaselli, 2002; Deschênes et al. 2008). This may suggest that a change in Na+ channel β subunit expression or function contributes to both the Na+ channel and A‐type K+ channel changes observed in fmr1−/y L2/3 neurons.
Physiological consequences
The synaptic and ion channel phenotypes in fragile X syndrome are known to be both cell‐type and brain region specific (Huber et al. 2002; Desai et al. 2006; Brager et al. 2012; Zhang et al. 2014; Contractor et al. 2015; Kalmbach et al. 2015). Here we show that action potentials in L2/3 neurons of fmr1−/y mouse prefrontal cortex are narrower compared to wild‐type action potentials. In contrast, action potentials in hippocampal CA3 neurons, L5 entorhinal and L5 somatosensory neurons are significantly broader in fmr1−/y mice compared to wild‐type (Deng et al. 2013; Zhang et al. 2014). In both of these studies, the broader action potentials were due in part to a loss of BK channel function. Although we did not directly test for changes in BK channels, several lines of evidence suggest that an increased BK channel activity did not contribute to the action potential phenotype in fmr1−/y L2/3 neurons. First, in hippocampal and cortical pyramidal neurons increasing or decreasing BK channel activity decreases or increases spike frequency accommodation, respectively (Gu et al. 2007). We found no significant difference in spike frequency adaptation across a range of current injections. Second, reduction in BK channel activity is accompanied by enhanced activity‐dependent broadening of action potentials (Shao et al. 1999; Faber & Sah, 2003). Indeed, in both CA3 and L5 somatosensory neurons of the fmr1−/y mouse, there was enhanced frequency‐dependent broadening of action potential width (Deng et al. 2013; Zhang et al. 2014). Although we observed activity‐dependent increases in action potential width during repetitive firing in L2/3 neurons (Fig. 5 F), there was no significant difference between fmr1−/y and wild‐type neurons. Lastly, BK channels contribute to the fast afterhyperpolarization (Poolos & Johnston, 1999). We found no significant difference in the fast afterhyperpolarization after either a single action potential (Fig. 4 H) or train of action potentials. A comparison between our experimental and modelling data suggest that additional conductances may be altered in fmr1−/y neurons and contribute to the elevated action potential firing phenotype.
Alterations to the action potential waveform have downstream effects on calcium influx and neurotransmitter release (Sabatini & Regehr, 1997). Deng et al. (2011) reported enhanced responses to high frequency stimulation, increased augmentation and reduced short‐term depression in fmr1−/y CA3 neurons. This was due in part to the enhanced activity‐dependent broadening of action potentials in fmr1−/y CA3 neurons leading to increased glutamate release. As we report that action potentials are narrower, the potential impact of narrower action potentials on synaptic function would be a reduction in excitatory synaptic strength between L2/3 neurons and their synaptic targets. This may also have implications for the induction of long‐term synaptic plasticity.
Impact on neuronal coding
Cortical neurons are able to encode time‐varying signals by phase locking the action potential output to high frequency inputs, and they respond very rapidly to change in the current stimulus (Köndgen et al. 2008; Higgs & Spain, 2009; Eyal et al. 2014; Testa‐Silva et al. 2014). Theoretical studies identified action potentials with fast onset dynamics as an essential component of these properties (Brunel et al. 2001; Fourcaud‐Trocmé et al. 2003). Experimental manipulations that reduce the onset dynamics of somatic action potentials, low concentrations of TTX or reducing extracellular Na+ concentration, severely impaired the encoding of high frequency input (Ilin et al. 2013). These manipulations also produced somatic action potentials that were lower in amplitude and broader in width. We found that somatic action potentials in fmr1−/y L2/3 neurons were larger in amplitude and narrower in width. We suggest that this is due in part to a larger somatic Na+ conductance. One prediction from these results is that L2/3 neurons in fmr1−/y mice would have improved phase locking to high frequency inputs. Indeed, L2/3 neurons in the somatosensory cortex of fmr1−/y show elevated firing rates and higher degree of AP synchrony in vivo compared to wild‐type mice (Gonçalves et al. 2013). We suggest that elevated Na+ current may contribute to these state‐dependent changes in network function.
