

Abstract
Two series of novel EPAC antagonists are designed, synthesized and evaluated in an effort to develop diversified analogues based on the scaffold of the previously identified high-throughput (HTS) hit 1 (ESI-09). Further SAR studies reveal that the isoxazole ring A of 1 can tolerate chemical modifications with either introduction of flexible electron-donating substitutions or structurally restrictedly fusing with a phenyl ring, leading to identification of several more potent and diversified EPAC antagonists (e.g., 10 (NY0617), 14 (NY0460), 26 (NY0725), 32 (NY0561), and 33 (NY0562)) with low micromolar inhibitory activities. Molecular docking studies on compounds 10 and 33 indicate that these two series of compounds bind at a similar site with substantially different interactions with the EPAC proteins. The findings may serve as good starting points for the development of more potent EPAC antagonists as valuable pharmacological probes or potential drug candidates.
Keywords: Exchange proteins directly activated by cAMP, EPAC, antagonist, molecular docking
Graphical abstract
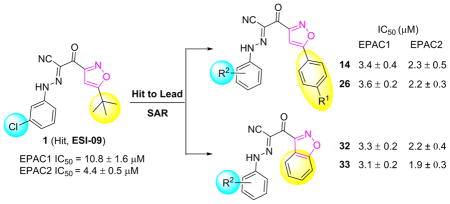
1. Introduction
Exchange proteins directly activated by cAMP (EPACs) were first identified as novel intracellular effector proteins of cyclic adenosine monophosphate (cAMP) by two independent groups in 1998 [1, 2]. Prior to the discovery of EPAC proteins, the major physiological effects of cAMP in mammalian cells are believed to be transduced by the classic protein kinase A/cAMP-dependent protein kinase (PKA/cAPK), and cyclic nucleotide-activated ion channels (CNG and HCN) in certain tissues [3–6]. Between two ubiquitously expressed intracellular cAMP receptor families, EPAC proteins, unlike PKA, have no kinase activity but act as guanine nucleotide exchange factors to catalyze the exchange of GDP with GTP for the down-stream small GTPases, Rap1 and Rap2, in response to intracellular cAMP [1, 2]. Two structurally homologous but functionally nonredundant isoforms of mammalian EPAC proteins have been identified, EPAC1 and EPAC2. EPAC1 is more ubiquitously expressed, whereas the expression of EPAC2 is relatively restricted, mainly found in brain, pancreatic islets and adrenal gland [2]. From nearly two decades of research on EPAC, accumulating studies, including those with the aid of small-molecule EPAC modulators [7, 8] such as various cAMP analogues (e.g. 007-AM [9]) and newly discovered EPAC-specific antagonists (e.g. ESI-09 [10–14]), have demonstrated that EPAC proteins play important roles in insulin secretion, energy homeostasis, cardiovascular response, pain sensing, osteoclast differentiation, neurotransmitter release, Treg-mediated immune suppression, integrin-mediated cell adhesion, cell migration and proliferation, cell exocytosis, and apoptosis as well as gene transcription and chromosomal integrity [15–19], and thus represent potential therapeutic targets for various human diseases such as cancer, bacterial and viral infections, chronic pain, diabetes, obesity, and heart failure.
Our previous high-throughput screening (HTS) campaign using automated, robust, and sensitive fluorescence based competition assay [10, 11] led to the identification of several EPAC specific inhibitors (ESIs), and was subsequently followed by extensive hit-to-lead optimizations [20–24]. Among these identified inhibitor hits, ESI-09 (1, Fig. 1) has been shown to selectively inhibit EPAC functions in vitro [12] and in vivo [13, 14]. With the aid of molecular docking studies of 1 into the cAMP binding domain B of active EPAC2 proteins, we hypothesized that binding interactions of inhibitors to EPAC2 proteins may primarily occur through two terminal hydrophobic pockets (P1 and P2) and the unique linker [7]. Later, systematic structure-activity–relationships (SARs) studies were performed, leading to the discovery of several more active EPAC antagonists (e.g., 2 (NY0123)) with low micromolar inhibitory activity and improved solubility [24].
Fig. 1.

Drug design strategy for the current work.
In a continuing effort to develop novel diversified analogues based on the scaffold of hit 1, we focus on our further chemical optimizations involving modifications of 5-tert-butyl group on the isoxazole ring A, meanwhile retaining favorable hydrophobic fragments of EPAC antagonists including fluorine-substitutions on the B-ring identified from our previous studies [24]. In order to explore the depth of the aforementioned hydrophobic pocket P2, as depicted in Fig. 1, series I was designed by inserting a rigid phenyl ring between the isoxazole A ring and its tert-butyl substitution at the 5-position. For comparison, we also attempted to make the molecular skeleton more compacted by fusing a phenyl ring with the isoxazole A (as depicted in series II, Fig. 1). Herein, we report such structural modifications of compound 1 with a focus on improving EPAC inhibitory activities and structural diversity of EPAC antagonists. The studies have resulted in the discovery of several novel potent EPAC antagonists such as 14 (NY0460), 26 (NY0725), 32 (NY0561), and 33 (NY0562), with low micromolar inhibitory activities for preclinical development.
2. Results and discussion
2.1. Chemistry
The synthesis of new derivatives based on 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazon-oyl cyanide scaffold with chemical optimizations of 5-tert-butyl group on the isoxazole is outlined in Scheme 1. Various ethyl isoxazole-3-carboxylates 3a–f were used as the key intermediates. 3a–f were prepared either from the commercially available pinacolones and oxalic acid diethyl esters in two steps as previously described by us [20, 24], or from ethyl esterification of the commercially available isoxazole-3-carboxylic acids. Ethyl esters 3a–f were first converted into the corresponding ketonitriles 4a–f by the treatment of MeLi and CH3CN using our previously reported protocols [20, 23]. By further modifications of Kowalsko’s reaction, NaH was used instead of MeLi as the base under a milder condition to generate the corresponding ketonitriles from ethyl esters 3a–f in good yields. On the other hand, aromatic amines 5a–h were treated with sodium nitrite and 2 N hydrochloric acid to give the corresponding aryldiazonium chlorides 6a–h. The aryldiazonium salts 6a–h with the crude cyanomethyl ketones 4a–f were then directly coupled in the presence of NaOAc as the catalyst at 0 °C afforded new derivatives of series I compounds 7–27 in 14–81% yields for two steps from 4a–f (Scheme 1). The desired products of series II 30–37 were accomplished from the commercially available ethyl benzo[d]isoxazole-3-carboxylate 28 with two steps in a similar fashion to those described for the synthesis of Series I.
Scheme 1.

Synthesis of the 2-oxo-N-phenyl-2-(5-phenylisoxazol-3-yl)acetohydrazonoyl cyanide analogues 7–27. Reagents and conditions: (a) CH3CN, MeLi, THF, -78 °C; or CH3CN, NaH, THF, 50 °C; (b) 2 N HCl, NaNO2, H2O, 0 °C; (c) 4a–f, NaOAc, EtOH, 14–81% for three steps.
