

ABSTRACT
The human CD99 protein is a 32-kDa glycosylated transmembrane protein that regulates various cellular responses, including cell adhesion and leukocyte extravasation. We previously reported that CD99 activation suppresses β1 integrin activity through dephosphorylation of focal adhesion kinase (FAK) at Y397. We explored a molecular mechanism underlying the suppression of β1 integrin activity by CD99 agonists and its relevance to tumor growth in vivo. CD99-Fc fusion proteins or a series of CD99-derived peptides suppressed β1 integrin activity by specifically interacting with three conserved motifs of the CD99 extracellular domain. CD99CRIII3, a representative CD99-derived 3-mer peptide, facilitated protein kinase A-SHP2 interaction and subsequent activation of the HRAS/RAF1/MEK/ERK signaling pathway. Subsequently, CD99CRIII3 induced FAK phosphorylation at S910, which led to the recruitment of PTPN12 and PIN1 to FAK, followed by FAK dephosphorylation at Y397. Taken together, these results indicate that CD99-derived agonist ligands inhibit fibronectin-mediated β1 integrin activation through the SHP2/ERK/PTPN12/FAK signaling pathway.
KEYWORDS: agonist ligands, β1 integrin, CD99, MCF-7 cells, PTPN12
INTRODUCTION
Integrins are transmembrane heterodimeric receptors composed of noncovalently bound α and β subunits that participate in cell-cell or cell-substrate interactions. The affinity or avidity of integrins is tightly regulated by the bidirectional transmission processes between intracellular and extracellular compartments. Binding of transmembrane integrins to the extracellular matrix (ECM) causes conformational changes in the extracellular domain, leading to increased ligand binding affinity. This increase then propagates a signal to the cytoplasmic domain to modify intracellular protein-interacting sites, thus providing a physical link to the intracellular cytoskeleton. This is known as outside-in signaling (1). Another process for integrin activation, called inside-out signaling, occurs when intracellular signals initiated by other cell surface receptors foster either adapter protein (talin or kindlin) binding to or dissociation from the cytoplasmic ends of the integrin β subunits; this subsequently regulates the binding capacity of integrin (2–4). The actin cytoskeleton could be linked to the extracellular environment through an inside-out and/or an outside-in signaling pathway, triggering several cellular responses. Therefore, it has been suggested that these two processes play a critical role in cell adhesion, spreading, migration, differentiation, and proliferation, thereby being essential for metazoan development, physiology, and pathology (5).
Integrins are among the major cell adhesion molecules involved in various stages of tumor progression (6–10). β1 integrins are the most commonly expressed integrins in tumor cells and are key players in tumor metastasis (11, 12). Extensive studies have established the relationship between β1 integrin expression levels and tumor metastatic potential in mouse models. For example, metastatic tumors showed much higher expression of β1 integrin than nonmetastatic tumors (13). Head and neck squamous cell carcinoma cells exhibiting low β1 integrin expression showed a significant reduction in lymph node and lung metastases in vivo. In addition, treatment of carcinoma cells with a monoclonal antibody (MAb) against human β1 integrin inhibited cell adhesion to ECM and acetone-fixed tissues and invasion through a basement membrane (14). Furthermore, the antibody treatment significantly inhibited liver metastasis of carcinoma cells in vivo. ATN-161, a peptide antagonist of α5β1, also notably decreased tumor growth and metastasis in a human breast cancer cell xenograft model (15). However, stable knockdown of β1 integrin and subsequent reconstitution with constitutively active β1 integrin in melanoma or breast cancer cell lines significantly increased hepatic colonization of tumor cells in chicken embryos or mice (16). Moreover, forced expression of β1 integrin significantly increased the metastasis of RAS-MYC-transformed fibroblasts with a disrupted β1 integrin gene (17). Thus, these important roles of β1 integrin in the hallmark of metastatic cancer suggest that it may represent a potential target for cancer therapy.
CD99 is a 32-kDa transmembrane glycoprotein ubiquitously expressed in almost all human cell types. CD99 is implicated in multiple cellular responses, including cell apoptosis, differentiation, transport of membrane proteins, and activation and proliferation of lymphocytes (18–23). The physiological significance of CD99 engagement has been clearly shown by an essential role for CD99 in the regulation of the transendothelial migration (TEM) of immune cells (24–26). The homophilic interaction of endothelial CD99 with leukocyte CD99 facilitates leukocyte TEM by promoting the recruitment of the lateral border recycling compartment to the site of TEM through protein kinase A (PKA) signaling (27). In addition, Ensemble's genomic database suggests that alternative splicing of CD99 mRNAs could produce the secretory isoform of CD99, which may play a significant physiological role as a CD99 ligand. Regulatory roles for CD99 have been implicated in pathological conditions as well. CD99 engagement reduces the malignant potential of Ewing sarcoma by inducing apoptosis or methuosis (21, 28, 29) and increased natural killer cell-mediated tumor lysis by inducing HSP70 expression (23). In addition, CD99 inhibits tumor metastasis through the suppression of C-SRC and ROCK2 activities in osteosarcoma (30–32). Notably, we reported that CD99 engagement blocks CD98-mediated β1 integrin signaling, which could suppress tumor progression by inhibiting a positive feedback loop of CD98/β1 integrin/focal adhesion kinase (FAK)/RHOA/ROCK (33).
In this study, we investigated the molecular mechanism through which CD99 ligands suppress fibronectin-mediated activation of β1 integrin in breast carcinoma MCF-7 cells. CD99-Fc fusion proteins or a series of CD99 peptides from conserved motifs of the CD99 extracellular domain function as ligands. CD99-derived agonist ligands activate PKA and the subsequent SHP2 recruitment. PKA-activated SHP2 then induces ERK1/2 phosphorylation via the HRAS/RAF1/MEK signaling pathway. Subsequently, they inversely regulate FAK phosphorylation at residues Y397 and S910 through intracellular tyrosine phosphatase PTPN12 and peptidyl-prolyl cis/trans PIN1. Collectively, our studies suggest that the CD99 agonist ligand may suppress β1 integrin activity via the PKA/SHP2/ERK1/2/PTPN12/FAK signaling pathway.
RESULTS
CD99-Fc fusion proteins suppress β1 integrin activity by specifically interacting with three conserved motifs in the CD99 extracellular domain.
Our previous results showed that cross-linking of CD99 with an anti-CD99 MAb suppresses β1 integrin activity (33, 34). Because CD99 interacts homophilically through its N-terminal extracellular domain (24, 25, 35), we tried to determine whether, similarly to the anti-CD99 MAb, CD99-derived agonist ligands could modulate β1 integrin activity. First, we generated plasmid fusions between human IgG Fc and intact extracellular and transmembrane domains or a series of deletion mutant proteins sequentially lacking three conserved motifs of the CD99 extracellular domain that were used for production of the CD99-Fc I, II, III, and IV proteins (Fig. 1A and B). Fluorescence-activated cell sorter (FACS) analysis showed that recombinant protein CD99-Fc I with an intact CD99 extracellular domain could bind to CD99 expressed on the MCF-7 cell surface (Fig. 1C). Remarkably, deletion mutant recombinant proteins CD99-Fc II and III maintained their ability to bind CD99, even though they possess only one or two conserved motifs. In contrast, the mutant recombinant protein CD99-Fc IV, completely lacking all three conserved motifs, failed to maintain this binding capacity. These results suggest that three conserved motifs within the CD99 extracellular domain play a critical role in the homophilic interaction of CD99.
FIG 1.

CD99 decreases β1 integrin activity through homotypic interactions mediated by three conserved motifs. (A) A series of chimeric proteins (CD99-Fc I, II, III, and IV) composed of the extracellular (EXT) and transmembrane (TM) domains of CD99 and human IgG-Fc fragments. S, signal sequence; CYT, cytoplasmic domain. (B) The expression levels and sizes of each purified fusion protein were determined by Western blotting with a mouse anti-human CD99 MAb (C) Flow cytometry analysis was performed to assess the ability of CD99-Fc fusion proteins to bind to CD99 expressed on the surface of MCF-7 cells. (D) In vitro cell-matrix adhesion assay performed to assess cell attachment to the ECM. PLL was used as a control substrate. Attached cells were counted with a hemocytometer. Lines indicate additional statistical comparisons, and significant differences from the control are shown by asterisks as follows: *, P < 0.05; **, P < 0.01. (E, F) MCF-7 cells were seeded into fibronectin-coated 35-mm dishes. After overnight serum starvation, cells were treated with either increasing concentrations of CD99-Fc I fusion protein or each of the purified proteins at 3 μg/ml for 1 h. Immunoblotting was performed to detect active or total β1 integrin. The graphs show the active β1 integrin intensities of bands normalized against that of the total form. β-Actin was used as a quantitative control.
Here we determined whether the purified recombinant proteins could function as specific ligands for the cell surface CD99 molecule. Recombinant proteins CD99-Fc I, II, and III, harboring at least one conserved motif, significantly inhibited cell adhesion to fibronectin, whereas CD99-Fc IV showed no inhibitory effect (Fig. 1D). In contrast, no inhibitory effects of the recombinant proteins were observed in cell adhesion to poly-l-lysine (PLL), which does not involve any participation by β1 integrin. To confirm these results, we performed a Western blot analysis to identify the change in β1 integrin activity. The CD99-Fc I, II, and III recombinant proteins significantly inhibited β1 integrin activity, while CD99-Fc IV failed to do so (Fig. 1E). Correspondingly, β1 integrin activity decreased in a dose-dependent manner after CD99-Fc I treatment (Fig. 1F). Taken together, these results suggest that CD99-Fc proteins containing conserved motifs induce the suppression of β1 integrin activity through homophilic interactions of conserved motifs in the extracellular domain of CD99.
CD99 conserved motif-derived peptides behave as ligands, similar to CD99-Fc fusion proteins.
To confirm the functional significance of conserved motifs in the extracellular domain of CD99, we synthesized three different 7-mer peptides, designated CD99CRI7, -II7, and -III7, displaying sequences that are similar to each other (Fig. 2A). We performed a competition assay with CD99 peptides and CD99-Fc I to verify whether they would compete to establish interactions with CD99. Treatment of MCF-7 cells with each of the three peptides partially blocked the binding of CD99-Fc I to CD99 on the cell surface (Fig. 2A). Remarkably, the combination of these three synthetic peptides resulted in stronger inhibition of the binding of CD99-Fc I to cell surface CD99. These results suggest that all three conserved regions are involved in the homophilic interaction of CD99.
FIG 2.