Additional information
Competing interests
The authors declare that there are no competing financial interests
Author contributions
B.E.K., D.J. and D.H.B. conceived and designed the experiments. B.N.R., M.E.B. and D.H.B. performed the electrophysiological experiments. R.K.R. designed and performed the neuronal simulations. B.N.R., R.K.R., M.E.B. and D.H.B. performed the data analysis. B.N.R., R.K.R., M.E.B., B.E.K., D.J. and D.H.B. interpreted the data, authored, revised and approved the final version of the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work supported by National Institutes of Health grants R01 MH100510 (D.H.B.) and R01 MH094839 (D.J.).
Authors’ present addresses
M. E. Baumgardner: University of Cambridge, Cambridge UK.
B. E. Kalmbach: Allen Institute for Brain Science, Seattle, WA, USA.
Acknowledgements
The authors wish to thank Komal Parikh for neuronal reconstructions.
References
- Anirudhan A & Narayanan R (2015). Analogous synaptic plasticity profiles emerge from disparate channel combinations. J Neurosci 35, 4691–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron AR, Robbins TW & Poldrack RA (2014). Inhibition and the right inferior frontal cortex: one decade on. Trends Cogn Sci 18, 177–185. [DOI] [PubMed] [Google Scholar]
- Bekkers JM (2000). Properties of voltage‐gated potassium currents in nucleated patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol 525, 593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM & Häusser M (2007). Targeted dendrotomy reveals active and passive contributions of the dendritic tree to synaptic integration and neuronal output. Proc Natl Acad Sci USA 104, 11447–11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR & Johnston D (2012). Impaired dendritic expression and plasticity of h‐channels in the fmr1–/y mouse model of fragile X syndrome. Cell Reports 1, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH & Johnston D (2014). Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res Bull 103, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel N, Chance FS, Fourcaud N & Abbott LF (2001). Effects of synaptic noise and filtering on the frequency response of spiking neurons. Phys Rev Lett 86, 2186–2189. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2012). Voltage‐gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590, 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ & Greenough WT (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci USA 94, 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA & Portera‐Cailliau C (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD & Darnell RB (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembrow N & Johnston D (2014). Subcircuit‐specific neuromodulation in the prefrontal cortex. Front Neural Circuits 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P‐Y & Klyachko VA (2016). Increased persistent sodium current causes neuronal hyperexcitability in the entorhinal cortex of fmr1 knockout mice. Cell Reports 16, 3157–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P‐Y, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS & Klyachko VA (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P‐Y, Sojka D & Klyachko VA (2011). Abnormal presynaptic short‐term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci 31, 10971–10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Casimiro TM, Gruber SM & Vanderklish PW (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol 96, 1734–1745. [DOI] [PubMed] [Google Scholar]
- Deschênes I, Armoundas AA, Jones SP & Tomaselli GF (2008). Post‐transcriptional gene silencing of KChIP2 and Navβ1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol 45, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes I & Tomaselli GF (2002). Modulation of Kv4.3 current by accessory subunits. FEBS Lett 528, 183–188. [DOI] [PubMed] [Google Scholar]
- Euston DR, Gruber AJ & McNaughton BL (2012). The role of medial prefrontal cortex in memory and decision making. Neuron 76, 1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyal G, Mansvelder HD, de Kock CPJ & Segev I (2014). Dendrites impact the encoding capabilities of the axon. J Neurosci 34, 8063–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ESL & Sah P (2003). Ca2+‐activated K+ (BK) channel inactivation contributes to spike broadening during repetitive firing in the rat lateral amygdala. J Physiol 552, 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaud‐Trocmé N, Hansel D, van Vreeswijk C & Brunel N (2003). How spike generation mechanisms determine the neuronal response to fluctuating inputs. J Neurosci 23, 11628–11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL (2003). Mechanisms of sodium channel inactivation. Curr Opin Neurobiol 13, 284–290. [DOI] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P & Portera‐Cailliau C (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci 16, 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC & Greenough WT (2006). Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature‐appearing profile of dendritic spines. Brain Res 1084, 158–164. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K & Storm JF (2007). BK potassium channels facilitate high‐frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol 580, 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Ono MY & Hagerman PJ (2005). Recent advances in fragile X: a model for autism and neurodegeneration. Curr Opin Psychiatry 18, 490–496. [DOI] [PubMed] [Google Scholar]