2.2. Biology
2.2.1 In Vitro Evaluation of EPAC1 Inhibition
To explore the SARs and examine how the modifications on the isoxazole ring affect biological activities of newly synthesized analogues, we first evaluated their ability to inhibit EPAC1-mediated Rap1b-bGDP exchange activity using purified recombinant full-length EPAC1 proteins. Previous hit 1 was used as the reference compound, with an IC50 value of 10.8 μM in inhibiting EPAC1 [25].
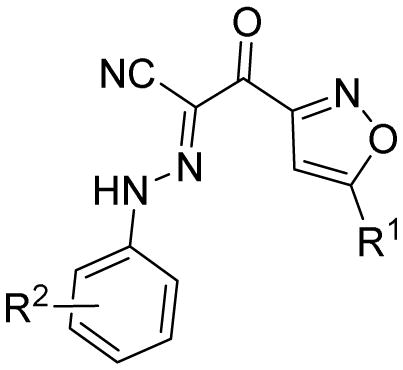
As shown in Table 1, we initially investigated the effect of the inserted phenyl ring between 5-tert-butyl group and isoxazole ring A of hit 1 in series I, leading to compound 7 with an IC50 value of 8.6 μM, which has a slight increase of inhibitory activity in comparison with that of 1. Given that fluorine substitutions may have an important contribution to the improvement of metabolic stability, solubility and bioactivity, as found in our previous publications [24, 26], selected fluorine substitutions on B ring were introduced to newly designed compounds 8–11. 3,5-di-CF3-substituted analogue 11 results in a slight loss of activity, while compound 10 with 3-CF3,4-Cl-substitution displays an enhanced potency, with an IC50 value of 7.3 μM. To evaluate the importance of 4-tert-butyl group on the inserted phenyl ring, we attempted the removal of this moiety, leading to new analogues 13–15. To our delight, compounds 13–15, not only have a better solubility than our previously reported compound 12 [24], but also exhibit an improved potency compared to that of 4-tBu-phenyl substituted analogues. This indicates that 4-tBu group on the inserted phenyl ring is dispensable for its activity. The most potent one of this series, 3-CF3,4-Cl-substituted compound 14 (Fig. 2), is about 5-fold increase in potency when compared to that of 1, with an IC50 value of 2.4 μM and as potent as previously reported lead compound 2. We next explored the electronic effect of substitutions at the 4-position of the inserted phenyl ring. Compounds 16–24 all result in a loss of activity. However, compounds 22–24 with other electron-donating groups such as methoxy, are more potent than corresponding compounds 16–21 with other electron-withdrawing groups such as fluoro and chloro. Particularly, 4-methoxy substituted analogues 23 and 24 display good activities, with the same IC50 values of 5.6 μM. As expected, replacement of 4-methoxy phenyl on the isoxazole ring A with its bioisostere, more electron-donating furan-2-yl group (as in compounds 25–27), also leads to an increase of activity. The most potent one of them, 25, shows an IC50 value of 3.6 μM. These results suggest that the 5-position of the isoxazole ring A is more suitable for chemical modifications and favorable with electron-donating groups.
Table 1.
Apparent IC50 values of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide scaffolds for inhibiting EPAC1 GEF activity.
| |||
|---|---|---|---|
| Compound | R1 | R2 | Rap1b-bGDP EPAC1 IC50 (μM)a |
| 1 | 10.8 ± 1.6 | ||
| 7 | 4-tBu-phenyl | 3-Cl | 8.6 ± 2.8 |
| 8 | 4-tBu-phenyl | 3-Cl, 5-CF3 | 10.2 ± 2.7 |
| 9 | 4-tBu-phenyl | 3-Cl, 4-CF3 | 9.0 ± 3.9 |
| 10 | 4-tBu-phenyl | 3-CF3, 4-Cl | 7.3 ± 2.3 |
| 11 | 4-tBu-phenyl | 3, 5-di-CF3 | 11.9 ± 6.0 |
| 12 | phenyl | 3-Cl | >150 |
| 13 | phenyl | 3-Cl, 5-CF3 | 9.5 ± 1.2 |
| 14 | phenyl | 3-CF3, 4-Cl | 2.4 ± 0.2 |
| 15 | phenyl | 3, 5-di-CF3 | 7.2 ± 0.7 |
| 16 | 4-F-phenyl | 3-Cl | 38.6 ± 7.7 |
| 17 | 4-F-phenyl | 3-Cl, 5-CF3 | 12.7 ± 1.1 |
| 18 | 4-F-phenyl | 3-CF3, 4-Cl | 10.2 ± 0.8 |
| 19 | 4-Cl-phenyl | 3-Cl | 77.6 ± 35.8 |
| 20 | 4-Cl-phenyl | 3-CF3, 4-Cl | 7.8 ± 2.1 |
| 21 | 4-Cl-phenyl | 3, 5-di-CF3 | 11.4 ± 2.7 |
| 22 | 4-OMe-phenyl | 3-Cl | 11.4 ± 2.8 |
| 23 | 4-OMe-phenyl | 3-CF3, 4-Cl | 5.6 ± 1.1 |
| 24 | 4-OMe-phenyl | 3, 5-di-CF3 | 5.6 ± 1.0 |
| 25 | furan-2-yl | 3-Cl | 9.9 ± 3.3 |
| 26 | furan-2-yl | 3-Cl, 5-CF3 | 3.6 ± 0.2 |
| 27 | furan-2-yl | 3-CF3, 4-Cl | 4.2 ± 0.9 |
The values are the mean ± SD of at least three independent experiments.
Fig. 2.

Relative inhibitory activity for EPAC1-mediated Rap1b-bGDP exchange. Dose-dependent inhibition of EPAC1 GEF activity by compound 1 (black), 8 (blue), 14 (red), 22 (green), 26 (brown) and 33 (purple), in the presence of 20 μM cAMP. Relative GEF activity were presented as normalized reaction rate constant (means ± SD, n = 3) described in the method.
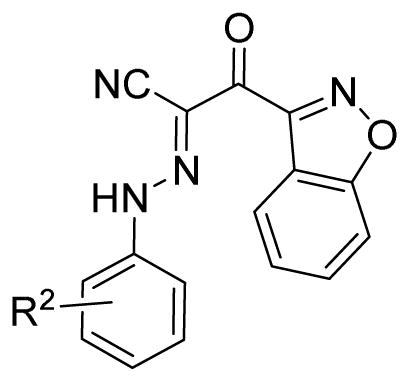
In series II as shown in table 2, replacement of isoxazole ring A of 1 with benzo[d]isoxazol moiety (as in compound 30), results in a slight loss of potency when compared to that of 1, with an IC50 value of 13.2 μM. However, further installation of fluorine-containing groups on its B ring quickly boosts the activity, except 3-Cl,5-F and 3,4,5-tri-F groups (as in compounds 34 and 37). Compounds 31–33 result in approximately 2~4-fold increase in potency when compared to that of 1, with IC50 values of about 2–4 μM (Fig. 2). Compound 33 has a substantially different new scaffold from that of previously described lead compound 2, but displays a comparable potency with an IC50 value of 2.7 μM.
