

Abstract
The lithium–sulfur battery has long been seen as a potential next generation battery chemistry for electric vehicles owing to the high theoretical specific energy and low cost of sulfur. However, even state-of-the-art lithium–sulfur batteries suffer from short lifetimes due to the migration of highly soluble polysulfide intermediates and exhibit less than desired energy density due to the required excess electrolyte. The use of sparingly solvating electrolytes in lithium–sulfur batteries is a promising approach to decouple electrolyte quantity from reaction mechanism, thus creating a pathway toward high energy density that deviates from the current catholyte approach. Herein, we demonstrate that sparingly solvating electrolytes based on compact, polar molecules with a 2:1 ratio of a functional group to lithium salt can fundamentally redirect the lithium–sulfur reaction pathway by inhibiting the traditional mechanism that is based on fully solvated intermediates. In contrast to the standard catholyte sulfur electrochemistry, sparingly solvating electrolytes promote intermediate- and short-chain polysulfide formation during the first third of discharge, before disproportionation results in crystalline lithium sulfide and a restricted fraction of soluble polysulfides which are further reduced during the remaining discharge. Moreover, operation at intermediate temperatures ca. 50 °C allows for minimal overpotentials and high utilization of sulfur at practical rates. This discovery opens the door to a new wave of scientific inquiry based on modifying the electrolyte local structure to tune and control the reaction pathway of many precipitation–dissolution chemistries, lithium–sulfur and beyond.
Short abstract
Achieving sparing solubility of polysulfides in electrolytes for Li−S cells induces fundamental changes in the reaction pathway that give rise to low polarization.
Introduction
Batteries as an electrochemical energy storage device have transformed portable electronics and launched the current generation of electric vehicles. Lithium ion (Li-ion batteries have improved tremendously since their commercialization in 1991, but will be limited in specific energy—and potentially cost—by the intercalation host materials that comprise their positive and negative electrodes. The use of chemical transformations rather than intercalation to store energy may provide a step change in specific energy and cost. Lithium sulfur (Li–S) is one such transformation chemistry that has been explored for decades with only modest success.1−4 Technoeconomic modeling of Li–S suggests that promising materials-only fundamentals can translate to long-term goals at the systems level, assuming that progress is made in reducing excess material burdens.5 While current Li–S cells are lighter than Li ion (350 vs 250 Wh kg–1), they have lower energy density and significantly poorer cycle life.6−9 Excess electrolyte is also required to overcompensate for its consumption in side reactions with lithium metal and to fully solvate the polysulfide intermediates. The fully solvating nature of conventional ether electrolytes enables facile reaction kinetics but concurrently promotes the migration of sulfur and polysulfides out of the positive electrode, resulting in the isolation of active material and/or unfavorable redistribution of reactants.10 Lithium metal must be protected to achieve long cycle life. This protection presumably would also mitigate the polysulfide shuttle mechanism; however, sulfur and polysulfide redistribution would still limit cycle life as has been demonstrated in many other precipitation–dissolution chemistries.11,12
The concept of sparingly solvating electrolytes has been recently proposed, in theory, as a pathway to control reactant distribution owing to the very low solubility of polysulfides in such systems, and thus their short residence time in the solvated state.13 Many well-known precipitation–dissolution secondary electrodes operate successfully by using the sparingly soluble concept, including Pb/PbSO4, Ag/AgCl, and Fe/FeCl2. Unlike conventional Li–S electrolytes, sparingly solvating electrolytes also break the coupling between the quantity of electrolyte required in the cell and the reaction mechanism. Solvates, ionic liquids, and superconcentrated electrolytes with reduced sulfur and polysulfide solubility have been used with varying degrees of success in Li–S cells.3,13−17 The electrochemistry reported in nearly all electrolytes exhibits the traditional voltage response under galvanostatic discharge. The initial capacity is delivered during a plateau in the 2.3–2.4 V vs Li/Li+ range before transition to a second, lower-voltage plateau around 2–2.1 V vs Li/Li+.18−20 The specific capacity associated with the transition to the second plateau varies, but often is near 300–400 mAh gs–1. The solubility of polysulfides in Li–S batteries can be suppressed through properly designed electrolyte systems. For example, very high salt concentrations in the electrolyte mean that most solvent molecules strongly interact with the Li salt, leaving minimal or none to solubilize polysulfides.21,22 A diluent, typically a hydrofluorinated ether (TTE),23 is often added to reduce the high viscosity of these electrolytes while maintaining or reducing polysulfide solubility further.14,22 High salt concentration electrolytes based on DOL:DME, glymes, and acetonitrile (ACN) exhibit good polysulfide solubility control and electrochemical performance in Li–S batteries.14,15,21,22,24 Such systems have been comprehensively reviewed.25,26 Another class of electrolyte composed of a mixture of a room temperature ionic liquid with low donor ability and lithium-bis(trifluoromethane)sulfonamide (LiTFSI) salt at a moderate concentration also exhibits selectively low solvation power for polysulfides of varied chain length.27−29 Li–S batteries using this class of electrolyte show promising performance.
A recent report of Li–S electrochemistry using an electrolyte with ACN2LiTFSI as the primary solvate and TTE as a diluent demonstrated a nontraditional reaction pathway.3 This electrolyte was classified as a nonsolvent, since it exhibited no solubility for typical polysulfides at room temperature. Using sulfur melt-infused into a mesoporous carbon host, sulfur utilization and Coulombic efficiency (CE) were enhanced. However, the electrochemistry at room temperature suffered from very significant polarization (overpotential) on both discharge and charge. Overcoming this factor is essential to move such an approach to more practical applications. Herein, we demonstrate that switching the nonsolvent electrolyte into a sparingly solvating electrolyte accomplishes this, confirming our original concept earlier theorized. We find that a moderate rise in operating temperature actually redirects the reaction pathway owing to a fundamental change in relative rates for competing reactions. This is evident from the unique electrochemical profile that is exhibited, which has not been observed before in Li–S batteries at elevated temperature.30 The underlying process is not based on a simple increase of polysulfide solubility with temperature. Mechanistic modeling—which reproduces the highly unusual electrochemical profile—shows that species are generated that act as internal redox mediators. We accomplish greatly reduced polarization even with micrometer-sized sulfur particles. This discovery opens the door to a new wave of scientific inquiry based on modifying the electrolyte local environment to tune and eventually control the reaction pathway of many precipitation–dissolution chemistries.
