

Abstract
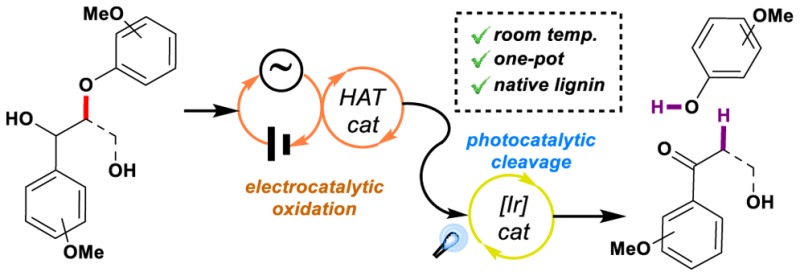
Lignin is a recalcitrant and underexploited natural feedstock for aromatic commodity chemicals, and its degradation generally requires the use of high temperatures and harsh reaction conditions. Herein we present an ambient temperature one-pot process for the controlled oxidation and depolymerization of this potent resource. Harnessing the potential of electrocatalytic oxidation in conjugation with our photocatalytic cleavage methodology, we have developed an operationally simple procedure for selective fragmentation of β-O-4 bonds with excellent mass recovery, which provides a unique opportunity to expand the existing lignin usage from energy source to commodity chemicals and synthetic building block source.
Short abstract
Selective β-O-4 linkage fragmentation for lignin depolymerization has been achieved by combining electro- and photocatalysis in a seamless batch to flow process at ambient temperature with excellent mass recovery.
Biomass is one of the most abundant renewable sources of carbon available and is considered the most sustainable alternative to petroleum.1,2 In parallel with its current exploitation as an energy source, biomass has the potential to be used as a source for production of low molecular weight chemicals.3,4 In particular, lignin is one of the most targeted components,5,6 as its distinctive aromatic backbone (Figure 1a) makes this biopolymer unique among its kind and considerably more valuable as a source for aromatic commodity chemicals7 (Figures 1b and 1d). Present technologies suffer from poor selectivity and usually provide the desired fragmentation products in low yields,8,9 a concern that has given rise to an increased interest of several research groups in developing novel and attractive strategies for fragmentation of lignin in order to fully harness this underexploited material. Efforts have been made in this context through the use of reductive,10,11 oxidative,12−15 and redox-neutral approaches,16−19 to target the β-O-4 linkage (Figure 1c, see Scheme S1 for more details) as it is the most abundant in native lignin (45–60% of all linkages).20,21
Figure 1.

Lignin structure and characteristics. (a) Schematic representation of lignin. The three main components (coumaryl, coniferyl, and sinapyl alcohol structures) and the linkage of interest (β-O-4) are highlighted.21 (b) Main characteristics of lignin. (c) Representative native lignin depolymerization strategies and typical structures observed. (d) Main and potential uses of lignin.
In this vein, a significant focus of our research program22 is to develop new approaches for the selective, catalytic depolymerization of lignin,5 for which we reported a strategy based on palladium and photoredox catalysis for the oxidation and fragmentation of lignin model dimers16,23,24 (Figure 2d, left). However, the intrinsic necessity of an oxidant in the reaction medium has so far limited our efforts on finding an oxidation method that would not interfere with our photochemical fragmentation conditions. For this reason we sought to investigate alternative avenues to chemical oxidation. Electrocatalytic oxidation was found to gather all the necessary requirements, as such an approach would allow for carrying out the overall transformation in a single reaction vessel due to the absence of chemical oxidants in the reaction media.
Figure 2.

General lignin oxidation strategies and main characteristics. (a) Chemical oxidation strategy. (b) Direct electrooxidation of the aromatic system. (c) Electrocatalytic oxidation via hydrogen atom transfer (HAT). (d) Depolymerization approaches developed in the Stephenson group. WE = working electrode. CE = counter electrode. RE = reference electrode.
Electrochemical oxidations and reductions have long been recognized as a more environmentally benign alternative tool for chemical reactions (Figure 2a), allowing for specific reactivity thanks in part to the ability to control the desired reaction potential.25,26 In the case of lignin, direct electroreduction of its backbone has been reported to be poorly efficient and to give complicated mixtures,27 and, in the same way, attempts of carrying out direct oxidation of its structure have proven to require high oxidation potentials,25 necessitating the use of expensive and toxic electrodes (IrO2,28 Pt,29 Ru/V oxides,30 Au,29 or Pb31). This can lead to diminished selectivity, since the oxidation of the lignin backbone via electron transfer can ultimately result in uncontrolled degradation of its structure (Figure 2b). Alternatively, these drawbacks can be easily overcome by the use of a mediator, which significantly reduces the necessary applied potential and thus offers milder oxidation conditions and enhanced efficiency and selectivity.26 In the case of lignin, the use of a hydrogen atom transfer (HAT) mediator was found to be a particularly attractive approach, as abstraction of the benzylic hydrogen would selectively provide benzylic oxidation (Figure 2c). Despite these notable advantages and even though electrocatalytic oxidations have found widespread applications, such as degradation of organic contaminants32 or synthesis of valuable organic molecules,33 they have found scarce application in lignin valorization.34−36 Herein, we present, to the best of our knowledge, the first sequential employment of electrocatalysis and photoredox catalysis in flow as a means to achieve efficient and highly selective fragmentation of β-O-4 lignin dimers and native lignin in a two-step one-pot process at ambient temperature (Figure 2d, right).