The ability of CD99 agonistic peptides to suppress β1 integrin activity is comparable to that of CD99-Fc proteins. (A) The sequences of 7-mer synthetic peptides are shown at the top. To assess the binding of CD99-derived peptides CD99CRI7, -II7, and -III7 to CD99 expressed on MCF-7 cells, a competition assay with a purified recombinant CD99-Fc I fusion protein was performed. The ability of CD99-Fc fusion proteins to bind to cell surface CD99 was measured by flow cytometry to evaluate the occupation of CD99 by CD99-derived peptides. Light solid line, negative control; heavy solid line, control peptide; dotted line, competition with peptide. (B, C) Cell attachment to the ECM was analyzed by adhesion assay on fibronectin as described above. Results were replicated in three independent experiments. Asterisks represent statistically significant differences from untreated cells as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) MCF-7 cells were treated with increasing concentrations of peptides (0 to 30 μM) for 1 h. Cell lysates were subjected to SDS-PAGE and analyzed by Western blotting with the antibodies indicated. The graphs show the active β1 integrin intensities of bands normalized against that of the total form. β-Actin was used as a loading control.
These results prompted us to test whether CD99 peptides could regulate cell attachment and β1 integrin activity similarly to CD99 protein ligands. MCF-7 cell adhesion to fibronectin was significantly decreased when MCF-7 cells were treated with each peptide, but adhesion to PLL was not affected (Fig. 2B). In contrast, a control peptide derived from the juxtatransmembrane region of CD99 failed to decrease cell adhesion to fibronectin. Similar results were obtained with human breast carcinoma MDA-MB-231 cells and human embryonic kidney 293T (HEK293T) cells (Fig. 2C). Consistently, immunoblotting revealed that these three peptides inhibit β1 integrin activation induced by fibronectin in a dose-dependent manner (Fig. 2D). To define the critical sequences required, we shortened the length of the peptide to a 4-mer or 3-mer on the basis of highly conserved sequences and carried out in situ proximity ligation assay (PLA) analysis to assess β1 integrin activation by measuring the physical interaction between β1 integrin and talin. All of the peptides inhibited the fibronectin-induced interaction of talin or kindlin with β1 integrin by more than 50% at a concentration of 10 μM (Fig. 3A and B), and the inhibitory effect was maximized at a concentration of 20 μM. Moreover, the shortest 3-mer peptide, CD99CRIII3, exerted a stronger inhibitory effect on the interaction between talin and β1 integrin than the 7-mer and 4-mer peptides. Consistently, CD99CRI3 and CD99CRII3, two other 3-mer peptides derived from the first and second conserved regions, exerted similar inhibitory effects (Fig. 3C). To further investigate the functional significance of each amino acid sequence of 3-mer peptide, we carried out in situ PLA with CD99 mutant peptides obtained by sequential amino acid substitution. Surprisingly, CD99CRIII3mut2(A96N) still maintained the ability to inhibit the interaction between β1 integrin and talin, while two other mutant peptides, CD99CRIII3mut1(L95N) and CD99CRIII3mut3(D97N), failed to inhibit this interaction (Fig. 3C).
FIG 3.


Functional analysis of highly conserved regions of CD99 in the regulation of β1 integrin activity. (A, B) A 7-mer peptide was shortened by either 4-mer or 3-mer. Cells were seeded onto PLL- or fibronectin (FN)-coated coverslips and treated with three different sizes of peptides for 1 h as indicated. In situ PLA was performed to determine the interaction of talin or kindlin with β1 integrin. (C) Cells were treated with three different 3-mer peptides (20 μM) that originated from three different conserved regions, as well as mutated forms of CD99CRIII3. The interaction between talin and β1 integrin was confirmed by in situ PLA. (D) To determine the receptor-specific binding of CD99CRIII3, wild-type, CD99 knockdown, or β1 integrin knockout MCF-7 cells were stained with FITC-conjugated CD99CRIII3 and then observed under a confocal microscope. Unconjugated peptides (control, CD99CRIII3, and CD99CRIII3mut1) were used as competitors. (E) Cells were treated with CD99CRIII3 for 1 h, and cell lysates were subjected to SDS-PAGE to analyze β1 integrin activity. (F) CD99-deficient human keratinocytes, HaCaT cells, were infected with a retroviral vector carrying wild-type CD99 cDNA and seeded into 35-mm dishes coated with fibronectin. After treatment with CD99CRIII3, β1 integrin activity was analyzed by flow cytometry with a BD Accuri C6 system. (G, H) Wild-type CD99 and a series of variant cDNA constructs of CD99. Each of the cDNA constructs was inserted into a pMSCV retroviral vector containing a neomycin resistance gene. The β1 integrin-talin interaction was examined by in situ PLA. (A to C, H) Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Red spots indicate the physical connection of the target molecules. PLA signals in cell populations (n = 8) were quantified by NIS Elements analysis. The average number of rolling-circle products (RCPs) per cell ± the standard error is shown. Asterisks represent statistically significant differences from untreated cells as follows: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Magnification, ×600. Scale bars = 10 μm. wt, wild type.
To verify the target specificity of 3-mer peptide CD99CRIII3, we carried out an immunofluorescence assay with fluorescein isothiocyanate (FITC)-conjugated CD99CRIII3. MCF-7 cells were infected with the lentivirus enabling delivery of CD99 short hairpin RNA (shRNA) or transiently transfected with a β1 integrin CRISPR/Cas9 knockout plasmid in vitro (Fig. 3D). CD99CRIII3-FITC bound to the surface of MCF-7 cells, but CD99 knockdown abolished the binding of the cells with CD99CRIII3-FITC. β1 integrin knockout did not affect the binding of CD99CRIII3-FITC to the MCF-7 cell surface, suggesting that a specific interaction occurs between CD99 and CD99CRIII3-FITC. To further confirm the specificity of the CD99-CD99CRIII3 interaction, we performed a peptide competition assay with control, CD99CRIII3, and CD99 mutant peptides as competing peptides. CD99CRIII3 abrogated the binding of CD99CRIII3-FITC to the cell surface, whereas the other competitors did not. These results show that CD99 is a specific receptor for 3-mer peptide CD99CRIII3. To determine the functional outcomes of target-specific binding of CD99CRIII3, we investigated the effect of CD99CRIII3 on β1 integrin activity in CD99 knockdown MCF-7 cells. As shown in Fig. 3E, the knockdown of CD99 prevented CD99CRIII3 from inactivating β1 integrin. Likewise, CD99CRIII3 suppressed fibronectin-mediated activation of β1 integrin in HaCaT cells expressing wild-type CD99 but not in wild-type HaCaT cells that rarely express endogenous CD99 (Fig. 3F). These results show that the minimal length of peptide required to maintain full activity is 3-mer. Next, to analyze the roles of the three conserved motifs of CD99 in the regulation of β1 integrin, we constructed wild-type CD99 and a series of mutant forms of CD99 carrying sequential substitutions of amino acids in each of the three conserved regions (Fig. 3G) and generated stable HaCaT cell lines expressing the empty vector, exogenous wild-type CD99, or seven different mutant forms of CD99 each displaying a substitution of amino acids in at least one of their conserved regions. In situ PLA analysis showed that CD99CRIII3 inhibited the interaction of β1 integrin with talin by approximately 55% in wild-type CD99-expressing cells (Fig. 3H). The β1 integrin activity inhibitory efficiency of the peptide in the cells expressing mutant CD99 carrying an amino acid residue substitution in one of the three conserved regions was lower than that observed in wild-type CD99-expressing cells. Consistently, the inhibitory efficiency was much lower in cells expressing mutant CD99 containing only one intact conserved region. Moreover, the inhibitory effect of the peptide was completely abrogated in cells with mutations in all three conserved regions of CD99 and in empty-vector-expressing cells. Together, these results indicate that the 3-mer peptide CD99CRIII3 specifically interacts with all three of the conserved regions and synergistically suppresses fibronectin-mediated β1 integrin activity.
The recruitment of SHP2 by CD99 activates the HRAS/ERK signaling pathway.
Previous studies showed that FAK, a cytoplasmic nonreceptor tyrosine kinase, is an essential mediator of integrin activation and focal adhesion dynamics (36–38). It is well known that SHP2 operates in integrin signaling and adhesion site dynamics by regulating FAK phosphorylation (39, 40). In addition, we previously reported that cross-linking of CD99 with an anti-CD99 MAb led to SHP2 recruitment, followed by FAK dephosphorylation at tyrosine 397 and β1 integrin inactivation (33). To determine whether SHP2 is engaged in the suppression of β1 integrin activity caused by CD99 peptide treatment, MCF-7 cells were transfected with a SHP2 small interfering RNA (siRNA) (Fig. 4A). Knockdown of SHP2 abolished the inactivation of β1 integrin and phosphorylation of SHP2 triggered by CD99CRIII3. To determine whether SHP2 and CD99 form a regulatory complex, we performed immunoprecipitation in the presence of either CD99-Fc I fusion protein or CD99 peptide. As expected, both CD99-Fc I fusion protein and CD99CRIII3 induced coprecipitation of CD99 with SHP2 (Fig. 4B).
FIG 4.

CD99 peptide suppresses β1 integrin activity via the signaling pathway involving SHP2, RASGAP1, HRAS, RAF1, and ERK1/2. (A, C, D, H) MCF-7 cells were treated with 20 μM CD99CRIII3 for 1 h. Western blot analysis was carried out with the antibodies indicated. β-Actin was used as a loading control. (B) Cells were treated with CD99-Fc I fusion protein or CD99CRIII3 for 1 h, and coimmunoprecipitation of SHP2 was performed with an anti-human CD99 MAb. Immunoprecipitates (IP) and whole-cell lysates (input) were separated by SDS-PAGE and analyzed by Western blotting. (E, F) Cells were dose-dependently treated with the MEK inhibitor PD98059 and incubated with CD99CRIII3 for 1 h. Western blotting was performed with the antibodies indicated. An in vitro cell-matrix adhesion assay was performed to determine the changes in cell adhesion to fibronectin. Statistically significant differences are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01. (G) MCF-7 cells were treated with 3-mer CD99 peptides for 1 h. ERK1/2 phosphorylation was examined by Western blotting.