Results
Electrochemistry
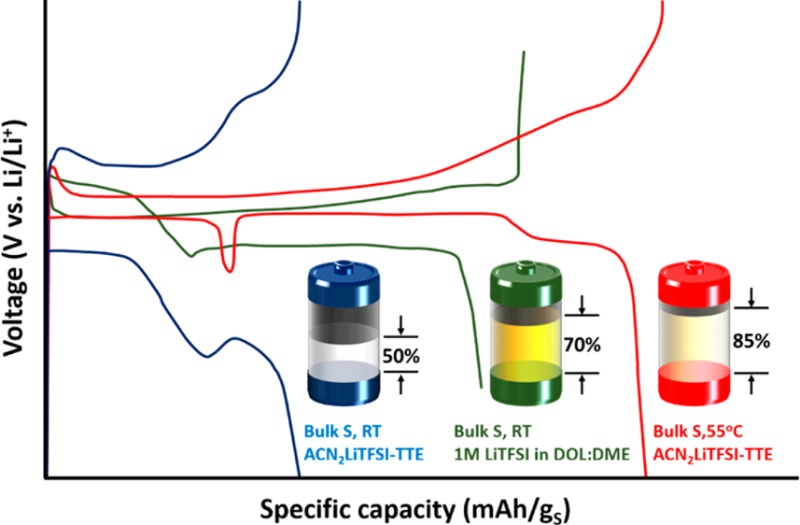
In Figure 1, the electrochemical behavior of Li–S cells using ACN2LiTFSI–TTE electrolyte (1:1 by volume, denoted as ACN–TTE cell) is compared to 1 M LiTFSI in DOL:DME (1:1 by volume) with 2 wt % LiNO3 electrolyte (denoted as DOL:DME cell). Cells were charged/discharged at a C/30 rate at different operating temperatures using various electrode architectures. In Figure 1a, the electrochemistry in the standard DOL:DME catholyte provides facile kinetics and reasonable sulfur utilization at 30 °C. In contrast, the cells with ACN–TTE electrolyte show a large overpotential. The use of melt-infused sulfur is typically necessary to obtain good sulfur utilization.31 However, micrometer-sized sulfur particles were used in this study. We believe that this can shed light on the nature of the electrochemistry that is truly intrinsic to sulfur, thereby providing mechanistic insights and guidance to overcome major stumbling blocks in solvate electrolyte systems. Figure 1b shows the voltage profiles at 55 °C. The DOL:DME cell shows electrochemical behavior similar to that at 30 °C, except with a higher discharge capacity of 1505 mAh gs–1. Therefore, increasing the temperature of the “catholyte” cell does not signficantly alter the behavior but promotes the transition to solid Li2S, thus increasing the capacity allocation in the second plateau. In sharp contrast, the electrochemistry of the ACN–TTE cell is remarkably improved by mild heating. At 30 °C, the initial capacity of the cell is only ∼580 mAh gs–1, and it exhibits poor energy efficiency resulting from the large voltage gap between discharge and charge (polarization). Conversely, the voltage profile at 55 °C shows minimal polarization and excellent sulfur utilization of 1435 mAh gs–1. The polarization for the ACN–TTE electrolyte at 55 °C is even lower than that observed in DOL:DME, in fact. Even at half the electrolyte volume, i.e., an electrolyte/sulfur (E/S) ratio of 5 mL gs–1, a very similar profile is observed, albeit with a little less capacity (see Figure S1). We note that the latter E/S value is close to the limit that can be sustained, but if the electrode is calendared to 20 μm thickness and if a single layer of separator is employed, the E/S ratio could be further lowered to 1.6 mLelectrolyte/gs: see calculations in Table S1. The CE during cycling is almost 100%, indicative of suppression of the polysulfide (PS) shuttle process even though no LiNO3 additive was added to passivate the negative electrode.32 These electrochemical characteristics of the ACN solvate system are particularly noteworthy as the positive electrode uses micrometer-sized (not mesoporous) sulfur as the active material.
Figure 1.

First cycle voltage profiles during galvanostatic cycling at a C/30 rate (1C = 1675 mA gs–1): (a) at 30 °C using bulk sulfur electrode in ACN–TTE compared to bulk sulfur electrode in DOL:DME and (b) at 55 °C using bulk sulfur electrodes in ACN–TTE compared with DOL:DME; specific discharge and charge capacities over multiple cycles for bulk sulfur electrodes in ACN−TTE compared to DOL:DME; (c) at 30 °C and (d) 55 °C. The DOL:DME electrolyte contains 2 wt % LiNO3 additive.
Three additional features of sulfur electrochemistry in the ACN−TTE electrolyte at 55 °C are striking due to their atypical nature. First, the 2.35 V plateau—characteristic of conversion of S8 to long-chain polysulfides in conventional catholyte electrolytes—is absent in the ACN–TTE system. This reveals that the mechanism of sulfur reduction in the ACN solvate is different from that of conventional electrolytes. Second, a noticeable voltage “dip” at ca. 450 mAh gs–1 during discharge separates the first and second plateaus. The dip is much larger in magnitude compared to what is commonly observed in catholyte systems. The presence of a voltage drop and recovery is often ascribed to a nucleation overpotential or supersaturation event preceding the onset of precipitation as will be discussed later. However, in our case, the voltage actually rises after the dip. On the second plateau, the charge is delivered at an electrode potential higher than during the first 400 mAh gs–1 of capacity. Open-circuit voltage (OCV) measurements—discussed at length later—confirm that a difference in equilibrium voltage is responsible for the voltage rise and not a difference in overpotential due to kinetic factors. These three characteristics of the sulfur electrochemistry in the ACN–TTE electrolyte, coupled with the excellent sulfur utilization and low polarization at 55 °C, result from a fundamentally different reaction pathway than what has been repeatedly reported in the literature over the past 20 years.