NHPI/Lutidine as the Optimal Catalyst
We decided to start investigating different HAT mediators for selective benzylic oxidation of the β-O-4 lignin dimers under electrocatalytic conditions. TEMPO-related derivatives were initially considered as these systems have been well studied for alcohol oxidations,37−39 but TEMPO was found to be too weak (BDETEMPO–H38 = 71 kcal·mol–1) to abstract the benzylic hydrogen of lignin-type alcohols (BDEC–H40 = 84 kcal·mol–1) (Figure S1 and Table S1).
On the other hand, in situ generated N-oxyl radicals, in particular phthalimide N-oxyl (PINO) from N-hydroxyphthalimide (NHPI), took our attention as it was found to have a suitable O–H bond strength (BDENHPI40 = 86 kcal·mol–1) for lignin electrocatalytic oxidation35,40−42 (Figure 2b). Furthermore, its combination with an appropriate base has been reported to dramatically lower the oxidation potential of the PINO/NHPI redox couple.42,43 Accordingly, upon addition of 1 equiv of 2,6-lutidine to a NHPI solution in acetonitrile, a dramatic shift from 0.85 to 0.38 V (vs Fc+/Fc),42 along with a more reversible redox couple (ΔENHPI = 155 mV vs ΔENHPI/2,6-lutidine = 82 mV) was observed, indicating improved catalytic reversibility of the system (Figure 3a). Interestingly, the NHPI/2,6-lutidine-catalyzed oxidation of 1-(3,4-dimethoxyphenyl)ethanol was found to be efficient, and the exposure of ethanol to this catalytic system resulted in no change of the cyclic voltammogram (CV), suggesting selective oxidation of the benzylic alcohol in lignin model substrates (Figure 3b and Figures S2 and S3).
Figure 3.

Redox behavior of NHPI/2,6-lutidine catalyst and oxidation optimization. Cyclic voltammograms (CVs) at 50 mV/s of a 0.1 M NaClO4 acetonitrile solution containing (a) 10 mM of NHPI (purple line), after addition of 10 mM of 2,6-lutidine (orange line), and (b) after addition of 100 mM 1-(3,4-dimethoxyphenyl)ethanol (green line) or 100 mM of ethanol (red line). (c) a3,5-Lutidine (10 mol %). bAdded in two portions. cCl4NHPI was used. dPotential not applied. eNo NHPI. (d) Difference on the colors observed in various mixtures. (e) Chronoamperometries of a solution containing 10 mM NHPI/2,6-lutidine and 100 mM 1-(3,4-dimethoxyphenyl)ethanol in N2-sparged (purple line) or O2-sparged (orange line) acetonitrile.
The catalytic system was further evaluated and optimized using 1-(3,4-dimethoxyphenyl)ethanol as the model substrate. Thus, 10 mol % of NHPI and 10 mol % of 2,6-lutidine was found to be the optimal combination for this reaction (Figures S4–S7). As opposed to literature precedents which typically require more than 1 equiv of base for alcohol oxidations,42 we found that addition of 10 mol % (1:1 NHPI/2,6-lutidine) afforded the greatest current densities. Indeed, cyclic voltammetry measurements revealed that addition of more than 10 mol % of base resulted in a decrease of the catalytic activity of NHPI/2,6-lutidine for alcohols of type 1, an effect that was more pronounced for diols of form 5 (Figures S5 and S6).
