

Abstract
Key points
Non‐muscle (NM) and smooth muscle (SM) myosin II are both expressed in smooth muscle tissues, however the role of NM myosin in SM contraction is unknown.
Contractile stimulation of tracheal smooth muscle tissues stimulates phosphorylation of the NM myosin heavy chain on Ser1943 and causes NM myosin filament assembly at the SM cell cortex.
Expression of a non‐phosphorylatable NM myosin mutant, NM myosin S1943A, in SM tissues inhibits ACh‐induced NM myosin filament assembly and SM contraction, and also inhibits the assembly of membrane adhesome complexes during contractile stimulation.
NM myosin regulatory light chain (RLC) phosphorylation but not SM myosin RLC phosphorylation is regulated by RhoA GTPase during ACh stimulation, and NM RLC phosphorylation is required for NM myosin filament assembly and SM contraction.
NM myosin II plays a critical role in airway SM contraction that is independent and distinct from the function of SM myosin.
Abstract
The molecular function of non‐muscle (NM) isoforms of myosin II in smooth muscle (SM) tissues and their possible role in contraction are largely unknown. We evaluated the function of NM myosin during contractile stimulation of canine tracheal SM tissues. Stimulation with ACh caused NM myosin filament assembly, as assessed by a Triton solubility assay and a proximity ligation assay aiming to measure interactions between NM myosin monomers. ACh stimulated the phosphorylation of NM myosin heavy chain on Ser1943 in tracheal SM tissues, which can regulate NM myosin IIA filament assembly in vitro. Expression of the non‐phosphorylatable mutant NM myosin S1943A in SM tissues inhibited ACh‐induced endogenous NM myosin Ser1943 phosphorylation, NM myosin filament formation, the assembly of membrane adhesome complexes and tension development. The NM myosin cross‐bridge cycling inhibitor blebbistatin suppressed adhesome complex assembly and SM contraction without inhibiting NM myosin Ser1943 phosphorylation or NM myosin filament assembly. RhoA inactivation selectively inhibited phosphorylation of the NM myosin regulatory light chain (RLC), NM myosin filament assembly and contraction, although it did not inhibit SM RLC phosphorylation. We conclude that the assembly and activation of NM myosin II is regulated during contractile stimulation of airway SM tissues by RhoA‐mediated NM myosin RLC phosphorylation and by NM myosin heavy chain Ser1943 phosphorylation. NM myosin II actomyosin cross‐bridge cycling regulates the assembly of membrane adhesome complexes that mediate the cytoskeletal processes required for tension generation. NM myosin II plays a critical role in airway SM contraction that is independent and distinct from the function of SM myosin.
Keywords: myosin II isoforms, myosin phosphorylation, nonmuscle myosin II, smooth muscle contraction
Key points
Non‐muscle (NM) and smooth muscle (SM) myosin II are both expressed in smooth muscle tissues, however the role of NM myosin in SM contraction is unknown.
Contractile stimulation of tracheal smooth muscle tissues stimulates phosphorylation of the NM myosin heavy chain on Ser1943 and causes NM myosin filament assembly at the SM cell cortex.
Expression of a non‐phosphorylatable NM myosin mutant, NM myosin S1943A, in SM tissues inhibits ACh‐induced NM myosin filament assembly and SM contraction, and also inhibits the assembly of membrane adhesome complexes during contractile stimulation.
NM myosin regulatory light chain (RLC) phosphorylation but not SM myosin RLC phosphorylation is regulated by RhoA GTPase during ACh stimulation, and NM RLC phosphorylation is required for NM myosin filament assembly and SM contraction.
NM myosin II plays a critical role in airway SM contraction that is independent and distinct from the function of SM myosin.
Abbreviations
- DTT
dithiothreitol
- EGFP
enhanced green fluorescent protein
- IPG
immobilized pH gradient
- RLC
regulatory light chain
- NM
non‐muscle
- PLA
proximity ligation assay
- PSS
physiological saline solution
- SM
smooth muscle
Introduction
Non‐muscle (NM) myosin II is ubiquitously expressed in all cell types and constitutes the primary motor for motility, migration and adhesion in non‐muscle cells. Although non‐muscle isoforms of myosin II are also expressed in muscle tissues, muscle‐specific isoforms of myosin II provide the molecular motors for mechanical force generation and contraction. The contractile stimulation of smooth muscle cells activates actomyosin cross‐bridge cycling by regulating phosphorylation of the 20 kDa regulatory light chain (RLC) of smooth muscle (SM) myosin. Although there is some evidence suggesting that NM myosin II can be regulated during smooth muscle contraction and that it also contributes to tension generation (Morano et al. 2000; Lofgren et al. 2003; Yuen et al. 2009), such evidence is controversial, and some investigators have concluded that NM myosin II plays no role in smooth muscle contraction (Eddinger & Meer, 2007). Consequently, the molecular function of NM myosin II in smooth muscle tissues and the nature of its role in contraction is unclear.
All isoforms of myosin II have a similar structure consisting of three pairs of peptides: two 230 kDa heavy chains with an α‐helical coiled‐coil rod backbone and a short non‐helical tail; two globular head domains that contain the ATP and actin binding sites; two 20 kDa RLCs that regulate the ATPase activity of the globular heads; and two essential light chains that stabilize the heavy chain structure (Vicente‐Manzanares et al. 2009). Non‐muscle and smooth muscle isoforms of myosin II exhibit a high degree of sequence similarity and are both regulated by phosphorylation of the 20 kDa RLC, which activates actomyosin cross‐bridge cycling (Bresnick, 1999; Eddinger & Meer, 2007; Yuen et al. 2009; Heissler & Sellers, 2016). Both SM and NM myosin II isoforms also contain a short non‐helical C‐terminal tailpiece that regulates filament assembly, which is variable in length and isoform‐specific.
SM and NM myosin II monomers in vitro can assume a folded inactive ‘assembly‐incompetent’ conformation (10S form) that is unable to assemble into filaments, and an open ‘assembly‐competent’ conformation (6S form) (Scholey et al. 1980; Trybus & Lowey, 1984; Vicente‐Manzanares et al. 2009; Dulyaninova & Bresnick, 2013; Heissler & Sellers, 2016). The folded inactive ‘10S’ myosin II interacts weakly with actin and does not compete with filamentous myosin for actin binding (Wendt et al. 1999; Jung et al. 2008). The phosphorylation of the 20 kDa RLC of SM and NM myosin II in vitro causes 10S (inactive folded) myosin II to unfold into its assembly‐competent (6S) conformation that can form filaments (Scholey et al. 1980; Smith et al. 1983; Trybus & Lowey, 1984; Vicente‐Manzanares et al. 2009; Dulyaninova & Bresnick, 2013). The regulated interconversion of myosin II between a folded inactive conformer and an assembly‐competent conformation has been documented in vitro but not in vivo (Trybus & Lowey, 1984; Kendrick‐Jones et al. 1987). There is evidence from several non‐muscle cell lines that a pool of NM myosin II in the assembly‐incompetent auto‐inhibited state is necessary for NM myosin II filament assembly and cell migration (Breckenridge et al. 2009; Kiboku et al. 2013). The existence of a pool of SM myosin in the 10S assembly‐incompetent conformation has been documented in cultured airway smooth muscle cells (Milton et al. 2011). However, in an earlier study of chicken gizzard tissue, only trace amounts of 10S myosin II could be detected using an antibody specific for the 10S conformation (Horowitz et al. 1994). There is no evidence as to whether NM myosin II filament assembly is regulated in muscle tissues.
Sequences within the myosin II heavy chain near the C‐terminal of the α‐helical coiled‐coil rod domain and the C terminal tailpiece are critical for the assembly of myosin II monomers into filaments (Ronen & Ravid, 2009; Dulyaninova & Bresnick, 2013; Heissler & Manstein, 2013; Beach & Hammer, 2015). In vitro protein assays using C terminal fragments of NM myosin IIA heavy chain have suggested that phosphorylation at Ser1943 within its tailpiece is associated with a reduction in NM myosin IIA filament assembly (Dulyaninova et al. 2005, 2007; Dulyaninova & Bresnick, 2013). Furthermore, phosphorylation at Ser1943 inhibits cell migration and motility and reduces lamellipod extension in vivo in several cancer cell lines (Dulyaninova et al. 2007). However, it is not known whether the phosphorylation of NM myosin II heavy chain and/or NM myosin II filament assembly is regulated in smooth muscle.
The contractile stimulation of airway smooth muscle and other smooth muscle tissues induces the polymerization of a small pool of actin at the cortex of the smooth muscle cell (Mehta & Gunst, 1999; Zhang et al. 2005; Gunst & Zhang, 2008; Kim et al. 2008; Zhang & Gunst, 2008; Lehman & Morgan, 2012). This actin polymerization is required for tension development in addition to actomyosin cross‐bridge cycling. In smooth muscle tissues, actin polymerization is regulated by protein complexes within integrin‐associated adhesion junctions (adhesome complexes) and is catalysed by distinct signalling pathways that are activated independently of SM myosin RLC phosphorylation and actomyosin cross‐bridge cycling (Opazo Saez et al. 2004; Zhang et al. 2005, 2012; Gunst & Zhang, 2008). Contractile stimulation triggers the recruitment of proteins to membrane adhesion junctions, where they assemble into adhesome signalling complexes that regulate signalling pathways to mediate actin polymerization and other cytoskeletal processes necessary for tension generation (Zhang & Gunst, 2008; Huang et al. 2011, 2014; Zhang et al. 2012, 2016; Wu & Gunst, 2015; Wu et al. 2016). Non‐muscle myosin II plays an integral role in the processes of cell adhesion and migration in non‐muscle cells, and it is indispensable for the assembly and disassembly of adhesions within the lamellipodium (Vicente‐Manzanares et al. 2009). We hypothesized that NM myosin II might regulate contraction in smooth muscle tissues by mediating the assembly of adhesome complexes in response to contractile stimuli.
In the present study, we aimed to determine whether NM myosin II activation is necessary for tension generation and also whether the assembly of NM myosin II is regulated during smooth muscle contraction. Accordingly, we investigated the role of NM myosin II heavy chain phosphorylation on Ser1943, which has been implicated in the regulation of NM myosin II filament assembly in non‐muscle cells. We also selectively inhibited the phosphorylation of the 20 kDa NM myosin II RLC during contractile stimulation to evaluate its effect on NM myosin filament assembly and function. Blebbistatin, a selective inhibitor of NM myosin, was used to determine the effects of inhibiting NM myosin cross‐bridge cycling. The results obtained demonstrate that the assembly and activation of NM myosin II is physiologically regulated during the contractile stimulation of smooth muscle tissues and that it requires NM myosin II heavy chain phosphorylation at Ser1943, as well as RhoA‐mediated NM myosin RLC phosphorylation. The results of the present study further demonstrate that NM myosin II regulates the assembly of adhesome complexes during smooth muscle contraction that mediate cytoskeletal processes required for tension generation. We conclude that NM myosin II plays a critical role in smooth muscle contraction that is independent and distinct from the function of smooth muscle myosin II.
Methods
Ethical approval
All procedures were conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee (IACUC) of Indiana University School of Medicine under the National Research Council's Guide for the Care and Use of Laboratory Animals. The Indiana University Laboratory Animal Resource Center (LARC) at Indiana University School of Medicine procured mongrel dogs (20–25 kg, either sex) from LBL Kennels (Reelsville, IN, USA). LARC personnel euthanized animals by i.v. injection of Fatal‐Plus (Vortech Pharmaceuticals, Ltd, Dearborn, MI, USA) (pentobarbital sodium 390 mg ml−1; propylene glycol, 0.01 mg ml−1; ethyl alcohol; 0.29 mg ml−1; benzyl alcohol (preservative), 0.2 mg ml−1) at a dose of ∼0.3 ml kg−1, in accordance with procedures approved by the IACUC of Indiana University School of Medicine. After euthanization, a tracheal segment was immediately removed and placed in physiological saline solution (PSS). All investigators understand the ethical principles under which the Journal of Physiology operates and all work complies with these principles.
