

Abstract
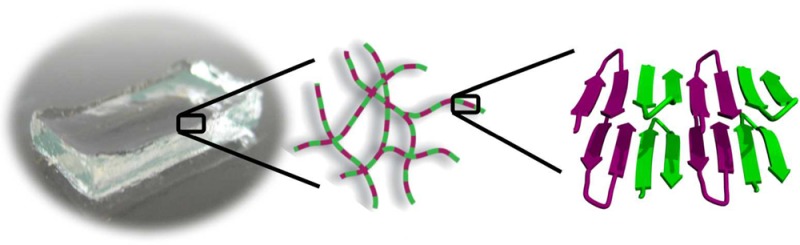
Hydrogels prepared from self-assembling peptides are promising materials for medical applications, and using both l- and d-peptide isomers in a gel’s formulation provides an intuitive way to control the proteolytic degradation of an implanted material. In the course of developing gels for delivery applications, we discovered that a racemic mixture of the mirror-image β-hairpin peptides, named MAX1 and DMAX1, provides a fibrillar hydrogel that is four times more rigid than gels formed by either peptide alone—a puzzling observation. Herein, we use transmission electron microscopy, small angle neutron scattering, solid state NMR, diffusing wave, infrared, and fluorescence spectroscopies, and modeling to determine the molecular basis for the increased mechanical rigidity of the racemic gel. We find that enantiomeric peptides coassemble in an alternating fashion along the fibril long axis, forming an extended heterochiral pleat-like β-sheet, a structure predicted by Pauling and Corey in 1953. Hydrogen bonding between enantiomers within the sheet dictates the placement of hydrophobic valine side chains in the fibrils’ dry interior in a manner that allows the formation of nested hydrophobic interactions between enantiomers, interactions not accessible within enantiomerically pure fibrils. Importantly, this unique molecular arrangement of valine side chains maximizes inter-residue contacts within the core of the fibrils resulting in their local stiffening, which in turn, gives rise to the significant increase in bulk mechanical rigidity observed for the racemic hydrogel.
Short abstract
An arsenal of microscopy, scattering, and spectroscopy reveals the molecular basis of enhanced mechanical rigidity of racemic peptide hydrogels. Peptides coassemble into a supersecondary structure predicted by Pauling and Corey in 1953.
Introduction
Self-assembling peptides can form a broad array of supramolecular structures including sheets,1−5 disks,6 spheres,7 barrels,8 tubes,9−12 and fibrils.13−22 Fibrils are privileged structures capable of higher order assembly leading to the formation of networks that constitute the formation of macroscopic hydrogels. The mechanical rigidities,23−25 enzymatic degradability,19,26,27 antibacterial activity,28−30 and cellular compatibility31−34 of peptide gels can be finely tuned through simple chemical and material-based strategies to enable targeted applications. For example, we recently reported the design of a UV-sensitive peptide gel that enables microanastomosis, the suturing of ultrasmall blood vessels.35
Chirality is now being employed as a design modality in the fabrication of hydrogels.36−39 Heterochiral systems, containing both l- and d-isomers, offer unique material morphologies40−43 as well as a control over hydrogel degradability44 as peptides composed of d-amino acids are resistant to proteolysis.45,46 As such, we initially sought to control the biodegradation of hydrogels formed by the peptide hydrogelator, MAX1, by preparing mixed gels of the parent peptide with its enantiomer. In the course of analyzing the rheological properties of the mixed hydrogels, a large, nonadditive, synergistic enhancement in material rigidity was unexpectedly observed compared to gels formed from enantiomerically pure peptides with the greatest increase found in the racemic hydrogel.47
MAX1 is a 20-amino acid peptide designed to undergo triggered folding and self-assembly into β-sheet rich fibrils that ultimately lead to the formation of a self-supporting hydrogel.24,48 The peptide’s primary sequence contains two segments of alternating hydrophobic (valine) and hydrophilic (lysine) residues that flank a tetrapeptide motif designed to adopt a reverse turn in the folded state, Table 1. In neutral pH and low ionic strength buffers, the peptide adopts an ensemble of random coil conformations due to the electrostatic repulsion between protonated lysine side chains affording a solution of monomeric peptide. Increasing the ionic strength of the solution by the addition of NaCl screens the lysine borne charges and allows the peptide to fold and self-assemble into fibrils.24 Increasing the temperature also favors folding and assembly by driving the hydrophobic effect and the burial of valine side chains into the dry interior of the fibrils.21 Solid state NMR shows that MAX1, in its folded and assembled state, adopts an amphiphilic β-hairpin structure that contains a type II′ β-turn as designed, and that the peptide assembles with high fidelity into monomorphic fibrils composed of a bilayered cross-β structure. Folded hairpins assemble in-register with a Syn arrangement of their β-turns aligned along a given fibril monolayer. However, across the bilayer, hairpins adopt an Anti orientation.49 The resulting network formed by these fibrils constitutes a mechanically rigid hydrogel.50,51
Table 1. Sequences of Peptides Utilized in These Studiesa.
| peptide | sequence |
|---|---|
| MAX1 | VKVKVKVKVDPLPTKVKVKVKV-NH2 |
| DMAX1 | VKVKVKVKVLPDPTKVKVKVKV-NH2 |
| MAX1-Azide | VKVKVKVKVDPLPTKVKVKVKVK(azide)-NH2 |
| DMAX1-Biotin | Biotin-KVKVKVKVKVLPDPTKVKVKVKV-NH2 |
| MAX1-EDANS | VKVKVE(Edans)VKVDPLPTKVKVKVKVK-NH2 |
| DMAX1-Dabcyl | VKVKVK(Dabcyl)VKVLPDPTKVKVKVKV-NH2 |
| MAX1-IR1 | VKVKVKVKVDPLPTKVKVKVKV-NH2 |
| MAX1-IR2 | VKVKVKVKVDPLPTKVKVKVKV-NH2 |
| DMAX1-NMR1 | VKVKVKVKVLPDPTKVKVKVKV-NH2 |
| MAX1-NMR1 | VKVKVKVKVDPLPTKVKVKVKV-NH2 |
| MAX1-NMR2 | VKVKVKVKVDPLPTKVKVKVKV-NH2 |
| MAX35 | IKVKIKVKVDPLPTKIKVKIKV-NH2 |
| DMAX35 | IKVKIKVKVLPDPTKIKVKIKV-NH2 |
| MARK4 | VRVKVRVKVDPLPTRVKVRVKV-NH2 |
| DMARK4 | VRVKVRVKVLPDPTRVKVRVKV-NH2 |
d-Amino acid residues are italicized. Uniformly 13C and 15N isotopically labeled residues are bold and underlined. 13C=18O isotopically labeled amino acid residues are in bold.
As stated earlier, when MAX1 is mixed with its enantiomer, DMAX1, a gel is formed that is significantly more rigid. When we initially reported this behavior the physical basis for this enhancement in material rigidity was unknown.47 According to theory for semiflexible biopolymer networks, hydrogel mechanical rigidity is dependent on both the number of cross-links made between fibrils in the network and the stiffness of the fibrils themselves.52−54 In the context of MAX1 and DMAX1, we envisioned that mixtures of these enantiomers could either self-sort to form an interpenetrating network of enantiomerically pure fibrils or coassemble into mixed fibrils, Figure 1. It had been unclear which scenario was more likely to occur and how either outcome would lead to an increase in network cross-link density, fibril stiffness, or both. Further, the molecular basis guiding the assembly process for these mirror-image peptides was not known. Herein, we use an arsenal of microscopy, scattering, spectroscopic techniques, rational peptide design, and modeling to uncover the physical basis for this material enhancement at the molecular, local-fibril, and hydrogel network length scales.
Figure 1.
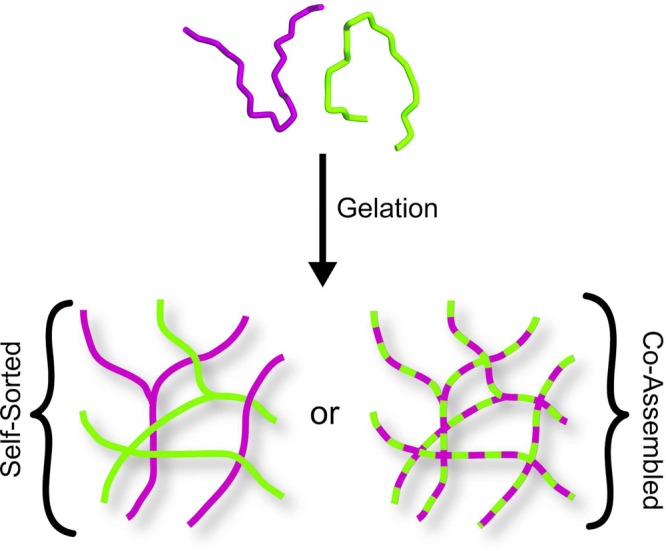
Possible modes of self-assembly for enantiomeric peptides MAX1 (green) and DMAX1 (purple) include self-sorting (solid-colored fibrils) or coassembly (dual-colored fibrils) into mixed fibrils.