From Micro to Bulk Electrolysis
With this promising lead in hand, bulk electrocatalytic oxidation on lignin model 1a was attempted. For this purpose, we decided to use reticulated vitreous carbon (RVC) panels as electrodes, avoiding the use of expensive glassy carbon or platinum.44 Unfortunately, the first attempt afforded only 31% conversion of alcohol 1a to ketone 2a using 10 mol % of NHPI/2,6-lutidine over 2 h in acetonitrile (0.1 M NaClO4) (Figure 3c, entry 1). Increasing the amount of base to 1.1 equiv barely improved the outcome, giving 38% conversion of alcohol 1a (Figure 3c, entry 2). Since the assumed mechanism in its non-electrochemical version implies the need for molecular oxygen to facilitate the oxidation,42 O2-sparged acetonitrile was used instead. Chronoamperometry measurements using O2-sparged versus N2-sparged acetonitrile solutions showed an improved catalytic activity (Figure 3e and Figure S12) resulting in 75% conversion to the desired oxidized product 2a even with the use of only 10 mol % of 2,6-lutidine (Figure 3c, entries 3 and 4). Other bases resulted in decreased reactivity, and attempts to increase the conversion of this reaction by adding the NHPI in two portions of 5 mol %45 resulted in no improvement (Figure 3c, entries 5 and 6). During the optimization, we observed that the addition of 2,6-lutidine to the electrochemical vessel always led to the formation of a red precipitate, whereas the NHPI/2,6-lutidine mixture in MeCN produces a soluble yellow complex. We realized that the supporting electrolyte, NaClO4, was the cause for the red discoloration (Figure 3d and Figure S14) and rationalized that the formation of the precipitate might be the reason for loss of catalytic activity. Alternative electrolytes were therefore evaluated, and KPF6 was found to produce a pale yellow homogeneous reaction mixture with no detrimental consequences on the catalytic efficiency (Figures S15 and S16). Of note, when using NaClO4 as electrolyte an unusual dependence in the order of addition of the reagents was observed, as the addition of 2,6-lutidine last to the reaction mixture resulted in a drastic decrease of the efficiency of the reaction, while the use of KPF6 as electrolyte ensured good catalytic efficiency independent of the order of addition of the reagents to the reaction mixture. Thus, using KPF6 as the electrolyte we observed a significant improvement in the reaction performance, and extending the reaction time from 2 to 4 h resulted in increased conversion of 1a, affording ketone 2a in 83% isolated yield (Figure 3c, entry 7). When the chlorinated NHPI derivative 3,4,5,6-tetrachloro-N-hydroxyphthalimide (Cl4NHPI)44 was tested, slightly lower conversion was observed (Figure 3c, entry 8, and Figure S42). Control experiments highlighted the crucial roles of NHPI, base, and the applied potential in this reaction (Figure 3c, entries 9–11, Table S2, and Figure S18).
En Route to a One-Pot Process
Evaluating the compatibility of this efficient electrocatalytic oxidation with our previously disclosed reductive C–O bond cleavage methodology16,24 was the crucial next step.46 As there are several benzylic oxidation methods that require reagents that would inhibit our photocatalytic reductive conditions, it was necessary to demonstrate that the presence of the NHPI, 2,6-lutidine, electrolyte, oxygen, or even H2O2 (generated in situ during the oxidation, Figure S13) could be tolerated. A series of control experiments testing the compatibility of these components with the reductive fragmentation methodology revealed complete conversion to the C–O bond cleavage products in all cases (Figure S19). With these encouraging results in hand, a one-pot reaction was performed by simply adding the [Ir(ppy)2(dtbbpy)](PF6) photocatalyst, diisopropylethylamine (DIPEA), and formic acid to the reaction vessel after oxidation. A flow reactor was selected in this case due to the demonstrated superior performance of these flow systems in photochemical transformations compared to batch reactions and their easier implementation in large scale processes.47,48 The use of a 4.6 mL flow reactor at 0.1 mL/min afforded 75% isolated yield of the cleaved ketone 3a accompanied by 30% of guaiacol (4a) (Figure 4). Only 18% of starting material 1a was recovered, but more importantly, no traces of the intermediate 2a were observed, highlighting the efficiency of our photocatalytic reductive methodology even in the presence of the additives from the electrocatalytic oxidation. When diol-type substrate 5a was submitted to the same procedure, only oxidation of the benzylic alcohol was observed, as reflected by our previous cyclic voltammetry measurements. After the two-step one-pot fragmentation sequence, the cleavage products β-hydroxyketone 7a and guaiacol (4a) were isolated along with 25% recovered diol 5a, observing no traces of byproducts derived from the oxidation of the primary alcohol. When the same procedure was applied to 1a using Cl4NHPI, 14% of intermediate 2a was recovered, potentially arising from interference of this mediator in the photocatalytic cleavage (Figure S43).
Figure 4.

Scope of the one-pot electrocatalytic oxidation–reductive cleavage. Degradation of substrates 1a, 1b, and 1c afforded minor detectable byproducts (Figures S20–S22). In the case of native lignin, HSQC, GPC, and GC–MS data are shown, acetone/DMSO (98:2) solvent mixture was used, HSQC was acquired in DMSO-d6, and tentative integrations based on β signals are shown (for complete HSQC see the Supporting Information). ID = internal diameter. HSQC = heteronuclear single quantum coherence spectroscopy.
Toward a Controlled Lignin Fragmentation
Encouraged by these results, the scope of the NHPI/2,6-lutidine-mediated one-pot methodology was examined on a variety of different lignin models. Cyclic voltammetry measurements demonstrated that the electrocatalytic oxidation of all the evaluated lignin model substrates (1 and 5) could be performed using the NHPI/2,6-lutidine system (Figures S35 and S36). Therefore, bulk electrocatalytic oxidation followed by photocatalytic fragmentation in flow was applied to coniferyl- and sinapyl-derived dimers 1b, 1c, 5b, and 5c, affording the cleaved products in good to excellent yields in all cases (Figure 4, conditions A). However, partial recovery of the starting lignin systems 1 and 5 was always observed, especially for alcohol 1c, where 37% of unreacted starting alcohol was recovered. We realized that extending the oxidation reaction time to 6 h gave complete conversion, obtaining excellent yields of the fragmentation products of dimers 1b, 1c, 5b, and 5c (Figure 4, conditions B). The improved reaction conditions were subsequently applied to coniferyl-derived alcohols 1d and 1e, diols 5d and 5e, and sinapyl derivative 5f, thus affording excellent yields of the fragmentation products. Not surprisingly, the less electron-rich alcohol 1f showed the lowest catalytic efficiency (Figure S35) and gave incomplete conversion in the electrocatalytic oxidation, resulting in 38% recovery of unreacted starting material (Figure 4). The two-step protocol was also compatible on large scale; conducting the electrocatalytic oxidation followed by reductive cleavage with 0.5 g of 1a afforded a 67% yield of ketone 3a and a 67% yield of guaiacol (4a) (Figure 4 and Figure S25).