Figures 1c and 1d show the capacity retention over 50 cycles of the cells utilizing ACN–TTE and DOL:DME electrolytes. As expected, the capacity retention of the DOL:DME cells is poor at this slow C/30 rate: a direct consequence of the highly active PS shuttle process in the absence of any physical confinement of sulfur in the electrode. Although a high initial capacity of ∼1500 mAh gs–1 is seen in DOL:DME at 55 °C, the PS shuttle effect is exacerbated at this temperature and the CE quickly drops. On the other hand, ACN–TTE cells exhibit improved cycling compared with DOL:DME cells, at both 30 and 55 °C. At 30 °C, the ACN–TTE cell requires a few initial activation cycles, but the capacity is delivered with >99% CE. At 55 °C, no activation is required, the sulfur utilization is excellent, and the ACN–TTE cell exhibits very high CE. Evidently, in the ACN solvate even at moderately high temperatures, the crossover of PS species from the positive to negative electrode is somewhat suppressed. However, capacity fade occurs, which could be related to other additional detrimental side reactions. To explore this, we isolated the lithium from the positive sulfur electrode with an Ohara glass membrane: a single-ion conductor (denoted as Ohara-ACN cell).33 Over 50 cycles after peak capacity, the Ohara-ACN cell shows 89% capacity retention while the ACN–TTE cell shows only 48% capacity retention (Figure S2). Future studies with protected lithium electrodes will probe the underlying mechanisms at play more closely, but are beyond the scope of this work.
To confirm the very significant improvement found at moderate temperatures with the ACN2LiTFSI–TTE solvate electrolyte, KB/S and CMK-3/S melt-infused sulfur composites with nanostructured carboneous materials were also examined at both 30 and 55 °C (Figure S3). The melt-infused sulfur composites, KB/S and CMK-3/S, behave similarly at 30 °C. Both appear to promote greater utilization of sulfur than the simple physical mixture of bulk sulfur and carbon black. However, all three sulfur electrodes exhibit striking similarities in the electrochemical discharge at 55 °C. Specifically, all cells show a plateau at the beginning of discharge; the characteristic dip at ∼400–450 mAh gs–1; and the subsequent increase in voltage on traversing the dip (Figure S3b). Encouraging is that the capacity and cycling stability of bulk sulfur electrodes are comparable to elaborately architectured sulfur electrodes at 55 °C (Figure S3d).
To fully understand the effect of elevated temperature on the electrochemistry, the first cycle in separate ACN–TTE cells was measured at several temperatures, from 20 to 70 °C. Figure 2a shows the remarkable change of the system with temperature. Lower than 30 °C, a very large IR drop is observed at the start of discharge and the capacity is highly limited. At 37 °C, polarization in the initial discharge is reduced, a broad voltage dip starts to take shape, and more capacity is extractable beyond the dip. Further increase in temperature leads to reduced polarization in both regions along with a concomitant increase in capacity. The voltage of the region beyond the dip becomes higher than the first plateau at 45 °C. In parallel, the sulfur utilization is also maximized to ∼1503 mAh gs–1 (∼90% of the theoretical capacity) at 70 °C. Similar evolution of the electrochemical profile is also observed as a function of C-rate for the first cycle (Figure 2b). This uncanny similarity—whether the temperature or the rate of charge/discharge is changed—strongly suggests that complex factors affect the electrochemical response, and particularly that of the second plateau.
Figure 2.

First cycle voltage profiles of separate ACN−TTE cells using bulk sulfur electrodes: (a) using C/30 rate and at indicated temperatures and (b) at a temperature of 55 °C but using various cycling rates (1C = 1675 mA gs–1); GITT profiles using ACN–TTE cells at (c) 55 °C and (d) 30 °C; the green lines indicate where the equilibrium voltages lies upon relaxing.
In summary, the Li–S discharge profile in ACN–TTE at moderate temperatures consists of a long plateau, interrupted by a prominent, unique dip in the profile around 400–500 mAh gs–1. Inspection reveals a small increase in electrode potential just past the dip. The fact that the lowest potential occurs for the first ∼25% of discharge is most unusual. This does not arise from impedance contributions but is actually thermodynamic in origin. In Figures 2c and 2d, the galvanostatic intermittent titration technique (GITT) is used to measure the quasi-OCV of the ACN–TTE cells at both 55 and 30 °C. In the region before the dip, the quasi-equilibrium voltage attains a constant value of 2.20 V at 55 °C. Interestingly, the exact same OCV was obtained at 30 °C, which was masked during discharge (Figure 1a) by a large overpotential (∼200 mV larger than at 55 °C). As alluded to earlier, a low OCV of 2.20 V measured early in discharge points to a distinctly different reduction pathway. Additionally, the presence of a flat plateau up to ca. 450 mAh gs–1 suggests that the system exhibits classic two phase behavior, i.e., direct conversion of S8 to another product. This process is largely independent of the temperature of the reaction. The equilibrium voltage profile at 55 °C does not show the dip, indicating that the underlying process is kinetically controlled. In the region past the dip the voltage response is complex: the OCV shows a small, continuous increase in the range of 400–1000 mAh gs–1, followed by a reduction toward the end of discharge. Additional studies that (a) explore the complexity of the reaction by interpretation of the GITT dynamics and (b) show that the addition of TTE improves the kinetics without altering the inherent thermodynamics of the Li–S cell are provided in Figures S4 and S5.
To investigate if the new electrochemical signature dip is a unique phenomenon of only ACN2LiTFSI, we performed sulfur electrochemistry using 1 M LiTFSI in ACN and another solvate electrolyte using THF, THF2LiTFSI–TTE. The 1 M LiTFSI in ACN electrolyte shows electrochemical behavior similar to that seen in other fully solvating electrolytes, such as in DOL:DME. In sharp contrast, the Li–S electrochemistry observed in THF2LiTFSI–TTE solvate closely resembled that of the ACN2LiTFSI–TTE electrolyte (see Figure S6 for details). Therefore, the presence of polar molecules with a 2:1 ratio of a functional group to lithium salt seems to enable the unique electrochemistry in these solvate electrolytes.
