

Abstract
Defining specific pathways for efficient heat transfer from protein–solvent interfaces to their active sites represents one of the compelling and timely challenges in our quest for a physical description of the origins of enzyme catalysis. Enzymatic hydrogen tunneling reactions constitute excellent systems in which to validate experimental approaches to this important question, given the inherent temperature independence of quantum mechanical wave function overlap. Herein, we present the application of hydrogen–deuterium exchange coupled to mass spectrometry toward the spatial resolution of protein motions that can be related to an enzyme’s catalytic parameters. Employing the proton-coupled electron transfer reaction of soybean lipoxygenase as proof of principle, we first corroborate the impact of active site mutations on increased local flexibility and, second, uncover a solvent-exposed loop, 15–34 Å from the reactive ferric center whose temperature-dependent motions are demonstrated to mirror the enthalpic barrier for catalytic C–H bond cleavage. A network that connects this surface loop to the active site is structurally identified and supported by changes in kinetic parameters that result from site-specific mutations.
Short abstract
Comparative analysis of thermal activation barriers of mass spectrometry-based hydrogen−deuterium exchange and catalytic proton-coupled electron transfer reveals a spatially resolved relationship between distal loop motions and active site catalysis.
One of the unmet challenges in protein engineering is the ability to produce highly active catalysts that achieve rate accelerations approaching those of native enzymes, upward of 1025-fold in relation to the uncatalyzed reaction.1 Despite promising improvements from the combined application of computation and directed evolution,2 designed biocatalysts are generally only weakly effective.3−5 The differences in reactivity and thermal stability between natural and artificial enzymes underscore the gaps in our understanding of the physical basis for enzymatic catalysis. The large size of enzymes, together with the frequent sequestration of their active sites from bulk solvent, raises the important question of the role of the entire protein in catalysis, in particular regions that are far from the active site and in contact with solvent.6
In the context of the inherent temperature independence of quantum mechanical wave function overlap, experimentally observed thermal excitation barriers in enzyme-based hydrogen tunneling reactions must arise from the heavy atoms of the protein.7 Such systems offer the opportunity to connect specific pathways within the protein matrix to the thermal transduction of the chemical step(s).8,9 In this context, both the ability to stably label C–H bonds with deuterium and the sensitivity of C–H activation reactions to isotopic labeling at the reactive bond of substrate provide additional kinetic parameters, including the kinetic isotope effect (Dkcat = kcat(H)/kcat(D)) and the temperature dependence of the kinetic isotope effect (ΔEa = Ea(D) – Ea(H)); together with the thermal activation barrier for proton transfer (Ea(H)), these parameters give further insight into the reaction coordinate and enable separation of the contributions from local, isotope-sensitive protein motions, related to ΔEa, and more global, isotope-insensitive motions that determine the catalytic Ea(H).9 Soybean lipoxygenase-1 (SLO-1) has emerged as a paradigmatic system for thermally activated, multidimensional tunneling models,7,9−11 in part because of a fully rate limiting hydrogen atom abstraction from linoleic acid substrate (Scheme 1) that displays a very large, nonclassical Dkcat (∼80) that is nearly temperature independent (ΔEa = Ea(D) – Ea(H) = 0.9 kcal/mol).12 An additional compelling feature of SLO-1 is the sensitivity of its defining kinetic parameters to a series of strategically placed site specific mutants.
Scheme 1. Rate-Limiting Hydrogen Atom Abstraction Process in SLO-1.

Hydrogen abstraction occurs at the active site via a PCET reaction,32 from the substrate, linoleic acid, to the ferric hydroxide cofactor. The relative positioning of residues discussed in the text, numbering of backbone carbons, and kinetic parameters for WT SLO are shown for reference.
Modern techniques with the potential to spatially resolve thermally activated heavy atom motions within a protein include NMR spectrometry,13 X-ray crystallography,14,99 and hydrogen–deuterium exchange (HDX15−17). SLO-1 is not readily amenable to NMR due to its large size (839 amino acid residues; 94.4 kDa) and paramagnetic active site. Additionally, previously published cryogenic X-ray structures of SLO-1 have failed to uncover a structural network for catalytically relevant thermal activation. We now present a study of hydrogen–deuterium exchange mass spectrometry (HDXMS) under conditions that enable a temperature-dependent analysis of protein flexibility in a spatially resolved manner. The HDX process involves chemical exchange of peptidyl backbone amide hydrogens for deuterons when the target protein is incubated in D2O. While there are site specific differences in intrinsic exchange at a given peptide bond attributed to inductive effects from amino acid content,18 these differences are expected to be minor in relation to the degree of exposure or protection at each position to solvent. Within the EX-2 regime, measured rate constants (kHDX) can be represented as the product of the intrinsic chemical exchange rate (kint) and the equilibrium constant for the interconversion between closed and open protein states (Kop = kopen/kclose). According to this formulation, the major determinant of spatial differences in HDX is the time that each segment of protein remains in an open, partially unfolded and solvent exposed state, as determined by the magnitude of the Kop. NMR and mass spectrometry (MS) are two conventional tools for detection of amide HDX. While NMR offers exceptional residue and temporal resolution, MS provides a cost-effective (e.g., low sample amounts, no isotope labeling), high throughput analytical tool, enabling examination of aggregate behavior within defined segments of the protein, including large (and even paramagnetic) systems, such as SLO-1 described herein.
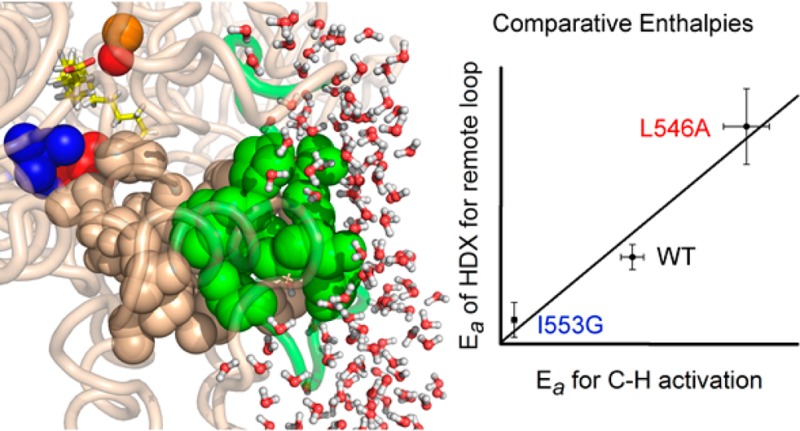
Previous HDXMS reports have demonstrated its structural/dynamic sensitivity to protein–protein interactions, enzyme activation, ligand/inhibitor binding, and/or long-range allosteric effects (for examples see references (19−25)). In contrast to these and numerous other hydrogen exchange studies, our approach interrogates the time and temperature dependencies as a function of protein mutation, uncovering a striking correlation between the activation enthalpies for H-tunneling26,27 and the apparent rates of HDXMS within a remote loop of the protein. These data reveal a hitherto undetected relationship between the motions of a spatially resolved, solvent accessible region of an enzyme and its active site, providing compelling experimental evidence in support of distal, likely solvent slaved,6 protein structural fluctuations that may modify the thermal barriers of catalytic bond cleavage.