Importantly, native lignin isolated from pine (Figure S26) was subjected to the one-pot reaction conditions. The isolated lignin was found to be insoluble in acetonitrile, but after evaluating different solvent systems, we realized that an acetone/DMSO (98:2) combination provided a homogeneous mixture, which was submitted to the optimal electrocatalytic oxidation conditions (Figures S27–S29). After 6 h, signals characteristic of oxidized lignin were observed by heteronuclear single quantum coherence (HSQC) spectroscopy (Figure 4, for complete HSQC see the Supporting Information), indicating successful oxidation of native lignin under the NHPI/2,6-lutidine catalytic system. In addition, the subsequent photocatalytic cleavage in flow was also compatible with the alternative solvent system (Figure S28) affording controlled fragmentation of native lignin as revealed by HSQC spectroscopy (Figure 4). Gel permeation chromatography (GPC) analysis indicated the formation of monomeric and oligomeric units with lower molecular weight than native lignin (∼43 wt % of oligomeric units, and up to 55 wt % of low molecular weight units), and this result was also supported by GC–MS, where three major peaks were identified (Figure 4 and Figures S30 and S31). The analysis of these three peaks revealed that monomeric units II and III observed in the fragmented lignin may arise from the same monomeric unit I, as the injection of pure material of I gave an additional two peaks corresponding to units II and III in the fragmented lignin sample. Using calibration curves, the entire area of peak II was calculated to directly derive from monomer I (Figure 4 and Figures S31–S33 for further details), which indicates that I and III are the major monomeric units obtained after the two-step one-pot procedure. Accordingly, the yields for monomers I and III were calculated to be 1.30 and 1.14 wt % respectively (Figure S33). The analysis of the structure of I and III shows that they can only arise from the cleavage of two consecutive β-O-4 linkages under the applied reaction conditions, which demonstrates that the developed electrocatalytic oxidation–photocatalytic cleavage is not only highly effective in fragmenting β-O-4 bonds but also highly selective, leaving intact other linkages found in native lignin. Compared to other methods reported in the literature for native lignin depolymerization,6,8,11,13,15,16 the yields reported for the observed monomeric units using our conditions might seem to be low. However, it is important to notice that due to the high selectivity of this method, nonconsecutive β-O-4 linkages that are also being cleaved are not being taken into account in the overall yield for the monomeric species (Figure S34).
While the methodology reported in this work does not represent the scale that would be required for the application of this procedure in industry, it is another clear example that illustrates the potential of both technologies, electro- and photocatalysis, to provide friendlier, alternative methods to more traditional processes. Further development of these technologies in industrial scale (especially flow electrochemistry49) would represent a viable renewable alternative to fossil fuels as a source for aromatic commodity chemicals.
Mechanistic Insights
We were intrigued about the mechanism of the electrocatalytic oxidation of alcohols via NHPI/base catalyst, as it has scarcely been discussed in the literature.41,42 For this reason kinetic isotope effect (KIE) experiments were performed for 1a and 1a-D1 using O2- or N2-sparged acetonitrile. The obtained values of 2.3 for individual runs and 5.6 for the intermolecular competition experiment using O2-sparged acetonitrile (Figure 5c, Figures S37–S39) indicate a primary KIE and suggest that the abstraction of the benzylic hydrogen might be the rate- or turnover-determining step. In the case of N2-sparged acetonitrile, similar values were obtained.
Figure 5.
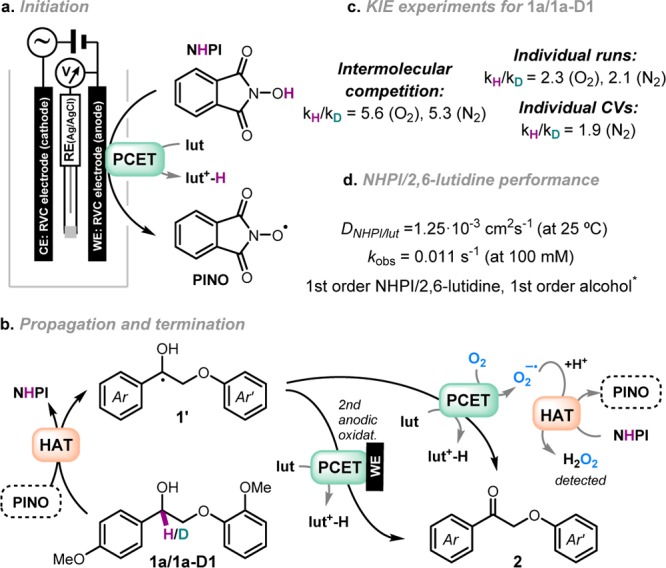
Mechanistic investigations of the electrocatalytic oxidation procedure. Proposed mechanism for NHPI/2,6-lutidine-mediated electrocatalytic oxidation of lignin systems under anaerobic and aerobic conditions. (a) Initiation step of NHPI anodic oxidation via PCET. (b) Propagation and termination pathways proposed under aerobic and anaerobic conditions. (c) Results from the kinetic isotope effect (KIE) experiments under O2- and N2-sparged acetonitrile. (d) Data of the NHPI/2,6-lutidine catalytic system performance. *Kinetic order of the chemical reaction rate. WE = working electrode; CE = counter or auxiliary electrode; RE = reference electrode; RVC = reticulated vitreous carbon; HAT = hydrogen atom transfer; PCET = proton-coupled electron transfer; lut = 2,6-lutidine.