Table 2.
Apparent IC50 values of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide scaffolds for inhibiting EPAC1 GEF activity.
| ||
|---|---|---|
| Compound | R2 | Rap1b-bGDP EPAC1 IC50 (μM)a |
| 30 | 3-Cl | 13.2 ± 3.6 |
| 31 | 3-Cl, 5-CF3 | 4.6 ± 0.8 |
| 32 | 3-Cl, 4-CF3 | 3.0 ± 0.3 |
| 33 | 3-CF3, 4-Cl | 2.7 ±0.3 |
| 34 | 3-Cl, 5-F | 18.9 ± 4.9 |
| 35 | 3-Cl, 4-F | 8.5 ± 3.5 |
| 36 | 3,5-di-CF3 | 6.7 ± 0.7 |
| 37 | 3,4,5-tri-F | 13.1 ± 2.5 |
The values are the mean ± SD of at least three independent experiments.
All these findings suggest that the isoxazole ring A of 1 can tolerate chemical modifications with either introduction of electron-donating substitutions or restrictedly fusing with a phenyl ring.
2.2.2 In Vitro Evaluation of EPAC2 Inhibition
From the biological results discussed above, compounds 10, 14–15, 23–24, 26–27, and 31–32 were identified as potent EPAC1 inhibitors with IC50 values lower than 8 μM and more potent than reference compound 1. Consistently, these selected compounds together with hit 1 were further evaluated for their ability to inhibit EPAC2-mediated Rap1b-bGDP exchange activity, rather than using previously described sensitive fluorescence based competition assay which may be interfered with their autoflorescence of these two series of compounds [24–25]. As shown in Table 3, the previous hit 1 is 2.5-fold more potent on EPAC2 inhibition than that on EPAC1, with an IC50 value of 4.4 μM. Interestingly, most of our selected, newly synthesized analogues exhibit significantly enhanced potency compared to that of 1, except compounds 23 and 24 (Fig. 3). Among these, compound 33 exhibits the best inhibitory activity for EPAC2, with an IC50 value of 1.9 μM (Fig. 3). Compounds such as 14, 26 and 32–33 with IC50 values lower than 4 μM for both EPAC1 and EPAC2 may serve as valuable pharmacological tools to probe the functions of EPAC in diseases or as potential drug candidates for further preclinical development.
Table 3.
Apparent IC50 values of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide scaffolds for inhibiting EPAC2 GEF activity.
| Compound | Rap1b-bGDP EPAC2 IC50 (μM)a |
|---|---|
| 1 | 4.4 ± 0.5 |
| 10 | 2.2 ± 0.3 |
| 14 | 2.3 ± 0.5 |
| 15 | 3.3 ± 0.8 |
| 23 | 4.5 ± 1.0 |
| 24 | 7.0 ± 0.9 |
| 26 | 2.2 ± 0.3 |
| 27 | 2.3 ± 0.2 |
| 31 | 2.2 ± 0.2 |
| 32 | 2.2 ± 0.4 |
| 33 | 1.9 ± 0.3 |
The values are the mean ± SD of at least three independent experiments.
Fig. 3.

Relative inhibitory activity for EPAC2-mediated Rap1b-bGDP exchange. Dose-dependent inhibition of EPAC2 GEF activity by compound 1 (black), 10 (blue), 24 (red), 27 (green), 33 (brown), in the presence of 20 μM cAMP. Relative GEF activity were presented as normalized reaction rate constant (means ± SD, n =3) described in the method.
2.3. Predicted Binding Modes of Compounds 10 and 33 with the cAMP Binding Domain B (CBD-B) of EPAC2 Proteins
Due to the lack of the X-ray cocrystal structures of our newly synthesized small molecules and their targeted proteins, molecular docking studies as useful methods may help us better understand the structure–activity relationships of these new compounds toward EPACs. Given the only available X-ray crystal structures of inactive and active EPAC2 proteins [27, 28], molecular docking studies of compounds 1, 10 and 33 at CBD-B of active EPAC2 protein (PDB Code 3CF6) were performed to investigate the predicted binding modes using the Schrödinger Small-Molecule Drug Discovery Suite [23]. Although this algorithm slightly differs from our previously employed AutoDock Vina [7, 24], the docking results are generally consistent with the previous studies. The current docking results also reveal that these new compounds fit well into the functional CBD-B binding pocket of active EPAC2 (Fig. 4A). As shown in Fig. 4B, the molecular docking studies of 10 comply with our previous results with hit 1 through the overlay analysis of two ligands. The isoxazolyl moiety and the 3-CF3-4-Cl-phenyl fragment of 10 respectively extend to two previously supposed hydrophobic pockets, while this binding mode is further stabilized by the occurrence of one hydrogen bond between the oxygen atom in carbonyl group of the linker and residue L406, as well as one halogen bond between the chloro atom in the 3-CF3-4-Cl-phenyl fragment and residue E451 (Figure 4C). Interestingly, compound 33 interacts with EPAC2 protein in a substantially different manner from that of compounds 1 and 10 (Figs. 4A and 4D). Its interactions with EPAC2 are dominated by the three strong hydrogen bonds and one halogen bond, including the oxygen atom in the benzo[d]isoxazol moiety with residue L406, the chloro group in phenyl fragment with residue N445, and the nitrogen atom in cyano group of the linker with K450 as well as its hydrogen atom with R448, in addition to the aforementioned hydrophobic interactions. These molecular docking studies could also reasonably explain why these two series of compounds with unique linkers might have better EPAC2 inhibitory activity (Figs. 4C and 4D). It is worth mentioning that this new scaffold as in compound 33, where the nitrogen atom on the heteroaryl ring forms a hydrogen bond with residue L406, may offer a good starting point for further drug design and structural optimizations.
Fig. 4.

(A) Overlay analysis of molecular docking poses of 1, 10 and 33 binding at the cAMP binding domain B (CBD-B) of EPAC2 protein (PDB Code 3CF6). cAMP is shown in red, 1 in yellow, 10 in magenta, and 33 in green. (B) Overlay of molecular docking poses of 1 (yellow) and 10 (magenta) binding at the CBD-B of EPAC2. (C) Predicted binding mode of 10 docked into the CBD-B of EPAC2. 10 is shown in magenta ball and stick representation. Key residues are displayed in sticks. Hydrogen bonds and halogen bond are shown in dotted purple lines. (D) Predicted binding mode of 33 docked into the CBD-B of EPAC2. 33 is shown in green ball and stick representation. Key residues are displayed in sticks. Hydrogen bonds and halogen bond are shown in dotted purple lines.