Physical and Chemical Characterization
As described earlier, the ACN–TTE cell shows a high capacity of ∼1430 mAh gs–1 at 55 °C, indicating a high sulfur utilization up to 86%. The enhanced sulfur utilization and lowered polarization compared to room temperature cells is significant and could be the result of enhanced reaction kinetics from increased polysulfide solvation, a solid-state reaction, or other factors. To further probe the reaction mechanism, operando XRD and XAS, polysulfide solubility, and other physicochemical measurements were carried out.
Ionic Conductivity and Polysulfide Solubility
The ionic conductivity of ACN–TTE electrolyte increases only marginally to 2.64 mS cm–1 at 55 °C from 1.59 mS cm–1 at 27 °C (Figure S7). It is unlikely that such a small increase can solely explain the much improved and complex electrochemistry at moderately high temperatures. We also measured the solubility of elemental sulfur and polysulfides (using Li2S6 as the representative species) by UV–vis spectroscopy at 70 °C. These results are shown in Table 1. The room temperature solubility of Li2S6 in ACN–TTE was below the detection limit of the UV–vis spectrometer. In contrast to the colorless solution at room temperature, the ACN–TTE electrolyte saturated with Li2S6 shows a pale yellow color at 70 °C, indicating some solubility (see details in Figures S8a and S8c). No precipitation occurred after the solution was cooled down to room temperature, even after 2 weeks. As quantified by UV–vis spectroscopy, the ACN–TTE electrolyte can dissolve 0.20 mg mL–1 of Li2S6, which corresponds to 0.2% of the total Li2S6 (if all the sulfur converted to Li2S6) in a cell with an electrolyte to sulfur ratio of 10 mL g–1, or 0.02% at the target ratio of electrolyte to sulfur of 1 mL g–1. In stark contrast, TEGDME solubilizes ca. 100% of the Li2S8 at an electrolyte to sulfur ratio of 4 mL g–1.26
Table 1. Solubility Limit of Elemental Sulfur (S8) and Li2S6 Determined Using UV–Vis Spectroscopy at an Elevated Temperature of 70 °C for Different Solvent/Electrolytes.
| solubility (mg/mL) |
||||
|---|---|---|---|---|
| TTE | ACN–TTE | G4-LiTFSI | G4-LiTFSI, rta | |
| Li2S6 | 0.11 | 0.20 | 4.90 | 0.57 (Li2S4) |
| S8 | 0.013 | 0.035 | 0.190 | |
Values taken from ref (14) (rt: room temperature).
Also, sulfur exhibits 1 order of magnitude lower solubility (Table 1), excluding its role in enhancing the reaction kinetics. These results confirm that ACN–TTE sparingly solvates polysulfides at elevated temperature. This small but essential effect acts as redox mediator for an otherwise solid-state reaction as we explain in the Discussion. Note that the ACN–TTE electrolyte shows almost double the solubility compared to pure TTE, which is likely due to the altered complexing behavior of the solvate at elevated temperatures: for instance, the presence of some free ACN solvent due to a shifted equilibrium.34 A TEGDME solvate (G4-LiTFSI) consisting of an equimolar ratio of TEGDME and LiTFSI, monitored using the same procedure, dissolves 25 times more Li2S6. Interestingly, however, G4-LiTFSI does not show major changes in its electrochemical behavior upon increasing temperature, much in the same way that DOL:DME does not.14 GITT studies show that the G4-LiTFSI solvate exhibits an OCV of 2.35 V in the initial discharge—characteristic of conversion of S8 to long-chain polysulfides—followed by a second plateau of a lower voltage (Figure S9). Clearly, increasing the solubility beyond a certain range might not necessarily be beneficial and/or only increasing the solubility is not sufficient to improve the electrochemistry. Fine details of the solvate chemistry and structure may play a preeminent role in determining the nature of the solubilized intermediates. In fact, Dunning et al. suggest that a reactant solubility of the order of 1 × 10–4 M (i.e., 0.02 mg mL–1 for Li2S6) is desired for long-lived and high-rate capability batteries based on precipitation/dissolution chemistry.35
X-ray Diffraction
Operando XRD was performed to understand the time-resolved evolution of crystalline species in the sparingly solvating electrolyte system. Although we observe the formation of crystalline Li2S upon discharge and re-formation of sulfur for both ACN–TTE and DOL:DME systems (Figures 3a and 3b respectively), their evolution of S8/Li2S is quite different. The individual diffraction patterns as a function of discharge/charge are shown in Figures 3c and 3e, respectively, as “waterfall” plots. The evolution of elemental sulfur/Li2S was quantified by integrating the respective major peak areas in these patterns, and are presented along with the corresponding electrochemical profiles in Figures 3d and 3f.
Figure 3.

Operando XRD characterization of sulfur electrochemistry in two different electrolytes using ground KB-bulk sulfur electrodes. The XRD patterns of electrodes (a) after first full discharge and (b) after first full charge in ACN–TTE and DOL:DME electrolytes at room temperature (rt) or elevated temperatures, along with that of the pristine electrode. (c, e) The waterfall diagrams showing the evolution of XRD patterns (22–33°) obtained operando as a function of discharge/charge states and (d, f) the peak-area quantified evolution of crystalline sulfur (α- or β-S8, blue dotted lines) and Li2S (green dotted lines) phases as a function of the capacity, during the first full cycle for cells in (c) the DOL:DME electrolyte at 60 °C and (e) the ACN–TTE solvate electrolyte at 70 °C. The red patterns in the central position of panels c and e indicate the end of discharge; the colors in panels d and f code distinct stages with different compositions in terms of the presence of S8 and Li2S (blue, only S8; green, only Li2S; yellow, neither S8 nor Li2S; red, both S8 and Li2S).