Preparation of smooth muscle tissues and measurement of force
A tracheal segment was immediately removed and immersed in PSS comprising (in mm): 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4 and 5.6 glucose). Strips of tracheal smooth muscle (1.0 × 0.2 to 0.5 × 15 mm) were dissected free of connective and epithelial tissues and maintained within a tissue bath in PSS at 37°C. Force was measured during isometric contractions by attaching the tissues to Grass force‐displacement transducers. Prior to the beginning of each experimental protocol, muscle length was increased to maintain a preload of ∼0.5–1.0 g and tissues were stimulated repeatedly with 10−5 m ACh until stable responses were obtained. The force of contraction in response to ACh was determined before and after treatment with plasmids or other reagents.
Immunoblots
For biochemical analysis, muscle tissues were rapidly frozen using liquid N2‐cooled tongs and pulverized in liquid N2 using a mortar and pestle. Pulverized muscle strips were mixed with extraction buffer containing: 20 mm Tris‐HCl at pH 7.4, 2% Triton X‐100, 0.4% SDS, 2 mm EDTA, phosphatase inhibitors (2 mm sodium orthovanadate, 2 mm molybdate and 2 mm sodium pyrophosphate) and protease inhibitors (2 mm benzamidine, 0.5 mm aprotinin and 1 mm phenylmethylsulphonyl fluoride). Each sample was centrifuged and the supernatant was then boiled in sample buffer (1.5% dithiothreitol, 2% SDS, 80 mm Tris‐HCl, pH 6.8, 10% glycerol and 0.01% bromphenol blue) for 5 min. Proteins were separated by SDS‐PAGE and transferred to nitrocellulose. The nitrocellulose membrane was blocked with 2–5% milk or Odyssey (Li‐Cor Biosciences, Lincoln, NE, USA) blocking buffer for 1 h and probed with primary antibodies against proteins of interest overnight followed by secondary antibodies for 1 h. Proteins were visualized by enhanced chemiluminescence using a ChemiDoc XRS detection system (Bio‐Rad, Hercules, CA, USA) or by infrared fluorescence using an Odyssey imager (Li‐Cor Biosciences).
Transfection of smooth muscle tissues
Plasmids were introduced into tracheal smooth muscle strips by the method of reversible permeabilization (Tang et al. 2003; Zhang et al. 2005, 2010, 2012). Tissues were equilibrated and the muscle length for the generation of maximal isometric force was determined. Muscle strips were then attached to metal mounts to maintain them at constant length. The strips were incubated successively in each of the following solutions: Solution 1 (at 4°C for 120 min) containing (in mm): 10 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2 and 20 N‐tris (hydroxymethyl) methyl‐2‐aminoethanesulphonic acid (TES); Solution 2 (at 4°C overnight) containing (in mm): 0.1 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2, 20 TES and 20 μg ml−1 plasmids. Solution 3 (at 4°C for 30 min) containing (in mm): 0.1 EGTA, 5 Na2ATP, 120 KCl, 10 MgCl2 and 20 TES; and Solution 4 (at 22°C for 90 min) containing (in mm): 110 NaCl, 3.4 KCl, 0.8MgSO4, 25.8 NaHCO3, 1.2 KH2PO4 and 5.6 dextrose. Solutions 1–3 were maintained at pH 7.1 and aerated with 100% O2. Solution 4 was maintained at pH 7.4 and was aerated with 95% O2/5% CO2. After 30 min in Solution 4, CaCl2 was added gradually to reach a final concentration of 2.4 mm. The strips were then incubated in a CO2 incubator at 37°C for 2 days in serum‐free Dulbecco's modified Eagle's medium containing 5 mm Na2ATP, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μg ml−1 kanamycin, 2.5 μg ml−1 anti‐fungal and 20 μg ml−1 plasmids to allow for expression of the recombinant proteins. Expression of recombinant proteins in transfected muscle tissues was determined by immunoblots and immunofluorescence.
Cell dissociation
Freshly dissociated primary cells were used for these studies to avoid the morphological changes in cytoskeletal organization and changes in phenotype that occur during the culture of smooth muscle cells. Smooth muscle cells were enzymatically dissociated from tracheal muscle strips (Opazo Saez et al. 2004; Zhang et al. 2005). Tracheal muscle strips were minced and transferred to 5 ml of dissociation solution (in mm): 130 NaCl, 5 KCl, 1.0 CaCl2, 1.0 MgCl2, 10 Hepes, 0.25 EDTA, 10 d‐glucose and 10 taurine (pH 7.0) with collagenase (type IV, 400 U ml−1), papain (type IV, 30 U ml−1), bovine serum albumin (1 mg ml−1) and dithiothreitol (DTT; 1 mm). All enzymes were obtained from Sigma (St Louis, MO, USA). The strips were then placed in a 37°C shaking water bath at 60 oscillations min−1 for 15–20 min, followed by three washes with a Hepes‐buffered saline solution (in mm): 130 NaCl, 5 KCl, 1.0 CaCl2, 1.0 MgCl2, 20 Hepes and 10 d‐glucose (pH 7.4) and triturated with a pipette to liberate individual smooth muscle cells from the tissue. The solution containing the dissociated cells was poured over glass coverslips and the cells were allowed to adhere to the coverslips for 30–60 min at room temperature. Cells were stimulated with 10−5 m ACh for 5 min at 37°C or left unstimulated and used as controls. Stimulated and unstimulated cells were fixed for 10 min in 4% paraformaldehyde (v/v) in PBS (in mm): 137 NaCl, 4.3 Na2HPO4, 1.4 KH2PO4 and 2.7 KCl (pH 7.4).
In situ proximity ligation assay
In situ proximity ligation assays (PLA) were performed as described previously to detect protein interactions (Huang et al. 2011; Zhang et al. 2012, 2016). PLA provides for the precise detection of protein–protein complexes (Soderberg et al. 2006, 2008). Target proteins are reacted with primary antibodies raised in different species, and a pair of oligonucleotide‐labelled secondary antibodies conjugated to + and − PLA probes is targeted to each pair of primary antibodies. The probes form circular DNA strands only when they are bound in very close proximity (< 40 nm). These DNA circles serve as templates for localized rolling circle amplification, generating a fluorescence signal (spot) that enables individual interacting pairs of the target protein molecules to be visualized. The PLA signal allows for the detection of a complex between two target proteins at a very high resolution.
Smooth muscle cells were dissociated from Sham‐treated or transfected canine tracheal smooth muscle tissues. Freshly dissociated smooth muscle cells were stimulated with 10−5 m ACh or left unstimulated and then fixed, permeabilized and incubated with primary antibodies against target proteins or epitopes followed by a pair of oligonucleotide‐labelled secondary antibodies conjugated to Duolink + and − PLA probes. PLA probe hybridization, ligation, amplification and detection media were administered in accordance with the manufacturer's instructions (Olink Bioscience, Uppsala, Sweden). Randomly selected cells from both unstimulated and ACh‐stimulated groups were analysed for protein interactions by visualizing PLA fluorescence spots using a LSM 510 confocal microscope (Carl Zeiss, Oberkochen, Germany).
The total number of PLA fluorescence spots/cell was counted using Image Tools (Olink Bioscience). In some experiments, the distribution of PLA fluorescence spots within each cell was quantified by separating the region of the cell cortex from the cytoplasm by a line ∼1 μm inside the cell border. The ratio of fluorescence intensity between the cortical compartment and the cytoplasmic compartment was calculated using Metamorph (Molecular Devices, Inc. Sunnyvale, CA, USA) as described previously (Zhang et al. 2012). The ratio of the area of cortical compartment to the area of the cytoplasmic compartment did not differ significantly in any of the groups.
Two‐dimensional electrophoresis for the analysis of NM and SM RLC phosphorylation
Canine tracheal smooth muscle tissues were stimulated with 10 μm ACh or left unstimulated for 5 min and frozen in liquid nitrogen. Frozen muscle strips were immersed in dry ice‐precooled acetone containing 10% (w/v) trichloroacetic acid and 10 mm DTT. Proteins were extracted in 7 m urea, 2 m thiourea, 2% (w/v) 3‐[(3‐cholamidopropyl) dimethylammonio]‐1‐propanesulphonate (CHAPS), 1% 3.9–5.1 immobilized pH gradient (IPG) buffer and 1× Roche Complete protease inhibitor. Suitable amounts of sample were resolved in the first dimension using the acidic half of 11 cm (pH 3.9–5.1) IPG dry strip gels. After separation in the first dimension, proteins were equilibrated in 6 m urea, 50 mm (pH 6.4) Bis‐Tris, 30% (v/v) glycerol, 2% SDS and 0.002% bromophenol blue, first containing 10 mm DTT and then containing 2.5% (w/v) iodoacetamide. Proteins were then separated in the second dimension by SDS‐PAGE electrophoresis, transferred to nitrocellulose, and then immunoblotted for total RLC and phospho‐Ser19 RLC. Phosphorylation was calculated from densitometry based on the intensity of the RLC spots.
Mass spectrometry
Mass spectrometry was used to identify the myosin SM and NM RLC proteins separated by two‐dimensional electrophoresis. The Indiana University School of Medicine Proteomics Core Laboratory performed all procedures for peptide identification. A database search for RLC sequences was carried out using SequestTM algorithms and the Canfa database (UniProt; www.uniprot.org). There are four unique peptides in which amino acid sequences differ between SM and NM myosin RLCs (Park et al. 2011). We used the unique peptides NAFACFDEEATGTIQEDYLR (NM myosin RLC) and NAFACFDEEASGFIHEDHLR (SM myosin RLC) to distinguish NM and SM myosin RLC isoforms. Briefly, the two‐dimensional gel spots were destained, reduced with 10 mm DTT in 10 mm ammonium bicarbonate and then alkylated with 55 mm iodoacetamide (prepared in 10 mm ammonium bicarbonate). Alkylated samples were digested with trypsin (Promega, Madison, WI, USA) overnight at 37°C. Digested peptides were extracted from the gel spots. Tryptic peptides were injected onto a Michrom Magic C18 packed tip capillary column (Michrom BioResources Inc., Auburn, CA, USA). Peptides were eluted with a linear gradient from 5% to 35% acetonitrile and developed over 60 min at room temperature, at a flow rate of 50 ul min−1, after which effluent was electrosprayed into the LTQ mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Blanks were run prior to the sample run to ensure there was no significant signal from solvents or the column.
Triton solubility assay
A Triton solubility assay was performed on extracts of smooth muscle tissues to separate cytoskeletal and Triton‐soluble proteins (Dulyaninova et al. 2007; Li et al. 2010; Kiboku et al. 2013). In brief, frozen and pulverized tracheal smooth muscle tissue samples were mixed with Triton X‐100 lysis buffer (150 mm KCl, 20 mm Pipes, 10 mm imidazole, pH 7.0, 0.05% Triton X‐100, 1 mm MgCl2, 1 mm EGTA, 1 mm DTT, 0.1 mm phenylmethane sulphonyl fluoride, 5 μg ml−1 aprotinin, 5 μg mL−1 leupeptin, 2 μg mL−1 pepstatin A, 1 mm Na3VO4 and 20 mm β‐glycerophosphate) at 4°C. After 5 min, the lysates were centrifuged at 8000 g for 5 min at 4°C, and the supernatant was removed as the soluble fraction. The pellet, the insoluble fraction, was added to equal volumes of Triton X‐100 lysis buffer with an additional 0.8% SDS and 2 mm EDTA, boiled for 5 min and rotated for 1 h, and then centrifuged at 16,000 g for 15 min at 4°C. The supernatant was transferred to another tube as the insoluble fraction. Equal volumes of soluble and insoluble fractions from the same sample were used for immunoblots.