A previous study reported that SHP2 regulates the HRAS/ERK signaling pathways through dephosphorylation of tyrosine residues on the Gab1 adapter (41). In this study, we found that the recruitment of SHP2 by CD99CRIII3 or CD99-Fc I fusion protein led to the specific activation of ERK1/2 (Fig. 4C and E). SHP2 can act as an upstream regulator of HRAS by interfering with the inactivation of HRAS catalyzed by RAS GTPase-activating protein 1 (RASGAP1) (42). To uncover the underlying mechanisms by which SHP2 recruited by CD99CRIII3 suppresses fibronectin-mediated β1 integrin activity, we evaluated the effects of RASGAP1 siRNA by immunoblotting. Transfection of RASGAP1 siRNA but not RASGAP3 siRNA abrogated the effect of CD99CRIII3 on the fibronectin-mediated activation of β1 integrin, as well as the phosphorylation of ERK1/2 (Fig. 4C). To confirm that CD99CRIII3 is involved in the regulation of β1 integrin activity through an HRAS-dependent pathway, HRAS siRNA was transfected into MCF-7 cells. Immunoblotting showed that transfection of HRAS siRNA disrupted the CD99CRIII3-induced suppression of β1 integrin activity (Fig. 4D). Moreover, ERK1/2 inactivation by PD98059 dramatically eliminated CD99CRIII3-facilitated inactivation of β1 integrin (Fig. 4E). Consistent with the activation level of β1 integrin, the rates of cell adhesion to fibronectin, which were decreased by CD99, were dose-dependently recovered through PD98059 treatment (Fig. 4F). To verify the target specificity of CD99-derived peptides, we compared the efficiency of 3-mer peptide phosphorylation of ERK1/2 in wild-type and CD99 knockdown MCF-7 cells (Fig. 4G). All three 3-mer peptides apparently increased ERK1/2 phosphorylation in wild-type MCF-7 cells but failed to do so in CD99 knockdown cells. In addition, we performed immunoblotting with either HRAS siRNA or dominant negative RAF1. Either knockdown of HRAS or forced expression of dominant negative RAF1 interrupted the activation of ERK1/2 that was induced by CD99CRIII3 (Fig. 4H). Collectively, our results suggest that CD99CRIII3-induced CD99 activation initiates the SHP2/HRAS/ERK-dependent pathway.
CD99-induced PKA activation is involved in the regulation of the SHP2/MAPK/ERK signaling pathway.
Recently, it was shown that endothelial CD99 forms a signaling complex with ezrin and soluble adenylyl cyclase (sAC) to promote PKA activation (27). Therefore, we determined whether CD99CRIII3 correspondingly activates the sAC/PKA signaling pathway in breast carcinoma cells and whether PKA acts as an upstream regulator of mitogen-activated protein kinase (MAPK)/ERK signaling in the CD99-mediated regulation of β1 integrin activity. MCF-7 cells were treated with the specific inhibitors indicated and CD99CRIII3. The activation of CD99 with CD99CRIII3 significantly increased CD99-ezrin and CD99-PKA interactions (Fig. 5A). Surprisingly, CD99CRIII3 treatment induced a strong interaction between PKA and SHP2. The interactions of ezrin and PKA with CD99 were not affected by treatment with NSC87877, a potential inhibitor of SHP2. Targeted inhibition of MEK by PD98059 had no effect on either the CD99-ezrin or the SHP2-PKA interaction. In contrast, treatment with the sAC inhibitor KH7 specifically inhibited the interactions of CD99 and SHP2 with PKA. Additionally, transfection of ezrin or PKA siRNA completely blocked the CD99CRIII3-induced interaction between CD99 and PKA. Contrarily, CD99 knockdown resulted in no changes in the interactions between these pairs of molecules. Consistently with the in situ PLA results, the immunoprecipitation assay showed that inhibitors of sAC or SHP2 strongly interrupted the binding of SHP2 to PKA that was stimulated by CD99CRIII3 (Fig. 5B). Physical binding between those two molecules was significantly reduced by knockdown of PKA. Furthermore, CD99CRIII3-induced phosphorylation of SHP2 was prominently suppressed by inhibition of sAC or knockdown of PKA. These results suggest that CD99 activated by CD99CRIII3 stimulates the formation of the ezrin-sAC-PKA signaling complex, leading to the recruitment and activation of SHP2. Next, we found that CD99CRIII3-induced ERK1/2 phosphorylation was significantly suppressed not only by inhibition of MEK or SHP2 but also by knockdown of PKA (Fig. 5C and D). In addition, the sAC inhibitor or PKA siRNA restored the level of FAK Y397 phosphorylation, which had been suppressed by CD99CRIII3 (Fig. 5E). Correspondingly, in situ PLA showed that the CD99CRIII3-induced interruption of the β1 integrin-talin interaction was abrogated by sAC, MEK inhibitors, or PKA siRNA (Fig. 5F). Taken together, these results demonstrate that CD99-derived agonistic ligand activates the sAC/PKA signaling complex, leading to activation of the SHP2/MAPK/ERK signaling pathway.
FIG 5.


CD99-induced PKA activation results in β1 integrin inactivation via regulation of the SHP2/MAPK/ERK signaling pathway. (A, F) In situ PLAs performed to assess the interactions between the pairs of molecules indicated in MCF-7 cells. Numerical values are the average intensities (±SD) of red spots in eight randomly selected fields. (B) Coimmunoprecipitation (Co-IP) assay performed to assess the physical interaction of PKA with SHP2. Immunoprecipitates (IP) and whole-cell lysates (input) were separated by SDS-PAGE and analyzed by Western blotting with the antibodies indicated. (C to E) Western blot analysis was carried out to assess the phosphorylation of ERK1/2 and FAK Y397 and the knockdown efficiency of PKA siRNA. β-Actin was used as a loading control. FN, fibronectin; RCPs, rolling-circle products.
CD99-induced ERK activation regulates the phosphorylation of FAK.
Next, we investigated how CD99 activation controls FAK phosphorylation. A previous study showed that activated HRAS induces FAK dephosphorylation at Y397 and FAK phosphorylation at S910 (43). MCF-7 cells were treated with CD99CRIII3 in a dose-dependent manner (Fig. 6A). Consistent with previous results, CD99CRIII3 inversely regulated FAK phosphorylation at Y397 and S910, suggesting that the CD99 peptide modulates FAK phosphorylation at Y397 and S910 in opposite manners. In addition, either phosphorylation of S910 or dephosphorylation of Y397 on FAK caused by treatment with CD99CRIII3 completely disappeared in HRAS siRNA transfectants (Fig. 6B). Similarly to the effect of HRAS siRNA, the forced expression of dominant negative RAF1 interrupted ERK1/2 activation, as well as the regulation of FAK phosphorylation at Y397 and S910. Finally, the results showed exactly the same pattern as those obtained with PD98059-treated cells regarding the regulation of FAK phosphorylation (Fig. 6C). These results demonstrate that CD99CRIII3 controls FAK phosphorylation via the HRAS/ERK1/2-dependent signaling pathway.
FIG 6.

CD99 peptide inversely regulates FAK phosphorylation at Y397 and S910 via a MAPK signaling cascade. (A) Cells were treated with CD99CRIII3 peptide in a dose-dependent manner. Levels of FAK phosphorylation at Y397 and S910 were assessed by Western blotting. (B) MCF-7 cells were transiently transfected with HRAS siRNA or dominant negative RAF1 and incubated with CD99CRIII3 for 1 h. Cell lysates were separated by SDS-PAGE and analyzed by Western blotting with the antibodies indicated. (C) Cells were dose-dependently treated with the MEK inhibitor PD98059 and incubated with CD99CRIII3 for 1 h. Western blotting was performed with the antibodies indicated. The graphs show the phosphorylated FAK intensities of bands normalized against the total FAK level. β-Actin was used as a loading control.
PTPN12 acts as a critical mediator in CD99-induced inactivation of β1 integrin.
On the basis of a previous report showing that activated HRAS induces ERK1- and ERK2-dependent phosphorylation of PTPN12, which subsequently induces FAK dephosphorylation at Y397 (43), we hypothesized that the CD99CRIII3-induced ERK1/2 signaling pathway could regulate the interaction between FAK and PTPN12. Treatment with CD99CRIII3 induced the interaction of ERK1/2 with FAK or PTPN12 (Fig. 7A). In addition, CD99CRIII3 stimulated FAK-PTPN12, FAK-PIN1, and PTPN12-PIN1 interactions. These CD99CRIII3-induced interactions between the signaling molecules were suppressed by treatment with the MEK inhibitor PD98059. To confirm these results, MCF-7 cell extracts were subjected to immunoprecipitation with an anti-PTPN12 MAb (Fig. 7B). Immunoblotting revealed that CD99CRIII3 increased the expression of PTPN12 and its interaction with FAK or β1 integrin but not SHP2 in a dose-dependent manner. Next, we investigated whether PKA regulates the interaction between PTPN12 and FAK. CD99CRIII3 treatment increased the interaction between PTPN12 and FAK, which was dose-dependently reduced by treatment with the sAC inhibitor or PKA siRNA (Fig. 7C). In contrast, the CD99CRIII3-induced interaction between those two molecules disappeared in CD99 knockdown cells. To determine the role of PTPN12 in the regulation of FAK activity, MCF-7 cells were transfected with a control or PTPN12 siRNA (Fig. 7D). FAK phosphorylation at Y397, which was suppressed by treatment with CD99CRIII3, was dose-dependently restored in the PTPN12 siRNA transfectants. Conversely, CD99CRIII3-induced phosphorylation of FAK at S910 was inhibited in the same cells. In contrast, no notable effects were observed in the control siRNA transfectants. Additionally, we found that CD99CRIII3 increased not only the phosphorylation of ERK1/2 but also the expression of PTPN12 in a dose-dependent manner (Fig. 7E). To further confirm the role of PIN1, as well as PTPN12, we performed immunoblotting with siRNAs targeting PTPN12 and PIN1 (Fig. 7F). Transfection with either PTPN12 or PIN1 siRNA disrupted CD99CRIII3-induced FAK dephosphorylation at Y397, while it suppressed the FAK phosphorylation at S910, which had been upregulated by CD99CRIII3. Double transfection with both PTPN12 and PIN1 siRNAs led to greater effects on FAK phosphorylation and dephosphorylation at Y397 and S910. No significant effects induced by either PTPN12 or PIN1 siRNA were detected in phosphate-buffered saline (PBS)-treated cells. To further investigate the role of PIN1 in the mediation of the interaction between PTPN12 and FAK, we transfected cells with siRNA to knock down PIN1 gene expression. As shown in Fig. 7G, knockdown of PIN1 abrogated the CD99CRIII3-induced interaction of PIN1 with FAK or PTPN12, showing that PIN1 expression was well downregulated by siRNA. PIN1 knockdown completely disrupted the interaction between FAK and PTPN12 that was induced by CD99CRIII3. In contrast, no effects on the interaction of FAK or PTPN12 with ERK1/2 were observed. These results suggest that PIN1 plays a critical role in the FAK dephosphorylation mediated by PTPN12 through its action downstream of ERK1/2.