Results and Discussion
Rheological, Microscopic, and Spectroscopic Evidence of Enantiomer Co-Assembly into Mixed Fibrils
Frequency-sweep rheology experiments measuring the storage modulus (G′, a measure of a gel’s mechanical rigidity) show that an equimolar mixture of MAX1 and DMAX1 self-assembles to form a hydrogel that is four times more stiff than gels formed by either pure enantiomer at identical peptide concentrations, Figure 2A. We previously reported that a racemic mixture of these peptides undergoes triggered folding to adopt β-hairpins that self-assemble into β-sheet rich fibrils where each fibril is comprised of a bilayer of hairpins.47 Thus, the local fibril morphology in the racemic network was identical to that of both pure MAX1 and DMAX1 fibrils, with measured fibril heights of ∼2.5 nm and widths of ∼3 nm. Although the local morphology of the fibrils comprising the racemic network was known, the composition and arrangement of enantiomers within those fibrils was a mystery. Interestingly, both assemblies (self-sorted or coassembled) illustrated in Figure 1 would lead to hydrogel networks composed of fibrils having identical local dimensions.
Figure 2.

Dynamic frequency-sweep oscillatory rheology measuring the storage moduli of 1 wt % hydrogels as a function of frequency. A 4-fold increase in storage modulus is achieved in the racemic hydrogel (■) with respect to either pure MAX1 (◇) or DMAX1 (●) gels (N = 3) (A). Time-sweeps of two racemic gels. The first prepared by allowing independent solutions of MAX1 and DMAX1 to self-assembly independently at 25 °C for 10 min, then mixed and further gelled at 37 °C (×). The second gel prepared by premixing solutions of monomeric unfolded MAX1 and DMAX1 and then triggering gelation at 37 °C (▲) (N = 3) (B).
Rheology was used to probe the nature of the racemic assembly process. We had previously determined that the rate of peptide folding, self-assembly, and thus fibril formation leading to gelation could be controlled by temperature.55 The higher the temperature, the faster the rate of gelation. Figure 2B shows experiments where temperature and self-assembly conditions were controlled to mimic the formation of a self-sorted interpenetrating network of fibrils to assess if this type of network would lead to an enhancement in material rigidity. Here, individual peptide solutions in separate containers were triggered to assemble at 25 °C. At this temperature, each peptide can slowly fold and assemble to form local regions of soluble fibrils in solution but not a fully percolated network. The two fibrillar solutions were then mixed in equal volumes, and the temperature was raised to 37 °C where the evolution of an interpenetrating network could be formed. The time-sweep measurement shown in panel B demonstrates that the gel resulting from this network realizes a storage modulus that is not significantly greater than gels prepared from pure enantiomers alone (G′ ≈ 300 Pa). A control experiment was performed in which an equimolar mixture of the enantiomers was initially prepared in the same container at 5 °C, where assembly is prohibited. The temperature of the mixed solution was then increased to 37 °C to trigger folding and self-assembly. Under these conditions, coassembly is possible. Data in panel B show that the gel resulting from these conditions displays the 4-fold increase in material rigidity (G′ ≈ 750 Pa). These data suggest that the enantiomers do not self-sort, but rather coassemble to form a network of mixed fibrils. Figure S1 shows CD data defining the temperature-dependent folding and assembly process.
We further investigated possible enantiomer coassembly by TEM. Here, two enantiomeric derivatives, namely, MAX1-Azide and DMAX1-Biotin, were designed to bind 10 and 5 nm gold nanoparticles (GNP) respectively while in the fibrillar state. Their sequences are shown in Table 1 along with the other peptides employed in this study. Peptides were prepared by standard Fmoc solid phase peptide synthesis and purified to homogeneity (Figures S2–S16). MAX1-Azide contains an azide moiety that can react with a dibenzylcyclooctyne (DBCO)-conjugated 10 nm GNP. DMAX1-Biotin is designed to bind to streptavidin-conjugated 5 nm GNPs. In the experiment depicted in Figure 3A, a fibrillar hydrogel was prepared from an equimolar mixture of these two peptides, diluted and deposited onto a TEM grid, and incubated with sequential additions of each gold nanoparticle. Should these peptides coassemble to form mixed fibrils, each individual fibril should be adorned by both 5 and 10 nm GNPs. Conversely, if the peptides self-sort into independent fibrils, distinct populations of fibrils decorated with only 5 nm or only 10 nm GNPs will exist. The micrograph shows fibrils that are decorated with both 5 and 10 nm nanoparticles. The inset shows a statistical evaluation of binding where it is clear that the frequency of fibrils observed bound by both nanoparticles is much greater than fibrils bound by a single-sized GNP, supporting a coassembly mechanism. Although the two enantiomeric derivatives are not strictly non-superimposible mirror images, the gel formed by an equimolar mixture of these derivatives also displays the 4-fold increase in mechanical rigidity (Figure S17). Additionally, TEM control experiments were performed on fibrils composed of either pure MAX1-Azide or, separately, pure DMAX1-Biotin which showed that these homogeneous fibrils preferentially bound 10 or 5 nm GNPs respectively (Figure S18).
Figure 3.
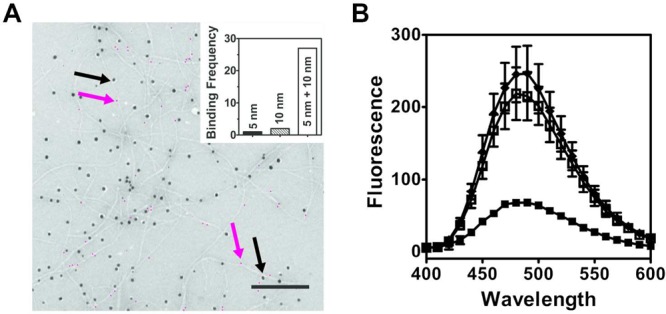
Coassembly of enantiomers MAX1 and DMAX1 is accessed by nanoparticle labeling of peptide fibrils using TEM (A) and fluorescence quenching (B). (A) DMAX1-Biotin and MAX1-Azide peptides were visualized within individual fibrils by the binding of 5 or 10 nm gold nanoparticles, respectively (scale bar = 200 nm). Five nanometer particles are false colored magenta for clarity. Magenta and black arrows indicate examples of bound 5 and 10 nm GNPs, respectively. The original noncolored micrograph is provided as Figure S18E. Inset: frequency of particles binding to distinct individual fibrils (N = 30). (B) Fluorescence spectra of MAX1-EDANS fibrils (◆), MAX1-EDANS fibrils and DMAX1-Dabcyl fibrils formed separately, then mixed (□) and a premixed solution of 1:1 MAX1-EDANS:DMAX1-Dabcyl that was allowed to assemble together (■) (N = 3).
The coassembly of the peptide enantiomers within racemic fibrils was further confirmed by fluorescence spectroscopy, Figure 3B. Here, derivatives of MAX1 and DMAX1, namely, MAX1-EDANS and DMAX1-Dabcyl respectively (Table 1), were prepared to obtain spectroscopic evidence of the peptide coassembly via fluorescence quenching. EDANS and Dabcyl are often exploited as a fluorophore/quencher pair in biological assays56−58 and have a distance-limited Förster’s radius of ∼3 nm. If these peptide derivatives coassemble, solutions containing the mixed fibrils should display diminished fluorescence compared to fibrils formed by pure MAX1-EDANS. As in the peptide pair used in the TEM experiments discussed above, the hydrogel produced from a 1:1 mixture of MAX1-EDANS and DMAX1-Dabcyl retains the 4-fold increase in storage modulus with respect to either MAX1-EDANS or DMAX1-Dabcyl hydrogels (Figure S19).
Solutions of pure MAX1-EDANS fibrils are highly fluorescent as shown in Figure 3B with an emission maximum centered at 494 nm. When a gel is formed from an equimolar mixtures of MAX1-EDANS and DMAX1-Dabcyl and diluted, the fluorescence of the resulting solution is highly attenuated, again suggesting that the peptides have coassembled into a given fibril where the enantiomeric pairs are in close proximity to each other. If the peptides were to self-sort, one would expect a solution of these fibrils to be highly fluorescent because the distance between the fluorophore and quenchers is greater than 3 nm. A control experiment was performed where separate gels of MAX1-EDANS and DMAX1-Dabcyl were prepared and mixed upon dilution mimicking the formation of a self-sorted fibrillar network. Figure 3B shows that the resulting solution is nearly as fluorescent as MAX1-EDANS fibrils alone. Taken together, the rheology, TEM, and fluorescence spectroscopy data show that the parent peptides MAX1 and DMAX1 coassemble into mixed fibrils where enantiomers are in close proximity within the solid state.