Results
For the conditions of the current study, EX-2 HDXMS behavior was both expected and corroborated by the patterns seen in the mass spectrometric analyses (see for examples, Figure S1, and references (15−17)). Of the more than 250 peptides, resulting from proteolytic cleavage of SLO-1, 46 reproducible nonoverlapping peptides of various lengths (average 16 amino acid residues) were selected for extensive quantitation (Figure S2). These peptides represent 89% coverage of the entire protein sequence. Time-resolved amide HDX experiments (between 10 s and 4 h) were first analyzed for the WT enzyme at 10, 20, 25, 30, and 40 °C. Experiments were conducted with Fe-containing protein, in the absence of substrate, to avoid trends that could arise from bound substrate such as site specific protection that may be dependent on temperature and mutation as well as to preclude contributions from reversible binding of substrate due to its moderately weak dissociation constant (Kd ∼ 5–10 μM27). Chromatographic retention times for each peptide were constant over all time points and temperatures and were, for the most part, independent of mutation (Table S1). Each trace was corrected for back-exchange values (Table S2). Controls with WT SLO-1, including activity assays and CD spectra, were conducted to demonstrate that the protein maintains its activity and structure under the conditions of the experiment (Supporting Information section I.k and Figure S3).
Visual Pattern Recognition Offers a Means To Characterize Large HDXMS Datasets
HDX traces of the WT SLO-1 were grouped into four classes of temperature effects, characterized on the basis of distinctly different visual patterns (Figure 1). There are few reports of the temperature response of HDXMS,28,29 and pattern recognition offers a means of readily classifying the large body of data that emerges from studies of proteins at variable temperature. In general, HDXMS is analyzed as a multiexponential kinetic process that encompasses fast, intermediate, and slow exchange rate constants with the last often invisible within the dynamic range of the performed experiments. From the patterns in Figure 1, four classes of behavior emerge that extend from full protection against exchange to rapid, temperature-independent exchange, encompassing Class I (Figure 1a), no detectable exchange (<10%) at the longest time and highest temperature; Class II (Figure 1b), HDX traces that are slowly exchanging and fail to reach an apparent plateau value at long times and high temperature; Class IIIA (Figure 1c), faster exchanging peptides that converge at similar values at 4 h; and Class IIIB (Figure 1d), traces that rapidly coalesce at their fullest extent of exchange at all temperatures. Among these four types of behavior, Class IIIA and Class II provide a measure of the temperature dependence of the fast or the fast and intermediate exchange processes, respectively, within a central swath of the protein. 44% of total peptides analyzed were categorized with Class II or IIIA behavior. When color coding, to indicate these four trends, is mapped onto the 3D model of SLO-1 (Figure 1e), the backbone of SLO-1 shows a highly nonuniform pattern with the region of fastest rates of exchange (red and pink) residing in the lower half of the protein (as visualized). We note that the lipoxygenase enzyme from plants and mammalian sources is composed of two domains: an N-terminal β-barrel of ca. 150 amino acids, which is responsible for lipid/membrane binding and protein stability but is not required for lipid peroxidation,30,31 and a larger, ca. 690 amino acid domain (in SLO-1) containing the primarily α-helical, catalytic C-terminal domain (displayed in Figure 1e); the latter is the focus of the current study.
Figure 1.

HDXMS temperature dependence classification for WT SLO-1. The left panels (a–d) display representatives for the four classes of temperature responsivity. HDXMS data, presented as percent of deuterium exchange, were collected over five temperatures and are represented by the color code in panels a–d as follows: 10 (blue), 20 (green), 25 (gray), 30 (orange), and 40 °C (red). The four classes were plotted onto the 1.4 Å SLO-1 model (PDB: 3PZW) in panel e, according to this color code: Class I (black), Class II (brown), Class IIIA (red), and Class IIIB (pink). The iron cofactor is represented as an orange sphere. The represented catalytic domain of SLO-1 (145–839) is the focus of this study. Gray sticks represent uncovered structure.
Mutation-Dependent HDXMS Effects
Having established a systematic method for HDX analysis using the WT protein, we proceeded to search for changes that result from single site mutants with known impacts on the properties of C–H activation. Reduction in the size of the side chain of I553, which resides one helix turn away from the active site L546 (cf. Figure 2a), has previously been shown to lead to a strong increase in the temperature dependence of the kinetic isotope effect (ΔEa; Table S3). We initiated HDXMS using the 553 mutant with the greatest kinetic deviations, I553G. As defined by WT, the same set of nonoverlapping peptides (Figure S2) was used to carry out a spatially resolved analysis of mutational effects on HDXMS behavior. The sequence coverages for the catalytic domain of WT and I553G are 92 and 77%, respectively. Important controls were conducted to demonstrate that the retention times and back-exchange values for each peptide analyzed are identical (with the exception of the peptide containing the mutation at position 553) regardless of time point or temperature (cf. Tables S1 and S2). As discussed in detail below, the impact of I553G is seen to manifest itself in a regionally defined manner, as changes either in the extent of HDX or in the temperature dependence of the weighted average rate constants for HDX, Figures 2 and 4, respectively.
Figure 2.

Active site (proximal) peptides (in yellow) that exhibit mutation-induced increases in the percent of deuterium exchange at 4 h. (a) The highlighted region of the protein is shown in the yellow box. The key active site residues, undergoing mutation in this study, are shown as space filling models: L546 (red) and I553 (blue). The residues participating in noncovalent interactions between 555–565 and 414–423 are shown as sticks. (b) HDX traces for the representative peptide, 555–565, at 30 °C with three I553 variants. (c) Percent exchange at 4 h from panel b is plotted versus the van der Waals volume of the 553 side chain. (d) Percent exchange at 4 h from panel b is plotted as a correlation to the temperature dependence of the catalytic Dkcat (ΔEa; Table S3). (e) Differences in percent exchange for 555–565 between WT and I553G at 4 h are observed at all temperatures.
Figure 4.