On discharge, compared to the DOL:DME electrolyte where rapid consumption of sulfur is completed before the supersaturation point (first purple region, Figure 3d), the ACN–TTE cell exhibits more gradual consumption of sulfur (purple region), which is complete only at the end of the second plateau (first pink region, Figure 3f). However, no other crystalline phase is detected. For ACN–TTE, after the first plateau, crystalline Li2S starts forming before the very end of the voltage dip (pink region, Figure 3f), which may indicate a nucleation overpotential for the crystallization of Li2S. As discussed in the Supporting Information, the dip has a complex response during extended cycling, and a definitive understanding is beyond the scope of the present study. The proposed reaction corresponding to the voltage rise after the dip is discussed later. In summary, the linear increase of Li2S and an almost constant amount of sulfur over the second plateau (pink region, Figure 3f) suggests a two-phase reaction to form Li2S from the disproportionation of Li2S2–4 that involves sulfur. At the very end of discharge (corresponding to the third plateau in GITT study, Figure 2c), sulfur is fully consumed, resulting in the voltage drop.
A salient contrasting feature of the two electrolyte systems is apparent on charge. While both commence discharge with orthorhombic α-S8, after charge, the ACN–TTE cell maintains the α-phase. However, a metastable monoclinic β-S8 is obtained for the catholyte DOL:DME cell, consistent with that previously reported.4 This strongly suggests that a different conversion pathway exists for the ACN–TTE solvate, although its exact nature is still unclear.36,37 Furthermore, for the DOL:DME system upon charge, sulfur formation begins only toward the final stage of charging (S82– → β-S8 + 2e–; purple region in Figure 3d); however, sulfur forms much earlier for the ACN–TTE cell, just before the voltage rise (pink region, Figure 3f). This early onset of sulfur formation indicates a more facile oxidation of short to mid chain length polysulfide intermediates directly to sulfur. We further note the copresence of solid S8 and Li2S in ACN–TTE (the red region in Figure 3f), in contrast to their mutually exclusive presence in the DOL:DME system (purple and yellow regions in Figure 3d). This demonstrates a quasi-solid-state reaction in the ACN–TTE system that is not dominated by polysulfide solution mediation as in the DOL:DME system. The S8/Li2S evolution at room temperature for the ACN solvate system shows a similar trend to that at elevated temperature (Figure S10), although more unreacted S8 is present on discharge. This means that sparing solvation at elevated temperature does not affect the products of discharge/charge, but only alters the speciation pathway, as described above in the GITT studies, and later in the Discussion.
X-ray Absorption Spectroscopy
In order to confirm that polysulfide species are formed prior to the dip even during elevated temperature operation, operando XAS experiments were carried out. The data are qualitative rather than quantitative owing to spectral distortion from self-absorption of the large sulfur particles. Nonetheless, in agreement with prior room temperature studies, lithium polysulfides are indeed clearly generated upon the initiation of discharge (Figure S11a).3 XRD experiments (Figure 3 and Figure S10) do not identify the formation of any new crystalline phase prior to the dip in ACN–TTE cells, irrespective of the temperature at which the electrochemistry is performed. Amorphous discharge products, however, are invisible by XRD. Evidently, the early formation of Li2S2–4 species results in the lower first-plateau voltage of the ACN–TTE cells. Spectral evolution during operando XAS is largely consistent with the consumption of sulfur and subsequent growth of polysulfides on the surface of sulfur particles (see Figure S11 for details). Ex situ SEM and EDX studies were used to analyze the sulfur morphology and distribution in the cathode and Li anode after cycling. The EDX sulfur mapping in the ACN–TTE system shows that the sulfur particles remain in a similar distribution as in the pristine electrode, upon cycling. Also, sulfur quantification in the lithium anode reveals that a factor of 10 less sulfur is found compared to a DOL:DME cell, which is in agreement with only minimal polysulfide shuttling (Figures S12 and S13).
Discussion
Proposed Reaction Scheme
Based on the observations reported above, we propose the following reaction scheme. Only two electrochemical reactions are utilized with equilibrium potentials suggested by observations made in the GITT study, though a slow relaxation process confounds the potential for reaction 2. The first reaction, eq 1, is hypothesized to be the direct formation of a polysulfide species Li2Sn·(4–n)S (i.e., Li2S4) in a two phase process. The nominal stoichiometry Li2S4 is suggested based on Faraday’s law; the observation of moderate- to short-chain polysulfides in XAS; and the absence of other crystalline species in XRD. Eq 2 corresponds to the reduction of a polysulfide with some small but perhaps essential solubility in the electrolyte, here referred to as Li2S6 although LiS3 would also be possible. The remaining reactions are chemical in nature and do not require an electron transfer from the conducting network to proceed.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
We believe that the role of increasing temperature is to raise the concentration of a key reactant in the electrolyte, eq 2. This increased reactant concentration may be the result of higher solubility and/or increased driving force for the cleavage of a divalent polysulfide into a monovalent radical such as shown in eq 6. Nonetheless the radical concentration is still low, i.e., below the detection limit by XAS (Figure S11).
| 6 |
Kinetic Modeling
To examine the feasibility of the proposed scheme, a kinetic model was created and solved numerically to generate a voltage profile, track temporal speciation, and quantify reaction rates (see Figure S14). The intermediate Li2S3 was assumed to undergo rapid chemical reaction by eqs 3–5 to minimize the number of unknown parameters to be fit. Figure 4 illustrates the results for one set of rate constants for discharge at the C/30 rate. The model is able to recreate the voltage dip and higher voltage during the middle third of discharge as reported experimentally. Key observations from this simulation are that sulfur lingers and in some conditions regenerates during the middle third of discharge where the electrode potential rises near or above the standard potential for reaction 1.
Figure 4.