Analysis of F‐actin and G‐actin
The relative proportions of F‐actin and G‐actin in smooth muscle tissues were analysed as described previously (Zhang et al. 2005, 2010). Briefly, each of the tracheal smooth muscle strips was homogenized in 200 μl of F‐actin stabilization buffer (50 mm PIPES, pH 6.9, 50 mm NaCl, 5 mm MgCl2, 5 mm EGTA, 5% glycerol, 0.1% Triton X‐100, 0.1% Nonidet P‐40, 0.1% Tween‐20,0.1% β‐mercaptoethanol, 0.001% anti‐foam, 1 mm ATP, 1 μg ml−1 pepstatin, 1 μg ml−1 leupeptin, 10 μg ml−1 benzamidine and 500 μg ml−1 tosyl arginine methyl ester). Supernatants of the protein extracts were collected after centrifugation (Optima MAX Ultracentrifuge; Beckman Coulter, Fullerton, CA, USA) at 150,000 g for 60 min at 37°C. The pellets were resuspended in 200 μl of ice‐cold water containing 10 μm cytochalasin D and then incubated on ice for 1 h to depolymerize F‐actin. The resuspended pellets were mixed gently every 15 min. Four microlitres of supernatant (G‐actin) and pellet (F‐actin) fractions were subjected to immunoblot analysis using anti‐actinantibody (clone AC‐40; Sigma). The ratios of F‐actin to G‐actin were determined using densitometry.
Reagents and antibodies
Sources of antibodies were: polyclonal rabbit anti‐human phospho‐myosin IIA (Ser1943) (catalogue number 5026; Cell Signaling Technology, Beverly, MA, USA); monoclonal mouse anti‐human non‐muscle myosin IIA (catalogue number ab55456; Abcam, Cambridge, MA, USA); monoclonal mouse anti‐human non‐muscle myosin IIB (catalogue number ab684; Abcam); polyclonal rabbit anti‐human non‐muscle myosin IIA (catalogue number M8064; Sigma); polyclonal rabbit anti‐human non‐muscle myosin IIB (catalogue number M7939; Sigma); monoclonal mouse anti‐human paxillin (catalogue number 610569; BD Transduction, Lexington, KT, USA); polyclonal rabbit anti‐human paxillin phospho‐tyrosine 118 (catalogue number 44‐722G; Invitrogen, Carlsbad, CA, USA); horseradish peroxidase‐conjugated IgG (catalogue number NA931 & NA934; Amersham Biosciences, Piscataway, NJ, USA); IRDye® 680RD Donkey‐anti‐Mouse IgG (catalogue number 926‐68070) and IRDye® 800CW Donkey‐anti‐Rabbit IgG (catalogue number 925‐32211; Li‐Cor Biosciences); polyclonal rabbit anti‐canine cardiac vinculin and polyclonal rabbit anti‐bovine smooth muscle myosin regulatory light chain were custom made by BABCO (Richmond, CA, USA). Odyssey® blocking buffer (catalogue number 927‐50000) was obtained from Li‐Cor Biosciences. All antibodies have been validated to confirm their reaction with the designated antigen in the smooth muscle tissue extracts, immunoblots or fixed cells. The Duolink in situ PLA (Olink Bioscience) was obtained from Sigma Chemical Co. Blebbistatin was also obtained from Sigma Chemical Co.
Plasmids encoding wild‐type and the Ser1943 point mutant of NM myosin heavy chain (pEGFP‐NMHC‐IIA and pEGFP‐NMHC‐IIA‐S1943A) (Dulyaninova et al. 2007) were generously provided by Dr Anne R. Bresnick (Albert Einstein College of Medicine, Bronx, New York, NY, USA). pEGFP‐NMHC‐IIA‐WT was generated by subcloning a DNA fragment encoding residues 1–1960 from human NM myosin‐IIA heavy chain into the HindIII and SalI sites of pEGFP‐C3 (Clontech, Palo Alto, CA, USA). In pEGFP‐NMHC‐IIA‐S1943A, Ser1943 was replaced by alanine.
Plasmids encoding human RhoA T19N have been described previously (Strassheim et al. 1999; Zhang et al. 2010). The cDNAs encoding the RhoA with hemagglutinin (HA)‐RhoA Asn19 mutant were subcloned into the mammalian expression vector pcDNA3.1.
Escherichia coli (Bluescript) transformed with these plasmids was grown in LB medium, and plasmids were purified by alkaline lysis with SDS using a purification kit from Qiagen Inc. (Valencia, CA, USA).
Statistical analysis
Comparisons between two groups were performed using a paired or unpaired two‐tailed Student's t test. Comparisons among multiple groups were performed by ANOVA. Values of ‘n’ refer to the number of cells or tissue strips used to obtain mean values. P < 0.05 was considered statistically significant.
Results
Contractile stimulation induces NM myosin II assembly in airway smooth muscle tissues
We evaluated the effect of stimulation with ACh on the assembly status of NM myosin II in tracheal smooth muscle tissues using a fractionation assay to quantify the ratio of NM myosin in the cytoskeletal and Triton X‐soluble fractions of tissue extracts at time points from 1 to 10 min after stimulation with 10−5 m ACh (Fig. 1 A) (Dulyaninova et al. 2005, 2007; Breckenridge et al. 2009; Milton et al. 2011). The proportion of NM myosin IIA and IIB isoforms in the cytoskeletal fraction of smooth muscle tissue extracts increased significantly within 1 min after stimulation with ACh, and rose by three‐ to four‐fold within 2 min with a time course similar to tension development (Fig. 1 B). These results suggest that stimulation of airway smooth muscle tissues with ACh triggers rapid NM myosin actomyosin assembly involving both A and B isoforms of NM myosin II.
Figure 1. Contractile stimulation induces the assembly of NM myosin II filaments in airway smooth muscle tissues.
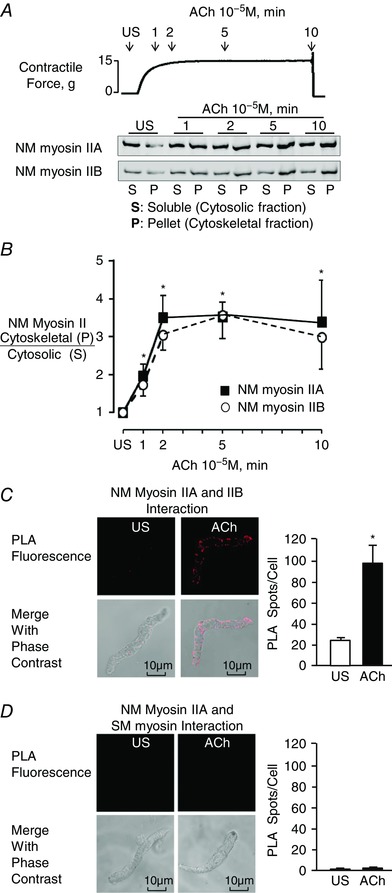
A, typical time course for force development during stimulation of muscle tissues with 10−5 ACh, illustrating the points at which muscle tissues were quick frozen for the Triton solubility assay (upper). The ratio of NM myosin IIA and IIB isoforms in the Triton X‐insoluble cytoskeletal (P) fraction vs. soluble (S) fraction of tissue extracts was assessed by immunoblot in five individual tracheal smooth muscle tissues frozen 1, 2, 5 or 10 min after contractile stimulation with ACh and unstimulated (US) (lower). B, P/S ratios of both A and B isoforms of NM myosin II increased significantly after ACh stimulation for 1 min or more relative to unstimulated tissues (n = 9). C, in situ PLA was used to determine the interaction of NM myosin IIA and NM myosin IIB in freshly dissociated differentiated tracheal smooth muscle cells. PLA fluorescence alone and merged with phase contrast images are shown for each cell. In US cells, very few spots were observed. Stimulation with ACh caused a significant increase in the number of PLA spots at the cell membrane (n = 17 cells). D, in situ PLA shows the interaction of NM myosin IIA and SM myosin in freshly dissociated smooth muscle cells. Very few spots were observed in either unstimulated or ACh stimulated cells (n = 31 for US; n = 34 for ACh). All values are the mean ± SEM. *Significant difference compared to US (P < 0.05).
We also used in situ PLA analysis to assess interactions between A and B isoforms of NM myosin II as an index of NM myosin II filament formation during the contractile stimulation of freshly dissociated tracheal smooth muscle cells. Because NM myosin II isoforms can coassemble into heterotypic filaments (Beach et al. 2014; Beach & Hammer, 2015), interactions between NM myosin IIA and IIB isoforms were quantified to assess the formation of NM myosin II polymers. We observed few interactions between NM myosin IIA and IIB in unstimulated cells, whereas the interaction of NM myosin IIA and IIB isoforms dramatically increased in cells stimulated with ACh (Fig. 1 C). Interactions between NM myosin IIA and IIB were localized to the cortical region of the smooth muscle cells (Fig. 1 C). We also used PLA to probe for interactions between NM myosin IIA and SM myosin to confirm the specificity of the PLA assay for NM myosin II isoform interactions (Fig. 1 D). As expected, no interactions between NM myosin IIA and SM myosin were observed in unstimulated or ACh‐stimulated smooth muscle cells. These results suggest that contractile stimulation triggers the assembly of NM myosin II into filaments in the cortical region of airway smooth muscle cells, and that these NM myosin II filaments remain stable for the 10 min period of contractile stimulation.
Stimulation of tracheal smooth muscle tissues with ACh induces phosphorylation of Ser1943 on the tail domain of the NM myosin II heavy chain
We investigated the effect of stimulation with 10−5 m ACh on the phosphorylation of Ser1943 within the non‐helical tail domain of NM myosin II in tracheal muscle tissues (Fig. 2 A). Muscle tissues were frozen at 1, 2 5 or 10 min after stimulation with ACh, and the Ser1943 phosphorylation of NM myosin II was probed in immunoblots using a site‐specific antibody (Fig. 2 A). NM myosin II Ser1943 phosphorylation increased significantly within 1 min of stimulation with ACh and continued to rise for the 10 min stimulation period. The time course of the increase in NM myosin heavy chain phosphorylation was consistent with that of force development and NM myosin filament assembly.
Figure 2. Stimulation of tracheal smooth muscle tissues with ACh induces phosphorylation of Ser1943 on the NM myosin II heavy chain at the cell cortex.