With these data in hand, the mechanism of the electrocatalytic oxidation is proposed to be initiated by the anodic oxidation of NHPI to PINO, a process facilitated by 2,6-lutidine via PCET43,50,51 (Figure 5a). HAT from the substrate 1 would regenerate the NHPI (rate-determining step) and provide the α-hydroxy radical 1′. Direct deprotonation to a ketyl radical anion followed by oxidation would be one route to 2, but as our previous results have shown,16 these intermediates should readily cleave the β-O-4 bond, a feature that is not observed under the electrocatalytic conditions. Alternatively, PCET-based oxidation of 1′ (2,6-lutidine as base) with O2 as oxidant (or the anode) would directly provide ketone 2 and superoxide. Additionally, dioxygen is known to trap α-hydroxy radicals at near diffusion-limited rates (k > 109 M–1 s–1 in water52), but oxygen adducts of this type are known to fragment to ketones (k > 106 s–1 in water52), a process that is greatly accelerated by base.53,54 This again indicates a PCET mechanism, one that would provide 2 equiv of superoxide per fragmentation. In either case, hydrogen peroxide would be formed from superoxide after protonation and HAT from NHPI (Figure 5b, see Figure S40 for further details). From this mechanism, the increased catalytic activity in the presence of molecular oxygen (31% with N2, 71% with O2 for the oxidation of 1a to 2a under the same reaction conditions, Figure 3c, entries 1 and 4) could thus be rationalized by the fact that two anodic oxidations are required under anaerobic conditions (2 mol of electrons per mol of substrate) compared to a single anodic oxidation event when oxygen is present in the reaction should the processes proceed via closed catalytic cycles. In addition, in the presence of oxygen an equivalent of PINO is also formed for each equivalent of reacted alcohol, suggesting that a propagative mechanism could be present. To further analyze the electrocatalytic system, the diffusion constant of the NHPI/2,6-lutidine catalyst derived from the Randles–Sevcik equation was calculated to be 1.25 × 10–3 cm2 s–1 at 25 °C (Figure S9) and the catalytic system exhibited a first-order dependence on the alcohol and a first-order dependence on the NHPI/2,6-lutidine catalyst (Figures S10 and S11) with an observed rate constant of 0.011 s–1 (v = kobsCNHPI/lutidine where kobs = kC1a0) (Figure 5d).
Conclusions
In conclusion, an efficient and selective one-pot method for fragmentation of the β-O-4 bond of lignin-type dimers and native lignin by combining electrocatalytic oxidation and photocatalytic fragmentation in a seamless batch to flow processing at ambient temperature has been described. Harnessing the potential of electrocatalysis to predict the initial behavior of electrocatalytic reactions enabled the optimization of a metal-free and inexpensive NHPI/2,6-lutidine system for bulk electrocatalytic oxidation of lignin-type substrates and native lignin. In addition, the use of photocatalytic and flow technologies, which are quickly being adopted for industrial applications due to their proven advantages, highlights the predictability and reliability of this one-pot protocol. We anticipate that this strategy will stimulate renewed interest in the application of electrochemistry to the depolymerization of lignin and its eventual utilization as a renewable carbon feedstock.
Methods
General Procedure for the One-Pot Electrocatalytic Oxidation–Reductive Cleavage in Flow
A mixture of lignin model 1 or 5 (0.32 mmol), NHPI (10 mol %, 5.2 mg), and 2,6-lutidine (10 mol %, 3.7 μL) in oxygen-sparged acetonitrile (0.05 M KPF6, 16 mL) was subjected to electrocatalytic oxidation for 6 h at 0.64 V (vs Fc+/Fc) (see Figure S17). Then, [Ir(ppy)2(dtbbpy)](PF6) (0.5 mol %, 1.4 mg) and a mixture of DIPEA (1.1 equiv, 61 μL) and formic acid (10 mol %, 1.2 μL) in acetonitrile (1.0 mL) were added and the reaction mixture was irradiated with blue LEDs in flow at 0.1 mL/min (10 m of PFA tubing, 0.03 in. internal diameter). The collected mixture was concentrated, extracted three times with CH2Cl2 from water, washed with water and brine, dried over Na2SO4, and concentrated. The crude mixture was purified by column chromatography to give the fragmentation products (see the Supporting Information for further details).