3. Conclusions
Two series of novel EPAC antagonists based on the scaffold of the previously identified high-throughput hit 1 (ESI-09) have been designed, synthesized, and biologically evaluated for their EPAC1 and EPAC2 inhibitory activities. The SAR results based on EPAC2 activity comply with our docking studies in general, indicating that the isoxazole ring B of 1 can tolerate chemical modifications with either introduction of flexible electron-donating substitutions or structurally restrictedly fusing with a phenyl ring. The new scaffold of series II, as in compound 33 interacting with EPAC2 in a novel binding mode, may offer a good starting point for further drug design and structural optimizations. All these modification efforts allow us to further tune the original hit 1 to achieve more potent and structurally diverse EPAC1 and EPAC2 inhibitors, such as 10 (NY0617), 14 (NY0460), 26 (NY0725), 32 (NY0561), and 33 (NY0562) with IC50 values in the low micromolar range. These compounds may hold promise as potential drug candidates toward novel therapeutics against human diseases, and serve as valuable pharmacological probes to elucidate the physiological functions of EPAC proteins. Currently, the in vitro and in vivo activities of these selected compounds in infectious disease models (e.g. rickettsiosis) are being investigated. Further systematic optimizations based upon identified new scaffolds of these two series toward EPAC subtype selectivity are also under way and the findings will be reported in due course.
4. Experimental section
4.1. Chemistry
All commercially available starting materials and solvents were reagent grade, and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 or Bruker-300 (1H, 600 & 300 MHz; 13C, 150 & 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; Capillary temperature was 275 °C and the resolution was 60,000; Ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal Digital Melting Point Apparatus and uncorrected. Purities of final compounds were established by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (0.1% TFA was added into both acetonitrile and water). All biologically evaluated compounds are > 95% pure.
4.1.1. N-(3-Chlorophenyl)-2-(5-(4-(tert-butyl)phenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (7)
To a solution of CH3CN (0.43 mL, 7.32 mmol) in anhydrous THF (10 mL) was added 1.6 M methyl lithium in diethyl ether (2.30 mL, 3.66 mmol) at −78 °C under nitrogen. The mixture was stirred at −78 °C for 0.5 h, and ethyl 5-(4-(tert-butyl)phenyl)isoxazole-3-carboxylate 3a (500 mg, 1.83 mmol) in THF (10 mL) was then added dropwise. The solution was stirred at −78 °C for 1 h and then quenched with acetic acid (0.21 mL, 3.66 mmol). The mixture was warmed to 0 °C and poured onto ice/water (10 mL) and extracted with ethyl acetate (20 mL). The organic lay was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue 4a (330 mg, 75%) was obtained as a white solid and directly used for next step without further purification. 1H NMR (300 MHz, CDCl3) δ 7.74 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 6.92 (s, 1H), 4.25 (s, 2H), 1.32 (s, 9H).
To a solution of 3-chloroaniline 5a (37 mg, 0.33 mmol) in H2O (10 mL cooled to −5 °C) was added 0.2 mL of 2 N HCl (aq.). To the resulting acidic aniline solution, 1 N solution of sodium nitrite (0.33 mL, 0.33 mmol) was added dropwise to generate the aryldiazonium salt solution 6a. To the aryldiazonium salt solution was added sodium acetate (54 mg, 0.66 mmol), followed by 1 mL solution of crude 3-oxo-3-(3-phenylisoxazol-5-yl)propanenitrile 4a (88 mg, 0.33 mmol) in ethanol. The reaction mixture was stirred at 0 °C for 5 min, and then poured onto H2O (10 mL) and extracted with ethyl acetate (20 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel, eluting with hexane/ethyl acetate (2/1) to provide the desired product 7 (67 mg, 50% for two steps from 3a) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.90 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 3H), 7.54 – 7.45 (m, 2H), 7.43 (s, 1H), 7.25 (d, J = 7.2 Hz, 1H), 1.32 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 179.41, 170.22, 161.42, 154.25, 144.10, 134.44, 131.73, 126.63, 126.17, 125.65, 124.03, 117.18, 116.11, 113.84, 101.39, 35.19, 31.32. HRMS (ESI) calcd for C22H20ClN4O2 407.1275 (M + H)+, found 407.1270.
4.1.2. N-(3-Chloro-5-(trifluoromethyl)phenyl)-2-(5-(4-(tert-butyl)phenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (8)
Compound 8 was prepared in 22% yield (two steps from 3a) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.88 (d, J = 8.4 Hz, 2H), 7.78 (s, 2H), 7.62 (s, 1H), 7.58 (d, J = 8.4 Hz, 2H), 7.42 (s, 1H), 1.32 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 179.13, 170.04, 166.63, 154.17, 135.45, 132.13 (q, J = 33.7 Hz), 126.57, 126.09, 125.33, 124.10, 121.42, 121.16, 113.32, 101.48, 35.17, 31.32. HRMS (ESI) calcd for C23H19F3ClN4O2 475.1149 (M + H)+, found 475.1145.
4.1.3. N-(3-Chloro-4-(trifluoromethyl)phenyl)-2-oxo-2-(5-(4-(tert-butyl)phenyl)isoxazol-3-yl)acetohydrazonoyl cyanide (9)
Compound 9 was prepared in 28% yield (two steps from 3a) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.92 – 7.86 (m, 3H), 7.71 (s, 1H), 7.59 (m, 3H), 7.39 (s, 1H), 1.33 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 179.17, 169.93, 161.89, 154.15, 132.22, 129.66, 126.61, 126.13, 124.14, 119.99, 116.73, 115.06, 101.43, 35.18, 31.33. HRMS (ESI) calcd for C23H19F3ClN4O2 475.1149 (M + H)+, found 475.1140.
4.1.4. N-(4-Chloro-3-(trifluoromethyl)phenyl)-2-oxo-2-(5-(4-(tert-butyl)phenyl)isoxazol-3-yl)acetohydrazonoyl cyanide (10)
Compound 10 was prepared in 41% yield (two steps from 3a) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.00 (d, J = 2.2 Hz, 1H), 7.88 (d, J = 8.5 Hz, 2H), 7.75 (m, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.41 (s, 1H), 1.33 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 179.21, 170.08, 161.68, 154.17, 133.34, 127.93 (q, J = 31.0 Hz), 126.59, 126.09, 124.79, 124.07, 122.48, 116.81, 114.22, 101.33, 35.18, 31.33. HRMS (ESI) calcd for C23H19F3ClN4O2 475.1149 (M + H)+, found 475.1147.
4.1.5. N-(3,5-Bis(trifluoromethyl)phenyl)-2-(5-(4-(tert-butyl)phenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (11)
Compound 11 was prepared in 26% yield (two steps from 3a) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.01 (s, 2H), 7.85 (d, J = 8.2 Hz, 2H), 7.78 (s, 1H), 7.57 (d, J = 8.4 Hz, 2H), 7.36 (s, 1H), 1.32 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 179.11, 169.61, 162.36, 153.99, 131.70 (dd, J = 65.6, 32.8 Hz), 126.50, 125.97, 125.44, 124.22, 121.82, 118.67, 117.72, 114.15, 101.45, 35.14, 31.32. HRMS (ESI) calcd for C24H19F6N4O2 509.1412 (M + H)+, found 519.1410.