A, immunoblot of phospho (P)‐NM myosin Ser1943 and NM myosin IIA from extracts of five muscle tissues stimulated with ACh for 1, 2, 5 or 10 min or unstimulated (US). Proteins were imaged by dual fluorescence. The ratio of phospho‐Ser1943 to NM myosin IIA was determined for each sample and normalized to the value for the US tissue. Non‐muscle myosin II heavy chain Ser1943 phosphorylation increased significantly after 1, 2, 5 or 10 min ACh stimulation compared to unstimulated tissues (n = 12). B, images of in situ PLA fluorescence (top) and PLA fluorescence merged with phase contrast (bottom) from freshly dissociated smooth muscle cells unstimulated (US) or stimulated with ACh. PLA shows interactions between NM myosin IIA and phospho‐NM myosin Ser1943 (left), NM myosin IIB and phospho‐NM myosin Ser1943 (middle) and SM myosin and phospho‐NM myosin Ser1943 (right). Very few interactions between NM myosin IIA and phospho‐NM myosin Ser1943 are observed in any of the unstimulated cells. ACh stimulation caused a significant increase in the number of PLA spots at the cell cortex for NM myosin IIA (n = 17, US; n = 28, ACh) and IIB (n = 16, US; n = 26, ACh) but did not result in any interactions between SM myosin and phospho‐NM myosin Ser1943 and NM myosin IIB (n = 13, US; n = 15, ACh). All values are the mean ± SEM. *Significant difference compared to unstimulated tissues (P < 0.05).
We evaluated the cellular localization of Ser1943 phosphorylated NM myosin IIA using in situ PLA in freshly dissociated differentiated tracheal smooth muscle cells to determine interactions between the phospho‐Ser1943 epitope and NM myosin IIA. We observed very few spots indicating Ser1943 phosphorylated NM myosin IIA in unstimulated cells, consistent with our immunoblot data showing little phosphorylation of NM myosin IIA Ser1943 in unstimulated smooth muscle. Stimulation with ACh caused a dramatic increase in the number of PLA spots near the cell membrane, suggesting an increase of NM myosin IIA Ser1943 phosphorylation in membrane complexes.
We also probed for interactions between NM myosin phospho‐Ser1943 and NM myosin IIB. No interactions were observed in unstimulated cells, although an increase in PLA spots was seen in ACh‐stimulated cells. NM myosin IIB has a serine phosphorylation site at residue 1952 that may be analogous to that at Ser1943 in NM myosin IIA (Dulyaninova & Bresnick, 2013); thus, these spots may be indicative of NM myosin IIB heavy chain phosphorylation at that site. Alternatively, the spots may indicate interactions between Ser1943‐phosphorylated NM myosin IIA and NM myosin IIB within heterotypic filaments.
Sequencing data for MYH11 have shown that the Ser1943 consensus sequence is not present on the smooth muscle isoform of myosin II (Eddinger & Meer, 2007) and so a SM myosin heavy chain specific antibody was used to confirm that the NM myosin II Ser1943 antibody did not react with SM myosin (Fig. 2 B). We observed negligible interactions between phospho‐NM myosin IIA Ser1943 and SM myosin heavy chain in either unstimulated or ACh‐stimulated cells, confirming that the PLA assay is specific for NM myosin II phospho‐Ser1943.
These results suggest that phosphorylated NM myosin IIA localizes to the cortical region of the smooth muscle cells and undergoes heavy chain phosphorylation on Ser1943 during contractile stimulation.
NM myosin II heavy chain Ser1943 phosphorylation regulates NM myosin filament assembly and tension development in smooth muscle tissues
We used a non‐phosphorylatable mutant of the NM myosin IIA isoform, EGFP‐NM myosin IIA S1943A (Dulyaninova et al. 2007) to determine whether phosphorylation at this site regulates NM myosin II assembly in smooth muscle tissues during contractile stimulation. We transfected tracheal smooth muscle tissues with pEGFP‐NMHC IIA S1943A using the method of reversible permeabilization. Expression of the recombinant protein was confirmed by immunoblot and also by visualizing enhanced green fluorescent protein (EGFP) fluorescence in cells dissociated from the transfected tissues (Fig. 3 A). The increase in the Ser1943 phosphorylation of endogenous NM myosin II in response to 5 min of stimulation with ACh was significantly inhibited in tissues expressing EGFP‐NM myosin IIA S1943A (Fig. 3 A).
Figure 3. NM myosin II heavy chain Ser1943 phosphorylation regulates NM myosin filament assembly and tension development in smooth muscle tissues.

A, EGFP fluorescence was observed in most cells dissociated from tissues expressing the non‐phosphorylatable NM myosin IIA mutant, EGFP‐NM IIA‐S1943A. The increase in NM myosin II Ser1943 phosphorylation in response to 5 min of stimulation with 10−5 m ACh was significantly inhibited in tissues expressing NM myosin S1943A (n = 18). B, ratio of NM myosin II in the cytoskeletal (P) vs. soluble (S) fractions increased in response to 5 min of stimulation with 10−5 m ACh in Sham‐treated tissues but was significantly inhibited by the expression of NM myosin II Ser1943A in ACh‐stimulated tissues (n = 8). C, in situ PLA fluorescence (top) and PLA fluorescence merged with phase contrast (bottom) from freshly dissociated smooth muscle cells unstimulated (US) or stimulated with ACh. PLA spots show interactions between NM myosin IIA and NM myosin IIB in cells dissociated from Sham‐treated tissues (left), and in cells dissociated from tissues expressing EGFP‐NM myosin IIA‐S1943A (right). ACh stimulation of Sham‐treated tissues resulted in a significant increase in the number of NM myosin IIA and IIB complexes at the cell cortex (n = 27, US; n = 30 ACh). By contrast, few NM myosin IIA and IIB complexes were observed in cells from tissues treated with the NM myosin S1943A (US, n = 27; ACh, n = 30). Very few NM myosin IIA and IIB complexes are observed in the unstimulated cells. D, contractile force in response to ACh is shown for tissues before and after they were transfected with plasmids encoding wild type (WT) EGFP‐NM myosin IIA heavy chain, EGFP‐NM myosin IIA S1943A or no plasmids (Sham) (n = 10). All force measurements were normalized to the Sham response. The expression of NM myosin S1943A mutant significantly inhibited tension development in response to 10−5 m ACh stimulation. All values are the mean ± SEM. *Significant difference between treatment groups (P < 0.05).
We evaluated the effect of inhibiting NM myosin Ser1943 phosphorylation on the assembly of NM myosin IIA in the tracheal smooth muscle tissues (Fig. 3 B). In tissues subjected to Sham transfections, the amount of NM myosin IIA in the cytoskeletal fraction of tracheal tissue extracts increased significantly in response to 5 min of stimulation with ACh. The increase in NM myosin IIA in the cytoskeletal fraction in ACh‐stimulated tissues was significantly inhibited by the expression of NM myosin S1943A in the tissues. These observations suggest that NM myosin Ser1943 phosphorylation is required for NM myosin II actomyosin assembly in response to contractile stimulation.
The role of NM myosin heavy chain Ser1943 phosphorylation on NM myosin II filament assembly was also assessed by in situ PLA in freshly dissociated tracheal smooth muscle cells (Fig. 3 C). Smooth muscle cells were dissociated from tissues expressing the NM myosin S1943A mutant and from Sham‐treated tissues. In cells from Sham‐treated tissues, ACh stimulation induced interactions between NM myosin IIA and IIB isoforms indicating NM myosin filament formation. By contrast, few interactions between NM myosin IIA and IIB isoforms were observed in cells from tissues treated with the NM myosin S1943A mutant. These observations provide additional evidence that NM myosin II heavy chain Ser1943 phosphorylation regulates NM myosin filament formation and actomyosin assembly in smooth muscle tissues stimulated by ACh.
In tissues expressing NM myosin S1943A, the contractile response to ACh was reduced to less than 40% of that in Sham‐treated tissues. Tissues expressing wild‐type NM myosin IIA or Sham‐treated tissues exhibited no deficits in force development in response to ACh (Fig. 3 D). These results suggest that NM myosin II filament formation and assembly contributes to contractile tension development in airway smooth muscle tissues.
NM myosin II regulates the assembly and activation of membrane adhesion junction signalling complexes during the contractile stimulation of smooth muscle tissues
We evaluated the role of NM myosin II filament assembly in the regulation of adhesion complex assembly and the activation of adhesome proteins in tracheal smooth muscle tissues during contractile stimulation with ACh. Adhesion junction proteins are recruited to integrin adhesomes during the contractile stimulation of intact airway smooth muscle tissues and differentiated freshly dissociated airway smooth muscle cells (Opazo Saez et al. 2004; Zhang & Gunst, 2008; Zhang et al. 2012, 2016). The assembly of these adhesome complexes is required for tension development in response to the contractile stimulation of airway smooth muscle.
The activation of tracheal smooth muscle tissues with a contractile agonist stimulates the recruitment of vinculin to adhesome complexes, where vinculin undergoes a conformational change that permits its binding to talin and F‐actin (Huang et al. 2011; Huang et al. 2014). We performed in situ PLA on smooth muscle cells freshly dissociated from tissues expressing NM myosin II S1943A to evaluate the role of NM myosin IIA in the recruitment of vinculin to adhesomes and its interaction with talin at the membrane (Fig. 4 A). In cells from Sham‐treated tissues, stimulation with ACh caused a dramatic increase in the interaction of vinculin and talin at cell adhesion junction sites, as indicated by a large increase in the number of PLA fluorescence spots at the cell membrane. The ACh‐induced increase in the interactions between vinculin and talin was completely inhibited in cells from tissues expressing NM myosin S1943A, indicating that NM myosin IIA filament assembly regulates the recruitment of vinculin to the cell cortex and/or its interaction with talin in adhesome complexes.
Figure 4. NM myosin II Ser1943 phosphorylation regulates the assembly and activation of membrane adhesion junction signalling complexes and actin polymerization.