FIG 7.


PTPN12 and PIN1 act as key mediators in the ERK1/2-mediated regulation of FAK phosphorylation. (A) MCF-7 cells were treated with CD99CRIII3 in the presence or absence of PD98059 for 1 h. In situ PLA was performed to determine the interactions between the protein pairs indicated. Magnification, ×600; scale bars = 10 μm. (B, E) PTPN12 is physically associated with the β1 integrin/FAK complex (B). Endogenous PTPN12 was coimmunoprecipitated (IP) with β1 integrin and FAK but not with SHP2. Lane C, control. Changes in PTPN12 expression and ERK1/2 phosphorylation were confirmed by Western blotting (E). (C) MCF-7 cells were transiently transfected with different amounts of PKA siRNA. Cells were treated with CD99CRIII3 in the presence or absence of KH7 for 1 h. The interaction between PTPN12 and FAK was analyzed by in situ PLA. RCPs, rolling-circle products. (D) Scrambled or PTPN12 siRNA was transiently transfected into MCF-7 cells in a dose-dependent manner. CD99CRIII3-induced modulation of FAK phosphorylation is restored in MCF-7 cells with transient siRNA knockdown of PTPN12. Phosphorylation of FAK at Y397 and S910 was detected with the antibodies indicated, and β-actin was used as a loading control. (F) Cells were transiently transfected with siRNA specifically targeting PTPN12 or PIN1 either alone or combined. Knockdown of endogenous PTPN12 and PIN1 and changes in FAK and ERK1/2 phosphorylation were detected by Western blotting. (G) The interactions between pairs of proteins were analyzed by in situ PLA as indicated. (C, G) Numerical values are the average intensity ± SD of red spots in eight randomly selected fields.
Finally, the participation of PTPN12 in the regulation of FAK phosphorylation prompted us to assess its effects on focal adhesion assembly. Mitra et al. suggested that phosphorylation of Y397 in FAK leads to the recruitment of adapter molecules such as paxillin, vinculin, and kindlin, resulting in the phosphorylation of other tyrosine residues on FAK and the formation of focal adhesion (44). As expected, fibronectin facilitated molecular interactions of FAK or β1 integrin with adapter proteins either individually or with each other. The inhibitory effect of CD99CRIII3 on the formation of focal adhesion was disrupted in the presence of PTPN12 siRNA or PD98059 (Fig. 8A and B). Treatment of MCF-7 cells with CD99CRIII3 decreased the clustering of β1 integrin and the recruitment of paxillin induced by fibronectin, whereas treatment with PD98059 or transfection with PTPN12 siRNA, done either individually or in combination with each other, restored them, which had been suppressed by CD99 peptide (Fig. 8C). These results are consistent with immunoblotting data showing that treatment with PD98059 or transfection with PTPN12 siRNA abrogated the inhibitory effects of CD99CRIII3 on the activation of β1 integrin and FAK (Fig. 8D). Quantitative flow cytometry showed consistent results by measuring the active conformation of β1 integrin (Fig. 8E). Taken together, our data indicate that PTPN12 plays a key role in the regulation of β1 integrin activation by disrupting the assembly of a focal adhesion complex via the ERK1/2 signaling pathway.
FIG 8.


PTPN12 functions as a key mediator in the disassembly of the focal adhesion complex and subsequent inactivation of β1 integrin. (A, B) In situ PLAs performed to examine paxillin-FAK, vinculin-FAK, FAK-β1 integrin, kindlin-β1 integrin, and talin-β1 integrin interactions. (C) To determine the clustering of β1 integrin/paxillin, cells were sequentially stained with a mouse antipaxillin MAb, rhodamine-conjugated goat anti-mouse IgG, and a mouse anti-human CD29–FITC MAb before analysis by confocal microscopy. (D) Western blotting was performed with the antibodies indicated to investigate either the effect of CD99CRIII3 or the role of PTPN12. β-Actin was used as a loading control. (E) FACS analysis was performed to detect the active form of β1 integrin with an anti-human β1 integrin active-conformation MAb (HUTS-4). Quantitative values represent the average ± SD from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t test). (A to C) Nuclei were stained with DAPI (blue). Magnification, ×600; scale bars = 10 μm.
DISCUSSION
The signaling mechanism of human CD99 is still unknown, despite the numerous studies describing its roles in various cellular responses, such as transmigration of leukocytes and activation, proliferation, and apoptosis of lymphocytes (22, 45–48). In this study, we showed that CD99-derived agonistic ligands function as novel suppressors of fibronectin-mediated β1 integrin activation in human breast carcinoma cells and uncovered for the first time the underlying mechanism by which CD99 regulates β1 integrin activity. This study is consistent with our previous study showing that antibody-mediated cross-linking of CD99 suppresses the mechanisms of β1 integrin activation by CD98 or ECMs such as collagen, fibronectin, and laminin (33). In addition, the activation of CD99 itself led to the activation of PKA and the subsequent recruitment of SHP2, resulting in the activation of the HRAS/MAPK signaling pathway. The activated HRAS/MAPK cascade enabled PIN1 to promote the binding of PTPN12 to FAK, which led to the suppression of fibronectin-induced β1 integrin activation through FAK dephosphorylation at Y397. Taken together, these results suggest that CD99 agonists suppress β1 integrin activity through the PKA/SHP2/ERK1/2/PTPN12/FAK signaling pathway.
Although a number of studies have shown that CD99 interacts homophilically via its extracellular domain, the mechanisms involved are not clearly understood (25, 49, 50). CD99 includes three highly conserved regions in its extracellular domain across multiple species (51). We hypothesized that the conserved regions CD99CRI, -II, and -III are critical for the homophilic interaction of CD99. Consistently with our expectation, purified CD99-human Fc fusion proteins with only one conserved region could bind effectively to CD99 expressed on the cell surface and suppress β1 integrin activation. Particularly, three amino sequences present within the conserved regions—Leu-Ser-Asp, Leu-Gly-Asp, and Leu-Ala-Asp—are critical for the homophilic interaction of CD99 and initiation of the intracellular signaling pathway regulating β1 integrin activity by targeting CD99 on the cell surface. In the peptide competition assay, the CD99 mutant peptides displaying leucine or aspartic acid replaced with arginine completely lost their inhibitory effects on the specific interaction between CD99CRIII3 and CD99. Furthermore, they lost their inhibitory effects on β1 integrin activity. Functionally, the inhibitory efficiency of CD99CRIII3 in HaCaT cells expressing mutant CD99 was inversely reduced with an increasing number of abnormal conserved regions. Taken together, these results indicate that leucine and aspartic acid, two highly conserved amino acids in the extracellular domain of CD99, provide a structural and functional basis for specific interactions for CD99 activation.
Our data showed that a CD99 agonist induces ERK1/2 phosphorylation through SHP2 and subsequent FAK dephosphorylation at Y397. Our results are consistent with a previous report showing that forced expression of CD99 induces ERK activation in osteosarcoma (20). SHP2 has been shown to activate the HRAS/RAF1/ERK signaling pathway through suppression of RASGAP1 (41). A previous study showed that activated HRAS regulates PTPN12 activity by ERK1/2-mediated phosphorylation and PIN1-dependent isomerization, resulting in increased PTPN12 binding to FAK (43, 52). The increased interaction of PTPN12 with FAK facilitated FAK dephosphorylation at Y397. Correspondingly, we found that CD99 stimulated the activation of HRAS and the phosphorylation of ERK1/2 and thereby induced the interaction between ERK1/2 and PTPN12, as well as ERK1/2 and FAK, leading to the phosphorylation of FAK S910 and subsequent binding of PTPN12 to FAK. PTPN12 is a cytosolic protein tyrosine phosphatase bearing a proline-glutamine-serine-threonine-rich motif (53, 54). It has been implicated in the regulation of focal adhesion disassembly and cell motility by regulating the dephosphorylation of several protein kinases and signaling molecules. Previously, we demonstrated the role of another cytosolic phosphatase, SHP2, in FAK dephosphorylation, but whether SHP2 dephosphorylates FAK directly or indirectly remains unknown (33). Our results showed that PTPN12 physically interacts with FAK, unlike SHP2, suggesting that PTPN12 directly contacts FAK and facilitates its dephosphorylation at Y397, whereas SHP2 is indirectly involved in the dephosphorylation of FAK by triggering the HRAS/MAPK signaling pathway. Notably, PIN1 knockdown induced by the transfection of exogenous siRNA completely disrupted the interaction between PTPN12 and FAK, but no inhibitory effects on the interaction of ERK1/2 with FAK or PTPN12 were observed. This result demonstrates that ERK1/2 independently interacts with FAK or PTPN12 upstream of the signaling cascade and that PIN1 then modulates the association of PTPN12 with FAK. Therefore, these results suggest that the interaction between PTPN12 and FAK is regulated by PIN1, resulting in the suppression of β1 integrin activation through the dephosphorylation of FAK at Y397.
A study recently reported on how CD99 acts as a signaling molecule during the TEM of leukocytes in the endothelium (27). Watson et al. suggested that endothelial CD99 interacts with leukocyte CD99 and activates PKA through a signaling mechanism involving a CD99-ezrin-sAC complex. That study prompted us to determine the relationship between the HRAS/MAPK and sAC/PKA signaling pathways triggered by CD99. The role of cyclic AMP (cAMP)/PKA in the ERK signaling pathway remains unresolved. Our study showed that CD99CRIII3 treatment induces a strong interaction between PKA and SHP2, leading to activation of the ERK signaling pathway and subsequent suppression of FAK Y397 phosphorylation via PTPN12 and β1 integrin-talin interaction. This is supported by several reports showing that cAMP/PKA plays an important role in the activation of ERK signaling (55, 56). On the contrary, it has been shown that PKA directly inhibits RAF1 through either phosphorylation of S43, 233, and 259 or dephosphorylation of S338 distributed throughout RAF1, which leads to inactivation of the HRAS/RAF1/MEK/ERK pathway (57–60). Recently, PKA has been shown to inhibit SHP2, which contradicts our data (61, 62). Although our results show that PKA positively acts upstream of SHP2 in the CD99CRIII3-mediated activation of the SHP2/HRAS/RAF1/ERK signaling pathway, how PKA activates SHP2 and whether PKA inhibits the function of RAF1 within this pathway remain unclear.