Why Do Co-Assembled Fibrils Afford a Stiffer Hydrogel Network?
Theory for semiflexible polymers52−54 predicts that the rigidity of a densely cross-linked gel is dependent on both the mesh size, ξ, and fibril bending modulus, κ, as in eq 1.
| 1 |
Thus, it is likely that changes in the mesh size and/or fibril bending modulus result in the enhancement in rigidity that is observed in the racemic hydrogel. Mesh size is directly related to the cross-link density: more cross-links lead to a smaller mesh size and a more rigid gel. With respect to the fibril bending modulus, if the fibrils comprising the network are themselves more stiff, the resulting gel will be more rigid.
Small angle neutron scattering (SANS) interrogates the hydrogel network structure at length scales relevant to the mesh size of a 1 wt % hydrogel, which was estimated to be in the tens of nanometers.50 This corresponds to a regime centered at q ≈ 0.01 Å–1 in the SANS experiment. If the racemic gel contains more cross-links and a smaller mesh, then qualitatively, one should expect the scattering intensity to be greater than the pure enantiomeric gels in this regime. Figure 4 shows SANS data for MAX1, DMAX1, and racemic hydrogels; panel D clearly shows that all the scattering profiles are superimposable, suggesting that the mesh sizes for these three gels are very similar. Further, each scattering profile (Figure 4A–C) was fit to a fractal model, where the scattering intensity I(q) = P(q)S(q), to determine the mesh size of the hydrogels.51 Here, the form factor, P(q), is calculated for spherical particles
| 2 |
where R0 is the radius of the spherical particles with volume, VP= 4/3πR03 and
| 3 |
Figure 4.

SANS scattering for 1 wt % hydrogels of pure MAX1, (◇) (A); pure DMAX1, (○) (B); and a mixture of 1:1 MAX1/DMAX1, (□) (C). Each scattering profile is an average of three independent experiments on separate gels performed on separate days. Error bars are smaller than the symbol where not evident. Colored lines represent fractal model fits for the individual hydrogels. (D) Scattering profiles of MAX1 (green), DMAX1 (purple), and racemic (blue) hydrogels are coplotted to emphasize that the enantiomeric assemblies have similar mesh sizes.
In this model, the peptide fibrils within the hydrogel network are approximated as an assembly of spherical building blocks that further associate to form fractal-like clusters with a defined average size, namely, the correlation length, λ, and self-similarity dimension, Df. These clusters are characterized by the structure factor
| 4 |
Fitting these data for MAX1 and DMAX1 hydrogels results in a fractal dimension of 1.33 and correlation length of 125 nm for both gels. The racemic gel has a very similar fractal dimension and correlation length of 1.36 and 118 nm. Thus, the SANS data show that the observed increase in mechanical rigidity enjoyed by the racemic gel is not a result of a more cross-linked network.
On the basis of eq 1 above, the alternative to a decrease in mesh size to bring about an increase in gel rigidity is an increase in the bending modulus of individual fibrils comprising the racemic gel. However, the small widths (∼3 nm) of the racemic fibrils make it incredibly difficult to measure their stiffness by established methods such as atomic force microscopy (AFM).59,60 Thus, we turned our attention to diffusing wave spectroscopy (DWS).61−63 DWS offers a noninvasive method to access the high frequency linear rheology of biomaterials, beyond the capabilities of conventional mechanical rheometers and particle tracking microrheology methods, enabling precise characterization of their mechanical properties.64 This technique has previously been applied to colloidal gels,65,66 emulsions,65 worm-like micelles,67−69 and F-actin solutions.70,71 Here, we use DWS for the first time to measure the stiffness of peptide fibrils, namely, those formed by pure MAX1, DMAX1, and their corresponding racemate.
In DWS experiments, the fibril stiffness is extracted by monitoring the mean-squared displacement, MSD ⟨Δr2(t)⟩ of polystyrene microspheres embedded within the hydrogels at high frequencies, Figure 5A. The MSD was fit to a stretched exponential of the form ⟨Δr2(τ)⟩ = δ2(1 – e(−τ/τc)a).66 Here, a is the short-time logarithmic slope, whose scaling (0.75) is characteristic of semiflexible polymer networks. τc is the characteristic relaxation time of the particle in the gel, and δ2 (defined by the plateau region) corresponds to the maximal particle displacement in the network (Table 2). The relaxation time for particles in the racemic gel is faster (62 μs) than that in either of the enantiomeric gels (∼118 μs), and the maximal displacement is correspondingly smaller for the racemate. This indicates that particle movement is significantly more restricted in the racemic gel. In panel B, MSD data are converted to the storage (G′) and loss (G′’, a measure of the material’s viscous response to stress) moduli numerically through calculation of the complex modulus, G*(ω), using the approximate form of the Generalized Stokes–Einstein Relation given in eqs S5–S7 in the Materials and Methods section. The 4-fold enhancement in material rigidity for the racemic hydrogel is observed in the low frequency plateau regime, which is in agreement with the bulk rheology measurements.
Figure 5.

Diffusing wave spectroscopy monitoring the mean squared displacement, ⟨Δr2(t)⟩, of particles embedded in 1 wt % MAX1 (◇), DMAX1 (●), and 1:1 MAX1/DMAX1 (□) hydrogels (N = 3) (A) (B) Corresponding storage, G′ (closed symbols) and loss, G″ (open symbols) moduli of 1 wt % MAX1 (green), DMAX1 (purple) and 1:1 MAX1/DMAX1 hydrogels (blue) obtained from DWS measurements. Nonlinear fit with logarithmic slope of 3/4 of early time regime in (A) and high frequency regime in (B) is indicated by colored lines. Loss moduli, G″, at high frequencies are fit to calculate persistence length. In panel C, the persistence length and G′ are coplotted against the mole fraction of DMAX1 used in the gel formulation (N = 3). Line is included to guide the eye.
Table 2. Fibril Parameters Derived from Diffusing Wave Spectroscopy.
| gel | τc (μs)a | δ2 (× 10–6 μm2)a | d (nm)b | L (nm)c | ρ (×104 μm–2)d | lp (nm)a | κ(pN·nm2)a |
|---|---|---|---|---|---|---|---|
| MAX1 | 118 (5) | 5.45 (0.70) | 3 | 125 | 2.36 | 7.5 (1.1) | 30.8 (4.5) |
| DMAX1 | 119 (18) | 5.66 (1.27) | 3 | 125 | 2.36 | 7.4 (0.8) | 30.4 (3.3) |
| racemate | 62 (14) | 2.21 (0.37) | 3 | 118 | 2.23 | 12.6 (0.8) | 51.8 (3.3) |
Measured from DWS; errors in parentheses.
Measured from TEM.
Determined by SANS.
Calculated from polymer concentration.
Importantly, at lag times <100 μs, corresponding to the high frequency regime in panel B, the probe particles are displaced on the order of 1 nm and their response is dictated by the single filament mechanics of the peptide network. This regime is used to determine the persistence length and bending modulus of the fibrils that comprise the hydrogel networks. Here, the theory for high frequency rheology of semiflexible polymers is invoked, and data are fit to eq 5 below:52,72,73
| 5 |
where ρ is the fibril length per unit volume, lp = κ/kT is the fibril persistence length, and ζ ≈ 4πη/ln(0.6L/d) is the lateral drag coefficient per unit length for filaments of length L and diameter d. Previous work (see Table 2) has determined all relevant parameters in eq 5 except the persistence length for MAX1, DMAX1, and racemic assemblies.47,50,51 This allows the loss modulus to be fit in the high frequency regime with the persistence length as the only adjustable parameter. All peptide samples exhibit the characteristic ω3/4 scaling as predicted by semiflexible polymer theory. Resulting values for fibril persistence length are shown in Table 2.
As expected, average persistence length (lp) values for fibrils formed from MAX1 and DMAX1 are nearly identical, 7.5 ± 1.1 nm and 7.4 ± 0.8 nm, respectively. However, the average persistence length of fibrils formed within the racemic network is nearly twice as long (12.6 ± 0.8 nm). The corresponding bending moduli for fibrils within each hydrogel can be calculated from κ = lpkT (Table 2), and it is clear that the fibrils in the racemic gel are significantly more stiff (51 vs 31 pN nm2). Further, the Job plot in Figure 5C nicely shows that the persistence length of the fibrils and the corresponding rigidity of their respective gels measured by bulk rheology track with the mole fraction of enantiomer used in the gel formulation. Taken together, the SANS and DWS show that the racemic hydrogel is mechanically more rigid because the fibrils that comprise the gel are, themselves, more rigid.