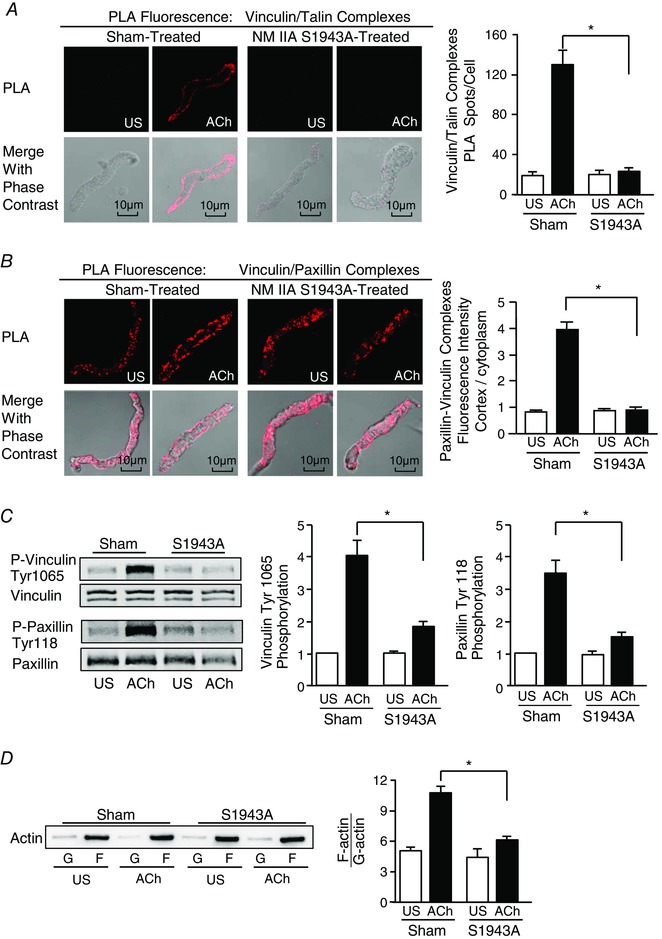
A and B, images of in situ PLA fluorescence (top) and PLA fluorescence merged with phase contrast (bottom) from freshly dissociated smooth muscle cells unstimulated (US) or stimulated with ACh. A, PLA shows interactions between vinculin and talin in cells dissociated from Sham‐treated tissues (left), and in cells dissociated from tissues expressing EGFP‐NM myosin IIA‐S1943A (right). ACh stimulation of Sham‐treated tissues resulted in a significant increase in the number of PLA spots at the cell cortex indicating interactions between vinculin and talin (n = 26, US; n = 29, ACh). By contrast, few spots were observed in cells from tissues treated with the NM myosin S1943A (US, n = 17; ACh, n = 25). Very few spots are observed in the unstimulated cells. B, PLA shows interactions between vinculin and paxillin in cells dissociated from Sham‐treated tissues (left), and in cells dissociated from tissues expressing EGFP‐NM myosin IIA‐S1943A (right). Paxillin/vinculin complexes are distributed throughout the cytoplasm of unstimulated cells from both Sham‐treated and EGFP‐NM myosin IIA‐S1943A treated tissues. ACh stimulation resulted the localization of paxillin/vinculin complexes to the cell cortex of Sham‐treated cells, although paxillin/vinculin complexes remained distributed throughout the cytoplasm of EGFP‐NM myosin IIA‐S1943A treated cells. The ratio of the cellular distribution of paxillin/vinculin complexes between the cortex and the cytoplasm was quantified for each cell. The ratio of the distribution of paxillin/vinculin complexes between the cortex/cytoplasm was significantly higher for Sham‐treated cells than for EGFP‐NM myosin IIA‐S1943A‐treated cells after ACh stimulation (n = 16 from each group). C, representative immunoblot from extracts of four muscle tissues treated with NM myosin S1943A or Sham‐treated and then stimulated with ACh or not stimulated (US) (left). Vinculin Tyr1065 phosphorylation and paxillin Tyr118 phosphorylation increased significantly in response to 10−5 m ACh in Sham treated tissues. Expression of NM myosin S1943A mutant inhibited the ACh‐induced increase in both vinculin Tyr 1065 phosphorylation (n = 13) and paxillin Tyr 118 phosphorylation (n = 10). D, immunoblot of soluble G‐actin (globular) and insoluble F‐actin (filamentous) in fractions from extracts of US or ACh stimulated muscle tissues treated with NM myosin S1943A mutant or Sham‐treated. Ratios of F‐actin to G‐actin were determined by quantitating F and G actin in extracts from each muscle strip (left). Expression of NM myosin S1943A prevented the increase in F‐actin/G‐actin ratio in response to ACh stimulation (n = 4) (right). Values are the mean ± SEM.*Significant difference between treatments (P < 0.05).
Vinculin forms a stable complex with paxillin that is maintained in both unstimulated and stimulated airway smooth muscle tissues (Opazo Saez et al. 2004; Huang et al. 2011; Zhang et al. 2012). The stimulation of tracheal smooth muscle tissues with ACh results in the recruitment of paxillin/vinculin complexes to membrane adhesion complexes (Zhang et al. 2012). We used PLA to determine the role of NM myosin II on the recruitment of paxillin/vinculin complexes to membrane adhesion junctions in response to ACh (Fig. 4 B). In Sham‐treated tissues, paxillin/vinculin complexes were observed throughout the cytoplasm of unstimulated cells, whereas, after contractile stimulation, they were localized predominantly within the submembranous cortex of ACh‐stimulated cells. In cells expressing NM myosin IIA S1943A, paxillin/vinculin complexes did not localize to the cortex of ACh‐stimulated cells; a diffuse distribution throughout the cytoplasm was observed in both ACh‐stimulated and unstimulated cells. These results suggest that NM myosin II assembly is necessary for the recruitment of paxillin/vinculin complexes to membrane adhesomes in response to stimulation with ACh. We conclude that NM myosin II regulates adhesome complex assembly during the contractile activation of smooth muscle.
Vinculin and paxillin are activated by tyrosine phosphorylation within membrane adhesion complexes in airway smooth muscle tissues, and their activation regulates the downstream signalling pathways that mediate actin polymerization and contraction (Tang et al. 2003; Huang et al. 2011; Zhang et al. 2012, 2016; Wu & Gunst, 2015). Vinculin phosphorylation on Tyr1065 at its C‐terminus regulates a conformational change that enables it to bind to F‐actin and talin (Huang et al. 2014), whereas paxillin phosphorylation at Tyr31 and Tyr118 regulates its coupling to signalling complexes that catalyse actin polymerization (Tang et al. 2005; Zhang et al. 2016). We analysed the effect of expressing NM myosin S1943A on the tyrosine phosphorylation of both paxillin and vinculin in response to contractile stimulation with ACh (Fig. 4 C). Vinculin Tyr1065 phosphorylation and paxillin Tyr118 phosphorylation in response to ACh stimulation were both inhibited by expression of the NM myosin S1943A mutant.
Contractile stimulation of airway smooth muscle elicits the polymerization of a small pool of actin, and this actin polymerization is required in addition to SM myosin RLC phosphorylation and cross‐bridge cycling for the development of active tension in airway smooth muscle (Gunst & Zhang, 2008; Zhang et al. 2015). The signalling proteins that catalyse the polymerization of actin during contractile activation localize to adhesion junction complexes (Zhang et al. 2005, 2012, 2015; Wu & Gunst, 2015). We therefore evaluated the role of NM myosin II Ser1943 phosphorylation and NM myosin II assembly in ACh‐stimulated actin polymerization.
Actin polymerization was measured in canine tracheal smooth muscle tissues using a fractionation assay to separate soluble (G‐actin) and insoluble (F‐actin) actin in the muscle extracts (Tang et al. 2005; Zhang et al. 2005, 2010, 2016). The expression of NM myosin II S1943A in the tissues significantly inhibited the increase in actin polymerization (increase in F‐actin/G‐actin ratio) in response to stimulation with ACh (Fig. 4 D). The results suggest that the NM myosin II assembly regulates cortical actin polymerization in response to a contractile stimulus by regulating the assembly of adhesome signalling complexes.
Actomyosin cross‐bridge cycling by NM myosin II is required for the assembly and activation of membrane adhesome signalling complexes and tension development during the contractile stimulation of tracheal smooth muscle tissues
We evaluated the role of cross‐bridge cycling by NM myosin II on tension development, NM myosin II filament formation, and adhesome protein recruitment and activation in tracheal smooth muscle tissues by treating tissues with blebbistatin. Blebbistatin prevents cross‐bridge cycling by non‐muscle myosin II by binding to its head domain and blocking it in an actin‐detached state. Although blebbistatin is a potent and selective inhibitor of actomyosin cross‐bridge cycling by NM myosin II isoforms, it does not inhibit cross‐bridge cycling by smooth muscle myosin II (Straight et al. 2003; Kovacs et al. 2004).
Tracheal smooth muscle tissues were incubated in 10 μm blebbistatin for 30 min and then contracted with 10−5 m ACh for 5 min for the determination of tension development and NM myosin II filament formation. Treatment with blebbistatin inhibited force development (Fig. 5 A), although it had no effect on NM myosin II filament formation as assessed by the Triton solubility assay (Fig. 5 B). Blebbistatin also did not inhibit the phosphorylation of NM myosin on Ser1943 in response to ACh (Fig. 5 C). We also assessed the effect of blebbistatin on NM myosin II filament formation using the PLA assay to assess interactions between NM myosin IIA and NM myosin IIB in freshly dissociated tracheal muscle cells. Cells were treated with 2 μm blebbistatin prior to stimulation with 5 min 10−5 m ACh (Fig. 5 D). Blebbistatin had no effect on the interaction of NM myosin IIA and IIB, suggesting that it did not inhibit NM myosin II filament formation in response to ACh. Thus, the results obtained using blebbistatin are consistent with the interpretation that NM myosin filament formation is a distinct step that occurs independently of actin‐activated cross‐bridge cycling by NM myosin II.
Figure 5. The NM myosin II cross‐bridge cycling inhibitor, blebbistatin suppresses ACh‐induced tension development and the assembly and activation of membrane adhesome complexes, although it does not inhibit NM myosin II assembly.
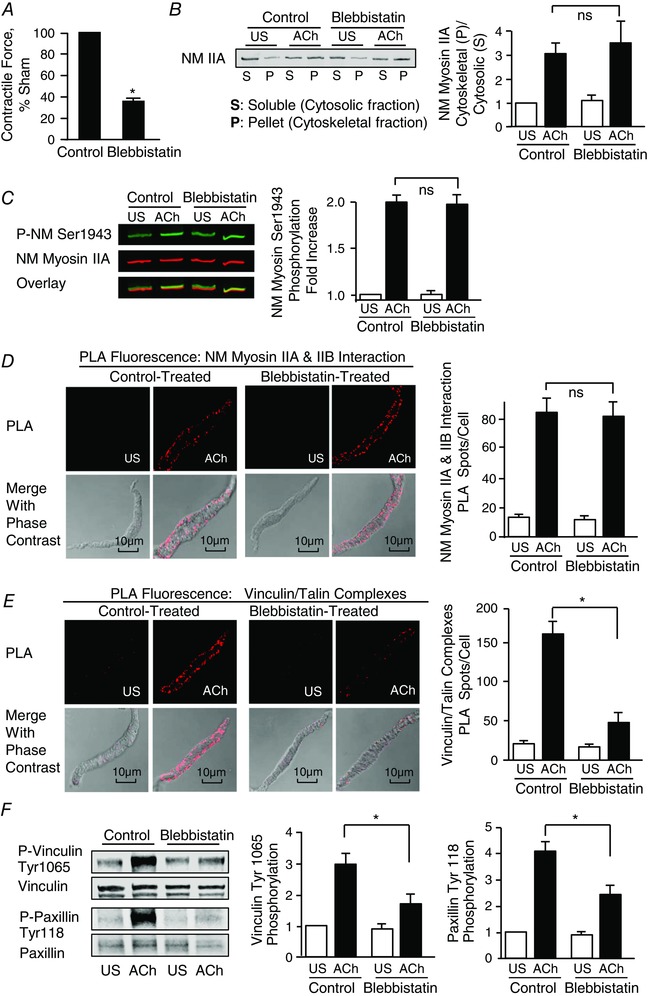
A, blebbistatin significantly inhibited force generation in response to 10−5 m ACh stimulation in tracheal smooth muscle (n = 8). B, immunoblot of cytosolic (soluble, S) and cytoskeletal (pellet, P) fractions from extracts of tracheal smooth muscle tissues treated with 10 μm blebbistatin or untreated (Control) and stimulated with ACh or unstimulated (US). Blebbistatin did not significantly affect the ACh‐induced increase in ratio of NM myosin II in the cytoskeletal (P) vs. soluble (S) fractions in response to 5 min of stimulation with 10−5 m ACh (n = 7). C, increase in NM myosin II Ser1943 phosphorylation in tracheal muscle tissues in response to 5 min of stimulation with 10−5 m. ACh was not significantly affected by blebbistatin (n = 12). D, blebbistatin had no significant effect on the increase in the number of NM myosin IIA and IIB complexes at the cell membrane in response to ACh (Control: US cells, n = 18; ACh stimulated cells, n = 28. Blebbistatin treated cells: US, n = 20; ACh, n = 29). Images show in situ PLA fluorescence (top) and PLA fluorescence merged with phase contrast (bottom) from freshly dissociated smooth muscle cells US or stimulated with ACh. PLA spots show interactions between NM myosin IIA and NM myosin IIB in freshly dissociated cells (left). E, PLA spots show interactions between talin and vinculin in freshly dissociated cells (left). ACh stimulation resulted in a significant increase in the number of talin‐vinculin complexes at the cell membrane in control cells (US, n = 24; ACh, n = 24). By contrast, few spots were observed in blebbistatin treated cells (US, n = 24; ACh, n = 25). Very few spots are observed in the unstimulated control cells and blebbistatin treated cells. F, representative immunoblot from extracts of two control muscle tissues and two blebbistatin treated tissues stimulated with ACh or not stimulated (US) (left). The increase in vinculin Tyr1065 phosphorylation and paxillin Tyr118 phosphorylation in response to 10−5 m ACh was significantly inhibited in tissues treated with blebbistatin (n = 7). Values are the mean ± SEM.*Significant difference between treatments (P < 0.05). ns, not significantly different.