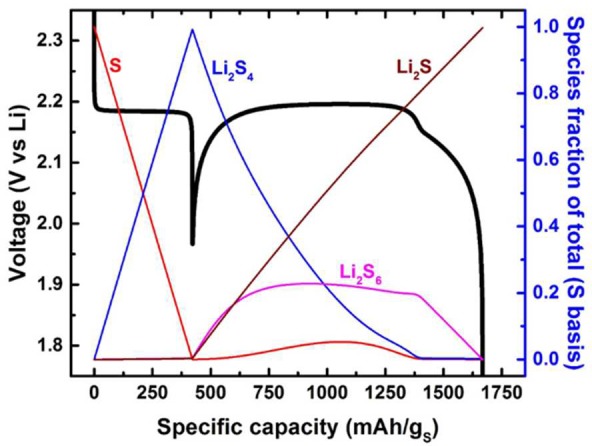
Simulated voltage profile using a kinetic model with a set of rate constants for discharge at the C/30 rate (1C = 1675 mAgs−1). The colored lines indicate the simulated evolution of each sulfur species as a function of specific capacity.
Herein lies the counterintuitive nature to this new pathway. The first reaction is more facile than the rest and thus controls the initial discharge voltage. This is seen in Figure 1 and Figure 2 where there is less polarization during the initial discharge. However, once reaction 1 has consumed a majority of sulfur, another reaction must carry the current. Our proposed scheme suggests that the equilibrium established by reactions 2 and 4 will generate a very small, but finite, quantity of a soluble polysulfide species from the existing Li2S4 generated by reaction 1. These polysulfides are referred to here as Li2S6, but others are also possible. The dip observed during discharge is the overpotential necessary to drive reactions 2 and 3. This then generates greater quantities of Li2S6, lowering the overpotential for further production. This autocatalytic type behavior results in a quick rise in the potential if the electrolyte can solubilize the important redox mediator. We believe that the solution concentration of the latter is the difference between room temperature and moderate temperature electrochemistry. Once sufficient polysulfide solubility is reached, the redox mediation of S8 and Li2S4 allows for high specific capacities to be accessed with minimal overpotential.
The question remains as to why eq 1 is favored in the ACN–TTE class of electrolytes over the formation of long-chain polysulfides observed in traditional catholyte electrolytes such as DOL:DME, eqs 7 and 8.38
| 7 |
| 8 |
We hypothesize that the dramatically reduced solubility of both elemental sulfur and polysulfides hinders the reaction kinetics of the traditional pathway to the point where eq 1 is favored. The competition between a solution pathway and a quasi-solid-state pathway likely always exists. In DOL:DME, the solution pathway kinetics are at least an order of magnitude more facile and thus the mixed potential is set by the solution pathway shown in eqs 7 and 8. Thus, a highly sloping regime is observed. Conversely in an electrolyte system that minimizes the solution reduction of S8 to Li2S8 and all the following redox mediation, we observe a classic two-phase reaction for the first 2.2 V plateau defined by eq 1. At the latter stages of reduction, the potential is again mixed and is therefore lower than the 2.3 V expected from eq 2 alone.
The variation in solution concentration of Li2S6 is observed as the voltage rise after the dip and the increased overpotential at the end of discharge. The voltage response in the regions dominated by S and Li2S4 suggests little dynamic change in solution concentration (i.e., rapid dissolution and precipitation at the solubility limit). Alternatively, the process proceeds in the solid-state via a chemical transformation absent of concentration gradients. We have used the term quasi-solid-state to describe this reaction. XRD and XAS studies leave unanswered questions to the exact speciation and mechanism. However, we believe that this unique pathway will be more completely elucidated through future complementary operando experiments.
Conclusions
In the Li–S battery, the use of sparingly solvating electrolytes provides a unique approach to control reactant distribution and hence the electrochemistry of the cell, while suppressing the solubility of polysulfides. While such sparingly solvating systems such as the ACN2TFSI–TTE examined here are effectively “nonsolvent” electrolytes at room temperature—which results in significant cell polarization—moderately raising the temperature promotes the solubility of key reaction intermediates. It enables operation at practical rates with minimal overpotential. More importantly, the electrochemical profile is profoundly changed by comparison to that observed in fully solvating electrolytes such as the well-known DOL:DME system. While lithium sulfide, Li2S, is still formed on discharge and consumed on charge as proven by operando XRD measurements, the very low but non-negligible solubility of polysulfides in the ACN2TFSI–TTE electrolyte fundamentally alters the pathway to form lithium sulfide. This gives rise to a quasi-solid-state reaction and minimal polysulfide shuttling. The initial formation of intermediate- to short-chain polysulfides is also in sharp contrast to a typical solvated electrolyte system where long-chain polysulfides are favored at the start. We propose that this owes to a change in relative kinetic rates for competing reactions, because the dramatically reduced solubility of both elemental sulfur and polysulfides hinders the reaction kinetics of the traditional pathway. Pairing sparingly solvating electrolytes with a protected Li-metal anode is the next step in the path to mitigate the polysulfide shuttle effect, and reach a high energy density and long-lived Li–S battery by minimizing electrolyte volume and polysulfide crossover. More generally, however, this discovery opens the door to tailored design based on modifying the electrolyte local environment to tune and ultimately control the reaction pathway of many precipitation–dissolution chemistries.
Acknowledgments
We gratefully acknowledge Dr. Larry Curtiss, Dr. Marine Cuisinier, Dr. Brian Adams, Dr. Tylan Watkins, Dr. Heng-Liang Wu, Dr. Kim See, Dr. Kah Chun Lau, Dr. Hao Wang, Mengyao Gao, and Dr. Zhengcheng Zhang for helpful discussions. The authors thank the staff at 9 BM for help with XAS studies. This work was supported as part of the Joint Center for Energy Storage Research, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences. The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan. http://energy.gov/downloads/doe-public-access-plan.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00123.