Peptides (in orange) remote from the active site that exhibit mutation-induced changes in the enthalpic barrier for HDX rates (EaHDX(avg)) (a). The enzyme structure is shown rotated 180° relative to Figure 2a. The pink colored peptides (297–305 and 306–316) have altered rates of exchange, but the EaHDX(avg) values are the same for all SLO-1 variants (Figure S8 and Table S5). Peptides displayed in orange, 317–334 (b and d) and 284–299 (c and e) are those that exhibit mutant variable EaHDX(avg). In panels b and c, Arrhenius-like plots of the weighted average exchange rates, ln(kHDX(avg)), for WT, I553G, and L546A are compared. The HDX traces for these peptides are found in Figure S7. In panels d and e, the relationship between EaHDX(avg) and the enthalpic barrier for hydrogen tunneling (Ea(H)) is shown.
Increased Active Site Flexibility upon Mutation
Focusing on the extent of exchange at 4 h for I553G at 30 °C, three peptides that deviate by 10% or greater from WT are identified; each of these is proximal to the active site: peptides 414–423, 541–554, and 555–565 (Figure 2a and Table S4). The largest deviation from WT is observed for peptide 555–565 (Figure 2b), which is linked to and structurally perpendicular to the peptide containing the mutation. To explore this behavior further, the study of I553G was extended to include I553L and I553A (Figures 2b and S4). An earlier, low temperature X-ray analysis of the same series of 553 variants led to the conclusion of little or no changes away from the site of mutation; no effort was made at that time to detect subtle structural changes.27 By contrast, using HDXMS there is a strong correlation between the extent of exchange at 30 °C and the volume of the side chain at position 553 (Figures 2c and S4). In light of the previously described trend between the bulk of the side chain and the magnitude of ΔEa,27 a relationship between the extent of HDX and ΔEa emerges, Figure 2d. The identified changes in the extent of HDX with I553G at 30 °C are retained at all temperatures (Figure 2e) and remain localized around the mutation site (i.e., at peptides 414–423 and 541–554 (Figure S4) and 555–565 (Figure 2)). As discussed extensively in the literature, enlarged ΔEa values that result from mutation can be explained by an elongation of hydrogen donor–acceptor distances (DADs) from the dominant equilibrium distance that precedes H-tunneling in WT (2.8 Å11,32) that is accompanied by enhanced DAD sampling to recapture catalytically efficient hydrogen transfer rates; successful modeling of this behavior focuses on local, active site motions on the nanosecond to picosecond time scale.26,27,33
We initially thought that some of the observed increase in HDX with decreasing side chain bulk at 553 might also reflect an increased hydration of the active site. To sensitively reveal changes in water occupancy, we therefore performed room temperature X-ray crystallography of the WT and I553G proteins, obtaining high resolution (1.7–1.8 Å) crystal structures (Figure S5). Room temperature data collection enables us to capture catalytically relevant alternate side chain conformations that are often absent under conventional cryogenic conditions. While there are no large backbone or side chain conformational differences when the room temperature X-ray structures of WT and I553G are compared, we did detect multiple conformations of some key hydrophobic active site residues that modulate the size and shape of the active site cavity (Figure S5). Subsequent modeling of the substrate into this I553G structure in the same position as docked for the WT structure shows a very significant enlargement of the active site volume, from 47.4 Å3 to 86.3 Å3. From these datasets, an isomorphous difference map (WT – I553G) was calculated using phases from the room temperature WT model (Figure 3); despite the near doubling in the active site volume for I553G, the isomorphous map reveals no significant difference in density that would implicate increased water occupancy, including diffuse water molecules, in the active site.34 We conclude that the changes in the extent of HDX arising from mutation at position 553 must be due to a loosening of active site constraints that enhances local flexibility. This is supported by an analysis of B-factors calculated for the I553G structure at room temperature, where only the region surrounding the active site displays significant differences between WT and this variant (Figure S6). The present combination of HDXMS and room temperature X-ray studies provides an important corroboration of the earlier kinetic evidence for altered active site dynamics among the series of hydrophobic side chain mutations introduced into position 553 of SLO-1.27
Figure 3.
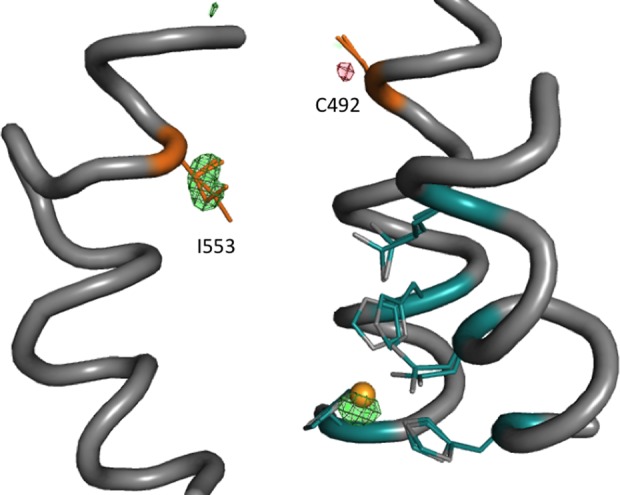
Isomorphous difference maps of WT – I553G room temperature structures. The difference map is generated from the individual room temperature crystal structures of WT and I553G (Figure S5). Shown in red (negative) and green (positive) is the WT – I553G isomorphous difference map contoured at 0.33 e–/Å3. Ligands to the iron (gold sphere) are in teal, and residue 553 is in orange. The isomorphous difference map shows slightly higher and shifted iron occupancy in the WT structure and a small rotamer change in cysteine 492.
Thermal Activation of HDX at a Solvent-Exposed Surface Loop Mirrors the Enthalpic Barrier for Active Site Chemistry
We next turned to a detailed inspection of the impact of temperature on the comparative weighted average HDX rate constants (kHDX(avg)) for WT vs I553G, detecting six peptides that display a ≥2-fold rate acceleration at each temperature following mutation at I553 (Table S5). Two of these peptides originate from the aforementioned proximal site, corresponding to positions 541–554 and 555–565. The remaining four peptides reside within a site remote from the active site (at 284–299, 297–305, 306-316, and 317–334; Figure 4a). The HDX traces (Figure S7) from this remote region of the protein show comparable values for the percent HDX at 30 °C (Table S4), clearly distinguishing the impact of mutation on rate behavior from the percent exchange effects observed near the active site (Figure 2). The unique and particularly revealing aspects of these data manifest from a comparative analysis of the temperature dependence of the averaged rate constants for exchange (EaHDX(avg)). No significant differences in EaHDX(avg) between WT and I553G are seen either for the two proximal peptides or for two of the four remote peptides (297–305 and 306-316) with altered rates nor, in fact, for any of the remaining peptides analyzed within WT and I553G (see Figure S8 and Table S6). In marked contrast, a new property emerges when interrogating the two remote peptides with altered rates for HDXMS upon mutation: peptides 317–334 and, to a lesser extent, 284−299. As illustrated in Figures 4b and 4d for 317–334, the EaHDX(avg) values (5.0 and 0.7 kcal/mol for WT and I553G, respectively) match the directionality of the variances in the enthalpies that control catalysis: Ea(H) = 2.1 and 0.03 kcal/mol, respectively. A corresponding, though less dramatic behavior is observed for peptide 284–299 (Figures 4c and 4e), where EaHDX(avg) is seen to decrease from 11.6 kcal/mol for WT to 4.0 kcal/mol for I553G.