We next evaluated the effect of blebbistatin on the assembly and activation of adhesome complexes in response to ACh. PLA was used to assess the effect of blebbistatin on the formation of vinculin/talin complexes in freshly dissociated cells (Fig. 5 E). Blebbistatin inhibited the formation of vinculin/talin complexes at the cell cortex. Blebbistatin also inhibited the tyrosine phosphorylation of both vinculin and paxillin in tissues stimulated with ACh for 5 min (Fig. 5 F).
The results obtained indicate that NM myosin II ATPase activity and cross‐bridge cycling are required for tension development in smooth muscle, and that the inhibition of NM myosin II cross‐bridge cycling inhibits tension development by preventing the assembly and activation of membrane adhesome complexes. By contrast, both the Triton solubility assay and the PLA assays for NM myosin IIA and IIB interaction indicate that blebbistatin does not inhibit NM myosin II filament formation in response to stimulation with ACh.
RhoA GTPase regulates NM myosin RLC phosphorylation during the contractile stimulation of smooth muscle tissues
The role of myosin RLC phosphorylation in the activation of actin‐activated myosin ATPase activity is well established for both SM and NM myosin II. We used two‐dimensional gel electrophoresis to evaluate the effect of contractile stimulation on the phosphorylation of NM myosin II RLC in tracheal smooth muscle tissues. NM and SM myosin RLC isoforms cannot be distinguished immunologically because their sequences differ only slightly (Gaylinn et al. 1989; Yuen et al. 2009).
NM and SM myosin RLCs extracted from ACh‐stimulated and unstimulated smooth muscle tissues were separated according to differences in isoelectric focusing. At the 20 kDa molecular weight, three distinct spots were observed in extracts from unstimulated tissues and four distinct spots in extracts from ACh stimulated tissues (Fig. 6 A). Mass spectrometry was performed on proteins extracted from these gels to confirm the sequences of these spots. The two less acidic spots (Spots 1 and 2) consisted of unphosphorylated SM myosin RLC (Spot 1) and Ser19‐phosphorylated SM myosin RLC (Spot 2). The two more acidic spots (Spots 3 and 4) contained unphosphorylated NM myosin RLC (Spot 3) and Ser19‐phosphorylated NM myosin RLCs (Spot 4). However, di‐phosphorylated (Thr18 and Ser19) SM myosin RLC was present in extracts from stimulated smooth muscle tissues and was found to co‐migrate with unphosphorylated NM myosin RLC (Spot 3).
Figure 6. ACh‐stimulated NM Myosin II RLC phosphorylation is regulated by RhoA GTPase.
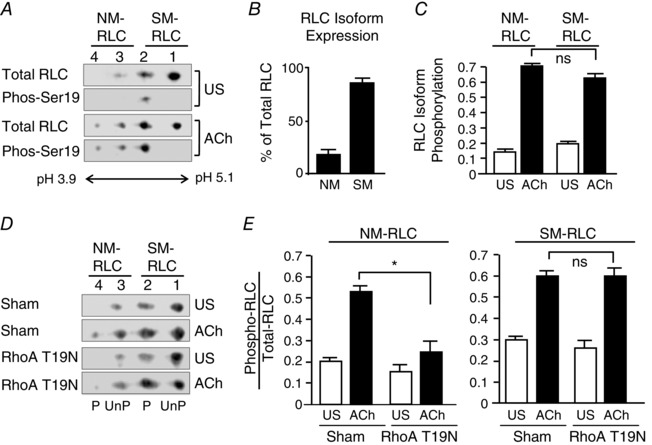
A, representative immunoblots of myosin RLCs from unstimulated (US) and ACh‐stimulated muscle tissues. RLCs were separated by two‐dimensional electrophoresis and immunoblotted for RLC and phospho‐Ser19 RLC using dual immunofluorescence. Unphosphorylated and Ser19 phosphorylated NM RLC migrated to Spots 3 and 4, respectively. Unphosphorylated SM RLC migrated to Spots 1, mono‐phosphorylated SM myosin RLC migrated to spot 2 and di‐phosphorylated (Ser 19 and Thr 18) SM RLC migrated to spot 3. In US tissues, Ser19 phosphorylation was only detected in spot 2; whereas it was detected in spots 2, 3 and 4 in ACh stimulated samples. B, proportion of NM and SM RLC in smooth muscle extracts, as quantified by two‐dimensional electrophoresis. NM RLCs represented 18.7 ± 2.2% of the total RLCs present in the tracheal smooth muscle tissue. C, RLC phosphorylation of both NM and SM myosin increased significantly and by a similar proportion in response to 5 min of stimulation with 10−5 m ACh (n = 8). D and E, representative immunoblots of myosin RLCs from four muscle tissues transfected with the RhoA T19N mutant or Sham‐treated and then stimulated with ACh or not stimulated (US). Expression of RhoA T19N significantly inhibited the increase of NM myosin II RLC phosphorylation, although it did not affect the increase in SM RLC phosphorylation in response to ACh stimulation (n = 5). Values are the mean ± SEM. *Significant difference between treatments (P < 0.05). ns, not significantly different.
Figure 6 A illustrates immunoblots of NM and SM RLCs from extracts of unstimulated and ACh‐stimulated smooth muscle tissues. Membranes were immunoblotted using antibody that reacts with both NM and SM RLC and with antibody against Ser19 RLC and then visualized by dual immunofluorescence. Immunoblots confirmed the presence of Ser19 phosphorylated RLC in Spot 2 from unstimulated samples and in Spots 2, 3 and 4 from ACh‐stimulated samples. The proportion of NM and SM RLCs was quantified from immunoblots from unstimulated smooth muscle tissues by comparing the proportion of RLCs and Spots 1 and 2 vs. Spots 3 and 4. NM RLCs represented 18.7 ± 2.2% of the total RLCs present in the tracheal smooth muscle tissues (Fig. 6 B).
The phosphorylation of SM myosin RLC was quantified based on the density of unphosphorylated SM myosin RLC (Spot 1) relative to the proportion of SM myosin RLC to total RLC (Fig. 6 C). The effect of ACh stimulation on the phosphorylation of NM myosin RLC was quantified based on the density of the phosphorylated NM myosin RLC (Spot 4) relative to the proportional NM myosin RLC to total RLC (Fig. 6 C). Both NM and SM myosin RLC exhibited low levels of phosphorylation in resting tissues. The phosphorylation of both isoforms increased by a similar proportion in response to stimulation of airway smooth muscle tissues with 10−5 m ACh for 5 min. These results demonstrate that stimulation with ACh activates both SM and NM myosin II similarly in airway smooth muscle.
RhoA inhibition suppresses actin polymerization and tension development in response to ACh, although it has only a small effect on total myosin RLC phosphorylation as assessed by one‐dimensional urea gel electrophoresis (Zhang et al. 2010). However, we previously showed that RhoA activation is critical for the recruitment of proteins to membrane adhesomes and the activation of adhesome signalling complexes during contractile stimulation of airway smooth muscle tissues (Zhang et al. 2012). We evaluated the role of RhoA in NM myosin RLC phosphorylation by expressing the RhoA inactive mutant, RhoA T19N, in tracheal smooth muscle tissues to inhibit endogenous RhoA activation (Zhang et al. 2010, 2012) (Fig. 6 D). The inactivation of RhoA almost completely suppressed ACh‐stimulated NM myosin II RLC phosphorylation, although it did not significantly affect SM myosin RLC phosphorylation (Fig. 6 E). These observations suggest that RhoA selectively regulates the activation of NM myosin II but not the SM myosin RLC isoforms.
The phosphorylation NM myosin RLC regulates NM myosin II filament assembly during the contractile stimulation of smooth muscle tissues
We assessed the role of RhoA in the regulation of NM myosin II filament assembly in airway smooth muscle. The expression of RhoA T19N in tracheal smooth muscle tissues inhibited the increase in NM myosin II into the cytoskeletal fraction in ACh stimulated tissues, which is consistent with the inhibition of NM myosin II filament assembly (Fig. 7 A). In situ PLA was also performed on freshly dissociated tracheal smooth muscle cells to evaluate the role of RhoA on NM myosin II filament formation. In smooth muscle cells dissociated from tissues expressing the RhoA T19N mutant, ACh did not induce interactions between NM myosin IIA and IIB isoforms, whereas many interactions were detected in ACh‐stimulated cells from Sham‐treated tissues (Fig. 7 B).
Figure 7. RhoA GTPase regulates NM myosin II assembly, NM myosin heavy chain Ser1943 phosphorylation and tension development during ACh stimulation.

A, left: immunoblot of cytoplasmic (soluble, S) and cytoskeletal (pellet, P) fractions of tracheal smooth muscle extracts from tissues treated with RhoA T19N or Sham‐treated, and stimulated with ACh or unstimulated (US). The ratio of NM myosin II in the cytoskeletal (P) vs. soluble (S) fractions increased in response to 5 min of stimulation with 10−5 m ACh in Sham‐treated tissues, although it was significantly inhibited by the expression of RhoA T19N in 10−5 m ACh‐stimulated tissues (n = 6). B, images of in situ PLA fluorescence (top) and PLA fluorescence merged with phase contrast (bottom) from freshly dissociated smooth muscle cells US or stimulated with ACh. PLA spots show interactions between NM myosin IIA and NM myosin IIB in cells dissociated from Sham‐treated tissues (left). ACh stimulation of Sham‐treated tissues resulted in a significant increase in the number of NM myosin IIA and IIB complexes at the cell membrane (n = 27, US; n = 30, ACh). By contrast, few NM myosin IIA and IIB complexes were observed in cells from tissues treated with the RhoA T19N (US, n = 26; ACh, n = 20). Very few NM myosin IIA and IIB complexes are observed in the unstimulated cells. C, increase in NM myosin II Ser1943 phosphorylation in response to 5 min of stimulation with 10−5 m ACh was significantly inhibited in tissues expressing RhoA T19N compared to Sham‐treated tissues (n = 8). Contractile force in response to ACh is shown for tissues before and after they were transfected with plasmids encoding RhoA T19N or no plasmids (Sham‐treated) (left). Expression of RhoA T19N significantly inhibited force generation in response to 10−5 m ACh stimulation in tracheal smooth muscle. All force measurements normalized to Sham response. Values are the mean ± SEM. *Significant difference between treatments (P < 0.05).
We evaluated the effect of RhoA inhibition on ACh‐induced NM myosin II heavy chain Ser1943 phosphorylation in tracheal smooth muscle tissues by expressing the RhoA T19N mutant in tracheal smooth muscle tissues (Fig. 7 C). RhoA inhibition prevented the increase in NM myosin II heavy chain Ser1943 phosphorylation in response to ACh stimulation. The inhibition of tension development by RhoA T19N was similar to that caused by expression of the NM myosin S1943A mutant (Figs 3 D and 7 C). The results are consistent with the possibility that activated RhoA mediates NM myosin II RLC phosphorylation and also that NM RLC phosphorylation regulates the conversion of NM myosin II to an assembly‐competent conformation that can then undergo Ser1943 phosphorylation on its heavy chain, thereby promoting filament formation. Thus, the results suggest that Rho‐mediated NM myosin RLC phosphorylation is a necessary prerequisite for NM myosin II filament assembly in airway smooth muscle tissues. In addition, the results indicate that RhoA regulates airway smooth muscle contraction by regulating NM myosin assembly and activation rather than by regulating SM myosin activation.