4.1.6. N′-(3-Chloro-5-(trifluoromethyl)phenyl)-2-oxo-2-(5-phenylisoxazol-3 yl)acetohydrazonoyl cyanide (13)
Compound 13 was prepared in 47% yield (two steps from 3b) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.96 (m, 2H), 7.78 (s, 2H), 7.63 (s, 1H), 7.58 (m, 3H), 7.51 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.11, 170.04, 161.55, 145.82, 135.49, 132.65 (q, J = 32.7 Hz), 131.38, 129.80, 126.64, 126.23, 125.27, 121.65, 121.60, 120.93, 114.72, 113.00, 111.19, 102.05. HRMS (ESI) calcd for C19H11ClF3N4O2 419.0523 (M + H)+, found 419.0516.
4.1.7. N-(3-Trifluromethyl-4-chlorophenyl)-2-oxo-2-(5-phenylisoxazol-3-yl)acetohydrazonoyl cyanide (14)
Compound 14 was prepared in 43% yield (two steps from 3b) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.96 (d, J = 7.5 Hz, 3H), 7.75 (d, J = 3.0 Hz, 2H), 7.62 – 7.52 (m, 3H), 7.47 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.16, 169.93, 161.76, 143.47, 133.29, 131.33, 129.80, 127.93 (q, J = 31.0 Hz), 126.69, 126.22, 124.78, 122.67, 121.16, 116.69, 114.16, 111.57, 101.88. HRMS (ESI) calcd for C19H11F3ClN4O2 419.0523 (M + H)+, found 419.0536.
4.1.8. N-(3,5-Bis(trifluoromethyl)phenyl)-2-oxo-2-(5-phenylisoxazol-3-yl)acetohydrazonoyl cyanide (15)
Compound 15 was prepared in 24% yield (two steps from ethyl 5-phenylisoxazole-3-carboxylate) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.03 (s, 2H), 7.93 (s, 2H), 7.81 (s, 1H), 7.56 (s, 3H), 7.48 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.09, 169.64, 166.63, 162.17, 131.74 (q, J = 32.8 Hz), 131.24, 129.75, 126.78, 126.13, 125.39, 121.78, 118.42, 117.91, 114.41, 102.06. HRMS (ESI) calcd for C20H11F6N4O2 453.0786 (M + H)+, found 453.0776.
4.1.9. N-(3-Chlorophenyl)-2-(5-(4-fluorophenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (16)
Compound 16 was prepared in 76% yield (two steps from 3c) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.10 – 8.02 (m, 2H), 7.57 (s, 1H), 7.52 – 7.39 (m, 5H), 7.26 (d, J = 7.5 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.29, 169.16, 163.89 (d, J = 249.2 Hz), 161.57, 144.28, 134.43, 131.72, 128.97, 128.85, 125.66, 123.42, 117.16, 116.87, 116.23, 113.73, 111.11, 101.90. HRMS (ESI) calcd for C18H11FClN4O2 369.0555 (M + H)+, found 369.0549.
4.1.10. N-(3-Chloro-5-(trifluoromethyl)phenyl)-2-(5-(4-fluorophenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (17)
Compound 17 was prepared in 41% yield (two steps from 3c) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.04 (dd, J = 8.6, 5.4 Hz, 2H), 7.76 (s, 2H), 7.61 (s, 1H), 7.49 (s, 1H), 7.43 (t, J = 8.8 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 179.04, 168.95, 163.85 (d, J = 249.1 Hz) 161.89, 135.44, 132.10 (q, J = 32.7 Hz), 128.87, 128.75, 125.32, 123.47, 121.45, 121.20, 117.11, 116.82, 114.45, 113.29, 111.64, 102.00. HRMS (ESI) calcd for C19H10F4ClN4O2 437.0428 (M + H)+, found 437.0420.
4.1.11. N-(4-Chloro-3-(trifluoromethyl)phenyl)-2-(5-(4-fluorophenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (18)
Compound 18 was prepared in 66% yield (two steps from 3c) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.08 – 7.96 (m, 3H), 7.82 – 7.71 (m, 2H), 7.49 (s, 1H), 7.43 (t, J = 8.8 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 179.15, 169.17, 163.87 (d, J = 247.5 Hz), 161.59, 142.56, 133.36, 128.90, 128.79, 126.75, 123.39, 122.34, 117.12, 116.82, 114.30, 111.13, 101.82. HRMS (ESI) calcd for C19H10F4ClN4O2 437.0428 (M + H)+, found 437.0422.
4.1.12. N-(3-Chlorophenyl)-2-(5-(4-chlorophenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (19)
Compound 19 was prepared in 52% yield (two steps from 3d) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO- d6) δ 7.99 (d, J = 6.6 Hz, 2H), 7.57 (m, 6H), 7.25 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.17, 168.92, 161.62, 144.36, 136.04, 134.43, 131.69, 129.95, 128.10, 125.66, 125.50, 117.18, 116.28, 113.68, 111.15, 102.49. HRMS (ESI) calcd for C18H11Cl2N4O2 385.0259 (M + H)+, found 385.0259.
4.1.13. N-(4-Chloro-3-(trifluoromethyl)phenyl)-2-oxo-2-(5-(4-chlorophenyl)isoxazol-3-yl)acetohydrazonoyl cyanide (20)
Compound 20 was prepared in 81% yield (two steps from 3d) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 7.98 (d, J = 8.1 Hz, 3H), 7.81–7.70 (m, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.54 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.04, 170.79, 168.96, 161.59, 142.49, 136.02, 133.35, 129.92, 128.17, 128.05, 127.93 (q, J = 31.0 Hz), 125.48, 122.29, 116.57, 114.28, 111.07, 102.44. HRMS (ESI) calcd for C19H10F3Cl2N4O2 453.0133 (M + H)+, found 453.0130.
4.1.14. N-(3,5-Bis(trifluoromethyl)phenyl)-2-(5-(4-chlorophenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (21)
Compound 21 was prepared in 38% yield (two steps from 3d) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 8.06 (s, 2H), 7.97 (d, J = 8.3 Hz, 2H), 7.84 (s, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.56 (s, 1H). 13C NMR (75 MHz, DMSO) δ 178.96, 168.60, 162.13, 135.91, 131.77 (q, J = 33.1 Hz), 129.88, 127.96, 125.60, 125.37, 121.75, 118.25, 118.01, 102.63. HRMS (ESI) calcd for C20H10F6ClN4O2 487.0396 (M + H)+, found 487.0390.