To further substantiate this unexpected and intriguing trend, we examined the time and temperature dependent analyses of HDX using a variant at L546, an active site residue that when mutated to alanine (i.e., L546A) generates many changes in its catalytic properties that include an elevated enthalpic barrier of catalysis (Ea(H)L546A = 4.1 kcal/mol; also see Extended Discussion in the Supporting Information).26 Using the same nonoverlapping peptides as defined in Figure S2 for WT, we achieve 88% coverage of the L546A catalytic domain. Inspection of kHDX(avg) for each of the L546A-derived peptides revealed four that are elevated ≥2-fold from WT at every temperature (Table S5). Significantly, not only do these four peptides correspond to the same remote peptides found to be altered in I553G but we observe values for EaHDX(avg) that are distinct from WT and map to the identical region highlighted above for I553G: namely, peptides 317–334 and 284–299. In both instances the magnitude of EaHDX(avg) (Figures 4b and 4c) is 14 kcal/mol, opposite in direction from I553G and greater than WT itself. Remarkably, once again the enthalpies derived from HDX (EaHDX(avg)) follow the same trend seen for Ea(H) (Figures 4d and 4e).
It is notable that the measured values of EaHDX(avg) for the surface loop (Figure 4) that contains peptides 317–334 and 284–299 are all smaller than the ca. 17 kcal/mol barrier reported for the intrinsic rate constant (kint) of deuterium exchange at a fully exposed peptide bond.18 This indicates that the temperature dependencies of the observed rates during HDXMS are being determined by (negative) enthalpic changes in Kop of varying magnitudes. Negative enthalpies of activation have been previously observed in the kinetic analysis of the temperature dependence of protein folding processes and rationalized via a dominance of the folding process by heat capacity effects in the unfolded ground state.35 A trend of enthalpic barriers of less than 17 kcal/mol during the analysis of HDXMS has already been documented, suggesting generalities in this type of behavior.29
It is also important to note that the loop including 317–334 and 284–299 resides in a region of SLO-1 remote from that ascribed to the substrate portal,36 indicating that the experimental correlations between EaHDX(avg) and Ea(H) are distinct from any motions that may be expected to accompany substrate binding. Temperature-dependent analyses of additional peptides in I553G and L546A that partially overlap with peptide 317–334 (Figure S8) parallel the trends in EaHDX(avg) and Ea(H) shown in Figure 4d. The cumulative data strongly support a relationship between the temperature dependence of the rate constants controlling HDX and those for catalysis within the specific region of protein represented by peptides 284–299 and 317–334. The atomic coordinates for side chains within these peptides lie 15 to 34 Å from the nearest atom of the L546 or I553 residues, signifying the long-range communication of mutations in the vicinity of the active site on the solvent exposed loop.
Identification of a Network for Communication between Peptides 284–299 and 317–334 and the Active Site of SLO-1
The WT SLO X-ray structure was examined, in an effort to locate a region of connectivity between the thermally activated remote loop (Figure 5a, orange) and the active site peptides that increase their percent exchange following mutation at position 553 (Figure 2a, yellow). We observe that the side chain of V750 is in van der Waals contact with I552, which further contacts the side chains of I553 and L546 (Figure 5a), whose previously characterized mutations form a basis for this study. Significantly, all of these positions (L546, I552, I553, and V750) adopt alternate conformations in the room temperature X-ray structure (Figure S5). Given the large number of hydrophobic interactions within the region of protein containing V750 and the remote loop 317–334, together with the fact that residues I552 and V750 are predicted to contact substrate (see ref (26) and Figure S5), we searched for a more isolated structural interaction to test for connectivity. A single hydrogen bonding pair composed of S749 and Y317 (Figure 5a) and located between V750 and the 317–334 loop was, thus, chosen to assess the impact of amino acid side chain changes in this region on catalytic activity.
Figure 5.
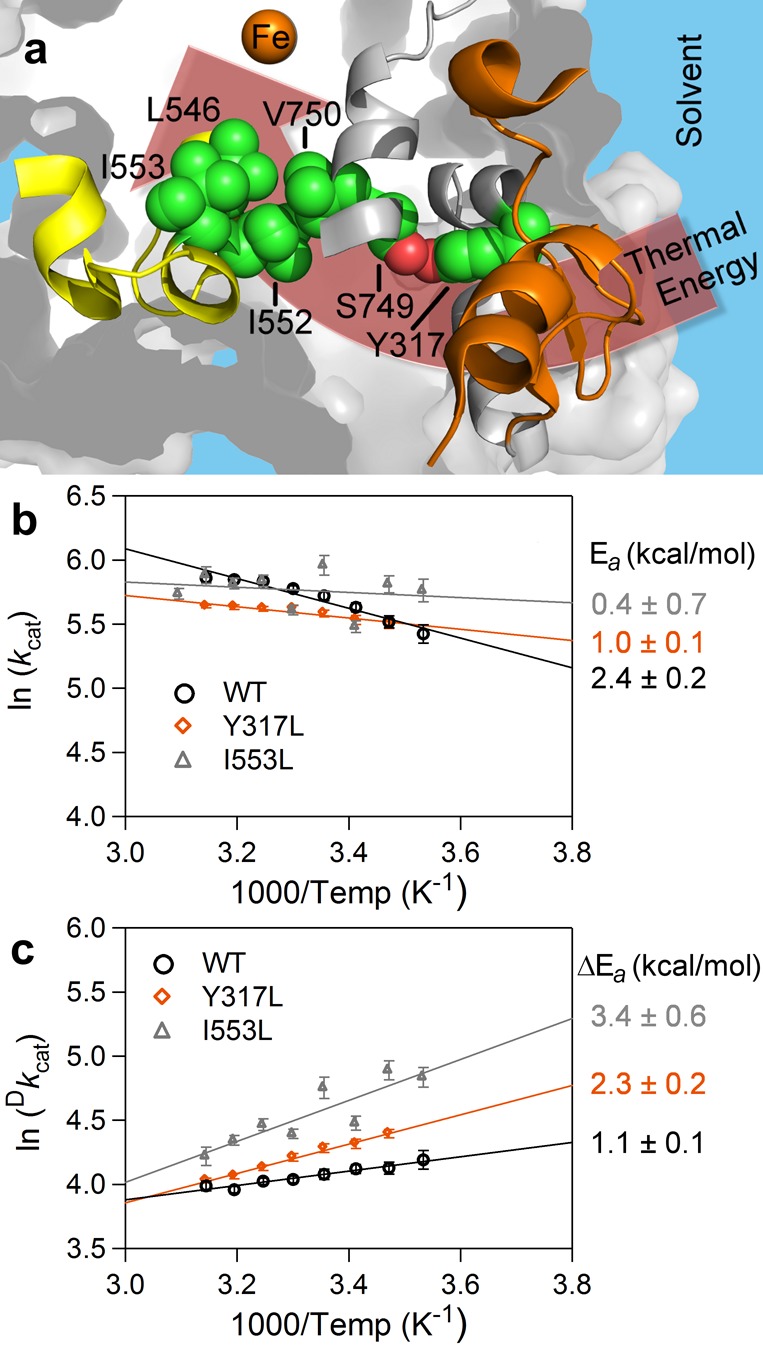