Discussion
The present study demonstrates that NM myosin II plays a critical role in the regulation of airway smooth muscle contraction. We find that the function of NM myosin II in airway smooth muscle is quite different from that of SM myosin II: smooth muscle myosin plays a well‐known role in generating cell shortening via actomyosin filament sliding, whereas NM myosin mediates the assembly of membrane adhesome complexes that connect actin filaments to the extracellular matrix for the transmission of tension to the outside of the cell (Fig. 8). The results suggest that the contractile stimulation of airway smooth muscle regulates the assembly of NM myosin II into filaments, and that NM myosin filament assembly is required for active contractile tension generation in smooth muscle tissues. Furthermore, the results suggest that the contractile activation of tracheal smooth muscle regulates NM myosin filament assembly by regulating the RhoA‐dependent phosphorylation of the NM myosin II RLC, and by regulating the phosphorylation of the NM myosin II heavy chain on Ser1943 (Fig. 8). Contractile stimulation activates proteins within adhesome complexes that mediate pathways catalysing cortical actin polymerization, which is also required for tension generation in smooth muscle. We conclude that NM myosin II plays an essential role in the physiological responses of airway smooth muscle tissues to extracellular contractile stimulation that is completely distinct from that of SM myosin (Fig. 8).
Figure 8. Proposed mechanism for the regulation of contraction by NM myosin II in airway smooth muscle.
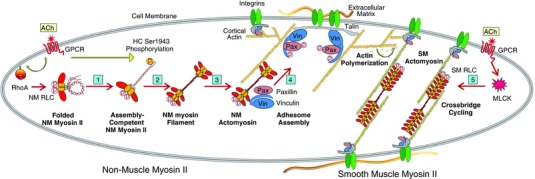
(1) ACh stimulation activates RhoA, which regulates phosphorylation of NM myosin regulatory light chain (RLC) and catalyses the conversion of inactive folded NM myosin II to its assembly‐competent conformation. (2) ACh catalyses the phosphorylation of NM myosin heavy chain (HC) on Ser1943, enabling it to form filaments. (3) NM myosin filaments interact with cortical actin filaments and undergo activation. (4) Activated NM actomyosin mediates the transport of inactive adhesome proteins to membrane adhesome complexes, where they form adhesion complexes that mediate force transmission and regulate cortical actin polymerization. (5) SM myosin RLCs are phosphorylated by MLC kinase resulting in SM myosin cross‐bridge cycling and filament sliding. This enables tension generation by the smooth muscle contractile apparatus.
The assembly of NM myosin II in response to extracellular stimulation has never been demonstrated in smooth muscle tissues, in which the predominant isoform of myosin II is SM myosin. We used several approaches to evaluate the assembly of NM myosin filaments during airway smooth muscle contraction. One method was to quantify the proportion of NM myosin II in the cytoskeletal vs. Triton‐soluble fractions of smooth muscle tissue extracts, a method previously used to document NM myosin filament assembly during adhesion and migration in several non‐muscle cancer cell lines (Dulyaninova et al. 2005, 2007; Breckenridge et al. 2009). The proportion of both isoforms of NM myosin II was higher in the cytoskeletal fraction of tissue extracts after stimulation with ACh than in unstimulated muscles, which is consistent with an increase in the formation of NM myosin II filaments that can interact with F‐actin and cytoskeletal proteins. Because the 10S myosin II monomer cannot form filaments or bind to actin (Wendt et al. 1999; Jung et al. 2008), it should remain in the soluble fraction, whereas the NM myosin found in the cytoskeletal fraction should be filamentous.
We further assessed NM myosin II filament formation by using in situ PLA to probe for interactions between NM myosin IIA and IIB (Fig. 1). NM myosin II can form mixed heterotypic NM myosin filaments in living cells (Beach et al. 2014; Beach & Hammer, 2015). In situ PLA detects protein associations that are less than 40 nm, a proximity that is usually indicative of binding between the target proteins (Soderberg et al. 2006, 2008). Thus, the PLA signals probably represent binding interactions between NM myosin II monomers. We detected very few interactions between NM myosin IIA and IIB isoforms in resting smooth muscle cells, although we observed a dramatic increase in NM myosin IIA and IIB interactions after contractile stimulation. These observations are consistent with the interpretation that NM myosin II exists in monomeric form in resting unstimulated muscles, possibly in a pool of assembly‐incompetent monomers (Milton et al. 2011) and also that contractile stimulation triggers the polymerization of NM myosin II monomers into filaments. In combination, our results provide strong evidence that contractile stimulation regulates the assembly of a pool of NM myosin II into filaments.
The effects of treating the tissues with the NM myosin II cross‐bridge cycling inhibitor, blebbistatin, provide further evidence for a role for NM myosin II in the regulation of airway SM contraction, and provide additional evidence that NM myosin filament assembly is triggered by contractile stimulation (Fig. 5). Blebbistatin binds to the myosin‐ADP‐Pi complex with high affinity and interferes with the phosphate release process, thus blocking myosin in an actin‐detached state and preventing actomyosin cross‐linking (Kovacs et al. 2004). It acts selectively on the NM myosin II by inhibiting the myosin ATPase, but it has no effect on myosin light chain kinase or smooth muscle myosin II (Straight et al. 2003; Kovacs et al. 2004). We found that blebbistatin had no effect on ACh‐induced NM myosin filament formation as assessed by either the Triton solubility assay or PLA for NM myosin IIA and IIB interactions (Fig. 5). However, blebbistatin inhibited force development and the assembly and activation of adhesome complexes in response to ACh. Because blebbistatin prevents NM myosin II from binding to actin filaments, our results are consistent with the interpretation that the Triton solubility assay assesses NM myosin II filament formation rather than the interaction of NM myosin II with actin filaments or the formation of cytoskeletal protein complexes containing NM myosin II. Our observations of few interactions between NM myosin IIA and IIB being observed in resting smooth muscle cells and blebbistatin not inhibiting interactions between NM myosin IIA and IIB in stimulated tissues further support the idea that NM myosin II forms filaments after ACh stimulation. In summary, our studies with blebbistatin provide additional evidence that NM myosin II filament formation and NM myosin cross‐bridge cycling are both necessary for the assembly and activation of adhesome complexes during the contractile stimulation of airway smooth muscle, and also that adhesome complex assembly is necessary for tension development.
The contractile stimulation of airway smooth muscle tissues with ACh resulted in the phosphorylation of NM myosin on its non‐helical tail domain on Ser1943. We inhibited the Ser1943 phosphorylation of endogenous NM myosin during contraction of the smooth muscle tissues by expressing the non‐phosphorylatable NM myosin IIA mutant, NM myosin S1943A. This inhibited airway smooth muscle contraction and prevented NM myosin II filament assembly, as assessed by both the Triton solubility assay and the PLA assay for interaction between NM myosin IIA and IIB. Blebbistatin inhibited adhesome complex assembly and the activation of adhesome proteins, presumably by inhibiting NM myosin II cross‐bridge cycling; however, it had no effect on NM myosin II Ser1943 phosphorylation or on filament assembly. These results are consistent with a role for NM myosin heavy chain phosphorylation at Ser1943 in promoting filament formation during contractile stimulation and support the conclusion that NM myosin II filament assembly is necessary for tension generation (Figs 2 and 3).
The NM myosin heavy chain has been reported to undergo phosphorylation at the Ser1943 site in several carcinoma cell lines in vivo (Dulyaninova et al. 2007; Dulyaninova & Bresnick, 2013; Heissler & Sellers, 2016). The in vivo expression of NM myosin S1943A in these cells reduced cell migration and lamellipod extension (Dulyaninova et al. 2007), which is consistent with our observations of reduced contractility in airway smooth muscle tissues. By contrast, in molecular studies using NM myosin rod C‐terminal tail fragments in vitro, the phosphorylation of NM myosin IIA at Ser1943 resulted in the disassembly of existing NM myosin II filaments or inhibited the formation of filaments by NM myosin II monomers (Murakami et al. 1995, 1998; Dulyaninova et al. 2005). The apparent differences in the effect of NM myosin II heavy chain phosphorylation on NM myosin filament assembly in the in vitro studies and in our in vivo studies may be a result of the exclusive use of NM myosin II tail fragments in the in vitro studies, which might not be reliable surrogates for full‐length NM myosin II. Furthermore, the effect of phosphorylation on the assembly of the NM myosin IIA isoform was shown to be highly dependent on the stoichiometric level of phosphate incorporation into the heavy chain (Dulyaninova et al. 2005), which could differ in vivo and in vitro. Other aspects of in vivo conditions, such as the presence of myosin‐binding proteins and other heavy chain phosphorylation events, might also alter the effect of Ser1943 phosphorylation on the molecular function of NM myosin II in vivo. It is also possible that NM myosin II filament assembly in living cells requires the disassembly of existing NM myosin II filaments to generate a pool of NM myosin II monomers that are reincorporated into de novo NM myosin II filaments. However, this explanation does not explain the marked increase in NM myosin IIA and IIB isoform interactions that occurred in response to ACh. The molecular function of NM myosin heavy chain Ser1943 phosphorylation on NM myosin II filament assembly has not been assessed previously in living cells.
We demonstrated that RhoA selectively regulates the phosphorylation of NM myosin RLC: RhoA inhibition had no significant effect on the RLC phosphorylation of SM myosin induced by ACh, although it caused a marked inhibition of the NM myosin RLC phosphorylation (Fig. 6). Phosphorylation of the RLC is widely recognized as a key step in the regulation of actin‐activated ATPase activity for both NM and SM myosin II. The results of the present study show that NM myosin II can be activated separately from SM myosin and the contractile apparatus and regulates the assembly of adhesomes that mediate signalling pathways critical for smooth muscle cell processes elicited by non‐contractile stimuli, as well as contractile stimuli (Desai et al. 2011; Wu et al. 2016). Thus, the activation of NM myosin II may be fundamental to the phenotypic and synthetic responses of the cell to extracellular stimuli that that do not activate contraction.
Myosin RLC chain phosphorylation is required for the inactive folded assembly‐incompetent conformer of monomeric myosin II to assume an open assembly‐competent conformation in vitro (Craig et al. 1983; Smith et al. 1983; Trybus & Lowey, 1984); however, there is no evidence regarding phosphorylation of the RLC being important for regulating either NM or SM myosin filament assembly in vivo. We found that NM myosin II filament assembly was inhibited in smooth muscle cells in which NM myosin II RLC phosphorylation was prevented by RhoA inactivation. The results are consistent with the possibility that NM myosin II RLC phosphorylation regulates the conversion of NM myosin monomers to an assembly‐competent conformation in smooth muscle. However, because we cannot directly measure the conformation of NM myosin monomers in the cells, it is possible that RhoA regulates NM myosin II assembly in airway smooth muscle through an alternative mechanism. Regardless of the mechanism, the ability to selectively inactivate NM myosin II assembly in airway smooth muscle by inactivating RhoA provides further evidence for the distinct cellular function of NM myosin during contraction. NM myosin inactivation prevents adhesome assembly, suppresses cytoskeletal signalling, and inhibits actin polymerization and tension development without affecting SM RLC phosphorylation and SM myosin activation.