Molecular Arrangement of Enantiomers within Racemic Fibrils
It is likely that unique molecular-level interactions, made between MAX1 and DMAX1 in the assembled state, are responsible for the enhanced stiffness of the racemic fibrils. Three arrangements can be envisioned for the coassembly of MAX1 and DMAX1, Figure 6. In the first arrangement (panel A), each enantiomer self-segregates into its respective monolayer. Here, MAX1 and DMAX1 make facial contact across the bilayer. However, within each monolayer, each enantiomer only makes lateral contact with itself as it traverses down the long axis of the fibril. In panel B, each enantiomer pairs with itself across the bilayer, but alternates down the long axis of the fibril. In the last arrangement (panel C), enantiomers pair across the bilayer and alternate down the fibril long axis.
Figure 6.
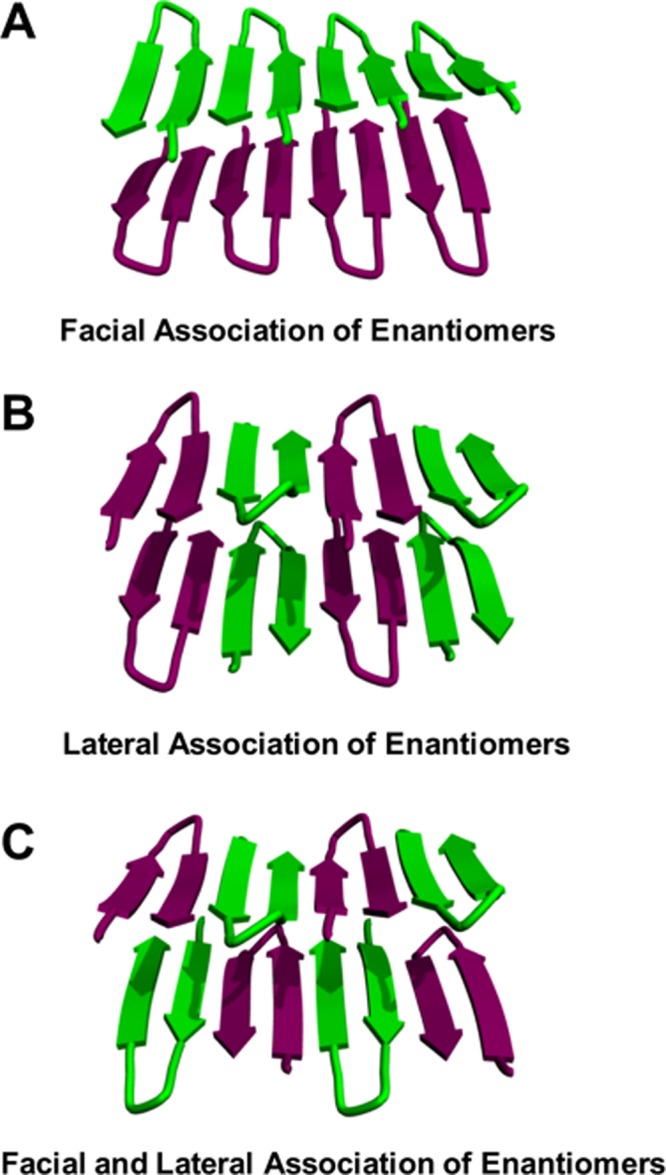
Possible arrangements of enantiomeric coassembly within racemic fibrils. (A) MAX1 and DMAX1 self-segregate into distinct monolayers. (B) Each enantiomer pairs with itself across the bilayer, but alternates down the long axis of the fibril. (C) MAX1 and DMAX1 pair across the bilayer and laterally within each monolayer.
The possibility of enantiomers assembling as depicted in Figure 6A was investigated using isotope-edited Fourier transform infrared spectroscopy (IE-FTIR). This technique has been utilized to monitor the assembly and alignment of β-sheet peptides.41,74−78 The fibrils that constitute the MAX1 and DMAX1 hydrogel networks exhibit a characteristic strong amide I stretch at ∼1615 cm–1 due to their large β-sheet secondary structure content.79 Spectra also included a weaker band centered at 1680 cm–1. This band can be assigned either to the intramolecular arrangement of antiparallel β-strands within each hairpin of the assembly or the β-turn secondary structure also contained with each hairpin.80 In the experiment presented in Figure 7A, 13C/18O-labeled amino acids were incorporated into MAX1 at Lys6 and Val14 (namely, MAX1-IR1, Table 1). A control gel prepared from pure MAX1-IR1 exhibits a red-shifted vibrational frequency stretch at 1571 cm–1 due to the heavy-atom labeled carbonyl groups81 (Figure 7A). The large magnitude of this frequency shift is indicative of the extended vibrational coupling between labeled amides that traverse uninterrupted along the long axis of the fibrils constituting the gel. Conceptually, if one prepares a racemic gel comprising equimolar mixtures of MAX1-IR1 and DMAX1 and the enantiomers self-segregate into monolayers, then a similar shift should be observed.
Figure 7.

Isotope edited FTIR spectra of MAX1-IR1 (A), MAX1-IR2 (B), and 1:1 MAX1-IR1/DMAX1 (C) hydrogels. Dotted line indicates the coupled stretch at 1571 cm–1 for reference.
A second control gel was prepared using MAX1-IR2 (Table 1). This peptide incorporates only one 13C=18O group at Val7. As such, a gel prepared from this peptide alone will consist of fibrils in which only alternating β-strands contain the label, and thus the extended coupling is lost. This results in a stretch at 1582 cm–1, blue-shifted relative to the pure MAX1-IR1 gel, Figure 7B. Again, conceptually, if one prepares a racemic gel comprised of MAX1-IR1 and DMAX1 and the peptides now alternate within each of the monolayers of a racemic fibril, the extended coupling would be lost. This would result in a blue-shift relative to that observed for a pure MAX1-IR1 gel.
Figure 7C shows the IE-FTIR spectrum of a hydrogel resulting from the equimolar mixture of MAX1-IR1 with unlabeled DMAX1. The lower vibrational frequency stretch at 1579 cm–1 is blue-shifted relative to that in pure MAX1-IR1 (dotted line), indicating that extended vibrational coupling does not occur. On the basis of these results, the arrangement depicted in Figure 6A is unlikely. Rather, MAX1 and DMAX1 enantiomers coassemble within each monolayer that defines the racemic fibril, as shown in Figure 6B or C. These two arrangements differ in the formation of facial associations made between the enantiomers across the bilayer. In panel B, homochiral facial contacts are made, whereas in panel C, MAX1 and DMAX1 make heterochiral facial contact. A further complication exists in that enantiomers within a given fibril monolayer can adopt a Syn orientation, where β-hairpin turns align along the monolayer, or an Anti arrangement, where turns alternate down the long axis of the fibril as shown in panels B and C.
To examine the arrangement of enantiomers within the fibril monolayer as well as across the fibril bilayer, solid state nuclear magnetic resonance (ssNMR) was employed. We recently determined the molecular structure of pure MAX1 in its fibrillar state (PDB file 2N1E) through a series of REDOR, PITHIRDS-CT, and two-dimensional (2D) 13C–13C radio frequency-assisted diffusion (RAD) measurements.49 MAX1 adopts a Syn/Anti arrangement where the β-turns of each hairpin align Syn along a given monolayer. Across the bilayer, hairpins are arranged Anti where β-turns occupy opposite fibril edges, much like the arrangement depicted in Figure 6A if all the peptides were MAX1. With the structural knowledge of pure MAX1 fibrils in hand, isotopically labeled peptides were designed to identify relative enantiomer orientation within the racemic fibril monolayer and across the bilayer. Peptides DMAX1-NMR1 and MAX1-NMR1 (Table 1, Figure 8) were designed with 13C-labeled DP11 and V1 respectively to allow for the determination of enantiomeric orientation within the fibril monolayer using PITHIRDS-CT measurements. MAX1-NMR2 was designed with uniformly labeled P11, T12, and V20 residues to enable the identification of bilayer partners through the absence or presence of through-space cross-peaks between isotopically labeled residues in 2D RAD measurements.
Figure 8.

ssNMR of racemic fibrils. (A, B) PITHIRDS-CT experiments indicate an Anti arrangement of enantiomers within each fibril monolayer employing isotopically labeled DMAX1-NMR1 and MAX1-NMR1 peptides. Color-coded lines in panel B are simulations of PITHIRDS-CT data for 13C spin pairs with the indicated distances. (C, D) Expected couplings between Val20 and Thr12 (shorter interaction at left) and between Val20 and Pro11 (longer interaction at right) if peptides are paired across the bilayer in a homochiral fashion. Spectra and arrows are defined in the main text.
To test if enantiomers adopt an Anti or Syn orientation within the racemic monolayer, a gel comprising a 1:1 mixture of MAX1-NMR1/DMAX1-NMR1 was prepared. PITHIRDS-CT measurements of this sample reveal signal decays consistent with a close proximity between isotopically labeled DP11 and V1, Figure 8B. This experiment affords a measured distance of approximately 6.5 Å between the labeled proline and valine, which can only be achieved if the enantiomers are oriented in an Anti arrangement where β-turns alternate along the fibril long-axis, Figure 8A.