4.1.15. N-(3-Chlorophenyl)-2-(5-(4-methoxyphenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (22)
Compound 22 was prepared in 56% yield (two steps from 3e) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 7.91 (d, J = 8.7 Hz, 2H), 7.57 (s, 1H), 7.52 – 7.42 (m, 2H), 7.33 (s, 1H), 7.25 (d, J = 7.3 Hz, 1H), 7.12 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H). 13C NMR (75 MHz, DMSO) δ 179.46, 170.12, 161.67, 161.46, 134.43, 131.69, 128.06, 125.61, 119.33, 117.17, 116.25, 115.25, 113.75, 100.39, 55.92. HRMS (ESI) calcd for C19H14ClN4O3 381.0754 (M + H)+, found 381.0760.
4.1.16. N-(4-Chloro-3-(trifluoromethyl)phenyl)-2-oxo-2-(5-(4-methoxyphenyl)isoxazol-3-yl)acetohydrazonoyl cyanide (23)
Compound 23 was prepared in 33% yield (two steps from 3e) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 8.00 (s, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.81 – 7.70 (m, 2H), 7.34 (s, 1H), 7.12 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H). 13C NMR (75 MHz, DMSO) δ 179.35, 170.15, 161.65, 161.48, 133.35, 128.01, 126.66, 124.76, 122.34, 119.32, 116.70, 116.62, 115.22, 114.32, 100.31, 55.91. HRMS (ESI) calcd for C20H13F3ClN4O3 449.0628 (M + H)+, found 381.0760.
4.1.17. N-(3,5-Bis(trifluoromethyl)phenyl)-2-(5-(4-chlorophenyl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (24)
Compound 24 was prepared in 14% yield (two steps from 3e) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 8.00 (s, 2H), 7.87 (d, J = 8.4 Hz, 2H), 7.78 (s, 1H), 7.28 (s, 1H), 7.10 (d, J = 8.5 Hz, 2H), 3.84 (s, 3H). 13C NMR (75 MHz, DMSO) δ 179.24, 169.56, 162.35, 161.51, 131.69 (q, J = 32.9 Hz), 127.86, 125.44, 121.83, 119.55, 118.63, 117.74, 115.14, 114.16, 100.46, 55.89. HRMS (ESI) calcd for C21H13F6N4O3 483.0892 (M + H)+, found 483.0890.
4.1.18. N-(3-Chlorophenyl)-2-(5-(furan-2-yl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (25)
To a solution of NaH (197 mg, 4.53 mmol) in anhydrous dioxane (3 mL) was added the solution of ethyl 5-(furan-2-yl)isoxazole-3-carboxylate 3f (350 mg, 1.81 mmol) in MeCN (3 mL) dropwise at 0 °C under nitrogen. The solution was stirred at 50 °C for 1 h, and then quenched with sat. NH4Cl (2 mL) at 0°C. The mixture was poured onto ice/water (10 mL) and extracted with ethyl acetate (20 mL). The organic lay was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue 4f (400 mg, quant.) was obtained as a yellow solid and directly used for next step without further purification.
Compound 25 was prepared in 40% yield (two steps from 3f) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 8.00 (s, 1H), 7.55 (s, 1H), 7.52 – 7.41 (m, 2H), 7.29 (d, J = 3.5 Hz, 1H), 7.25 (d, J = 6.7 Hz, 1H), 7.19 (s, 1H), 6.78 (dd, J = 3.3, 1.7 Hz, 1H). 13C NMR (75 MHz, DMSO) δ 179.03, 161.57, 161.08, 146.46, 144.03, 141.98, 134.47, 131.72, 125.70, 117.12, 116.10, 113.80, 113.02, 112.68, 110.94, 100.98. HRMS (ESI) calcd for C16H10ClN4O3 341.0441 (M + H)+, found 341.0444.
4.1.19. N-(3-Chloro-5-(trifluoromethyl)phenyl)-2-(5-(furan-2-yl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (26)
Compound 26 was prepared in 38% yield (two steps from 3f) by a procedure similar to that used to prepare compound 25. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 7.99 (d, J = 1.7 Hz, 1H), 7.76 (s, 2H), 7.62 (s, 1H), 7.27 (d, J = 3.5 Hz, 1H), 7.20 (s, 1H), 6.77 (dd, J = 3.5, 1.8 Hz, 1H). 13C NMR (75 MHz, DMSO) δ 178.78, 161.50, 161.21, 146.42, 141.99, 135.52, 132.19 (d, J = 32.9 Hz), 125.26, 121.65, 120.94, 114.68, 112.98, 112.56, 101.10. HRMS (ESI) calcd for C17H9F3ClN4O3 409.0315 (M + H)+, found 409.0310.
4.1.20. N-(4-Chloro-3-(trifluoromethyl)phenyl)-2-(5-(furan-2-yl)isoxazol-3-yl)-2-oxoacetohydrazonoyl cyanide (27)
Compound 27 was prepared in 36% yield (two steps from 3f) by a procedure similar to that used to prepare compound 25. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO) δ 8.00 (d, J = 1.7 Hz, 1H), 7.96 (d, J = 2.1 Hz, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.76 – 7.69 (m, 1H), 7.27 (d, J = 3.6 Hz, 1H), 7.19 (s, 1H), 6.78 (dd, J = 3.5, 1.8 Hz, 1H). 13C NMR (75 MHz, DMSO) δ 178.89, 161.52, 161.24, 146.41, 142.86, 142.01, 133.37, 128.00 (q, J = 31.1 Hz), 126.73, 124.75, 122.44, 121.14, 116.65, 116.58, 114.30, 112.99, 112.58, 111.19, 100.96. HRMS (ESI) calcd for C17H9F3ClN4O3 409.0315 (M + H)+, found 409.0312.
4.1.21. 2-(Benzo[d]isoxazol-3-yl)-N-(3-chlorophenyl)-2-oxoacetohydrazonoyl cyanide (30)
To a solution of CH3CN (0.46 mL, 8.80 mmol) in anhydrous THF (8 mL) was added 1.6 M methyl lithium in diethyl ether (2.75 mL, 4.40 mmol) at −78 °C under nitrogen. The mixture was stirred at −78 °C for 0.5 h, and ethyl benzo[d]isoxazole-3-carboxylate 28 (420 mg g, 2.20 mmol) in THF (10 mL) was then added dropwise. The solution was stirred at −78 °C for 1 h and then quenched with acetic acid (0.26 mL, 4.40 mmol). The mixture was warmed to 0 °C and poured onto ice/water (10 mL) and extracted with ethyl acetate (20 mL). The organic lay was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue 29 (400 mg, 98%) was obtained as a yellow solid and directly used for next step without further purification. 1H NMR (300 MHz, CDCl3) δ 8.24 (d, J = 8.0 Hz, 1H), 7.77 – 7.66 (m, 2H), 7.57 – 7.49 (m, 1H), 4.40 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 182.38, 164.73, 131.23, 126.16, 123.23, 118.36, 112.60, 110.18, 30.34.