CD99 is known to function as a tumor suppressor in osteosarcoma (30–32). Transfection of CD99 inhibits tumor metastasis through the suppression of C-SRC and ROCK2 activities. These results are consistent with our data showing that CD99 agonists inhibit β1 integrin activity through the suppression of a focal complex, including FAK and C-SRC, which could downregulate cell migration through the integrin-FAK-RHOA-ROCK axis. As described above, our study proposes that SHP2 plays an important role in CD99-mediated suppression of β1 integrin activity, which may lead to the suppression of tumorigenic and metastatic ability. On the contrary, many previous studies have shown that SHP2 acts as an oncogene because it facilitates cell proliferation in various human cancers through HRAS/ERK signaling (63, 64). However, SHP2 suppresses the development of hepatocellular carcinoma by suppressing inflammatory signaling through the STAT3 pathway (65). Likewise, our results show that SHP2 activates the tumor suppressor PTPN12 through ERK signaling, suggesting a tumor-suppressive role for SHP2 through the CD99 signaling axis (66–68). The influence of the cellular context and tumor microenvironment on the regulation of SHP2's role in tumor progression remains unclear.
In conclusion, we propose a schematic model to illustrate the CD99 signaling pathway and its role in the regulation of β1 integrin activity (Fig. 9). CD99-derived agonist ligands inhibit fibronectin-induced activation of β1 integrin through the PKA/SHP2/ERK1/2/PTPN12/FAK signaling pathway by specifically interacting with the three conserved motifs in the CD99 extracellular domain. They facilitate SHP2 recruitment to the plasma membrane via cAMP-dependent PKA activation and sequentially activate HRAS by inhibiting GTPase activation by RASGAP1. Activated HRAS induces ERK1/2-dependent phosphorylation of FAK at S910 and PTPN12 at S571, facilitating the recruitment of PIN1 to both FAK and PTPN12. cis-trans isomerization of PTPN12 and FAK, mediated by PIN1, leads to an increased interaction of PTPN12 with FAK, dephosphorylation of FAK at Y397, and subsequent inhibition of focal adhesion assembly. Dissociation of the FAK-talin-β1 integrin complex results in reduced β1 integrin activity. In this study, we propose that PTPN12 plays a key role in CD99-mediated negative regulation of β1 integrin activity and tumor progression. This assumption is consistent with recent reports showing that PTPN12 functions as a tumor suppressor (67–69). Taken together, our results suggest that CD99-derived agonist ligands have potential as novel therapeutic reagents to suppress tumor progression via negative regulation of β1 integrin activity.
FIG 9.
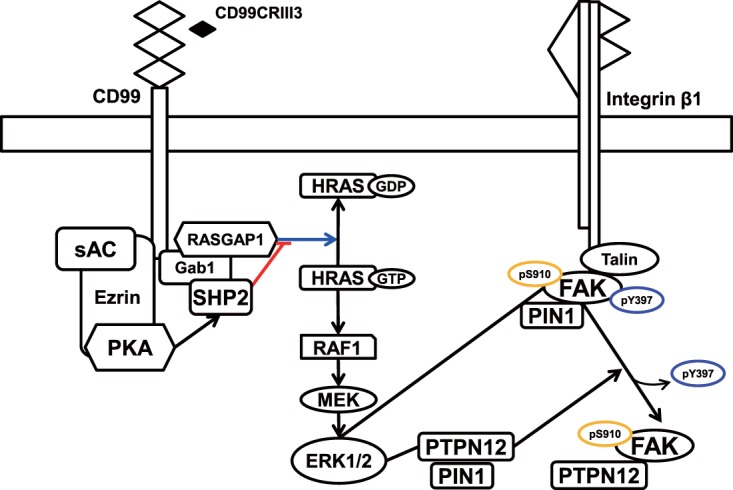
Schematic model describing the CD99 peptide-induced signal transduction pathway. CD99 engagement by agonistic ligands elicits the recruitment of SHP2 through the sAC/PKA signaling pathway, resulting in activation of the HRAS/MAPK signaling pathway. MAPK activation leads to the isomerization of FAK and PTPN12 mediated by PIN1, followed by PTPN12 binding to FAK and subsequent dephosphorylation of FAK at Y397, resulting in the inactivation of β1 integrin.
MATERIALS AND METHODS
Reagents and antibodies.
Fibronectin, PLL, and all culture wares and reagents were purchased from Invitrogen (Carlsbad, CA). Immun-Blot polyvinylidene difluoride membranes for protein blotting were purchased from Bio-Rad Laboratories (Hercules, CA). An antibody detection kit (West-Zol plus) was obtained from iNtRON Biotechnology, Inc. (Seongnam, South Korea). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). PD98059 and Lipofectamine LTX with Plus Reagent were from Invitrogen (Life Technologies, Grand Island, NY). Protein A/G-agarose was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The SHP1 and SHP2 inhibitor NSC87877 was obtained from Tocris Bioscience (Bristol, United Kingdom). The sAC inhibitor KH7 was purchased from Cayman Chemical (Ann Arbor, MI). An anti-human CD99 MAb (YG32), mouse IgG, a rhodamine-conjugated anti-mouse IgG antibody, an FITC-conjugated anti-mouse IgG antibody, and a horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody were purchased from DiNonA (Seoul, South Korea). A mouse anti-human β1 integrin MAb (JB1A), a mouse anti-β1 integrin preservative-free MAb (B44), a rabbit anti-ERK1/2 (MAPK) polyclonal antibody, a mouse anti-PECAM-1 MAb, and an HRP-conjugated goat anti-rabbit IgG antibody were purchased from Chemicon (Temecula, CA). Antibodies against HRAS, RAF1, phospho-FAK (Ser910), GFP, phospho-SHP2 (Tyr542), paxillin, SHP2, PTPN12, PIN1, β1 integrin, RASGAP3, phospho-PKA (Thr 198), PKA, and ezrin and an HRP-conjugated donkey anti-goat IgG antibody were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against kindlin, phospho-Akt1 (Ser473), Akt1, phospho-FAK (Tyr397), FAK, and phospho-p44/42 MAPK (Thr202/Tyr204) (E10) were purchased from Cell Signaling (Danvers, MA). An anti-human p120 GTPase-activating protein MAb was obtained from Upstate Biotechnology (Lake Placid, NY). An anti-human β-actin MAb and mouse IgG purified immunoglobulin and anti-human IgG (Fc specific) antibodies were from Sigma-Aldrich Co. (St. Louis, MO). An R-phycoerythrin-conjugated anti-human CD29 antibody was obtained from Cymbus Biotechnology Ltd. (Southampton, United Kingdom). An anti-ERK1/2 (3A1) MAb was obtained from Abfrontier (Seoul, South Korea). An FITC-conjugated mouse anti-human CD29 MAb was purchased from SouthernBiotech (Birmingham, AL). A mouse anti-human vinculin MAb and a mouse anti-human talin1 MAb were purchased from Millipore (Temecula, CA).
Cell culture.
Human breast carcinoma cells (MCF-7 and MDA-MB-231) and HEK293T cells were obtained from the American Type Culture Collection (ATCC). Human epidermal keratinocytes (HaCaT) were from Invitrogen (Life Technologies, Grand Island, NY). C57BL mouse Lewis lung carcinoma (LLc1) cells were purchased from Sigma-Aldrich Co. (St. Louis, MO). Human umbilical vein endothelial cells were isolated from a human umbilical cord vein by collagenase treatment. Cells were cultured as described previously (70, 71).
Preparation of CD99-Fc fusion proteins.
Full-length human CD99 cDNA was purchased from ATCC. To prepare CD99 variants that partially lack the extracellular domain, PCR was carried out with several pairs of primers. Sense primer I (5′-GATGGTGGTTTCGATTTATCTGATG-3′) was for the cDNA of a CD99 I fragment variant with the signal peptide sequence deleted, sense primer II (5′-GGAGAAAATGACGACCCACGACC-3′) was for the cDNA of a CD99 II fragment lacking the first conserved region, sense primer III (5′-AGCTTTTCAGATGCTGACCTTGCG-3′) was for the cDNA of a CD99 III fragment lacking two conserved regions, sense primer IV (5′-GGCAGTGATGGTGGAGGCAGC-3′) was for the cDNA of a CD99 IV fragment lacking every conserved region, and primer V (5′-GCCATCTCTAGCTTCATTGCT-3′) was commonly used as an antisense primer. To obtain mutant CD99-Fc fusion constructs, each CD99 variant cDNA was subcloned into the EcoRI site of vector pET28a(+) carrying cDNA coding for the Fc domain of the human IgG1 antibody. All mutant CD99-Fc constructs were confirmed by DNA sequencing. Each plasmid DNA was transformed into BL21(DE3). Isopropyl-β-d-thiogalactopyranoside (IPTG) at 1.4 mM was added to induce protein expression. To purify recombinant proteins, 8 M urea buffer (0.01 M Tris-Cl, 0.1 M NaH2PO4) was prepared at various pH values (8.0, 6.3, and 4.5). The cells were lysed with 8 M urea buffer (pH 8.0) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 1 μg/ml, pepstatin, and 1 μg/ml aprotinin) and centrifuged at 12,000 × g for 20 min at 4°C. The supernatants were mixed with histidine (His)-binding resins (Ni-nitrilotriacetic acid [NTA] His Binding Resin; Novagen) in a 1-ml Eppendorf tube, and the mixtures were incubated at 4°C for 16 h to facilitate the binding of His-tagged proteins to the NTA resin. The protein-resin suspensions were centrifuged, and the resulting supernatants were discarded. The pellets were washed with 8 M urea buffer (pH 6.3). The CD99-Fc fusion proteins were then eluted with 8 M urea buffer (pH 4.5) and dialyzed in 1× ice-cold PBS. CD99-Fc protein expression was analyzed by SDS-PAGE. The eluted protein aliquots were prepared and stored in a cold chamber.
DNA constructs and retrovirus infection.