Next, the relative enantiomer pairing across the bilayer was examined by preparing a gel comprising an equimolar mixture of labeled MAX1-NMR2 and unlabeled DMAX, Table 1. If MAX1-NMR2 makes homochiral interfacial contact as depicted in Figures 6B and 8C, intermolecular coupling between two MAX1-NMR2 hairpins should be observed across the bilayer. The absence of interfacial-residue coupling would argue against this arrangement and argue for heterochiral pairing as depicted in Figure 6C, where the distances between spin labels are too long (>8 Å) to observe intermolecular coupling. Figure 8D shows one-dimensional (1D) slices at the Cα (green) and Cβ (blue) chemical shifts of labeled valine 20 (V20) derived from 2D RAD 13C–13C spectra of fibrils comprised of a 1:1 MAX1-NMR2/DMAX mixture. Careful inspection of the 2D spectrum with a 500 ms mixing period suggests the possibility of weak Val20-Thr12 and Val20-Pro11 cross-peaks, which are not observed in the 2D RAD spectrum with a 25 ms mixing period (Figure S20). Although these observations support the arrangement depicted in Figure 6B, we cannot definitively rule out heterochiral pairing as shown in panel C. This is because the Val20 signals in the spectra are not fully resolved from Pro11 and Thr12 signals, making it difficult to clearly distinguish these weak intermolecular cross-peaks from stronger intraresidue Pro11-Thr12 cross-peaks. At any rate, the IR and NMR studies support the formation of racemic fibrils comprised of an Anti arrangement of alternating enantiomers in each monolayer, and the possible homochiral pairing of enantiomers across the bilayer as depicted in Figure 6B.
Molecular Basis for Enhanced Fibril Stiffness: Predictions from Pauling and Corey
Thus far, the data indicate that the racemic gel of MAX1 and DMAX1 is more mechanical rigid because the individual fibrils that comprise the gel are themselves more rigid. Further, these fibrils contain both enantiomers arranged in a unique alternating molecular orientation within each monolayer of a fibril’s bilayered β-sheet rich structure. The question remains as to why this molecular arrangement leads to the formation of fibrils that are more stiff than fibrils comprised of either enantiomer alone. Using the structural insights gained from the spectroscopic data, an energy-minimized model of the racemic assembly was constructed and compared to a model of an assembly formed by pure MAX1 based on ssNMR.49
Shown in the top panel of Figure 9A is a dimer formed by two enantiomers within a monolayer of the racemic fibrillar assembly. Although each individual hairpin contains two intramolecular β-strands arranged in an antiparallel fashion, the β-sheet interface between the two hairpins comprises β-strands aligned in a parallel manner. Thus, MAX1 and DMAX1 form an extended network of intermolecular parallel β-strands that traverse the long axis of a given fibril. In contrast, hairpins within enantiomerically pure MAX1 fibrils are defined by an extended array of anti-parallel hydrogen bonded β-strands as shown in Figure 9B. Both the racemic and pure enantiomeric fibrils maximize the hydrogen-bonding potential of every strand in their respective systems. Although the exact geometry of the H-bonds is different in each assembly (bent versus linear), the number of bonds formed is identical. Thus, it is unlikely that enthalpic contributions from H-bonding are responsible for the large enhancement in fibril stiffness for the racemate. The central image in panel A shows a tetrameric portion of a fibril monolayer. The formation of bent H-bonds between enantiomeric strands is evident and is responsible for defining the alignment of strands within the β-sheet assembly. Importantly, these bent H-bonds not only define strand alignment but also dictate the exact placement of the hydrophobic valine side chains into the dry interior core of the fibril. As shown in the CPK rendering, valine side chains form intimate nested hydrophobic interactions where the entire isopropyl group of one residue nestles between the isopropyl groups of its valine neighbors. This facilitates tight packing and maximizes side-side/side-chain van der Waals contacts. The bottom panel in Figure 9A shows the entire fibril viewed axially down its long axis showing the interior of the bilayer. The staggered arrangement of the nested valines is clearly evident with the side chains of each successive enantiomer translated relative to its mirror image neighbor. These nested interactions are not realized in enantiomerically pure fibrils, Figure 9B (center and bottom images). Here, the antiparallel arrangement of β-strands, with its linear H-bonds, dictates that the valine side chains are arranged in a head-to-head manner within the core of the fibril. Although packing can occur between the valines where one Cγ atom of a valine side chain can penetrate the fork of a neighboring isopropyl group, this interaction does not pack the interior of the fibril as efficient as does nesting of an entire isopropyl side chain. This is especially apparent when comparing the axial views of the fibril interiors, Figure 9 (bottom panels). Interestingly, nested hydrophobic interactions between β-branched residues in sheet-rich proteins frequently occur, as first noted by Curmi at el.82 As in naturally occurring proteins where effective packing of side chains stabilizes their folded state, the well-packed interior of the racemic fibril is also stabilizing and results in its local stiffening (Table 2).
Figure 9.

(A) Model of MAX1/DMAX1 in their coassembled, racemic fibrillar state. (B) Model of pure MAX1 in its self-assembled state. ChemDraw figures at top define strand orientation in each assembly. Central images highlight the relative orientation of hairpins within a single monolayer of each fibril type with the valine side chains rendered in CPK (magenta). Bottom images view the racemic and pure enantiomeric fibrils along their long axes. The central and bottom images illustrate the differential packing of the valine residues within the racemic and pure enantiomeric fibrils.
Remarkably, the arrangement of β-strands afforded by the racemic assembly results in a supersecondary structure predicted by Pauling and Corey over 60 years ago, which they termed the rippled sheet.40 They proposed models of both antiparallel and parallel extended rippled sheets formed by the alternating assembly of linear β-strands of opposing chirality. These models were based directly on their earlier predictions of the pleated sheet formed by homochiral β-strands and were generated simply by “the reflection of alternate chains into their enantiomers”. Although the Corey and Pauling models comprise β-strands as opposed to the β-hairpins studied here, the similarity with respect to overall sheet topology formed by both is striking. Further, since our initial report,47 Nilsson et al. observed rippled sheet formation via the assembly of fibril forming linear β-strand amphiphiles, suggesting that this structural topology may be more prevalent than previously appreciated for sheet forming heterochiral peptides.41 Importantly, the few models of rippled sheets that have been reported40,41 are largely derived based on hydrogen-bonding arguments with little mention of side chain packing. In fact, the models drawn by Pauling and Corey only show the main chain atoms of the peptide’s backbone, omitting all side chains entirely. Our work demonstrates that side-chain packing is indeed important to the overall formation of this unique sheet topology and is responsible for the enhanced material properties enjoyed by the hydrogels formed by the racemic fibrils described here.
The model shown in Figure 9A, which underscores the importance of side chain interactions, makes several predictions. The first is that design can be used to modulate the bulk mechanical properties of the gel by incorporating amino acids that either enhance or diminish side-chain interactions between enantiomers. This prediction was first tested by preparing MAX35 and DMAX35, enantiomers that incorporate four isoleucine residues on their hydrophobic faces among a background of valines, Table 1. When assembled into racemic fibrils, both Ile–Ile and Val–Val pairwise interactions can form between neighboring enantiomers. Ile–Ile cross-pairs within β-sheets are generally rare in natural proteins especially when compared to Ile–Val or Va–Val pairs,82 suggesting that they are less energetically favorable. The rheological time-sweep experiment in Figure 10A shows that the MAX35/DMAX35 racemic gel displays only a 2-fold enhancement in rigidity compared to either of the pure enantiomeric gels. Importantly, the enhancement displayed by the MAX35/DMAX35 gel is 2-fold less than that displayed by the MAX1/DMAX1 gel, suggesting weaker inter-enantiomer interactions in the former.
Figure 10.

Dynamic time sweep oscillatory rheology measuring storage moduli of 1 wt % hydrogels formed from MAX35 and DMAX35 (A) and MARK4 and DMARK4 (B). The rigidity of each racemic gel (□) is greater than their respective pure enantiomeric gels (◇, ○), but by varying degrees based on the design of each enantiomeric peptide pair.
To strengthen the enantiomeric interaction, MARK4 and DMARK4 were designed with residue changes on the hydrophilic face of the hairpin, Table 1. Here, arginine residues were incorporated to enhance inter-enantiomer interactions in the fibrillar state through the formation of inter-guanidino hydrogen bonds. These interactions are not possible in the pure enantiomer assemblies. Figure 10B shows that the racemic gel displays an impressive 11-fold enhancement in its mechanical rigidity. Thus, modulating side-chain–side-chain interactions between enantiomers, whether in the fibril’s core or on its solvent exposed exterior, provide a means to tune the gel’s bulk properties via molecular-level engineering.