- Harris KD & Shepherd GMG (2015). The neocortical circuit: themes and variations. Nat Neurosci 18, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilman KM, Voeller KK & Nadeau SE (1991). A possible pathophysiologic substrate of attention deficit hyperactivity disorder. J Child Neurol 6 Suppl, S76–S81. [DOI] [PubMed] [Google Scholar]
- Higgs MH & Spain WJ (2009). Conditional bursting enhances resonant firing in neocortical layer 2‐3 pyramidal neurons. J Neurosci 29, 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs MH & Spain WJ (2011). Kv1 channels control spike threshold dynamics and spike timing in cortical pyramidal neurones. J Physiol 589, 5125–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines ML & Carnevale NT (1997). The NEURON simulation environment. Neural Comput 9, 1179–1209. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL & Huxley AF (1952a). Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo . J Physiol 116, 449–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL & Huxley AF (1952b). A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA & Johnston D (1998). Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci 18, 3521–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST & Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Hamill OP & Prince DA (1988). Developmental changes in Na+ conductances in rat neocortical neurons: appearance of a slowly inactivating component. J Neurophysiol 59, 778–795. [DOI] [PubMed] [Google Scholar]
- Ilin V, Malyshev A, Wolf F & Volgushev M (2013). Fast computations in cortical ensembles require rapid initiation of action potentials. J Neurosci 33, 2281–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach BE, Johnston D & Brager DH (2015). Cell‐type specific channelopathies in the prefrontal cortex of the fmr1‐/y mouse model of fragile X syndrome eNeuro 2, ENEURO.0114‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köndgen H, Geisler C, Fusi S, Wang X‐J, Lüscher H‐R & Giugliano M (2008). The dynamical response properties of neocortical neurons to temporally modulated noisy inputs in vitro. Cerebral Cortex 18, 2086–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DD, Osterweil EK, Chen SP, Tye LD & Bear MF (2011). Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA 108, 2587–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF & Sejnowski TJ (1996). Influence of dendritic structure on firing pattern in model neocortical neurons. Nature 382, 363–366. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kazen‐Gillespie K, Hortsch M & Isom LL (2000). Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell‐cell contact. J Biol Chem 275, 11383–11388. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Koopmann MC, Kazen‐Gillespie KA, Fettman N, Hortsch M & Isom LL (2002). Structural requirements for interaction of sodium channel β1 subunits with ankyrin. J Biol Chem 277, 26681–26688. [DOI] [PubMed] [Google Scholar]
- Martin HGS, Lassalle O, Brown JT & Manzoni OJ (2015). Age‐dependent long‐term potentiation deficits in the prefrontal cortex of the Fmr1 knockout mouse model of fragile X syndrome. Cerebral Cortex 26, 2084–2092. [DOI] [PubMed] [Google Scholar]
- McCormick KA, Isom LL, Ragsdale D, Smith D, Scheuer T & Catterall WA (1998). Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit. J Biol Chem 273, 3954–3962. [DOI] [PubMed] [Google Scholar]
- Meredith RM, Holmgren CD, Weidum M, Burnashev N & Mansvelder HD (2007). Increased threshold for spike‐timing‐dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–638. [DOI] [PubMed] [Google Scholar]
- Migliore M, Hoffman DA, Magee JC & Johnston D (1999). Role of an A‐type K+ conductance in the back‐propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J Comput Neurosci 7, 5–15. [DOI] [PubMed] [Google Scholar]
- Miller EK & Cohen JD (2001). An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24, 167–202. [DOI] [PubMed] [Google Scholar]