Experimental details, electrochemical studies (E/S ratio calculation, charge/discharge profiles, cycle performance and GITT study), ionic conductivity, polysulfide solubility measurement, operando XAS/XRD studies, SEM/EDX study, and a kinetic model (PDF)
Author Contributions
‡ These authors (C.-W.L. and Q.P.) contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Bruce P. G.; Freunberger S. A.; Hardwick L. J.; Tarascon J.-M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 2012, 11, 19–29. 10.1038/nmat3191. [DOI] [PubMed] [Google Scholar]
- Manthiram A.; Fu Y.; Chung S.-H.; Zu C.; Su Y.-S. Rechargeable Lithium–Sulfur Batteries. Chem. Rev. 2014, 114, 11751–11787. 10.1021/cr500062v. [DOI] [PubMed] [Google Scholar]
- Cuisinier M.; Cabelguen P. E.; Adams B. D.; Garsuch A.; Balasubramanian M.; Nazar L. F. Unique behaviour of nonsolvents for polysulphides in lithium-sulphur batteries. Energy Environ. Sci. 2014, 7, 2697–2705. 10.1039/C4EE00372A. [DOI] [Google Scholar]
- Urbonaite S.; Poux T.; Novák P. Progress Towards Commercially Viable Li–S Battery Cells. Adv. Energy Mater. 2015, 5, 1500118. 10.1002/aenm.201500118. [DOI] [Google Scholar]
- Eroglu D.; Zavadil K. R.; Gallagher K. G. Critical Link between Materials Chemistry and Cell-Level Design for High Energy Density and Low Cost Lithium-Sulfur Transportation Battery. J. Electrochem. Soc. 2015, 162, A982–A990. 10.1149/2.0611506jes. [DOI] [Google Scholar]
- Nelson J.; Misra S.; Yang Y.; Jackson A.; Liu Y.; Wang H.; Dai H.; Andrews J. C.; Cui Y.; Toney M. F. In Operando X-ray Diffraction and Transmission X-ray Microscopy of Lithium Sulfur Batteries. J. Am. Chem. Soc. 2012, 134, 6337–6343. 10.1021/ja2121926. [DOI] [PubMed] [Google Scholar]
- Diao Y.; Xie K.; Xiong S.; Hong X. Analysis of Polysulfide Dissolved in Electrolyte in Discharge-Charge Process of Li-S Battery. J. Electrochem. Soc. 2012, 159, A421–A425. 10.1149/2.060204jes. [DOI] [Google Scholar]
- Nazar L. F.; Cuisinier M.; Pang Q. Lithium-sulfur batteries. MRS Bull. 2014, 39, 436–442. 10.1557/mrs.2014.86. [DOI] [Google Scholar]
- Hagen M.; Hanselmann D.; Ahlbrecht K.; Maça R.; Gerber D.; Tübke J. Lithium–Sulfur Cells: The Gap between the State-of-the-Art and the Requirements for High Energy Battery Cells. Adv. Energy Mater. 2015, 5, 1401986. 10.1002/aenm.201401986. [DOI] [Google Scholar]
- Mikhaylik Y. V.; Akridge J. R. Polysulfide Shuttle Study in the Li/S Battery System. J. Electrochem. Soc. 2004, 151, A1969–A1976. 10.1149/1.1806394. [DOI] [Google Scholar]
- Yu X.; Pan H.; Zhou Y.; Northrup P.; Xiao J.; Bak S.; Liu M.; Nam K.-W.; Qu D.; Liu J.; Wu T.; Yang X.-Q. Direct Observation of the Redistribution of Sulfur and Polysufides in Li–S Batteries During the First Cycle by In Situ X-Ray Fluorescence Microscopy. Adv. Energy Mater. 2015, 5, 1500072. 10.1002/aenm.201500072. [DOI] [Google Scholar]
- Peng H.-J.; Huang J.-Q.; Liu X.-Y.; Cheng X.-B.; Xu W.-T.; Zhao C.-Z.; Wei F.; Zhang Q. Healing High-Loading Sulfur Electrodes with Unprecedented Long Cycling Life: Spatial Heterogeneity Control. J. Am. Chem. Soc. 2017, 10.1021/jacs.6b12358. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Curtiss L. A.; Zavadil K. R.; Gewirth A. A.; Shao Y.; Gallagher K. G. Sparingly Solvating Electrolytes for High Energy Density Lithium–Sulfur Batteries. ACS Energy Lett. 2016, 1, 503–509. 10.1021/acsenergylett.6b00194. [DOI] [Google Scholar]
- Dokko K.; Tachikawa N.; Yamauchi K.; Tsuchiya M.; Yamazaki A.; Takashima E.; Park J.-W.; Ueno K.; Seki S.; Serizawa N.; Watanabe M. Solvate Ionic Liquid Electrolyte for Li–S Batteries. J. Electrochem. Soc. 2013, 160, A1304–A1310. 10.1149/2.111308jes. [DOI] [Google Scholar]
- Ueno K.; Park J. W.; Yamazaki A.; Mandai T.; Tachikawa N.; Dokko K.; Watanabe M. Anionic Effects on Solvate Ionic Liquid Electrolytes in Rechargeable Lithium-Sulfur Batteries. J. Phys. Chem. C 2013, 117, 20509–20516. 10.1021/jp407158y. [DOI] [Google Scholar]
- Seo D. M.; Borodin O.; Han S.-D.; Boyle P. D.; Henderson W. A. Electrolyte Solvation and Ionic Association II. Acetonitrile-Lithium Salt Mixtures: Highly Dissociated Salts. J. Electrochem. Soc. 2012, 159, A1489–A1500. 10.1149/2.035209jes. [DOI] [Google Scholar]
- Yamada Y.; Furukawa K.; Sodeyama K.; Kikuchi K.; Yaegashi M.; Tateyama Y.; Yamada A. Unusual Stability of Acetonitrile-Based Superconcentrated Electrolytes for Fast-Charging Lithium-Ion Batteries. J. Am. Chem. Soc. 2014, 136, 5039–5046. 10.1021/ja412807w. [DOI] [PubMed] [Google Scholar]
- Yamin H.; Gorenshtein A.; Penciner J.; Sternberg Y.; Peled E. Lithium Sulfur Battery: Oxidation/Reduction Mechanisms of Polysulfides in THF Solutions. J. Electrochem. Soc. 1988, 135, 1045–1048. 10.1149/1.2095868. [DOI] [Google Scholar]