Compound 30 was prepared in 53% yield (two steps from 28) by a procedure similar to that used to prepare compound 7. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.05 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 8.6 Hz, 1H), 7.77 (t, J = 7.7 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.44 – 7.33 (m, 3H), 7.26 – 7.19 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.64, 162.99, 155.16, 144.19, 134.42, 131.65, 131.50, 125.70, 125.65, 123.74, 120.52, 116.96, 116.15, 113.98, 111.13, 110.42. HRMS (ESI) calcd for C16H10ClN4O2 325.0492 (M + H)+, found 325.0483.
4.1.22. 2-(Benzo[d]isoxazol-3-yl)-N-(3-chloro-5-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide (31)
Compound 31 was prepared in 33% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.04 (d, J = 8.0 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.60 (s, 3H), 7.49 (t, J = 7.6 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.40, 163.00, 155.37, 145.94, 135.46, 132.12 (q, J = 32.8 Hz), 131.44, 125.63, 123.76, 121.59, 120.85, 120.56, 114.90, 112.83, 111.28, 110.39. HRMS (ESI) calcd for C17H9F3ClN4O2 393.0366 (M + H)+, found 393.0376.
4.1.23. 2-(Benzo[d]isoxazol-3-yl)-N-(3-chloro-4-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide (32)
Compound 32 was prepared in 22% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.02 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.7 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.56 – 7.42 (m, 3H). 13C NMR (75 MHz, DMSO-d6) δ 179.41, 162.96, 132.27, 132.25, 131.44, 129.75, 129.68, 125.61, 123.72, 120.61, 119.55, 116.47, 115.39, 110.39. HRMS (ESI) calcd for C17H9F3ClN4O2 393.0366 (M + H)+, found 393.0378.
4.1.24. 2-(Benzo[d]isoxazol-3-yl)-N-(4-chloro-3-(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide (33)
Compound 33 was prepared in 55% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.02 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.81 – 7.69 (m, 3H), 7.63 (dd, J = 9.0, 2.2 Hz, 1H), 7.48 (t, J = 7.5 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.43, 163.01, 155.19, 142.68, 133.29, 131.43, 127.96 (q, J = 31.2 Hz), 126.77, 125.61, 124.68, 123.66, 122.34, 121.06, 120.53, 116.45, 116.37, 114.51, 111.19, 110.36. HRMS (ESI) calcd for C17H9F3ClN4O2 393.0366 (M + H)+, found 393.0374.
4.1.25. 2-(Benzo[d]isoxazol-3-yl)-N-(3-chloro-5-fluorophenyl)-2-oxoacetohydrazonoyl cyanide (34)
Compound 34 was prepared in 57% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.04 (d, J = 7.9 Hz, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.24 – 7.16 (m, 2H), 7.11 (d, J = 10.3 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.43, 162.99, 162.98 (d, J = 245.0 Hz), 155.13, 146.02, 135.30 (d, J = 5.9 Hz), 131.47, 125.66, 123.77, 120.54, 114.60, 113.59, 112.99, 112.65, 111.16, 110.43, 103.49 (d, J = 26.6 Hz). HRMS (ESI) calcd for C16H9FClN4O2 343.0398 (M + H)+, found 343.0388.
4.1.26. 2-(Benzo[d]isoxazol-3-yl)-N-(3-chloro-4-fluorophenyl)-2-oxoacetohydrazonoyl cyanide (35)
Compound 35 was prepared in 44% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.04 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.80 – 7.72 (m, 1H), 7.54–7.36 (m, 4H). 13C NMR (75 MHz, DMSO-d6) δ 179.48, 162.99, 155.29 (d, J = 240. 0 Hz), 155.07, 140.15, 131.46, 125.64, 123.70, 121.00, 120.74, 120.53, 118.95, 118.38, 118.08, 117.90, 117.80, 113.78, 111.15, 110.40. HRMS (ESI) calcd for C16H9FClN4O2 343.0398 (M + H)+, found 343.0387.
4.1.27. 2-(Benzo[d]isoxazol-3-yl)-N-(3,5-bis(trifluoromethyl)phenyl)-2-oxoacetohydrazonoyl cyanide (36)
Compound 36 was prepared in 31% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.02 (d, J = 7.9 Hz, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.88 (s, 2H), 7.81 (s, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 179.35, 162.99, 155.56, 146.13, 132.83 (q, J = 32.9 Hz), 131.37, 125.55, 125.24, 123.70, 121.62, 120.59, 117.75, 115.06, 111.39, 110.34. HRMS (ESI) calcd for C18H9F6N4O2 427.0630 (M + H)+, found 427.0617.
4.1.28. 2-(Benzo[d]isoxazol-3-yl)-N-(3,4,5-trifluorophenyl)-2-oxoacetohydrazonoyl cyanide (37)
Compound 37 was prepared in 25% yield (two steps from 28) by a procedure similar to that used to prepare compound 30. The title compound was obtained as a brown solid. 1H NMR (300 MHz, DMSO-d6) δ 8.02 (d, J = 7.7 Hz, 1H), 7.91 (d, J = 8.6 Hz, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.17 (dd, J = 9.1, 6.8 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 179.20, 170.81, 162.92, 155.28, 151.14 (ddd, J = 246.5, 10.5, 5.0 Hz), 140.66, 138.18, 135.11, 131.39, 125.58, 123.71, 120.64, 114.17, 111.60, 110.41, 102.22 (d, J = 24.6 Hz). HRMS (ESI) calcd for C16H8F3N4O2 345.0599 (M + H)+, found 345.0590.
4.2. In vitro guanine nucleotide exchange factor (GEF) activity assay of EPAC proteins
In vitro EPAC GEF activity was acquired as previously described [25]. Briefly, the assay was performed using 500 nM Rap1b-BODIPY-GDP and 200 nM EPAC proteins in buffer containing 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 50 mM GDP and the indicated concentrations of test compounds at room temperature using half-area 96-well plates (Corning Costar 3915). The exchange reaction was monitored using a Spectramax M2 Plate Reader (Molecular Devices) with the excitation/emission wavelengths set at 485/515 nm. The reaction rate constant (kobs) was determined by globally fitting the experimental data to a single exponential equation. Quantification was processed by nominalizing the observed kobs in the presence of inhibitor with the rate constant in the presence of 20 μM cAMP (no inhibitor) (kcAMP) and the rate constant without cAMP or inhibitor (k0) using equation: Relative GEF activity = (kobs – k0)/(kcAMP – k0) × 100.
4.3. Molecular docking studies
The docking study was performed with Schrödinger Small-Molecule Drug Discovery Suite [29]. The crystal structure of EPAC2 (PDB code: 3CF6) was downloaded from RCSB PDB Bank and prepared with Protein Prepared Wizard [30]. During this step, hydrogens were added, crystal waters were removed, and partial charges were assigned using the OPLS-2005 force field. The 3D structures of 1 (ESI-09), 10 (NY0617) and 33 (NY0562) were created with Schrödinger Maestro [31] and the initial lowest energy conformations were calculated with LigPrep [32]. For all dockings, the grid center was chosen on the centroid of included ligand of PDB structure CBD-B site and a 24 × 24 × 24 Å grid box size was used. All dockings were employed with Glide [33] using the XP protocol. Docking poses were incorporated into Schrödinger Maestro for a visualization of ligand-receptor interactions and overlay analysis.