Network connecting the peptides affected by active site mutation in SLO-1 (a) and impact of mutation at Y317 on enthalpic parameters of catalysis (b and c). The active site and surface loop are color coded as yellow and orange, respectively, and the putative connectivity for thermal activation from solvent interactions at 284–299 and 317–334 (orange) to active site via a hydrogen bond between S749 and Y317 is also displayed. Notable side chains are represented in space-filling mode with carbon atoms colored in green and oxygen atoms colored in red. Note that I552 is in contact with V750; the latter is on the opposite side of the peptide backbone containing S749. S749 and V750 are adjacent to peptide 751–761 (gray ribbon), which also shows trends in EaHDX(avg) and Ea(H) for the lowest three temperatures (10, 20, and 25 °C) studied (Figure S8e and S8f). In panel b, ln kcat values are plotted versus 1000/temperature. The enthalpy of activation, Ea(H), for protio substrate, H31-linoleic acid, is determined by multiplying the slope of the linear fits by −R, where R is the ideal gas constant. The data, collected here, are WT (black circle) and Y317L (orange diamond). The data plotted for mutant, I553L (gray triangle), are reproduced from a previous report.27 The corresponding Ea values are listed in matching color to the right of the figure. In c, ln Dkcat values are plotted versus 1000/temperature. The data are represented as described in panel b. Kinetic data for other S749 and Y317 mutants are summarized in Table 1.
We first examined the impact of this serine–tyrosine hydrogen bond via generation of S749A and S749G, both of which lack the hydroxyl group of serine. The ca. 20% reduction in kcat exhibited in these S749 mutants is considered a minor effect, and the remaining parameters are almost within experimental error of WT (Table 1). S749 is located within a long α-helical domain spanning residues 740–755. While alanine has a high propensity for stabilizing α-helical structure, glycine is considered a “helix-breaker” in water,37 and altered kinetics were anticipated in this case. However, as indicated above, there are extensive hydrophobic interactions that lie between the helix 740–755 of the catalytic core and the surface loop (Figure S9a and S9b), and this feature may explain the retention of WT-like behavior in S749G. Glycine has previously been shown to sustain helical conformation in hydrophobic-rich environments, such as those found in transmembrane helices.38,39
Table 1. Kinetic Parameters for S749 and Y317 SLO-1 Mutants Reported Here.
| enzyme | kcat(H)a (s–1) | Ea(H)b (kcal/mol) | Dkcatc | ΔEad (kcal/mol) |
|---|---|---|---|---|
| WTe | 359 (7) | 2.4 (0.2) | 57 (2) | 1.1 (0.1) |
| S749A | 289 (7) | 1.8 (0.2) | 59 (2) | 1.3 (0.2) |
| S749G | 296 (10) | 1.7 (0.2) | 63 (3) | 1.0 (0.1) |
| Y317S | 330 (8) | 2.3 (0.2) | 64 (2) | 1.2 (0.2) |
| Y317L | 368 (8) | 1.0 (0.1) | 68 (2) | 2.3 (0.2) |
| Y317F | 331 (10) | 2.5 (0.3) | 63 (2) | 0.2 (0.1) |
Parameters recorded at 30 °C. All kcat values have been corrected for iron content, determined by the ferrozine assay (see the Supporting Information for details).
Determined from kcat values across 7 to 8 (depending on mutant) temperatures (10–45 °C); see Table S7 for complete kcat data.
Dkcat is the kinetic isotope effect on kcat at 30 °C.
ΔEa = Ea(D) – Ea(H). This is the temperature dependence on the kinetic isotope effect.
The parameters reported for WT here are slightly different from previous reports (see Table S3) due to a modification in the protein purification procedure.
We next turned to the introduction of mutations at position Y317 that eliminate the possibility of hydrogen bonding to S749. A range of variants was generated that included Y317F, Y317L, Y317A, and Y317G. In support of a critical role for a space filling side chain at position 317, substitution with Ala and Gly failed to produce detectable levels of expressed protein, precluding further characterization. Using Phe as a non-hydrogen bonding place holder, it can be seen that both kcat and Ea are similar to WT (Table 1); however, the ΔEa is found to be reduced by a significant amount, to 0.2 kcal/mol from 1.1 kcal/mol for this preparation of WT. Given the high sensitivity of the temperature dependence of the KIE to the initial positioning between bound substrate and the active site Fe(III)–OH and subsequent distance sampling,9,11,33 we consider this decrease in ΔEa as evidence for transmission of a structural and dynamical defect emanating from Phe at 317 to the catalytically competent enzyme–substrate complex. A minor displacement of the hydrophobic Phe side chain into its surrounding hydrophobic environment (Figure S9c) may be sufficient to produce the measured effect. The variant Y317L was then chosen as a compromise substitution that could retain sufficient bulk to stabilize the protein sufficiently for characterization, while minimizing its displacement into the surrounding hydrophobic milieu. Kinetic analysis of this variant reveals changes in both Ea (reduced from 2.4 (WT) to 1.0 kcal/mol) and ΔEa (increased from 1.1 (WT) to 2.3 kcal/mol) (Table 1 and Figures 5b and 5c). The trends in these kinetic parameters are very similar to those observed with an active site variant (I553L), 15 Å removed from Y317 (Figure 5a). Despite their distance from one another, both mutations (Y317L and I553L) appear equally capable of controlling long-range and active site flexibility, reflected in Ea and ΔEa, respectively.