Full-length wild-type CD99 or mutant CD99 variants with amino acid substitutions in three conserved regions were produced by PCR. The coding sequence of human CD99 was obtained with 10 pmol of sense primer 5′-CGGAATGCATGCATCATCATCATCATCACGCCCGCGGGGCTGCCCTG-3′ and antisense primer 5′-GCTCGAGCTATTTCTCTAAAAGAGTACGCTGAAC-3′. Several pairs of primers were used to make mutant CD99 cDNA fragments, i.e., sense primer I (5′-GGTGGTTTCGATAACTCCAACGCCCTTCCT-3′] and antisense primer I (5′-AGGAAGGGCGTTGGAGTTATCGAAACCACC-3′) for the amino acid substitutions in the first conserved region, sense primer II (5′-GATGACTTTGACAACGGAAACGCTGTTGTTGATGG-3′) and antisense primer II (5′-CCATCAACAACAGCGTTTCCGTTGTCAAAGTCATC-3′) for those in the second conserved region, and sense primer III (5′-GATGCTGACAACGCGAACGGCGTTTC-3′) and antisense primer III (5′-GAAACGCCGTTCGCGTTGTCAGCATC-3′) for those in the third conserved region. As shown in Fig. 3, an asparagine residue was substituted for the leucine or aspartic acid residue in the conserved regions. His-tagged CD99 variant cDNAs were subcloned into EcoRI and XhoI sites of pMSCV-neo vectors (BD Biosciences Clontech, San Jose, CA). The sequences of the pMSCV-CD99 constructs were confirmed by DNA sequencing. Retroviral particles were harvested from retrovirus producer GP2-293 cells. MCF-7 or HaCaT cells were transfected with the viral supernatant and selected by treatment with 0.6 mg/ml G418 (Invitrogen Corp.) for 2 weeks. Resistant clones were selected, and changes in CD99 expression levels were confirmed by FACS or Western blot analysis.
Synthesis of polypeptides.
All polypeptides were synthesized with an automatic peptide synthesizer (PeptrEx-R48; Peptron, Daejeon, South Korea) by the 9-fluorenylmethoxy carbonyl solid-phase method. Some of them were labeled with FITC. The polypeptides synthesized were purified and analyzed by reverse-phase (RP) high-performance liquid chromatography (Prominence LC-20AB; Shimadzu, Japan) with a C18 analytical RP column (Capcell Pak column; Shiseido Co., Ltd., Japan) and a mass spectrometer (HP1100 Series LC/MSD; Hewlett-Packard, Roseville, CA).
Plasmid DNAs and RNA interference.
The construction of siRNAs was carried out with the Silencer siRNA construction kit in accordance with the manufacturer's instruction manual (Ambion, Austin, TX). The target sequences were human CD99 (sense primer I [5′-AAGATTGCAGTGGGTTTCTTGCCTGTCTC-3′] and antisense primer I [5′-AACAAGAAACCCACTGCAATCCCTGTCTC-3′]), human SHP2 (sense primer I [5′-AAATGTGGGTGACAGCTCCATCCTGTCTC-3′], antisense primer I [5′-AAATGGAGCTGTCACCCACATCCTGTCTC-3′], sense primer II [5′-AATCCTCCACCAACGTCGTATCCTGTCTC-3′], and antisense primer II [5′-AAATACGACGTTGGTGGAGGACCTGTCTC-3′]), human HRAS (sense primer I [5′-AATACGACCCCACTATAGAGGCCTGTCTC-3′], antisense primer I [5′-AACCTCTATAGTGGGGTCGTACCTGTCTC-3′], sense primer II [5′-AAGTGGATGTCCTCAAAAGACCCTGTCTC-3′], and antisense primer II [5′-AAGTCTTTTGAGGACATCCACCCTGTCTC-3′]), human RASGAP1 (sense primer I [5′-AACCACCTCTTCTACTAGGTCCCTGTCTC-3′], antisense primer I [5′-AAGACCTAGTAGAAGAGGTGGCCTGTCTC-3′], sense primer II [5′-AAGTCCGGAAGTTCAGGTACACCTGTCTC-3′], and antisense primer II [5′-AATGTACCTGAACTTCCGGACCCTGTCTC-3′]), and human RASGAP3 (sense primer I [5′-AAGTAAAACGGGCAGAGTGACCCTGTCTC-3′], antisense primer I [5′-AAGTCACTCTGCCCGTTTTACCCTGTCTC-3′], sense primer II [5′-AAGAGAATGTCGATCCAGTCCCCTGTCTC-3′], and antisense primer II [5′-AAGGACTGGATCGACATTCTCCCTGTCTC-3′]). siRNAs targeting PTPN12 and PIN1 were purchased from Genolution Pharmaceuticals Inc. (Seoul, South Korea). Control siRNA was purchased from Bioneer Corp. (Daejeon, South Korea). Dominant negative DNA or siRNA duplexes were transfected into cells with Lipofectamine reagent. After transfection, the medium was changed and cells were maintained in medium with 10% fetal bovine serum for 24 h. Knockdown of each of the target molecules or expression of dominant negative DNA was confirmed by Western blot analysis as described above. The CD99 lentiviral shRNA was purchased from Thermo Scientific (Waltham, MA). The CD99 knockdown transfectants were selected by treatment with 0.6 mg/ml puromycin, and knockdown of CD99 was confirmed by FACS or Western blot analysis.
Competitive ligand binding assay.
MCF-7 cells were pretreated with 30 μg/ml CD99-derived peptides in serum-free medium (SFM) and incubated for 30 min at 4°C. Cells were resuspended in 1 ml of SFM including 10 μg of human CD99-Fc I fusion protein with an intact extracellular domain and then incubated for 30 min at 4°C. Unbound CD99-Fc I fusion protein was removed by washing with SFM. The cells were resuspended in 200 μl of SFM including 1 μg of goat anti-human Fc-specific antibody and then incubated for 30 min at 4°C. Cells were washed with SFM to remove unbound antibodies. FITC-conjugated anti-goat IgG antibody was added to the cells, and they were incubated for 30 min at 4°C. After being washed twice with SFM, the cells were fixed with 1% formalin in 1× ice-cold PBS. Fluorescence intensity was measured with a FACScan flow cytometer (Becton Dickinson, San Jose, CA).
Flow cytometry.
Flow cytometry was performed as previously described (70). MCF-7 cells were incubated with 10 μg of CD99-Fc fusion protein or 1 μg of anti-human CD99 MAb for 30 min at 4°C. Cells were washed with SFM and resuspended in 200 μl of SFM containing 1 μg of FITC-conjugated anti-mouse IgG antibody. The cells were finally fixed in 400 μl of 1× ice-cold PBS containing 1% formalin. To detect the active form of β1 integrin, cells were seeded into fibronectin-coated six-well plates and then transfected with PTPN12 siRNA. The following day, cells were treated with CD99 peptide in the presence or absence of the MEK inhibitor PD98059. After 1 h of incubation at 37°C, cells were harvested and incubated in SFM with mouse anti-β1 integrin antibodies, an activated (B44) and FITC-conjugated secondary antibody, or a Milli-Mark anti-β1 integrin–FITC (HUTS-4) antibody to detect active β1 integrin (1 μg/100 μl) for 30 min at 4°C and then washed twice with ice-cold SFM. Fluorescence intensity was measured with a FACScan flow cytometer or a BD Accuri C6 system (Becton Dickinson, San Jose, CA).
Adhesion assay.
Cells were treated with each reagent (CD99-Fc fusion proteins or CD99 peptides) as described in the figure legends. After 1 h of incubation in 5% CO2 at 37°C, 1.5 × 105 cells were transferred to 96-well plates coated with fibronectin. Cells were sent to the bottom by centrifugation (1,200 rpm, 1 min) of the plate, allowed to attach to fibronectin, and incubated for 1 h in 5% CO2 at 37°C under static conditions. Unbound cells were removed by being washed twice with 1× PBS. Cells bound to the fibronectin were examined either by manual counting with a hemocytometer under a microscope (Olympus CK 40; Olympus, Tokyo, Japan) or by measurement of the absorbance at 450 nm with CCK-8 (Dojindo Laboratories, Kumamoto, Japan).
Confocal microscopy.
To determine the target specificity of a CD99-derived peptide, CD99CRIII3, MCF-7 cells were seeded onto coverslips. The next day, cells were incubated with FITC-conjugated CD99CRIII3 (for 1 h at room temperature [RT]) in the presence or absence of competing peptides. To determine the changes in β1 integrin activity, MCF-7 cells were seeded onto fibronectin-coated coverslips. PLL was used as a control substrate. The following day, cells were transfected with either a control or a PTPN12 siRNA and incubated for 24 h in 5% CO2 at 37°C. Cells were treated with CD99CRIII3 in the presence or absence of 10 μM PD98059 and incubated for 1 h in 5% CO2 at 37°C. Cells were washed twice with PBS and fixed in 4% paraformaldehyde in 1× PBS. Subsequently, cells were washed twice with PBS, permeabilized for 5 min in 0.1% Triton X-100 in PBS, and washed with PBS. Cells were incubated with 1 μg/ml anti-β1 integrin MAb conjugated with FITC for 30 min and washed twice with PBS for 5 min. Cells were then stained with an antipaxillin MAb (1 μg/ml, 30 min) and a rhodamine-conjugated anti-IgG antibody (5 μg/ml). Fluorescence images were acquired with an Olympus FluoView FV1000 confocal microscope (Olympus, Tokyo, Japan).
Western blotting and immunoprecipitation.
Western blot analysis was carried out as described previously (70). A nonreducing condition lacking β-mercaptoethanol was used to detect the active form of β1 integrin. For immunoprecipitation, cells were collected and lysed with lysis buffer (1% Triton X-100, 0.15 M NaCl, 0.05 M Tris [pH 8.0], 5 mM EDTA, 0.02% NaN3) containing 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, 10 μg/ml leupeptin, 1 μg/ml aprotinin, and 5 mM sodium orthovanadate. After centrifugation at 13,000 rpm and 4°C for 20 min, supernatants were collected and then incubated overnight with the appropriate antibodies at 4°C. The immunoprecipitates were incubated with protein A/G-agarose beads for 1 h at 4°C on a rotating instrument and washed with lysis buffer containing proteinase inhibitors. The precipitates were eluted with 1× sample buffer (50 mM Tris-HCl [pH 6.8], 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol). Western blot analysis was carried out with the antibodies indicated to detect the proper target proteins.
In situ PLA.
In situ PLA studies of fixed MCF-7 cells were performed with Duolink In Situ reagents (O-LINK Bioscience, Uppsala, Sweden) in accordance with the manufacturer's instructions. Briefly, approximately 1 × 105 MCF-7 cells were plated on glass coverslips in 24-well cell culture plates and grown in Dulbecco's modified Eagle's medium. The next day, cells were treated with fibronectin (20 μg/ml) in the presence or absence of peptides and each reagent for 1 h in an atmosphere containing 5% CO2 at 37°C and then washed twice with 1× PBS. Cells were fixed in 2% formaldehyde in PBS for 15 min at RT. Subsequently, cells were washed twice with 1× PBS, permeabilized for 5 min in 0.1% Triton X-100 in PBS, and then washed twice with wash buffer A for 5 min. Cells were incubated with blocking solution at 37°C for 30 min and then washed twice with wash buffer A for 5 min. Protein-protein interactions were analyzed with an Olympus FluoView FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan).