The second prediction our model makes is that the hairpins comprising the “racemic” fibrils need not be related as true enantiomers (e.g., non-superimposable mirror images) to realize the enhancement in fibril stiffness and corresponding gel rigidity. As long as important side-chain/side-interactions are not disrupted, alterations that break mirror image symmetry are tolerated. In fact, the fluorophore-, azide-, and biotin-labeled derivatives used in this study assembled readily with their nonmirror image partners. These hairpins were chemically modified at their strand termini or on their hydrophilic faces; regions of the amphiphile that do not participate in the formation of critical pairwise interactions that stabilize the fibril.
Conclusion
Heterochiral peptide assembly affords materials with unique macroscopic traits that are dependent on the sequence and composition of monomers used in their preparation. A racemic mixture of enantiomeric peptides MAX1 and DMAX1 assembled into a fibrillar hydrogel that is 4-fold more rigid than enantiomerically pure gels formed from either peptide alone. Herein, we thoroughly characterize the racemic hydrogel across molecular, local-fibril, and hydrogel network length scales using an arsenal of spectroscopy, microscopy, and scattering techniques to undercover the physical basis for this enhancement in mechanical rigidity. MAX1 and DMAX1 coassemble into fibrils comprised of a bilayer of peptides that H-bond along the long axis of a given fibril. Within each monolayer, enantiomers coassemble in an alternating fashion forming an extended heterochiral rippled sheet, a structure predicted by Pauling and Corey in 1953. Importantly, valine residues display their side chains into the fibrils’ dry interior, forming nested hydrophobic interactions between enantiomers, interactions not accessible within enantiomerically pure fibrils. This arrangement maximizes inter-residue hydrophobic contacts within the interior of fibrils that results in their local stiffening, which in turn, gives rise to the significant increase in bulk mechanical rigidity observed for the racemic hydrogel. We go on to show that modulation of inter-enantiomer interactions within fibrils can be used as a design tool to control the bulk mechanical properties of gels.
Acknowledgments
This work was supported by the Intramural Research Programs of the National Cancer Institute and National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. The authors would like to acknowledge Dr. Norman Wagner and Dr. Eric Yearly for their assistance with SANS experiments and data analysis. We also thank Dr. Daniel Smith for his assistance in the synthesis of isotopically labeled amino acids used in IE-FTIR experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00115.
Experimental methods, temperature-dependent circular dichroism of MAX1, analytical HPLC and mass spectra of all peptides, oscillatory rheology of hydrogels, micrographs of gold nanoparticle controls, solid NMR spectra of peptide gels and NMR characterization of labeled amino acids (PDF)
Author Present Address
# Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, HCI H 515, 8093 Zürich, Switzerland.
Author Present Address
∥ Bruker BioSpin Corporation, Billerica, MA 01821-3991.
The authors declare no competing financial interest.
Supplementary Material
References
- Hughes M.; Xu H. X.; Frederix P.; Smith A. M.; Hunt N. T.; Tuttle T.; Kinloch I. A.; Ulijn R. V. Biocatalytic self-assembly of 2D peptide-based nanostructures. Soft Matter 2011, 7, 10032–10038. 10.1039/c1sm05981e. [DOI] [Google Scholar]
- Jang H.-S.; Lee J.-H.; Park Y.-S.; Kim Y.-O.; Park J.; Yang T.-Y.; Jin K.; Lee J.; Park S.; You J. M.; Jeong K.-W.; Shin A.; Oh I.-S.; Kwon M.-K.; Kim Y.-I.; Cho H.-H.; Han H. N.; Kim Y.; Chang Y. H.; Paik S. R.; Nam K. T.; Lee Y.-S. Tyrosine-mediated two-dimensional peptide assembly and its role as a bio-inspired catalytic scaffold. Nat. Commun. 2014, 5 (3655), 1–11. 10.1038/ncomms4665. [DOI] [PubMed] [Google Scholar]
- Jiang T.; Vail O. A.; Jiang Z. G.; Zuo X. B.; Conticello V. P. Rational Design of Multilayer Collagen Nanosheets with Compositional and Structural Control. J. Am. Chem. Soc. 2015, 137, 7793–7802. 10.1021/jacs.5b03326. [DOI] [PubMed] [Google Scholar]
- Segman-Magidovich S.; Lee M. R.; Vaiser V.; Struth B.; Gellman S. H.; Rapaport H. Sheet-Like Assemblies of Charged Amphiphilic alpha/beta-Peptides at the Air-Water Interface. Chem. - Eur. J. 2011, 17, 14857–14866. 10.1002/chem.201101775. [DOI] [PubMed] [Google Scholar]
- Xu F.; Khan I. J.; McGuinness K.; Parmar A. S.; Silva T.; Murthy N. S.; Nanda V. Self-Assembly of Left- and Right-Handed Molecular Screws. J. Am. Chem. Soc. 2013, 135, 18762–18765. 10.1021/ja4106545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla D. E.; Chmielewski J. Metal-Triggered Collagen Peptide Disk Formation. J. Am. Chem. Soc. 2010, 132, 7866–7867. 10.1021/ja103148t. [DOI] [PubMed] [Google Scholar]
- Przybyla D. E.; Perez C. M. R.; Gleaton J.; Nandwana V.; Chmielewski J. Hierarchical Assembly of Collagen Peptide Triple Helices into Curved Disks and Metal Ion-Promoted Hollow Spheres. J. Am. Chem. Soc. 2013, 135, 3418–3422. 10.1021/ja307651e. [DOI] [PubMed] [Google Scholar]
- Thomson A. R.; Wood C. W.; Burton A. J.; Bartlett G. J.; Sessions R. B.; Brady R. L.; Woolfson D. N. Computational design of water-soluble alpha-helical barrels. Science 2014, 346, 485–488. 10.1126/science.1257452. [DOI] [PubMed] [Google Scholar]
- Adler-Abramovich L.; Marco P.; Arnon Z. A.; Creasey R. C. G.; Michaels T. C. T.; Levin A.; Scurr D. J.; Roberts C. J.; Knowles T. P. J.; Tendler S. J. B.; Gazit E. Controlling the Physical Dimensions of Peptide Nanotubes by Supramolecular Polymer Coassembly. ACS Nano 2016, 10, 7436–7442. 10.1021/acsnano.6b01587. [DOI] [PubMed] [Google Scholar]
- Ashkenasy N.; Horne W. S.; Ghadiri M. R. Design of self-assembling peptide nanotubes with delocalized electronic states. Small 2006, 2, 99–102. 10.1002/smll.200500252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Mehta A. K.; Sidorov A. N.; Orlando T. M.; Jiang Z. G.; Anthony N. R.; Lynn D. G. Design of Asymmetric Peptide Bilayer Membranes. J. Am. Chem. Soc. 2016, 138, 3579–3586. 10.1021/jacs.6b00977. [DOI] [PubMed] [Google Scholar]
- Thomas F.; Burgess N. C.; Thomson A. R.; Woolfson D. N. Controlling the Assembly of Coiled-Coil Peptide Nanotubes. Angew. Chem., Int. Ed. 2016, 55, 987–991. 10.1002/anie.201509304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit M.; Appel S.; Cohen R.; Cheng G.; Hamley I. W.; Ashkenasy N. Hybrid Proton and Electron Transport in Peptide Fibrils. Adv. Funct. Mater. 2014, 24, 5873–5880. 10.1002/adfm.201401111. [DOI] [Google Scholar]
- Fukunaga K.; Tsutsumi H.; Mihara H. Self-Assembling Peptide Nanofibers Promoting Cell Adhesion and Differentiation. Biopolymers 2013, 100, 731–737. 10.1002/bip.22309. [DOI] [PubMed] [Google Scholar]
- Hudalla G. A.; Sun T.; Gasiorowski J. Z.; Han H. F.; Tian Y. F.; Chong A. S.; Collier J. H. Gradated assembly of multiple proteins into supramolecular nanomaterials. Nat. Mater. 2014, 13, 829–836. 10.1038/nmat3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner K.; Taraban M. B.; Feng Y.; Yu Y. B. An Interplay Between Electrostatic and Polar Interactions in Peptide Hydrogels. Biopolymers 2013, 100, 174–183. 10.1002/bip.22194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King P. J. S.; Saiani A.; Bichenkova E. V.; Miller A. F. A de novo self-assembling peptide hydrogel biosensor with covalently immobilised DNA-recognising motifs. Chem. Commun. 2016, 52, 6697–6700. 10.1039/C6CC01433J. [DOI] [PubMed] [Google Scholar]