One final variant, Y317S, was generated to assess the impact of a hydrophilic side chain of reduced size. In silico modeling of Y317S (Figure S9d) supported an increased cavity volume that would be sufficient to accommodate at least one new water molecule capable of sustaining a hydrogen bonded network between S749 and Y317S. The observed rescue of the thermal activation parameters by Y317S to that for WT (Table 1) supports the prediction of the restoration of a hydrogen bond between Y317S and S749. Similar water rescue has been previously observed, via a serine for tyrosine substitution, within the active site of ketosteroid isomerase.40
These extensive mutagenesis data highlight the importance of both local hydrophobic packing within the helix 740–755 and loop 317–334 and hydrogen bonding between Y317 and S749 (Figure S9e) in generating a pathway of thermal connectivity between the SLO-1 surface loop and its active site. We note that the generation of Y317L, 19 Å from the catalytic iron, is the first reported, non-substrate binding site mutation that influences active site dynamics in SLO-1, in support of the postulated protein network (Figure 5a).
Discussion
An important ongoing goal in the pursuit of the physical origins of enzymatic catalysis has been to identify, in as precise a manner as possible, local and remote protein motions that modulate the reaction barrier. The present work shows how a combined study of the time-, temperature-, and mutation-dependent properties of HDXMS can uncover regional differences in protein motions that can be correlated with the enthalpic barriers for kcat (Ea(H)) or for kinetic isotope effects (ΔEa). Of particular importance, this methodology has uncovered an unanticipated relationship between the enthalpic barrier (Ea(H)) for the hydrogen tunneling step in SLO-1 and the energetic barrier for activation of heavy atoms toward HDX within a distal protein loop region that is in direct contact with solvent (Figure 4).
In an early study, Tolman described the empirical Ea in chemical reactions as the difference in the total energy of all reactive states relative to the total energy of all available states.41 This interpretation has been extended42 to invoke a stochastic sampling of available protein states via a multidimensional conformational landscape,13,43,44 with catalysis resulting from the successful achievement of a subset of uniquely competent conformers.10 In evaluating the extent to which a statistical thermodynamic picture can explain experimentally derived values of activation energies in enzymatic C–H activation, it is important to consider the full range of factors that may contribute to temperature sensitivity. From the vibronically nonadiabatic proton-coupled electron transfer (PCET) theory,7,45 which describes the mechanism of H transfer in SLO-1 and many other C–H activation enzymes, the stochastic sampling of protein motions responsible for proper alignment of active site electrostatics and geometries, prior to hydrogen atom transfer, constitutes the reaction barrier. SLO-1, as a paradigmatic enzyme12 for modeling multidimensional, ground state hydrogen tunneling, thus offers an excellent system in which to interrogate the molecular origins of the enthalpic PCET reaction barrier. The rate expression for an enzymatic reaction, which is controlled by both a stochastic protein conformational sampling term and tunneling probability, can be formulated at a given temperature as
| 1 |
where Fconf represents the fraction of enzyme substates that lead to catalysis and ktun is the nonadiabatic, multidimensional barrier shown to be appropriate for SLO-1:26
| 2 |
We note that, according to this formalism, all protein motions, whether contributing to Fconf or ktun, are stochastic in nature, relative to the virtually instantaneous isotope-dependent wave function overlap (discussed in several previous publications from this laboratory, for example, refs (9, 10, 12), and (27)).
Equation 2, one of the described formalisms for ktun,26 represents the primary coordinates as three exponential terms with distinctive mass and temperature dependencies. As shown, the second exponent, representing the hydrogenic wave function overlap between donor and acceptor atoms, is mass dependent, but temperature independent. The temperature dependence arises from the properties of the first and third exponents, with the first Marcus-like exponent expected to dominate PCET for WT SLO-1, given the evidence of only minor donor–acceptor distance sampling (exponent 3) in the case of the evolutionarily optimized native enzyme. As described within the Marcus formalism for electron transfer, both thermodynamic, ΔG°, and kinetic, λ, terms are potentially temperature dependent. Thus, the observed catalytic enthalpic barrier will be the sum of the following thermodynamic, ΔH°conf + ΔH°ΔG°, and kinetic, ΔH⧧λ, contributions, eq 3:46
| 3 |
where ΔH°conf represents the enthalpic differences between reactive and nonreactive PCET protein conformational substates. The kinetic barrier, ΔH⧧λ, is composed of both inner-sphere (λin) and outer-sphere (λout) contributions, λ = λin + λout. For the present study of WT, λout may be the critical kinetic contributor to λ, given the prior evidence of (temperature independent) tunneling within the substrate backbone that accompanies the tunneling of the transferred particle in SLO-1.47
In the context of the theoretical framework of eq 3, we consider which terms will contribute dominantly to changes in Ea when SLO-1 has undergone mutation at positions 553 and 546 (Figure 4). Given that the temperature dependence of ΔG° may not be greatly perturbed by the generation of altered, aliphatic hydrophobic side chains at these positions, the major contributing terms are expected to originate from the sum of ΔH°conf, ΔH⧧λout, and any additional contribution to ΔH⧧ from the DAD sampling term (exponent 3 in eq 2). In general, the impact of the latter is seen largely in the course of deuterium rather than protium transfer, leading to significant increases in ΔEa, rather than Ea(H).10 Overall, the two dominant terms within the temperature dependence of eq 3 for PCET are expected to be ΔH°conf and ΔH⧧λout, fully compatible with the observation of correlations between Ea(H) (for PCET at the active site) and EaHDX(avg) (within a remote loop). Because of the differential origins of the activation enthalpies for HDX and catalysis, it is not surprising that the absolute values of Ea(H) and EaHDX(avg), as shown in Figures 4d and 4e, are divergent. The key finding is that mutational trends in EaHDX(avg) are found to track, in a spatially resolved and distal manner, with mutational trends in catalytic Ea(H). Combining this remarkable finding with the results from site-directed mutagenesis at Y317, a protein network connecting the solvent-exposed loop to the site of C–H cleavage in SLO-1 has also emerged (Figure 5). Specific pathways for heat transfer in proteins are not unexpected, given the highly anisotropic nature of most folded protein structures.48 While this area of investigation is still in its infancy, a number of recently pursued experimental studies have demonstrated either asymmetric pathways for the dissipation of heat released during enzyme-catalyzed bond cleavage processes49 or the presence of thermal networks from remote subunit interfaces to the active sites of a family of prokaryotic alcohol dehydrogenases that have adapted to either high or low temperatures.50
An increasing amount of evidence illustrates that loop flexibility can influence many facets of protein function that include an enzyme’s rate, specificity, and stability.51,52 However, there has been a lack of generic and easily accessible physical measurements capable of resolving such remote, stochastic protein motions relevant to an enzyme’s catalytic parameters. The surface loop, identified herein as exhibiting thermally activated motions that correlate with the enthalpic barrier for catalytic PCET, showcases HDXMS as a potentially powerful tool for the spatial resolution of distal regions of an enzyme that may play a role in catalytic fitness.