Supplementary Material
Scheme 2.

Synthesis of 2-(benzo[d]isoxazol-3-yl)-2-oxo-N-phenylacetohydrazonoyl Cyanide Analogues 30–37. Reagents and conditions: (a) CH3CN, MeLi, THF, −78 °C; (b) 6a–h, NaOAc, EtOH, 22–57% for two steps.
Highlights.
Further structural modifications and SAR studies based on ESI-09 are presented.
Two series of novel diversified analogues have been designed and synthesized.
14, 32 and 33 identified as potent EPAC antagonists with low micromolar activities.
Molcular dockings on ligand-EPAC2 protein binding interactions are explored.
Benzo[d]isoxazol analogues offer new lead scaffolds for further optimization.
Acknowledgments
This work was supported by grants R01 GM106218, R01 AI111464, and R01 GM066170 from the National Institutes of Health. We want to thank Drs. Lawrence C. Sowers and Cheryl F. Lichti at the Department of Pharmacology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance.
Abbreviations
- EPAC
exchange proteins directly activated by cAMP
- SAR
structure-activity relationship
- cAMP
cyclic adenosine monophosphate
- 8-NBD-cAMP
8-(2-[7-nitro-4- benzofurazanyl]aminoethylthio)adenosine-3′,5′-cyclic monophosphate
- GDP
guanosine diphosphate
- PKA
protein kinase A
- GEF
guanine nucleotide exchange factor
- GTP
guanosine triphosphate
- Rap
Ras-related protein
- HTS
high-throughput screening
- TLC
thin layer chromatography
- UV
ultraviolet
- TMS
tetramethylsilane
- HRMS
high-resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- DCM
dichloromethane
- EtOAc
ethyl acetate
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- DDT
dichlorodiphenyltrichloroethane
- ADP
adenosine diphosphate
- CBD
cAMP binding domain
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.xxxx.xx.xxx.
Footnotes
Notes
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 2.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P. Protein kinases--the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 4.Zambon AC, Zhang L, Minovitsky S, Kanter JR, Prabhakar S, Salomonis N, Vranizan K, Dubchak I, Conklin BR, Insel PA. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc Natl Acad Sci U S A. 2005;102:8561–8566. doi: 10.1073/pnas.0503363102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biel M. Cyclic nucleotide-regulated cation channels. J Biol Chem. 2009;284:9017–9021. doi: 10.1074/jbc.R800075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biel M, Michalakis S. Cyclic nucleotide-gated channels. Handb Exp Pharmacol. 2009:111–136. doi: 10.1007/978-3-540-68964-5_7. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Wild C, Zhou X, Ye N, Cheng X, Zhou J. Recent advances in the discovery of small molecules targeting exchange proteins directly activated by cAMP (EPAC) J Med Chem. 2014;57:3651–3665. doi: 10.1021/jm401425e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Liu Z, Chen H, Ye N, Cheng X, Zhou J. Exchange proteins directly activated by cAMP (EPACs): Emerging therapeutic targets. Bioorg Med Chem Lett. 2017;27:1633–1639. doi: 10.1016/j.bmcl.2017.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. ChemBioChem. 2008;9:2052–2054. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 10.Tsalkova T, Mei FC, Cheng X. A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS One. 2012;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A. 2012;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83:122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong B, Shelite T, Mei FC, Ha T, Hu Y, Xu G, Chang Q, Wakamiya M, Ksiazek TG, Boor PJ, Bouyer DH, Popov VL, Chen J, Walker DH, Cheng X. Exchange protein directly activated by cAMP plays a critical role in bacterial invasion during fatal rickettsioses. Proc Natl Acad Sci U S A. 2013;110:19615–19620. doi: 10.1073/pnas.1314400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almahariq M, Mei FC, Wang H, Cao AT, Yao S, Soong L, Sun J, Cong Y, Chen J, Cheng X. Exchange protein directly activated by cAMP modulates regulatory T-cell-mediated immunosuppression. Biochem J. 2015;465:295–303. doi: 10.1042/BJ20140952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc’h F. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell Signalling. 2011;23:1257–1266. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 18.Almahariq M, Chao C, Mei FC, Hellmich MR, Partikeev I, Motamedi M, Cheng X. Pharmacological inhibition and genetic knockdown of EPAC1 reduce pancreatic cancer metastasis in vivo. Mol Pharmacol. 2015;87:142–149. doi: 10.1124/mol.114.095158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65:670–709. doi: 10.1124/pr.110.003707. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Ding C, Wild C, Liu H, Wang T, White MA, Cheng X, Zhou J. Efficient synthesis of ESI-09, a novel non-cyclic nucleotide EPAC antagonist. Tetrahedron Lett. 2013;54:1546–1549. doi: 10.1016/j.tetlet.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Tsalkova T, Chepurny OG, Mei FC, Holz GG, Cheng X, Zhou J. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J Med Chem. 2013;56:952–962. doi: 10.1021/jm3014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Tsalkova T, Mei FC, Hu Y, Cheng X, Zhou J. 5-Cyano-6-oxo-1, 6-dihydro-pyrimidines as potent antagonists targeting exchange proteins directly activated by cAMP. Bioorg Med Chem Lett. 2012;22:4038–4043. doi: 10.1016/j.bmcl.2012.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wild CT, Zhu Y, Na Y, Mei F, Ynalvez MA, Chen H, Cheng X, Zhou J. Functionalized N, N-diphenylamines as potent and selective EPAC2 inhibitors. ACS Med Chem Lett. 2016;7:460–464. doi: 10.1021/acsmedchemlett.5b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye N, Zhu Y, Chen H, Liu Z, Mei FC, Wild C, Chen H, Cheng X, Zhou J. Structure-activity relationship studies of substituted 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide analogues: identification of potent exchange proteins directly activated by cAMP (EPAC) antagonists. J Med Chem. 2015;58:6033–6047. doi: 10.1021/acs.jmedchem.5b00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye N, Chen CH, Chen T, Song Z, He JX, Huan XJ, Song SS, Liu Q, Chen Y, Ding J, Xu Y, Miao ZH, Zhang A. Design, synthesis, and biological evaluation of a series of benzo[de][1, 7]naphthyridin-7(8H)-ones bearing a functionalized longer chain appendage as novel PARP1 inhibitors. J Med Chem. 2013;56:2885–2903. doi: 10.1021/jm301825t. [DOI] [PubMed] [Google Scholar]
- 27.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- 28.Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455:124–127. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 29.Small-Molecule Drug Discovery Suite 2016–4. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 30.Schrödinger Release 2016–4: Schrödinger Suite 2016–4 Protein Preparation Wizard. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 31.Schrödinger Release 2016–4: Maestro. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 32.Schrödinger Release 2016–4: LigPrep. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 33.Schrödinger Release 2016–4: Glide. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.