- Kumar V. A.; Shi S.; Wang B. K.; Li I. C.; Jalan A. A.; Sarkar B.; Wickremasinghe N. C.; Hartgerink J. D. Drug-Triggered and Cross-Linked Self-Assembling Nanofibrous Hydrogels. J. Am. Chem. Soc. 2015, 137, 4823–4830. 10.1021/jacs.5b01549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Ou Y. C.; Cheetham A. G.; Cui H. G. Rational Design of MMP Degradable Peptide-Based Supramolecular Filaments. Biomacromolecules 2014, 15, 1419–1427. 10.1021/bm500020j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z. L.; Wang S. K.; Zhang S. G. Fabrication of self-assembling D-form peptide nanofiber scaffold d-EAK16 for rapid hemostasis. Biomaterials 2011, 32, 2013–2020. 10.1016/j.biomaterials.2010.11.049. [DOI] [PubMed] [Google Scholar]
- Micklitsch C. M.; Medina S. H.; Yucel T.; Nagy-Smith K. J.; Pochan D. J.; Schneider J. P. Influence of Hydrophobic Face Amino Acids on the Hydrogelation of beta-Hairpin Peptide Amphiphiles. Macromolecules 2015, 48, 1281–1288. 10.1021/ma5024796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Du X. W.; Yamagata N.; Xu B. Enzyme-Instructed Self-Assembly of Small D-Peptides as a Multiple-Step Process for Selectively Killing Cancer Cells. J. Am. Chem. Soc. 2016, 138, 3813–3823. 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J. P.; Jones J. L.; Cronier S. A.; Collier J. H. Modulating the mechanical properties of self-assembled peptide hydrogels via native chemical ligation. Biomaterials 2008, 29, 2143–2151. 10.1016/j.biomaterials.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbas B.; Kretsinger J.; Rajagopal K.; Schneider J. P.; Pochan D. J. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules 2004, 37, 7331–7337. 10.1021/ma0491762. [DOI] [Google Scholar]
- Rajagopal K.; Lamm M. S.; Haines-Butterick L. A.; Pochan D. J.; Schneider J. P. Tuning the pH Responsiveness of beta-Hairpin Peptide Folding, Self-Assembly, and Hydrogel Material Formation. Biomacromolecules 2009, 10, 2619–2625. 10.1021/bm900544e. [DOI] [PubMed] [Google Scholar]
- Giano M. C.; Pochan D. J.; Schneider J. P. Controlled biodegradation of Self-assembling beta-hairpin Peptide hydrogels by proteolysis with matrix metalloproteinase-13. Biomaterials 2011, 32, 6471–6477. 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y. F.; Hudalla G. A.; Han H. F.; Collier J. H. Controllably degradable beta-sheet nanofibers and gels from self-assembling depsipeptides. Biomater. Sci. 2013, 1, 1037–1045. 10.1039/c3bm60161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga A. S.; Sinthuvanich C.; Gaspar D.; Franquelim H. G.; Castanho M.; Schneider J. P. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials 2012, 33, 8907–8916. 10.1016/j.biomaterials.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salick D. A.; Kretsinger J. K.; Pochan D. J.; Schneider J. P. Inherent antibacterial activity of a peptide-based beta-hairpin hydrogel. J. Am. Chem. Soc. 2007, 129, 14793–14799. 10.1021/ja076300z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S.; Das P. K. Antibacterial hydrogels of amino acid-based cationic amphiphiles. Biotechnol. Bioeng. 2008, 100, 756–764. 10.1002/bit.21803. [DOI] [PubMed] [Google Scholar]
- Haines-Butterick L.; Rajagopal K.; Branco M.; Salick D.; Rughani R.; Pilarz M.; Lamm M. S.; Pochan D. J.; Schneider J. P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 7791–7796. 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinthuvanich C.; Haines-Butterick L. A.; Nagy K. J.; Schneider J. P. Iterative design of peptide-based hydrogels and the effect of network electrostatics on primary chondrocyte behavior. Biomaterials 2012, 33, 7478–7488. 10.1016/j.biomaterials.2012.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude S.; Ingham E.; Aggeli A. Biomimetic self-assembling peptides as scaffolds for soft tissue engineering. Nanomedicine 2013, 8, 823–847. 10.2217/nnm.13.65. [DOI] [PubMed] [Google Scholar]
- Szkolar L.; Guilbaud J. B.; Miller A. F.; Gough J. E.; Saiani A. Enzymatically triggered peptide hydrogels for 3D cell encapsulation and culture. J. Pept. Sci. 2014, 20, 578–584. 10.1002/psc.2666. [DOI] [PubMed] [Google Scholar]
- Smith D. J.; Brat G. A.; Medina S. H.; Tong D. D.; Huang Y.; Grahammer J.; Furtmuller G. J.; Oh B. C.; Nagy-Smith K. J.; Walczak P.; Brandacher G.; Schneider J. P. A multiphase transitioning peptide hydrogel for suturing ultrasmall vessels. Nat. Nanotechnol. 2016, 11, 95–102. 10.1038/nnano.2015.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards W.; Smith D. K. Enantioselective Component Selection in Multicomponent Supramolecular Gels. J. Am. Chem. Soc. 2014, 136, 1116–1124. 10.1021/ja411724r. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Easton C. D.; Styan K. E.; Waddington L. J.; Kushkaki F.; Goodall L.; McLean K. M.; Forsythe J. S.; Hartley P. G. Chirality effects at each amino acid position on tripeptide self-assembly into hydrogel biomaterials. Nanoscale 2014, 6, 5172–5180. 10.1039/c3nr06752a. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Waddington L.; Easton C. D.; Winkler D. A.; Goodall L.; Forsythe J.; Hartley P. G. Unzipping the role of chirality in nanoscale self-assembly of tripeptide hydrogels. Nanoscale 2012, 4, 6752–6760. 10.1039/c2nr32006a. [DOI] [PubMed] [Google Scholar]
- Taraban M. B.; Feng Y.; Hammouda B.; Hyland L. L.; Yu Y. B. Chirality-Mediated Mechanical and Structural Properties of Oligopeptide Hydrogels. Chem. Mater. 2012, 24, 2299–2310. 10.1021/cm300422q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L.; Corey R. B. Rippled-sheet configurations of polypeptide chains, and a note about the pleated sheets. Proc. Natl. Acad. Sci. U. S. A. 1953, 39, 253–256. 10.1073/pnas.39.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanekamp R. J.; DiMaio J. T. M.; Bowerman C. J.; Nilsson B. L. Coassembly of Enantiomeric Amphipathic Peptides into Amyloid-Inspired Rippled beta-Sheet Fibrils. J. Am. Chem. Soc. 2012, 134, 5556–5559. 10.1021/ja301642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbuch I.; Illos R. A.; Bolbach G.; Lahav M. Racemic beta-Sheets as Templates of Relevance to the Origin of Homochirality of Peptides: Lessons from Crystal Chemistry. Acc. Chem. Res. 2009, 42, 1128–1140. 10.1021/ar900033k. [DOI] [PubMed] [Google Scholar]
- Mitsui Y.; Iitaka Y.; Tsuboi M. Side-chain interation between alpha-helices of poly-gamma-benyzl-L- and D- glutamates. J. Mol. Biol. 1967, 24, 15–26. 10.1016/0022-2836(67)90085-X. [DOI] [Google Scholar]
- Luo Z. L.; Zhao X. J.; Zhang S. G. Self-organization of a chiral D-EAK16 designer peptide into a 3D nanofiber scaffold. Macromol. Biosci. 2008, 8, 785–791. 10.1002/mabi.200800003. [DOI] [PubMed] [Google Scholar]
- Li X. M.; Du X. W.; Li J. Y.; Gao Y.; Pan Y.; Shi J. F.; Zhou N.; Xu B. Introducing D-Amino Acid or Simple Glycoside into Small Peptides to Enable Supramolecular Hydrogelators to Resist Proteolysis. Langmuir 2012, 28, 13512–13517. 10.1021/la302583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Y.; Fan Z. Y.; Chen Y. Z.; Fang X. L.; Sha X. Y. Retro-Inverso Carbohydrate Mimetic Peptides with Annexin1-Binding Selectivity, Are Stable In Vivo, and Target Tumor Vasculature. PLoS One 2013, 8, e80390. 10.1371/journal.pone.0080390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy K. J.; Giano M. C.; Jin A.; Pochan D. J.; Schneider J. P. Enhanced Mechanical Rigidity of Hydrogels Formed from Enantiomeric Peptide Assemblies. J. Am. Chem. Soc. 2011, 133, 14975–14977. 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J. P.; Pochan D. J.; Ozbas B.; Rajagopal K.; Pakstis L.; Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- Nagy-Smith K.; Moore E.; Schneider J.; Tycko R. Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9816–9821. 10.1073/pnas.1509313112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbas B.; Rajagopal K.; Schneider J. P.; Pochan D. J. Semiflexible chain networks formed via self-assembly of beta-hairpin molecules. Phys. Rev. Lett. 2004, 93, 268106. 10.1103/PhysRevLett.93.268106. [DOI] [PubMed] [Google Scholar]