We note that lipoxygenases from sources other than plants, e.g., from mammals, fungi, and prokaryotes, contain a surface topology that is distinct from SLO-1.31 In fact, representatives from these alternate lipoxygenases routinely display considerably higher catalytic enthalpy barriers (ca. 8–12 kcal/mol)53−55 than that of SLO-1, while their reported ΔEa values can be small and close to zero. Plant lipoxygenases have been implicated in several critical physiological responses56 that include pathogenic defense mechanisms and seed development and germination, where the latter takes place at reduced temperatures (≤20 °C). Conversely, the mammalian lipoxygenases play key roles in normal homeostasis and inflammatory responses57 that occur within a small temperature range centered at 37 °C. In the context of emerging models that attribute elevated enthalpic barriers to perturbed protein conformational landscapes,46,58 the enlarged loops in plant-derived lipoxygenases may play a key role in minimizing trapped protein substates that would necessitate a significant input of energy to reach the catalytically relevant active site configurations. This property would allow the maintenance of high enzyme activity over a much wider temperature range than is operative with lipoxygenases from other sources
Conclusions
The experimental protocols developed herein, of an interrogation of HDXMS as a function of time, temperature, and catalytic PCET-altering mutations, offer a previously unexplored roadmap for relating equilibrium thermal motions of a protein to its active site catalysis. The presented results suggest that the incorporation of specific networks of protein motions, originating from solvent-exposed loops, may provide a new design principle for optimizing thermally activated processes in de novo enzymes and biomimetic catalysts.
Acknowledgments
Financial support was provided by the National Institutes of Health (GM113432 (F32) to A.R.O., 1S10OD020062-01 to A.T.I., and GM025765 and GM118117 to J.P.K.). The authors thank Prof. Natalie Ahn for initial HDX experiments and discussions. The authors also thank Dr. Ian Barr for assistance with circular dichroism experiments. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD006234.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00142.
Experimental methods and details, supporting figures, extended data tables, extended discussion on SLO mutants, and all HDX traces (PDF)
Author Present Address
⊥ C.A.M.C.: Riffyn, Oakland, California 94612, United States. D.P.: Molecular Immunity Unit, Department of Medicine, University of Cambridge, Cambridge, CB2 0QH, United Kingdom. A.D.S.: Sauce Laboratories, San Francisco, California 94107, United States.
The authors declare no competing financial interest.
Author Status
† T.A. is deceased.
Supplementary Material
References
- Wolfenden R.; Snider M. J. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001, 34, 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Goldsmith M.; Tawfik D. S. Directed enzyme evolution: Beyond the low-hanging fruit. Curr. Opin. Struct. Biol. 2012, 22, 406–412. 10.1016/j.sbi.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Rothlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J. L.; Tanaka F.; Barbas C. F. III; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss G.; Çelebi-Ölçüm N.; Moretti R.; Baker D.; Houk K. N. Computational enzyme design. Angew. Chem., Int. Ed. 2013, 52, 5700–5725. 10.1002/anie.201204077. [DOI] [PubMed] [Google Scholar]
- Kries J.; Blomberg R.; Hilvert D. De novo enzymes by computational design. Curr. Opin. Chem. Biol. 2013, 17, 221–228. 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H.; Chen G.; Berendzen J.; Fenimore P. W.; Jansson H.; McMahon B. H.; Stroe I. R.; Swenson J.; Young R. D. A unified model of protein dynamics. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5129–5134. 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Hydrogen tunneling and protein motion in enzyme reactions. Acc. Chem. Res. 2006, 39, 93–100. 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- Sutcliffe M. J.; Scrutton N. S. A new conceptual framework for enzyme catalysis. Eur. J. Biochem. 2002, 269, 3096–3102. 10.1046/j.1432-1033.2002.03020.x. [DOI] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P. An integrated model for enzyme catalysis emerges from studies of hydrogen tunneling. Chem. Phys. Lett. 2009, 471, 179–193. 10.1016/j.cplett.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudackov A. V.; Hammes-Schiffer S. Proton-coupled electron transfer reactions: analytical rate constants and case study of kinetic isotope effects in lipoxygenase. Faraday Discuss. 2016, 195, 171–189. 10.1039/C6FD00122J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P. Importance of protein dynamics during enzymatic C-H bond cleavage. Biochemistry 2013, 52, 2068–2077. 10.1021/bi301504m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Fraser J.; Clarkson M. W.; Degnan S. C.; Erion R.; Kern D.; Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature 2009, 462, 669–673. 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekstra D. R.; White K. I.; Socolich M. A.; Henning R. W.; Šrajer V.; Ranganathan R. Electric-field-stimulated protein mechanics. Nature 2016, 540, 400–405. 10.1038/nature20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Ahn N. G. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 1–25. 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- Englander S. W. Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom. 2006, 17, 1481–1489. 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T. E.; Engen J. R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Milne J. S.; Mayne L.; Englander S. W. Primary structure effects on peptide group hydrogen exchange. Proteins: Struct., Funct., Genet. 1993, 17, 75–86. 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Goldsmith E. J.; Ahn N. G. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 956–961. 10.1073/pnas.98.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Garrod S. M.; Deal M. S.; Anand G. S.; Woods V. L. Jr.; Taylor S. Allosteric network of cAMP-dependet protein kinase revealed by mutation Tyr204 in the P+1 loop. J. Mol. Biol. 2005, 346, 191–201. 10.1016/j.jmb.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Anand G. S.; Law D.; Mandell J. G.; Snead A. N.; Tsigelny I.; Taylor S. S.; Eyck L. F. T.; Komives E. A. Identification of the protein kinase A regulatory R1a-catalytic subunit interface by amide H/2H exchange and protein docking. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13264–13269. 10.1073/pnas.2232255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantom P. A.; Zhang H.-M.; Emmett M. R.; Marshall A. G.; Blanchard J. S. Mapping of the allosteric network in the regulation of a-isopropylmalate synthase from Mycobacterium tuberculosis by the feedback inhibitor L-leucine: solution phase H/D exchange monitored by FT-ICR mass spectrometry. Biochemistry 2009, 48, 7457–7464. 10.1021/bi900851q. [DOI] [PubMed] [Google Scholar]