Acknowledgments
Financial support from the NSF (CHE-1565782), the Camille and Henry Dreyfus Foundation, and the University of Michigan is gratefully acknowledged. I.B. gratefully acknowledges the Ramón Areces Foundation (Becas para Estudios Postdoctorales) for a postdoctoral fellowship. G.M. would like to thank the University of Michigan for a Rackham Merit fellowship. M.D.K. would like to thank the Swedish Research Council (637-2013-7314) and the Royal Swedish Academy of Agriculture and Forestry (Kungliga Skogs- och Lantbruksakademien) for a postdoctoral fellowship. The authors would like to thank Dr. Bryan S. Matsuura and Dr. Daryl Staveness for their helpful suggestions and Prof. Derek A. Pratt from the University of Ottawa for his help with the discussion of the electrocatalytic oxidation mechanism.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00140.
Experimental procedures, additional figures, schemes, and tables, and compound characterizations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Alonso D. M.; Bond J. Q.; Dumesic J. A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12, 1493–1513. 10.1039/c004654j. [DOI] [Google Scholar]
- Löfstedt J.; Dahlstrand C.; Orebom A.; Meuzelaar G.; Sawadjoon S.; Galkin M. V.; Agback P.; Wimby M.; Corresa E.; Mathieu Y.; Sauvanaud L.; Eriksson S.; Corma A.; Samec J. S. Green diesel from kraft lignin in three steps. ChemSusChem 2016, 9, 1392–1396. 10.1002/cssc.201600172. [DOI] [PubMed] [Google Scholar]
- Vennestrøm P.; Osmundsen C.; Christensen C.; Taarning E. Beyond petrochemicals: the renewable chemicals industry. Angew. Chem., Int. Ed. 2011, 50, 10502–10509. 10.1002/anie.201102117. [DOI] [PubMed] [Google Scholar]
- Collinson S. R.; Thielemans W. The catalytic oxidation of biomass to new materials focusing on starch, cellulose and lignin. Coord. Chem. Rev. 2010, 254, 1854–1870. 10.1016/j.ccr.2010.04.007. [DOI] [Google Scholar]
- Kärkäs M. D.; Matsuura B. S.; Monos T. M.; Magallanes G.; Stephenson C. R. J. Transition-metal catalyzed valorization of lignin: the key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. 10.1039/C5OB02212F. [DOI] [PubMed] [Google Scholar]
- Deuss P. J.; Barta K. From models to lignin: Transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016, 306, 510–532. 10.1016/j.ccr.2015.02.004. [DOI] [Google Scholar]
- Zakzeski J.; Bruijnincx P.; Jongerius A.; Weckhuysen B. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- Shuai L.; Amiri M. T.; Questell-Santiago Y. M.; Héroguel F.; Li Y.; Kim H.; Meilan R.; Chapple C.; Ralph J.; Luterbacher J. S. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 2016, 354, 329–333. 10.1126/science.aaf7810. [DOI] [PubMed] [Google Scholar]
- Pandey M. P.; Kim C. S. Lignin depolymerization and conversion: a review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41. 10.1002/ceat.201000270. [DOI] [Google Scholar]
- Sergeev A. G.; Hartwig J. F. Selective, nickel-catalyzed hydrogenolysis of aryl ethers. Science 2011, 332, 439–443. 10.1126/science.1200437. [DOI] [PubMed] [Google Scholar]
- Galkin M. V.; Samec J. S. M. Selective route to 2-propenyl aryls directly from wood by a tandem organosolv and palladium-catalysed transfer hydrogenolysis. ChemSusChem 2014, 7, 2154–2158. 10.1002/cssc.201402017. [DOI] [PubMed] [Google Scholar]
- Wang M.; Li L. H.; Lu J. M.; Li H. J.; Zhang X. C.; Liu H. F.; Luo N. C.; Wang F. Acid promoted C–C bond oxidative cleavage of β-O-4 and β-1 lignin models to esters over a copper catalyst. Green Chem. 2017, 19, 702–706. 10.1039/C6GC02970A. [DOI] [Google Scholar]
- Zhu C.; Ding W.; Shen T.; Tang C.; Sun C.; Xu S.; Chen Y.; Wu J.; Ying H. Metallo-deuteroporphyrin as a biomimetic catalyst for the catalytic oxidation of lignin to aromatics. ChemSusChem 2015, 8, 1768–1778. 10.1002/cssc.201500048. [DOI] [PubMed] [Google Scholar]
- Sedai B.; Díaz-Urrutia C.; Baker R. T.; Wu R.; Silks L. A.; Hanson S. K. Aerobic oxidation of β-1 lignin model compounds with copper and oxovanadium catalysts. ACS Catal. 2013, 3, 3111–3122. 10.1021/cs400636k. [DOI] [Google Scholar]
- Rahimi A.; Ulbrich A.; Coon J. J.; Stahl S. S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252. 10.1038/nature13867. [DOI] [PubMed] [Google Scholar]