The expression of NM myosin II in smooth muscle has long been recognized; however, its physiological function in muscle tissues has never been established (Eddinger & Meer, 2007). In non‐muscle cells, the functions of NM myosin II have been studied extensively and its roles in adhesion and cell crawling are widely documented (Vicente‐Manzanares et al. 2009). By contrast, there are few studies of the function of NM myosin II in smooth muscle. Studies investigating the physiological properties of bladder smooth muscle tissues from SM myosin knockout mice demonstrated that the contractility of bladder strips from these mice was markedly diminished (Morano et al. 2000; Lofgren et al. 2003); however, the tissues were still capable of generating slow contractions that produced very low levels of tonic force. Although these studies suggested a possible role for NM myosin II in smooth muscle contraction, they were not definitive because it is impossible to rule out compensatory changes in the expression or function of other proteins in tissues from these knockout animals. Yuen et al. (2009) demonstrated that the activation of NM myosin II is regulated by Ca2+‐calmodulin activated myosin light chain kinase in mouse aortic tissues during smooth muscle contraction. They also found that force maintenance was slightly reduced in the aortas from NM myosin IIB knockout mice and suggested that NM myosin IIB plays a role in force maintenance.
The results of the present study demonstrate that the function of NM myosin II is critical to force development in airway smooth muscle: when the assembly or activation of NM myosin II is inhibited, force development is reduced by 60%. Furthermore, we show that contractile stimuli can regulate the activation of NM myosin II by regulating NM myosin heavy chain phosphorylation, as well as NM myosin II regulatory light chain phosphorylation. These results suggest a central role for NM myosin II in airway smooth muscle contraction that is probably broadly relevant to other smooth muscle tissue types.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SJG conceived the idea for the project. WZ and SJG designed the experiments. WZ conducted the experiments. WZ analysed the results. SJG supervised the data analysis. WZ and SJG wrote the paper. Both authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Heart, Lung, and Blood Institute Grants HL029289, HL048522 and HL109629.
References
- Beach JR & Hammer JA 3rd (2015). Myosin II isoform co‐assembly and differential regulation in mammalian systems. Exp Cell Res 334, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach JR, Shao L, Remmert K, Li D, Betzig E & Hammer JA, 3rd (2014). Nonmuscle myosin II isoforms coassemble in living cells. Curr Biol 24, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge MT, Dulyaninova NG & Egelhoff TT (2009). Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells. Mol Biol Cell 20, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick AR (1999). Molecular mechanisms of nonmuscle myosin‐II regulation. Curr Opin Cell Biol 11, 26–33. [DOI] [PubMed] [Google Scholar]
- Craig R, Smith R & Kendrick‐Jones J (1983). Light‐chain phosphorylation controls the conformation of vertebrate non‐muscle and smooth muscle myosin molecules. Nature 302, 436–439. [DOI] [PubMed] [Google Scholar]
- Desai LP, Wu Y, Tepper RS & Gunst SJ (2011). Mechanical stimuli and IL‐13 interact at integrin adhesion complexes to regulate expression of smooth muscle myosin heavy chain in airway smooth muscle tissue. Am J Physiol Lung Cell Mol Physiol 301, L275–L284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulyaninova NG & Bresnick AR (2013). The heavy chain has its day: regulation of myosin‐II assembly. Bioarchitecture 3, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulyaninova NG, House RP, Betapudi V & Bresnick AR (2007). Myosin‐IIA heavy‐chain phosphorylation regulates the motility of MDA‐MB‐231 carcinoma cells. Mol Biol Cell 18, 3144–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulyaninova NG, Malashkevich VN, Almo SC & Bresnick AR (2005). Regulation of myosin‐IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry 44, 6867–6876. [DOI] [PubMed] [Google Scholar]
- Eddinger TJ & Meer DP (2007). Myosin II isoforms in smooth muscle: heterogeneity and function. Am J Physiol Cell Physiol 293, C493–C508. [DOI] [PubMed] [Google Scholar]
- Gaylinn BD, Eddinger TJ, Martino PA, Monical PL, Hunt DF & Murphy RA (1989). Expression of nonmuscle myosin heavy and light chains in smooth muscle. Am J Physiol 257, C997–C1004. [DOI] [PubMed] [Google Scholar]
- Gunst SJ & Zhang W (2008). Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295, C576–C587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissler SM & Manstein DJ (2013). Nonmuscle myosin‐2: mix and match. Cell Mol Life Sci 70, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissler SM & Sellers JR (2016). Various themes of myosin regulation. J Mol Biol 428, 1927–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz A, Trybus KM, Bowman DS & Fay FS (1994). Antibodies probe for folded monomeric myosin in relaxed and contracted smooth muscle. J Cell Biol 126, 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Day RN & Gunst SJ (2014). Vinculin phosphorylation at Tyr1065 regulates vinculin conformation and tension development in airway smooth muscle tissues. J Biol Chem 289, 3677–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang W & Gunst SJ (2011). Activation of vinculin induced by cholinergic stimulation regulates contraction of tracheal smooth muscle tissue. J Biol Chem 286, 3630–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Komatsu S, Ikebe M & Craig R (2008). Head‐head and head‐tail interaction: a general mechanism for switching off myosin II activity in cells. Mol Biol Cell 19, 3234–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick‐Jones J, Smith RC, Craig R & Citi S (1987). Polymerization of vertebrate non‐muscle and smooth muscle myosins. J Mol Biol 198, 241–252. [DOI] [PubMed] [Google Scholar]
- Kiboku T, Katoh T, Nakamura A, Kitamura A, Kinjo M, Murakami Y & Takahashi M (2013). Nonmuscle myosin II folds into a 10S form via two portions of tail for dynamic subcellular localization. Genes Cells 18, 90–109. [DOI] [PubMed] [Google Scholar]
- Kim HR, Gallant C, Leavis PC, Gunst SJ & Morgan KG (2008). Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol 295, C768–C778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs M, Toth J, Hetenyi C, Malnasi‐Csizmadia A & Sellers JR (2004). Mechanism of blebbistatin inhibition of myosin II. J Biol Chem 279, 35557–35563. [DOI] [PubMed] [Google Scholar]
- Lehman W & Morgan KG (2012). Structure and dynamics of the actin‐based smooth muscle contractile and cytoskeletal apparatus. J Muscle Res Cell Motil 33, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZH, Dulyaninova NG, House RP, Almo SC & Bresnick AR (2010). S100A4 regulates macrophage chemotaxis. Mol Biol Cell 21, 2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren M, Ekblad E, Morano I & Arner A (2003). Nonmuscle Myosin motor of smooth muscle. J Gen Physiol 121, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D & Gunst SJ (1999). Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol 519, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton DL, Schneck AN, Ziech DA, Ba M, Facemyer KC, Halayko AJ, Baker JE, Gerthoffer WT & Cremo CR (2011). Direct evidence for functional smooth muscle myosin II in the 10S self‐inhibited monomeric conformation in airway smooth muscle cells. Proc Natl Acad Sci USA 108, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano I, Chai GX, Baltas LG, Lamounier‐Zepter V, Lutsch G, Kott M, Haase H & Bader M (2000). Smooth‐muscle contraction without smooth‐muscle myosin. Nat Cell Biol 2, 371–375. [DOI] [PubMed] [Google Scholar]
- Murakami N, Chauhan VP & Elzinga M (1998). Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry 37, 1989–2003. [DOI] [PubMed] [Google Scholar]
- Murakami N, Singh SS, Chauhan VP & Elzinga M (1995). Phospholipid binding, phosphorylation by protein kinase C, and filament assembly of the COOH terminal heavy chain fragments of nonmuscle myosin II isoforms MIIA and MIIB. Biochemistry 34, 16046–16055. [DOI] [PubMed] [Google Scholar]
- Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD & Gunst SJ (2004). Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol 286, C433–C447. [DOI] [PubMed] [Google Scholar]
- Park I, Han C, Jin S, Lee B, Choi H, Kwon JT, Kim D, Kim J, Lifirsu E, Park WJ, Park ZY, Kim DH & Cho C (2011). Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem J 434, 171–180. [DOI] [PubMed] [Google Scholar]
- Ronen D & Ravid S (2009). Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. J Biol Chem 284, 24948–24957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey JM, Taylor KA & Kendrick‐Jones J (1980). Regulation of non‐muscle myosin assembly by calmodulin‐dependent light chain kinase. Nature 287, 233–235. [DOI] [PubMed] [Google Scholar]
- Smith RC, Cande WZ, Craig R, Tooth PJ, Scholey JM & Kendrick‐Jones J (1983). Regulation of myosin filament assembly by light‐chain phosphorylation. Philos Trans R Soc Lond B Biol Sci 302, 73–82. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG & Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG & Landegren U (2008). Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR & Mitchison TJ (2003). Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science 299, 1743–1747. [DOI] [PubMed] [Google Scholar]
- Strassheim D, May LG, Varker KA, Puhl HL, Phelps SH, Porter RA, Aronstam RS, Noti JD & Williams CL (1999). M3 muscarinic acetylcholine receptors regulate cytoplasmic myosin by a process involving RhoA and requiring conventional protein kinase C isoforms. J Biol Chem 274, 18675–18685. [DOI] [PubMed] [Google Scholar]
- Tang DD, Turner CE & Gunst SJ (2003). Expression of non‐phosphorylatable paxillin mutants in canine tracheal smooth muscle inhibits tension development. J Physiol 553, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DD, Zhang W & Gunst SJ (2005). The adapter protein CrkII regulates neuronal Wiskott–Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J Biol Chem 280, 23380–23389. [DOI] [PubMed] [Google Scholar]
- Trybus KM & Lowey S (1984). Conformational states of smooth muscle myosin. Effects of light chain phosphorylation and ionic strength. J Biol Chem 259, 8564–8571. [PubMed] [Google Scholar]
- Vicente‐Manzanares M, Ma X, Adelstein RS & Horwitz AR (2009). Non‐muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt T, Taylor D, Messier T, Trybus KM & Taylor KA (1999). Visualization of head‐head interactions in the inhibited state of smooth muscle myosin. J Cell Biol 147, 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y & Gunst SJ (2015). Vasodilator‐stimulated phosphoprotein (VASP) regulates actin polymerization and contraction in airway smooth muscle by a vinculin‐dependent mechanism. J Biol Chem 290, 11403–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Huang Y & Gunst SJ (2016). Focal adhesion kinase (FAK) and mechanical stimulation negatively regulate the transition of airway smooth muscle tissues to a synthetic phenotype. Am J Physiol Lung Cell Mol Physiol 311, L893–L902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen SL, Ogut O & Brozovich FV (2009). Nonmuscle myosin is regulated during smooth muscle contraction. Am J Physiol Heart Circ Physiol 297, H191–H199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Du L & Gunst SJ (2010). The effects of the small GTPase RhoA on the muscarinic contraction of airway smooth muscle result from its role in regulating actin polymerization. Am J Physiol Cell Physiol 299, C298–C306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W & Gunst SJ (2008). Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc 5, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang Y & Gunst SJ (2012). The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J Biol Chem 287, 33996–34008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang Y & Gunst SJ (2016). p21‐Activated kinase (Pak) regulates airway smooth muscle contraction by regulating paxillin complexes that mediate actin polymerization. J Physiol 594, 4879–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang Y, Wu Y & Gunst SJ (2015). A novel role for RhoA GTPase in the regulation of airway smooth muscle contraction. Can J Physiol Pharmacol 93, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Du L, Tang DD & Gunst SJ (2005). Activation of the Arp2/3 complex by N‐WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol 288, C1145–C1160. [DOI] [PubMed] [Google Scholar]