Statistical analysis.
Data are expressed as the average of the mean values obtained ± the standard deviation (SD). Statistical significance was determined by the Student t test with the statistical software Prism (version 4.0; GraphPad, San Diego, CA), and P < 0.05 was considered statistically significant. All experiments were conducted twice or more to obtain reproducible results. Representative data are shown in the figures.
ACKNOWLEDGMENTS
This work was supported by a grant from the Korea Research Foundation (C1006649) and the National Research Foundation grants funded by the Korean government (MEST) (Regional Core Research Program C1006636 and Leaders in Industry-University Cooperation Project C1008913) to J.-H.H.
J.-H. Hahn and K.-J. Lee proposed the concept, conceived the entire study, and wrote the paper. K.-J. Lee and Y. Kim performed most of the experiments with assistance from Y. H. Yoo, S.-H. Lee, C.-G. Kim, and M.-S. Kim for some experiments. K. Park, D. Jeoung, H. Lee, and I. Y. Ko provided suggestions and advice.
We have no conflict of interest to declare.
REFERENCES
- 1.Zhu J, Carman CV, Kim M, Shimaoka M, Springer TA, Luo BH. 2007. Requirement of alpha and beta subunit transmembrane helix separation for integrin outside-in signaling. Blood 110:2475–2483. doi: 10.1182/blood-2007-03-080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim C, Ye F, Ginsberg MH. 2011. Regulation of integrin activation. Annu Rev Cell Dev Biol 27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, Liu J, Jiang X, Haydar N, Zhang C, Shan H, Zhu J. 2013. Modulation of integrin activation and signaling by alpha1/alpha1′-helix unbending at the junction. J Cell Sci 126:5735–5747. doi: 10.1242/jcs.137828. [DOI] [PubMed] [Google Scholar]
- 4.Springer TA, Dustin ML. 2012. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol 24:107–115. doi: 10.1016/j.ceb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwamoto DV, Calderwood DA. 2015. Regulation of integrin-mediated adhesions. Curr Opin Cell Biol 36:41–47. doi: 10.1016/j.ceb.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, Eklund L, Ivaska J, Alitalo K, Saharinen P. 2015. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat Commun 6:5962. doi: 10.1038/ncomms6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hongu T, Funakoshi Y, Fukuhara S, Suzuki T, Sakimoto S, Takakura N, Ema M, Takahashi S, Itoh S, Kato M, Hasegawa H, Mochizuki N, Kanaho Y. 2015. Arf6 regulates tumour angiogenesis and growth through HGF-induced endothelial beta1 integrin recycling. Nat Commun 6:7925. doi: 10.1038/ncomms8925. [DOI] [PubMed] [Google Scholar]
- 8.Vitorino P, Yeung S, Crow A, Bakke J, Smyczek T, West K, McNamara E, Eastham-Anderson J, Gould S, Harris SF, Ndubaku C, Ye W. 2015. MAP4K4 regulates integrin-FERM binding to control endothelial cell motility. Nature 519:425–430. doi: 10.1038/nature14323. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, Frye M, Zeuschner D, Nakayama M, Vestweber D, Adams RH. 2015. Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat Commun 6:6429. doi: 10.1038/ncomms7429. [DOI] [PubMed] [Google Scholar]
- 10.Vellon L, Menendez JA, Lupu R. 2005. AlphaVbeta3 integrin regulates heregulin (HRG)-induced cell proliferation and survival in breast cancer. Oncogene 24:3759–3773. doi: 10.1038/sj.onc.1208452. [DOI] [PubMed] [Google Scholar]
- 11.Paschos KA, Canovas D, Bird NC. 2009. The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis. Cell Signal 21:665–674. doi: 10.1016/j.cellsig.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 12.Desgrosellier JS, Cheresh DA. 2010. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Muller S, Amin AR, Huang D, Su L, Hu Z, Rahman MA, Nannapaneni S, Koenig L, Chen Z, Tighiouart M, Shin DM, Chen ZG. 2012. The pivotal role of integrin beta1 in metastasis of head and neck squamous cell carcinoma. Clin Cancer Res 18:4589–4599. doi: 10.1158/1078-0432.CCR-11-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujita S, Suzuki H, Kinoshita M, Hirohashi S. 1992. Inhibition of cell attachment, invasion and metastasis of human carcinoma cells by anti-integrin beta 1 subunit antibody. Jpn J Cancer Res 83:1317–1326. doi: 10.1111/j.1349-7006.1992.tb02764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khalili P, Arakelian A, Chen G, Plunkett ML, Beck I, Parry GC, Donate F, Shaw DE, Mazar AP, Rabbani SA. 2006. A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol Cancer Ther 5:2271–2280. doi: 10.1158/1535-7163.MCT-06-0100. [DOI] [PubMed] [Google Scholar]
- 16.Kato H, Liao Z, Mitsios JV, Wang HY, Deryugina EI, Varner JA, Quigley JP, Shattil SJ. 2012. The primacy of beta1 integrin activation in the metastatic cascade. PLoS One 7:e46576. doi: 10.1371/journal.pone.0046576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brakebusch C, Wennerberg K, Krell HW, Weidle UH, Sallmyr A, Johansson S, Fassler R. 1999. Beta1 integrin promotes but is not essential for metastasis of ras-myc transformed fibroblasts. Oncogene 18:3852–3861. doi: 10.1038/sj.onc.1202770. [DOI] [PubMed] [Google Scholar]
- 18.Waclavicek M, Majdic O, Stulnig T, Berger M, Sunder-Plassmann R, Zlabinger GJ, Baumruker T, Stockl J, Ebner C, Knapp W, Pickl WF. 1998. CD99 engagement on human peripheral blood T cells results in TCR/CD3-dependent cellular activation and allows for Th1-restricted cytokine production. J Immunol 161:4671–4678. [PubMed] [Google Scholar]
- 19.Sohn HW, Shin YK, Lee IS, Bae YM, Suh YH, Kim MK, Kim TJ, Jung KC, Park WS, Park CS, Chung DH, Ahn K, Kim IS, Ko YH, Bang YJ, Kim CW, Park SH. 2001. CD99 regulates the transport of MHC class I molecules from the Golgi complex to the cell surface. J Immunol 166:787–794. doi: 10.4049/jimmunol.166.2.787. [DOI] [PubMed] [Google Scholar]
- 20.Sciandra M, Marino MT, Manara MC, Guerzoni C, Grano M, Oranger A, Lucarelli E, Lollini PL, Dozza B, Pratelli L, Renzo MF, Colombo MP, Picci P, Scotlandi K. 2014. CD99 drives terminal differentiation of osteosarcoma cells by acting as a spatial regulator of ERK 1/2. J Bone Miner Res 29:1295–1309. doi: 10.1002/jbmr.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sohn HW, Choi EY, Kim SH, Lee IS, Chung DH, Sung UA, Hwang DH, Cho SS, Jun BH, Jang JJ, Chi JG, Park SH. 1998. Engagement of CD99 induces apoptosis through a calcineurin-independent pathway in Ewing's sarcoma cells. Am J Pathol 153:1937–1945. doi: 10.1016/S0002-9440(10)65707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Husak Z, Printz D, Schumich A, Potschger U, Dworzak MN. 2010. Death induction by CD99 ligation in TEL/AML1-positive acute lymphoblastic leukemia and normal B cell precursors. J Leukoc Biol 88:405–412. doi: 10.1189/jlb.0210097. [DOI] [PubMed] [Google Scholar]
- 23.Husak Z, Dworzak MN. 2012. CD99 ligation upregulates HSP70 on acute lymphoblastic leukemia cells and concomitantly increases NK cytotoxicity. Cell Death Dis 3:e425. doi: 10.1038/cddis.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. 2002. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol 3:143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- 25.Lou O, Alcaide P, Luscinskas FW, Muller WA. 2007. CD99 is a key mediator of the transendothelial migration of neutrophils. J Immunol 178:1136–1143. doi: 10.4049/jimmunol.178.2.1136. [DOI] [PubMed] [Google Scholar]
- 26.Bixel G, Kloep S, Butz S, Petri B, Engelhardt B, Vestweber D. 2004. Mouse CD99 participates in T-cell recruitment into inflamed skin. Blood 104:3205–3213. doi: 10.1182/blood-2004-03-1184. [DOI] [PubMed] [Google Scholar]
- 27.Watson RL, Buck J, Levin LR, Winger RC, Wang J, Arase H, Muller WA. 2015. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J Exp Med 212:1021–1041. doi: 10.1084/jem.20150354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scotlandi K, Baldini N, Cerisano V, Manara MC, Benini S, Serra M, Lollini PL, Nanni P, Nicoletti G, Bernard G, Bernard A, Picci P. 2000. CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res 60:5134–5142. [PubMed] [Google Scholar]
- 29.Manara MC, Terracciano M, Mancarella C, Sciandra M, Guerzoni C, Pasello M, Grilli A, Zini N, Picci P, Colombo MP, Morrione A, Scotlandi K. 2016. CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1R/RAS/Rac1 signaling. Oncotarget 7:79925–79942. doi: 10.18632/oncotarget.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manara MC, Bernard G, Lollini PL, Nanni P, Zuntini M, Landuzzi L, Benini S, Lattanzi G, Sciandra M, Serra M, Colombo MP, Bernard A, Picci P, Scotlandi K. 2006. CD99 acts as an oncosuppressor in osteosarcoma. Mol Biol Cell 17:1910–1921. doi: 10.1091/mbc.E05-10-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scotlandi K, Zuntini M, Manara MC, Sciandra M, Rocchi A, Benini S, Nicoletti G, Bernard G, Nanni P, Lollini PL, Bernard A, Picci P. 2007. CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene 26:6604–6618. doi: 10.1038/sj.onc.1210481. [DOI] [PubMed] [Google Scholar]
- 32.Zucchini C, Manara MC, Pinca RS, De Sanctis P, Guerzoni C, Sciandra M, Lollini PL, Cenacchi G, Picci P, Valvassori L, Scotlandi K. 2014. CD99 suppresses osteosarcoma cell migration through inhibition of ROCK2 activity. Oncogene 33:1912–1921. doi: 10.1038/onc.2013.152. [DOI] [PubMed] [Google Scholar]
- 33.Lee KJ, Yoo YH, Kim MS, Yadav BK, Kim Y, Lim D, Hwangbo C, Moon KW, Kim D, Jeoung D, Lee H, Lee JH, Hahn JH. 2015. CD99 inhibits CD98-mediated beta1 integrin signaling through SHP2-mediated FAK dephosphorylation. Exp Cell Res 336:211–222. doi: 10.1016/j.yexcr.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Lee KJ, Lee SH, Yadav BK, Ju HM, Kim MS, Park JH, Jeoung D, Lee H, Hahn JH. 2012. The activation of CD99 inhibits cell-extracellular matrix adhesion by suppressing beta(1) integrin affinity. BMB Rep 45:159–164. doi: 10.5483/BMBRep.2012.45.3.159. [DOI] [PubMed] [Google Scholar]