- Morishima M & Kawaguchi Y (2006). Recurrent connection patterns of corticostriatal pyramidal cells in frontal cortex. J Neurosci 26, 4394–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Morita K, Kubota Y & Kawaguchi Y (2011). Highly differentiated projection‐specific cortical subnetworks. J Neurosci 31, 10380–10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R & Johnston D (2010). The h current is a candidate mechanism for regulating the sliding modification threshold in a BCM‐like synaptic learning rule. J Neurophysiol 104, 1020–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM & Svoboda K (2001). Abnormal development of dendritic spines in FMR1 knock‐out mice. J Neurosci 21, 5139–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka T & Kawaguchi Y (2011). Cell diversity and connection specificity between callosal projection neurons in the frontal cortex. J Neurosci 31, 3862–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YY, Johnston D & Gray R (2013). Slowly inactivating component of Na+ current in peri‐somatic region of hippocampal CA1 pyramidal neurons. J Neurophysiol 109, 1378–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA & Isom LL (2010). Electrophysiology and beyond: multiple roles of Na+ channel β subunits in development and disease. Neurosci Lett 486, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirazi P, Brannon T & Mel BW (2003). Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron 37, 977–987. [DOI] [PubMed] [Google Scholar]
- Poolos NP & Johnston D (1999). Calcium‐activated potassium conductances contribute to action potential repolarization at the soma but not the dendrites of hippocampal CA1 pyramidal neurons. J Neurosci 19, 5205–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz AA, Billimoria CP & Marder E (2003). Alternative to hand‐tuning conductance‐based models: construction and analysis of databases of model neurons. J Neurophysiol 90, 3998–4015. [DOI] [PubMed] [Google Scholar]
- Prinz AA, Bucher D & Marder E (2004). Similar network activity from disparate circuit parameters. Nat Neurosci 7, 1345–1352. [DOI] [PubMed] [Google Scholar]
- Qin M, Entezam A, Usdin K, Huang T, Liu Z‐H, Hoffman GE & Smith CB (2011). A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol Dis 42, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathour RK & Narayanan R (2012a). Inactivating ion channels augment robustness of subthreshold intrinsic response dynamics to parametric variability in hippocampal model neurons. J Physiol 590, 5629–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathour RK & Narayanan R (2012b). Influence fields: a quantitative framework for representation and analysis of active dendrites. J Neurophysiol 107, 2313–2334. [DOI] [PubMed] [Google Scholar]
- Rathour RK & Narayanan R (2014). Homeostasis of functional maps in active dendrites emerges in the absence of individual channelostasis. Proc Natl Acad Sci USA 111, E1787–E1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh BN, Johnston D & Brager DH (2013). Loss of functional A‐type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J Neurosci 33, 19442–19450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell VA (2002). Hypodopaminergic and hypernoradrenergic activity in prefrontal cortex slices of an animal model for attention‐deficit hyperactivity disorder–the spontaneously hypertensive rat. Behav Brain Res 130, 191–196. [DOI] [PubMed] [Google Scholar]
- Sabatini BL & Regehr WG (1997). Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci 17, 3425–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg‐Graham L & Storm JF (1999). The role of BK‐type Ca2+‐dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol 521, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens EB, Cox PJ, Shah BS, Dixon AK, Richardson PJ, Pinnock RD & Lee K (2001). Tissue distribution and functional expression of the human voltage‐gated sodium channel β3 subunit. Pflugers Arch 441, 481–488. [DOI] [PubMed] [Google Scholar]
- Testa‐Silva G, Loebel A, Giugliano M, de Kock CPJ, Mansvelder HD & Meredith RM (2012). Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb Cortex 22, 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa‐Silva G, Verhoog MB, Linaro D, de Kock CPJ, Baayen JC, Meredith RM, De Zeeuw CI, Giugliano M & Mansvelder HD (2014). High bandwidth synaptic communication and frequency tracking in human neocortex. PLoS Biol 12, e1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueta Y, Hirai Y, Otsuka T & Kawaguchi Y (2013). Direction‐ and distance‐dependent interareal connectivity of pyramidal cell subpopulations in the rat frontal cortex. Front Neural Circuits 7, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen GL & Durand D (1991). Reconstruction of hippocampal granule cell electrophysiology by computer simulation. Neuroscience 41, 411–423. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, Sans N, Rossier J, Oostra B, LeMasson G & Frick A (2014). Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1–/y mice. Nat Neurosci 17, 1701–1709. [DOI] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM & Bennett V (1998). AnkyrinG is required for clustering of voltage‐gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]