- Chung K. Y.; Rasmussen S. G. F.; Liu T.; Li S.; DeVree B. T.; Chae P. S.; Calinski D.; Kobilka B. K.; Woods V. L. Jr.; Sunahara R. K. Conformational changes in the G protein Gs induced by the b2 adrenergic receptor. Nature 2011, 477, 611–615. 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underbakke E. S.; Iavarone A. T.; Marletta M. A. Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 6777–6782. 10.1073/pnas.1301934110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deredge D.; Li J.; Johnson K. A.; Wintrode P. L. Hydrogen/deuterium exchange kinetics demonstrate long range allosteric effects of thumb site 2 inhibitors of hepatitis C viral RNA-dependent RNA polymerase. J. Biol. Chem. 2016, 291, 10078–10088. 10.1074/jbc.M115.708370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp M. J.; Rickert K.; Klinman J. P. Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 2002, 124, 3865–3874. 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- Meyer M. P.; Tomchick D. R.; Klinman J. P. Enzyme structure and dynamics affect hydrogen tunneling: The impact of a remote side chain (I553) in soybean lipoxygenase. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1146–1151. 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z.-X.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9556–9561. 10.1073/pnas.0403337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyeyemi O. A.; Sours K. M.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Temperature dependence of protein motions in a thermophilic dihydrofolate reductase and its relationship to catalytic efficiency. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 10074–10079. 10.1073/pnas.1003678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Venere A.; Salucci M. L.; van Zadelhoff G.; Veldink G.; Mei G.; Rosato N.; Finazzi-Agro A.; Maccarrone M. Structure-to-function relationship of mini-lipoxygenase, a 60-kDa fragment of soybean lipoxygenase-1 with lower stability but higher enzymatic activity. J. Biol. Chem. 2003, 278, 18281–18288. 10.1074/jbc.M212122200. [DOI] [PubMed] [Google Scholar]
- Newcomer M. E.; Brash A. R. The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 2015, 24, 298–305. 10.1002/pro.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E.; Soudackov A. V.; Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase. J. Am. Chem. Soc. 2004, 126, 5763–5775. 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- Edwards S. J.; Soudackov A. V.; Hammes-Schiffer S. Impact of distal mutation on hydrogen transfer interface and substrate conformation in soybean lipoxygenase. J. Phys. Chem. B 2010, 114, 6653–6660. 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. W.; Liu L. A review about nothing: Are apolar cavities in proteins really empty?. Protein Sci. 2009, 18, 494–502. 10.1002/pro.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveberg M.; Tan Y. J.; Fersht A. R. Negative activation enthalpies in the kinetics of protein folding. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 8926–8929. 10.1073/pnas.92.19.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney B. J. Connecting lipoxygenase function to structure by electron paramagnetic resonance. Acc. Chem. Res. 2014, 47, 3588–3595. 10.1021/ar500290r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthanarayanan V. S.; Andreatta R. H.; Poland D.; Scheraga H. A. Helix-coil stability constants for the naturally occurring amino acids in water. III. Glycine parameters from random poly(hydroxybutylglutamine-co-glycine). Macromolecules 1971, 4, 417–424. 10.1021/ma60022a009. [DOI] [Google Scholar]
- Li S.-C.; Deber C. M. Influence of glycine residues on peptide conformation in membrane environments. Int. J. Pept. Protein Res. 1992, 40, 243–248. 10.1111/j.1399-3011.1992.tb00297.x. [DOI] [PubMed] [Google Scholar]
- Bright J. N.; Sansom M. S. P. The flexing/twirling helix: Exploring the flexibility about molecular hinges formed by proline and glycine motifs in transmembrane helices. J. Phys. Chem. B 2003, 107, 627–636. 10.1021/jp026686u. [DOI] [Google Scholar]
- Kraut D. A.; Sigala P. A.; Fenn T. D.; Herschlag D. Dissecting the paradoxical effects of hydrogen bond mutations in the ketosteroid isomerase oxyanion hole. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1960–1965. 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman R. C. Statistical mechanics applied to chemical kinetics. J. Am. Chem. Soc. 1920, 42, 2506–2528. 10.1021/ja01457a008. [DOI] [Google Scholar]
- Truhlar D. G. Interpretation of the activation energy. J. Chem. Educ. 1978, 55, 309–311. 10.1021/ed055p309. [DOI] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes G. G.; Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry 2008, 47, 3317–3321. 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- Soudackov A. V.; Hammes-Schiffer S. Probing nonadiabaticity in the proton-coupled electron transfer reaction catalyzed by soybean lipoxygenase. J. Phys. Chem. Lett. 2014, 5, 3274–3278. 10.1021/jz501655v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Dong M.; Bahnson B. J.; Klinman J. P. Impaired protein conformational landscapes as revealed in anomalous Arrhenius prefactors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10520–10525. 10.1073/pnas.1104989108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M. P.; Klinman J. P. Investigating inner-sphere reorganization via secondary kinetic isotope effects in the C-H cleavage reaction catalyzed by soybean lipoxygenase: Tunneling in the substrate backbone as well as the transferred hydrogen. J. Am. Chem. Soc. 2011, 133, 430–439. 10.1021/ja1050742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W.; Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 1999, 286, 295–299. 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Riedel C.; Gabizon R.; Wilson C. A. M.; Hamadani K.; Tsekouras K.; Marqusee S.; Pressé S.; Bustamante C. The heat released during catalytic turnover enhances the diffusion of an enzyme. Nature 2015, 517, 227–230. 10.1038/nature14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Cun S.; Klinman J. P. Identification of a long-range protein network that modulates active site dynamics in extremophilic alcohol dehydrogenases. J. Biol. Chem. 2013, 288, 14087–14097. 10.1074/jbc.M113.453951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestl B. M.; Hauer B. Engineering of flexible loops in enzymes. ACS Catal. 2014, 4, 3201–3211. 10.1021/cs500325p. [DOI] [Google Scholar]
- Papaleo E.; Saladino G.; Lambrughi M.; Lindorff-Larsen K.; Gervasio F. L.; Nussinov R. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 2016, 116, 6391–6423. 10.1021/acs.chemrev.5b00623. [DOI] [PubMed] [Google Scholar]
- Su C.; Oliw E. H. Manganese lipoxygenase. Purification and characterization. J. Biol. Chem. 1998, 273, 13072–13079. 10.1074/jbc.273.21.13072. [DOI] [PubMed] [Google Scholar]
- Carr C. A. M.; Klinman J. P. Hydrogen tunneling in a prokaryotic lipoxygenase. Biochemistry 2014, 53, 2212–2214. 10.1021/bi500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segraves E. N.; Holman T. R. Kinetic investigations of the rate-limiting step in human 12- and 15-lipoxygenase. Biochemistry 2003, 42, 5236–5243. 10.1021/bi0273462. [DOI] [PubMed] [Google Scholar]
- Feussner I.; Wasternack C. The lipoxygenase pathway. Annu. Rev. Plant Biol. 2002, 53, 275–297. 10.1146/annurev.arplant.53.100301.135248. [DOI] [PubMed] [Google Scholar]
- Haeggstrom J. Z.; Funk C. D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- Sharma S. C.; Klinman J. P. Kinetic detection of orthogonal protein and chemical coordinates in enzyme catalysis: double mutants of soybean lipoxygenase. Biochemistry 2015, 54, 5447–5456. 10.1021/acs.biochem.5b00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.