- 35.Sullivan DP, Muller WA. 2014. Neutrophil and monocyte recruitment by PECAM, CD99, and other molecules via the LBRC. Semin Immunopathol 36:193–209. doi: 10.1007/s00281-013-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michael KE, Dumbauld DW, Burns KL, Hanks SK, Garcia AJ. 2009. Focal adhesion kinase modulates cell adhesion strengthening via integrin activation. Mol Biol Cell 20:2508–2519. doi: 10.1091/mbc.E08-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schober M, Raghavan S, Nikolova M, Polak L, Pasolli HA, Beggs HE, Reichardt LF, Fuchs E. 2007. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol 176:667–680. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serrels B, Serrels A, Brunton VG, Holt M, McLean GW, Gray CH, Jones GE, Frame MC. 2007. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol 9:1046–1056. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- 39.Lacalle RA, Mira E, Gomez-Mouton C, Jimenez-Baranda S, Martinez AC, Manes S. 2002. Specific SHP-2 partitioning in raft domains triggers integrin-mediated signaling via Rho activation. J Cell Biol 157:277–289. doi: 10.1083/jcb.200109031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Wichert G, Haimovich B, Feng GS, Sheetz MP. 2003. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J 22:5023–5035. doi: 10.1093/emboj/cdg492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montagner A, Yart A, Dance M, Perret B, Salles JP, Raynal P. 2005. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J Biol Chem 280:5350–5360. doi: 10.1074/jbc.M410012200. [DOI] [PubMed] [Google Scholar]
- 42.Agazie YM, Hayman MJ. 2003. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol 23:7875–7886. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y, Yang W, Xia Y, Hawke D, Liu DX, Lu Z. 2011. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol Cell Biol 31:4258–4269. doi: 10.1128/MCB.05547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitra SK, Hanson DA, Schlaepfer DD. 2005. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 45.Moricoli D, Muller WA, Carbonella DC, Balducci MC, Dominici S, Watson R, Fiori V, Weber E, Cianfriglia M, Scotlandi K, Magnani M. 2014. Blocking monocyte transmigration in in vitro system by a human antibody scFv anti-CD99. Efficient large scale purification from periplasmic inclusion bodies in E coli expression system. J Immunol Methods 408:35–45. doi: 10.1016/j.jim.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muller WA. 2014. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol 184:886–896. doi: 10.1016/j.ajpath.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park HJ, Ban YL, Byun D, Park SH, Jung KC. 2010. Interaction between the mouse homologue of CD99 and its ligand PILR as a mechanism of T cell receptor-independent thymocyte apoptosis. Exp Mol Med 42:353–365. doi: 10.3858/emm.2010.42.5.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung KC, Kim NH, Park WS, Park SH, Bae Y. 2003. The CD99 signal enhances Fas-mediated apoptosis in the human leukemic cell line, Jurkat. FEBS Lett 554:478–484. doi: 10.1016/S0014-5793(03)01224-9. [DOI] [PubMed] [Google Scholar]
- 49.Pliyev BK, Shepelev AV, Ivanova AV. 2013. Role of the adhesion molecule CD99 in platelet-neutrophil interactions. Eur J Haematol 91:456–461. doi: 10.1111/ejh.12178. [DOI] [PubMed] [Google Scholar]
- 50.Dufour EM, Deroche A, Bae Y, Muller WA. 2008. CD99 is essential for leukocyte diapedesis in vivo. Cell Commun Adhes 15:351–363. doi: 10.1080/15419060802442191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suh YH, Shin YK, Kook MC, Oh KI, Park WS, Kim SH, Lee IS, Park HJ, Huh TL, Park SH. 2003. Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs. Gene 307:63–76. doi: 10.1016/S0378-1119(03)00401-3. [DOI] [PubMed] [Google Scholar]
- 52.Byun HJ, Hong IK, Kim E, Jin YJ, Jeoung DI, Hahn JH, Kim YM, Park SH, Lee H. 2006. A splice variant of CD99 increases motility and MMP-9 expression of human breast cancer cells through the AKT-, ERK-, and JNK-dependent AP-1 activation signaling pathways. J Biol Chem 281:34833–34847. doi: 10.1074/jbc.M605483200. [DOI] [PubMed] [Google Scholar]
- 53.Zhang P, Liu X, Li Y, Zhu X, Zhan Z, Meng J, Li N, Cao X. 2013. Protein tyrosine phosphatase with proline-glutamine-serine-threonine-rich motifs negatively regulates TLR-triggered innate responses by selectively inhibiting IkappaB kinase beta/NF-kappaB activation. J Immunol 190:1685–1694. doi: 10.4049/jimmunol.1202384. [DOI] [PubMed] [Google Scholar]
- 54.Yang Q, Co D, Sommercorn J, Tonks NK. 1993. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J Biol Chem 268:17650. [PubMed] [Google Scholar]
- 55.Li Y, Dillon TJ, Takahashi M, Earley KT, Stork PJ. 2016. Protein kinase A-independent Ras protein activation cooperates with Rap1 protein to mediate activation of extracellular signal-regulated kinases (ERK) by cAMP. J Biol Chem 291:21584–21595. doi: 10.1074/jbc.M116.730978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stork PJ, Schmitt JM. 2002. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12:258–266. doi: 10.1016/S0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 57.Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. 2002. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J 21:64–71. doi: 10.1093/emboj/21.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. 2002. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol 22:3237–3246. doi: 10.1128/MCB.22.10.3237-3246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dumaz N, Marais R. 2003. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem 278:29819–29823. doi: 10.1074/jbc.C300182200. [DOI] [PubMed] [Google Scholar]
- 60.Edin ML, Juliano RL. 2005. Raf-1 serine 338 phosphorylation plays a key role in adhesion-dependent activation of extracellular signal-regulated kinase by epidermal growth factor. Mol Cell Biol 25:4466–4475. doi: 10.1128/MCB.25.11.4466-4475.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burmeister BT, Wang L, Gold MG, Skidgel RA, O'Bryan JP, Carnegie GK. 2015. Protein kinase A (PKA) phosphorylation of Shp2 protein inhibits its phosphatase activity and modulates ligand specificity. J Biol Chem 290:12058–12067. doi: 10.1074/jbc.M115.642983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burmeister BT, Taglieri DM, Wang L, Carnegie GK. 2012. Src homology 2 domain-containing phosphatase 2 (Shp2) is a component of the A-kinase-anchoring protein (AKAP)-Lbc complex and is inhibited by protein kinase A (PKA) under pathological hypertrophic conditions in the heart. J Biol Chem 287:40535–40546. doi: 10.1074/jbc.M112.385641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, Dobson JR, Deng Z, Fei F, Firestone B, Fodor M, Fridrich C, Gao H, Grunenfelder D, Hao HX, Jacob J, Ho S, Hsiao K, Kang ZB, Karki R, Kato M, Larrow J, La Bonte LR, Lenoir F, Liu G, Liu S, Majumdar D, Meyer MJ, Palermo M, Perez L, Pu M, Price E, Quinn C, Shakya S, Shultz MD, Slisz J, Venkatesan K, Wang P, Warmuth M, Williams S, Yang G, Yuan J, Zhang JH, Zhu P, Ramsey T, Keen NJ, et al. 2016. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535:148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 64.Gu J, Han T, Ma RH, Zhu YL, Jia YN, Du JJ, Chen Y, Jiang XJ, Xie XD, Guo X. 2014. SHP2 promotes laryngeal cancer growth through the Ras/Raf/Mek/Erk pathway and serves as a prognostic indicator for laryngeal cancer. Int J Oncol 44:481–490. doi: 10.3892/ijo.2013.2191. [DOI] [PubMed] [Google Scholar]
- 65.Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, Fang DD, Han T, Bailly-Maitre B, Poli V, Varki NM, Wang H, Feng GS. 2011. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 19:629–639. doi: 10.1016/j.ccr.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Su Z, Tian H, Song HQ, Zhang R, Deng AM, Liu HW. 2013. PTPN12 inhibits oral squamous epithelial carcinoma cell proliferation and invasion and can be used as a prognostic marker. Med Oncol 30:618. doi: 10.1007/s12032-013-0618-4. [DOI] [PubMed] [Google Scholar]
- 67.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, Nguyen DX, Pavlova NN, Botero M, Huang J, Bernardi RJ, Schmitt E, Hu G, Li MZ, Dephoure N, Gygi SP, Rao M, Creighton CJ, Hilsenbeck SG, Shaw CA, Muzny D, Gibbs RA, Wheeler DA, Osborne CK, Schiff R, Bentires-Alj M, Elledge SJ, Westbrook TF. 2011. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 144:703–718. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J, Davidson D, Martins Souza C, Zhong MC, Wu N, Park M, Muller WJ, Veillette A. 2015. Loss of PTPN12 stimulates progression of ErbB2-dependent breast cancer by enhancing cell survival, migration, and epithelial-to-mesenchymal transition. Mol Cell Biol 35:4069–4082. doi: 10.1128/MCB.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo RZ, Cai PQ, Li M, Fu J, Zhang ZY, Chen JW, Cao Y, Yun JP, Xie D, Cai MY. 2014. Decreased expression of PTPN12 correlates with tumor recurrence and poor survival of patients with hepatocellular carcinoma. PLoS One 9:e85592. doi: 10.1371/journal.pone.0085592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee KJ, Kim HA, Kim PH, Lee HS, Ma KR, Park JH, Kim DJ, Hahn JH. 2004. Ox-LDL suppresses PMA-induced MMP-9 expression and activity through CD36-mediated activation of PPAR-g. Exp Mol Med 36:534–544. doi: 10.1038/emm.2004.68. [DOI] [PubMed] [Google Scholar]
- 71.Lee KJ, Lim D, Yoo YH, Park EJ, Lee SH, Yadav BK, Lee YK, Park JH, Kim D, Park KH, Hahn JH. 2016. Paired Ig-like type 2 receptor-derived agonist ligands ameliorate inflammatory reactions by downregulating beta1 integrin activity. Mol Cells 39:557–565. doi: 10.14348/molcells.2016.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]