- Lancefield C. S.; Ojo O. S.; Tran F.; Westwood N. J. Isolation of functionalized phenolic monomers through selective oxidation and C–O bond cleavage of the β-O-4 linkages in lignin. Angew. Chem., Int. Ed. 2015, 54, 258–262. 10.1002/anie.201409408. [DOI] [PubMed] [Google Scholar]
- Nguyen J. D.; Matsuura B. S.; Stephenson C. R. J. A photochemical strategy for lignin degradation at room temperature. J. Am. Chem. Soc. 2014, 136, 1218–1221. 10.1021/ja4113462. [DOI] [PubMed] [Google Scholar]
- Chan J. M. W.; Bauer S.; Sorek H.; Sreekumar S.; Wang K.; Toste F. D. Studies on the Vanadium-Catalyzed Nonoxidative Depolymerization of Miscanthus giganteus-Derived Lignin. ACS Catal. 2013, 3, 1369–1377. 10.1021/cs400333q. [DOI] [Google Scholar]
- Galkin M. V.; Sawadjoon S.; Rohde V.; Dawange M.; Samec J. S. M. Mild heterogeneous palladium-catalyzed cleavage of β-O-4′-ether linkages of lignin model compounds and native lignin in air. ChemCatChem 2014, 6, 179–184. 10.1002/cctc.201300540. [DOI] [Google Scholar]
- Ralph J.; Lundquist K.; Brunow G.; Lu F.; Kim H.; Schatz P. F.; Marita J. M.; Hatfield R. D.; Ralph S. A.; Christensen J. H.; Boerjan W. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenyl-propanoids. Phytochem. Rev. 2004, 3, 29–60. 10.1023/B:PHYT.0000047809.65444.a4. [DOI] [Google Scholar]
- Vanholme R.; Demedts B.; Morreel K.; Ralph J.; Boerjan W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. 10.1104/pp.110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staveness D.; Bosque I.; Stephenson C. R. J. Free radical chemistry enabled by visible light-induced electron transfer. Acc. Chem. Res. 2016, 49, 2295–2306. 10.1021/acs.accounts.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkäs M. D.; Bosque I.; Matsuura B. S.; Stephenson C. R. J. Photocatalytic oxidation of lignin model systems by merging visible-light photoredox and palladium catalysis. Org. Lett. 2016, 18, 5166–5169. 10.1021/acs.orglett.6b02651. [DOI] [PubMed] [Google Scholar]
- Monos T. M.; Magallanes G.; Sebren L. J.; Stephenson C. R. J. Visible light mediated reductions of ethers, amines and sulfides. J. Photochem. Photobiol., A 2016, 328, 240–248. 10.1016/j.jphotochem.2016.05.014. [DOI] [Google Scholar]
- Weinberg N. L.; Weinberg N. R. Electrochemical oxidation of organic compounds. Chem. Rev. 1968, 68, 449–523. 10.1021/cr60254a003. [DOI] [Google Scholar]
- Horn E. J.; Rosen B. R.; Baran P. S. Synthetic organic electrochemistry: an enabling and innately sustainable method. ACS Cent. Sci. 2016, 2, 302–308. 10.1021/acscentsci.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromskaya G. I.; Chupka E. I. Electrochemical reduction of lignin in liquid ammonia. 1. Change in the chromophore and molecular-weight composition during electrochemical reduction in liquid ammonia. Koksnes Kimija 1983, 2, 60–63. [Google Scholar]
- Tolba R.; Tian M.; Wen J.; Jiang Z.-H.; Chen A. Electrochemical oxidation of lignin at IrO2-based oxide electrodes. J. Electroanal. Chem. 2010, 649, 9–15. 10.1016/j.jelechem.2009.12.013. [DOI] [Google Scholar]
- Parpot P.; Bettencourt A. P.; Carvalho A. M.; Belgsir E. M. Biomass conversion: attempted electrooxidation of lignin for vanillin production. J. Appl. Electrochem. 2000, 30, 727–731. 10.1023/A:1004003613883. [DOI] [Google Scholar]
- Reichert E.; Wintringer R.; Volmer D. A.; Hempelmann R. Electro-catalytic oxidative cleavage of lignin in a protic ionic liquid. Phys. Chem. Chem. Phys. 2012, 14, 5214–5221. 10.1039/c2cp23596j. [DOI] [PubMed] [Google Scholar]
- Bailey A.; Brooks H. M. Electrolytic oxidation of lignin. J. Am. Chem. Soc. 1946, 68, 445–446. 10.1021/ja01207a029. [DOI] [PubMed] [Google Scholar]
- Liu M.; Xia H.; Lu W.; Xu T.; Zhu Z.; Chen W. Electrocatalytic degradation of organic contaminants using carbon fiber coupled with cobalt phthalocyanine electrode. J. Appl. Electrochem. 2016, 46, 583–592. 10.1007/s10800-016-0939-z. [DOI] [Google Scholar]
- Francke R.; Little R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- Masui M.; Ueshima T.; Ozaki S. N-Hydroxyphthalimide as an effective mediator for the oxidation of alcohols by electrolysis. J. Chem. Soc., Chem. Commun. 1983, 479–480. 10.1039/c39830000479. [DOI] [Google Scholar]