- Branco M. C.; Nettesheim F.; Pochan D. J.; Schneider J. P.; Wagner N. J. Fast Dynamics of Semiflexible Chain Networks of Self-Assembled Peptides. Biomacromolecules 2009, 10, 1374–1380. 10.1021/bm801396e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackintosh F. C.; Kas J.; Janmey P. A. Elasticity of semiflexible biopolymer networks. Phys. Rev. Lett. 1995, 75, 4425–4428. 10.1103/PhysRevLett.75.4425. [DOI] [PubMed] [Google Scholar]
- Morse D. C. Viscoelasticity of concentrated isotropic solutions of semiflexible polymers. 1. Model and stress tensor. Macromolecules 1998, 31, 7030–7043. 10.1021/ma9803032. [DOI] [Google Scholar]
- Morse D. C. Viscoelasticity of concentrated isotropic solutions of semiflexible polymers. 2. Linear response. Macromolecules 1998, 31, 7044–7067. 10.1021/ma980304u. [DOI] [Google Scholar]
- Larsen T. H.; Branco M. C.; Rajagopal K.; Schneider J. P.; Furst E. M. Sequence-Dependent Gelation Kinetics of beta-Hairpin Peptide Hydrogels. Macromolecules 2009, 42, 8443–8450. 10.1021/ma901423n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi E. D.; Wang G. T.; Krafft G. A.; Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy-transfer. Science 1990, 247, 954–958. 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- Garcia-Echeverria C.; Rich D. H. New intramolecularly quenched fluorogenic peptide-substrates for the study of the kinetic specificity of papain. FEBS Lett. 1992, 297, 100–102. 10.1016/0014-5793(92)80336-F. [DOI] [PubMed] [Google Scholar]
- Gulnik S. V.; Suvorov L. I.; Majer P.; Collins J.; Kane B. P.; Johnson D. G.; Erickson J. W. Design of sensitive fluorogenic substrates for human cathepsin D. FEBS Lett. 1997, 413, 379–384. 10.1016/S0014-5793(97)00886-7. [DOI] [PubMed] [Google Scholar]
- Sweers K. K. M.; Bennink M. L.; Subramaniam V. Nanomechanical properties of single amyloid fibrils. J. Phys.: Condens. Matter 2012, 24, 243101. 10.1088/0953-8984/24/24/243101. [DOI] [PubMed] [Google Scholar]
- Adamcik J.; Mezzenga R. Study of amyloid fibrils via atomic force microscopy. Curr. Opin. Colloid Interface Sci. 2012, 17, 369–376. 10.1016/j.cocis.2012.08.001. [DOI] [Google Scholar]
- Oelschlaeger C.; Cota Pinto Coelho M.; Willenbacher N. Chain Flexibility and Dynamics of Polysaccharide Hyaluronan in Entangled Solutions: A High Frequency Rheology and Diffusing Wave Spectroscopy Study. Biomacromolecules 2013, 14, 3689–3696. 10.1021/bm4010436. [DOI] [PubMed] [Google Scholar]
- Pine D. J.; Weitz D. A.; Zhu J. X.; Herbolzheimer E. Diffusing-wave spectroscopy - dynamic light-scattering in the multiple-scattering limit. J. Phys. (Paris) 1990, 51, 2101–2127. 10.1051/jphys:0199000510180210100. [DOI] [Google Scholar]
- Pine D. J.; Weitz D. A.; Chaikin P. M.; Herbolzheimer E. Difussing-wave spectroscopy. Phys. Rev. Lett. 1988, 60, 1134–1137. 10.1103/PhysRevLett.60.1134. [DOI] [PubMed] [Google Scholar]
- Willenbacher N.; Oelschlaeger C. Dynamics and structure of complex fluids from high frequency mechanical and optical rheometry. Curr. Opin. Colloid Interface Sci. 2007, 12, 43–49. 10.1016/j.cocis.2007.03.004. [DOI] [Google Scholar]
- Mason T. G.; Gang H.; Weitz D. A. Diffusing-wave-spectroscopy measurements of viscoelasticity of complex fluids. J. Opt. Soc. Am. A 1997, 14, 139–149. 10.1364/JOSAA.14.000139. [DOI] [Google Scholar]
- Krall A. H.; Weitz D. A. Internal dynamics and elasticity of fractal colloidal gels. Phys. Rev. Lett. 1998, 80, 778–781. 10.1103/PhysRevLett.80.778. [DOI] [Google Scholar]
- Willenbacher N.; Oelschlaeger C.; Schopferer M.; Fischer P.; Cardinaux F.; Scheffold F. Broad bandwidth optical and mechanical rheometry of wormlike micelle solutions. Phys. Rev. Lett. 2007, 99, 068302. 10.1103/PhysRevLett.99.068302. [DOI] [PubMed] [Google Scholar]
- Oelschlaeger C.; Schopferer A.; Scheffold F.; Willenbacher N. Linear-to-Branched Micelles Transition: A Rheometry and Diffusing Wave Spectroscopy (DWS) Study. Langmuir 2009, 25, 716–723. 10.1021/la802323x. [DOI] [PubMed] [Google Scholar]
- Oelschlaeger C.; Suwita P.; Willenbacher N. Effect of Counterion Binding Efficiency on Structure and Dynamics of Wormlike Micelles. Langmuir 2010, 26, 7045–7053. 10.1021/la9043705. [DOI] [PubMed] [Google Scholar]
- Le Goff L.; Amblard F.; Furst E. M. Motor-driven dynamics in actin-myosin networks. Phys. Rev. Lett. 2002, 88, 018101. 10.1103/PhysRevLett.88.018101. [DOI] [PubMed] [Google Scholar]
- Huh J. Y.; Furst E. M. Colloid dynamics in semiflexible polymer solutions. Phys. Rev. E 2006, 74, 031802. 10.1103/PhysRevE.74.031802. [DOI] [PubMed] [Google Scholar]
- Gittes F.; MacKintosh F. C. Dynamic shear modulus of a semiflexible polymer network. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 1998, 58, R1241–R1244. 10.1103/PhysRevE.58.R1241. [DOI] [Google Scholar]
- Mackintosh F. C.; John S. Diffusing-wave spectroscopy and multiple-scattering of light in correlated random-media. Phys. Rev. B: Condens. Matter Mater. Phys. 1989, 40, 2383–2406. 10.1103/PhysRevB.40.2383. [DOI] [PubMed] [Google Scholar]
- Silva R.; Barber-Armstrong W.; Decatur S. M. The organization and assembly of beta-sheet formed by a prion peptide in solution: An isotope-edited FTIR study. J. Am. Chem. Soc. 2003, 125, 13674–13675. 10.1021/ja036725v. [DOI] [PubMed] [Google Scholar]
- Petty S. A.; Decatur S. M. Intersheet rearrangement of polypeptides during nucleation of beta-sheet aggregates. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14272–14277. 10.1073/pnas.0502804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty S. A.; Decatur S. M. Experimental evidence for the reorganization of beta-strands within aggregates of the a beta(16–22) peptide. J. Am. Chem. Soc. 2005, 127, 13488–13489. 10.1021/ja054663y. [DOI] [PubMed] [Google Scholar]
- Decatur S. M. Elucidation of residue-level structure and dynamics of polypeptides via isotope-edited infrared spectroscopy. Acc. Chem. Res. 2006, 39, 169–175. 10.1021/ar050135f. [DOI] [PubMed] [Google Scholar]
- Nagarajan S.; Taskent-Sezgin H.; Parul D.; Carrico I.; Raleigh D. P.; Dyer R. B. Differential Ordering of the Protein Backbone and Side Chains during Protein Folding Revealed by Site-Specific Recombinant Infrared Probes. J. Am. Chem. Soc. 2011, 133, 20335–20340. 10.1021/ja2071362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubelka J.; Keiderling T. A. Differentiation of beta-sheet-forming structures: Ab initio-based simulations of IR absorption and vibrational CD for model peptide and protein beta-sheets. J. Am. Chem. Soc. 2001, 123, 12048–12058. 10.1021/ja0116627. [DOI] [PubMed] [Google Scholar]
- Surewicz W. K.; Mantsch H. H.; Chapman D. Determination of protein secondary structure by Fourier-transformed infrared spectroscopy- a critical assessment. Biochemistry 1993, 32, 389–394. 10.1021/bi00053a001. [DOI] [PubMed] [Google Scholar]
- Marecek J.; Song B.; Brewer S.; Belyea J.; Dyer R. B.; Raleigh D. P. A simple and economical method for the production of C-13, O-18-labeled Fmoc-amino acids with high levels of enrichment: Applications to isotope-edited IR studies of proteins. Org. Lett. 2007, 9, 4935–4937. 10.1021/ol701913p. [DOI] [PubMed] [Google Scholar]
- Wouters M. A.; Curmi P. M. G. An analysis of side-chain interactionsand pair correlations within antiparallel beta-sheets - the differences between backbone hydrogen-bonded and non-hydrogen bonded residue pairs. Proteins: Struct., Funct., Genet. 1995, 22, 119–131. 10.1002/prot.340220205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.