- Cheon S.-E.; Ko K.-S.; Cho J.-H.; Kim S.-W.; Chin E.-Y.; Kim H.-T. Rechargeable Lithium Sulfur Battery: I. Structural Change of Sulfur Cathode During Discharge and Charge. J. Electrochem. Soc. 2003, 150, A796–A799. 10.1149/1.1571532. [DOI] [Google Scholar]
- Lu Y.-C.; He Q.; Gasteiger H. A. Probing the Lithium–Sulfur Redox Reactions: A Rotating-Ring Disk Electrode Study. J. Phys. Chem. C 2014, 118, 5733–5741. 10.1021/jp500382s. [DOI] [Google Scholar]
- Suo L. M.; Hu Y. S.; Li H.; Armand M.; Chen L. Q. A new class of Solvent-in-Salt electrolyte for high-energy rechargeable metallic lithium batteries. Nat. Commun. 2013, 4, 1481. 10.1038/ncomms2513. [DOI] [PubMed] [Google Scholar]
- Pang Q.; Kundu D.; Cuisinier M.; Nazar L. F. Surface-enhanced redox chemistry of polysulphides on a metallic and polar host for lithium-sulphur batteries. Nat. Commun. 2014, 5, 4759. 10.1038/ncomms5759. [DOI] [PubMed] [Google Scholar]
- Weng W.; Pol V. G.; Amine K. Ultrasound assisted design of sulfur/carbon cathodes with partially fluorinated ether electrolytes for highly efficient Li/S batteries. Adv. Mater. 2013, 25, 1608–1615. 10.1002/adma.201204051. [DOI] [PubMed] [Google Scholar]
- Shin E. S.; Kim K.; Oh S. H.; Cho W. I. Polysulfide dissolution control: the common ion effect. Chem. Commun. 2013, 49, 2004–2006. 10.1039/C2CC36986A. [DOI] [PubMed] [Google Scholar]
- Yamada Y.; Yamada A. Review-Superconcentrated Electrolytes for Lithium Batteries. J. Electrochem. Soc. 2015, 162, A2406–A2423. 10.1149/2.0041514jes. [DOI] [Google Scholar]
- Zhang S.; Ueno K.; Dokko K.; Watanabe M. Recent Advances in Electrolytes for Lithium-Sulfur Batteries. Adv. Energy Mater. 2015, 5, 1500117. 10.1002/aenm.201500117. [DOI] [Google Scholar]
- Park J. W.; Ueno K.; Tachikawa N.; Dokko K.; Watanabe M. Ionic Liquid Electrolytes for Lithium-Sulfur Batteries. J. Phys. Chem. C 2013, 117, 20531–20541. 10.1021/jp408037e. [DOI] [Google Scholar]
- Yuan L.; Feng J.; Ai X.; Cao Y.; Chen S.; Yang H. Improved dischargeability and reversibility of sulfur cathode in a novel ionic liquid electrolyte. Electrochem. Commun. 2006, 8, 610–614. 10.1016/j.elecom.2006.02.007. [DOI] [Google Scholar]
- Park J. W.; Yamauchi K.; Takashima E.; Tachikawa N.; Ueno K.; Dokko K.; Watanabe M. Solvent Effect of Room Temperature Ionic Liquids on Electrochemical Reactions in Lithium-Sulfur Batteries. J. Phys. Chem. C 2013, 117, 4431–4440. 10.1021/jp400153m. [DOI] [Google Scholar]
- Huang J.-Q.; Liu X.-F.; Zhang Q.; Chen C.-M.; Zhao M.-Q.; Zhang S.-M.; Zhu W.; Qian W.-Z.; Wei F. Entrapment of sulfur in hierarchical porous graphene for lithium–sulfur batteries with high rate performance from – 40 to 60 °C. Nano Energy 2013, 2, 314–321. 10.1016/j.nanoen.2012.10.003. [DOI] [Google Scholar]
- Ji X.; Lee K. T.; Nazar L. F. A highly ordered nanostructured carbon-sulphur cathode for lithium-sulphur batteries. Nat. Mater. 2009, 8, 500–506. 10.1038/nmat2460. [DOI] [PubMed] [Google Scholar]
- Zhang S. S. Effect of Discharge Cutoff Voltage on Reversibility of Lithium/Sulfur Batteries with LiNO3-Contained Electrolyte. J. Electrochem. Soc. 2012, 159, A920–A923. 10.1149/2.002207jes. [DOI] [Google Scholar]
- Gotoh N.; Hayasaka M.. Glass-ceramics. U.S. Patent 5028567 A, Jul 2, 1991.
- See K. A.; Wu H. L.; Lau K. C.; Shin M.; Cheng L.; Balasubramanian M.; Gallagher K. G.; Curtiss L. A.; Gewirth A. A. Effect of Hydrofluoroether Cosolvent Addition on Li Solvation in Acetonitrile-Based Solvate Electrolytes and Its Influence on S Reduction in a Li-S Battery. ACS Appl. Mater. Interfaces 2016, 8, 34360–34371. 10.1021/acsami.6b11358. [DOI] [PubMed] [Google Scholar]
- Ruetschi P. Review on the lead—acid battery science and technology. J. Power Sources 1977, 2, 3–120. 10.1016/0378-7753(77)85003-9. [DOI] [Google Scholar]
- Waluś S.; Barchasz C.; Bouchet R.; Leprêtre J.-C.; Colin J.-F.; Martin J.-F.; Elkaïm E.; Baehtz C.; Alloin F. Lithium/Sulfur Batteries Upon Cycling: Structural Modifications and Species Quantification by In Situ and Operando X-Ray Diffraction Spectroscopy. Adv. Energy Mater. 2015, 5, 1500165. 10.1002/aenm.201500165. [DOI] [Google Scholar]
- Lowe M. A.; Gao J.; Abruna H. D. Mechanistic insights into operational lithium-sulfur batteries by in situ X-ray diffraction and absorption spectroscopy. RSC Adv. 2014, 4, 18347–18353. 10.1039/c4ra01388c. [DOI] [Google Scholar]
- Kumaresan K.; Mikhaylik Y.; White R. E. A Mathematical Model for a Lithium–Sulfur Cell. J. Electrochem. Soc. 2008, 155, A576–A582. 10.1149/1.2937304. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.