- Shiraishi T.; Takano T.; Kamitakahara H.; Nakatsubo F. Studies on electro-oxidation of lignin and lignin model compounds. Part 2: N-Hydroxyphthalimide (NHPI)-mediated indirect electro-oxidation of non-phenolic lignin model compounds. Holzforschung 2012, 66, 311–315. 10.1515/hf.2011.140. [DOI] [Google Scholar]
- Nguyen B. H.; Perkins R. J.; Smith J. A.; Moeller K. D. Solvolysis, electrochemistry, and development of synthetic building blocks from sawdust. J. Org. Chem. 2015, 80, 11953–11962. 10.1021/acs.joc.5b01776. [DOI] [PubMed] [Google Scholar]
- Rafiee M.; Miles K. C.; Stahl S. S. Electrocatalytic Alcohol oxidation with TEMPO and bicyclic nitroxyl derivatives: driving force trumps steric effects. J. Am. Chem. Soc. 2015, 137, 14751–14757. 10.1021/jacs.5b09672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey D. P.; Schiedler D. A.; Matanovic I.; Doan P. V.; Atanassov P.; Minteer S. D.; Sigman M. S. Predicting electrocatalytic properties: modeling structure–activity relationships of nitroxyl radicals. J. Am. Chem. Soc. 2015, 137, 16179–16186. 10.1021/jacs.5b11252. [DOI] [PubMed] [Google Scholar]
- Badalyan A.; Stahl S. S. Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators. Nature 2016, 535, 406–410. 10.1038/nature18008. [DOI] [PubMed] [Google Scholar]
- d’Acunzo F.; Baiocco P.; Fabbrini M.; Galli C.; Gentili P. The radical rate-determining step in the oxidation of benzyl alcohols by two N–OH-type mediators of laccase: the polar N-oxyl radical intermediate. New J. Chem. 2002, 26, 1791–1794. 10.1039/B206928H. [DOI] [Google Scholar]
- Coseri S. Phthalimide-N-oxyl (PINO) radical, a powerful catalytic agent: its generation and versatility towards various organic substrates. Catal. Rev.: Sci. Eng. 2009, 51, 218–292. 10.1080/01614940902743841. [DOI] [Google Scholar]
- Recupero F.; Punta C. Free radical functionalization of organic compounds catalyzed by N-hydroxyphthalimide. Chem. Rev. 2007, 107, 3800–3842. 10.1021/cr040170k. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 2010, 110, 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn E. J.; Rosen B. R.; Chen Y.; Tang J.; Chen K.; Eastgate M. D.; Baran P. S. Scalable and sustainable electrochemical allylic C–H oxidation. Nature 2016, 533, 77–81. 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda C.; Noyama M.; Ohmori H.; Masui M. Reactivity of phthalimide-N-oxyl: a kinetic study. Chem. Pharm. Bull. 1987, 35, 1372–1377. 10.1248/cpb.35.1372. [DOI] [Google Scholar]
- At this point, any electroreduction attempts have been unsuccessful.
- Garlets Z. J.; Nguyen J. D.; Stephenson C. R. J. The Development of visible-light photoredox catalysis in flow. Isr. J. Chem. 2014, 54, 351–360. 10.1002/ijch.201300136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noël T. Applications of continuous-flow photochemistry in organic synthesis, material science, and water treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- Kabeshov M. A.; Musio B.; Murray P. R. D.; Browne D. L.; Ley S. V. Expedient preparation of nazlinine and a small library of indole alkaloids using flow electrochemistry as an enabling technology. Org. Lett. 2014, 16, 4618–14621. 10.1021/ol502201d. [DOI] [PubMed] [Google Scholar]
- Gentry E. C.; Knowles R. R. Synthetic applications of proton-coupled electron transfer. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both sequential transfer mechanisms are not feasible, since the base (2,6-lutidine, pKa 12 (in MeCN)) is not able to deprotonate NHPI (pKa NHPI 23.5 (in MeCN)) to any extent. See ref (43) for more details.
- Konya K. G.; Paul T.; Lin S.; Lusztyk J.; Ingold K. U. Laser flash photolysis studies on the first superoxide thermal source. First direct measurements of the rates of solvent-assisted 1,2-hydrogen atom shifts and a proposed new mechanism for this unusual rearrangement. J. Am. Chem. Soc. 2000, 122, 7518–7527. 10.1021/ja993570b. [DOI] [Google Scholar]
- Bothe E.; Behrens G.; Schulte-Frohlinde D. Mechanism of the first order decay of 2-hydroxy-propyl-2-peroxyl radicals and of O2–· formation in aqueous solution. Z. Naturforsch., B: J. Chem. Sci. 1977, 32b, 886–889. 10.1515/znb-1977-0811. [DOI] [Google Scholar]
- Valgimigli L.; Amorati R.; Fumo M. G.M; DiLabio G. A.; Pedulli G. F.; Ingold K. U.; Pratt D. A. The unusual reaction of semiquinone radicals with molecular oxygen. J. Org. Chem. 2008, 73, 1830–1841. 10.1021/jo7024543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.