

Abstract
Neuroblastoma (NB) originates from neural crest-derived precursors and represents the most common childhood extracranial solid tumour. MicroRNAs (miRNAs), a class of small non-coding RNAs that participate in a wide variety of biological processes by regulating gene expression, appear to play an essential role within the NB context. High-throughput next generation sequencing (NGS) was applied to study the miRNA transcriptome in a cohort of NB tumours with and without MYCN-amplification (MNA and MNnA, respectively) and in dorsal root ganglia (DRG), as a control. Out of the 128 miRNAs differentially expressed in the NB vs. DRG comparison, 47 were expressed at higher levels, while 81 were expressed at lower levels in the NB tumours. We also found that 23 miRNAs were differentially expressed in NB with or without MYCN-amplification, with 17 miRNAs being upregulated and 6 being downregulated in the MNA subtypes. Functional annotation analysis of the target genes of these differentially expressed miRNAs demonstrated that many mRNAs were involved in cancer-related pathways, such as DNA-repair and apoptosis as well as FGFR and EGFR signalling. In particular, we found that miR-628-3p negatively affects MYCN gene expression. Furthermore, we identified a novel miRNA candidate with variable expression in MNA vs. MNnA tumours, whose putative target genes are implicated in the mTOR pathway. The present study provides further insight into the molecular mechanisms that correlate miRNA dysregulation to NB development and progression.
Keywords: neuroblastoma, NGS, microRNAs, bioinformatics, isomiRs, novel miRNA molecules, cancer pathways
Introduction
Neuroblastoma (NB) is a paediatric solid tumour arising from neural crest-derived precursors, and it accounts for 8–10% of all malignancies and 12–15% of cancer-related deaths in children (1). It is characterized by marked clinical heterogeneity, and the assessment of NB-related genetic abnormalities has been helpful in predicting clinical outcome, stratifying patients into different risk classes and designing appropriate treatments (2,3). MYCN amplification at 2p24 is a well-known genetic alteration associated with NB development and progression (4–8), irrespective of age at diagnosis and clinical stage, whose examination is required in NB tumours before treatment begins (9). Indeed, approximately half of high-risk aggressive tumours are characterized by amplification of the MYCN gene, an oncogene of the MYC family of transcription factors that controls the expression of many target genes, which in turn regulates fundamental cellular processes including proliferation, cell growth, apoptosis and differentiation (10).
MicroRNAs (miRNAs) are a class of small non-coding RNAs that regulate gene expression at the post-transcriptional level, by binding to the complementary sites in the 3-untranslated region (3-UTR) of target messenger RNAs (11–13). Currently, more than 1,800 miRNA genes have been identified in the human genome (miRBase release 21.0) (14), and bioinformatic predictions indicate that up to 60% of protein-coding genes may be regulated by miRNAs (15). miRNAs play a central role in many cellular processes and their aberrant expression has been reported in childhood solid tumours, including NB (16,17). In the latter, by applying miRNA microarrays and high-throughput sequencing technologies, a series of miRNAs have been reported to play a role as oncogenes or tumour suppressors, as reviewed by Mei et al (18). For example, dysregulation of miR-542-5p is highly correlated with tumour aggressiveness and poor survival (19), while miRNA-34a was found to be a potent tumour-suppressor molecule (20). Moreover, some miRNA levels appear to have the potential to increase the efficacy of treatment by regulating the sensitivity of NB cells to cisplatin (21,22).
In the present study, we investigated the expression of miRNAs in NB by using next generation sequencing (NGS) technology, which allows for a comprehensive assessment of the miRNA transcriptome and has the potential to detect miRNA length variations or sequence modifications (isomiRs) as well as novel miRNA sequences. The SOLiD System was used to sequence small RNA (sRNA) libraries from NB tumours with and without MYCN-amplification and from human dorsal root ganglia (DRG) as a control, in order to detect the genome-wide differences in miRNA levels in these two subtypes of NB. The analysis showed that 128 known miRNAs were significantly dysregulated in the NB tumours in comparison to DRG, utilised as a normal control. Notably, 23 miRNAs were differentially expressed in MYCN-amplified vs. MYCN-non amplified tumours. Furthermore, 18 new miRNA candidates were detected in NB samples: 10 in the MNAs, 6 in the MNnA samples and 2 in both tumour subtypes; 2 putative novel miRNAs were found in the DRG. Notably, the Gene Ontology (GO) analysis of the potential targets of the upregulated and downregulated miRNAs indicated that many genes belong to biological processes and cellular components that are involved in cell growth, cell shape, protein activity and genomic instability necessary for cancer initiation and progression. Furthermore, analysis with Kyoto Encyclopaedia of Genes and Genomes (KEGG) web tools revealed significant enrichment in several cancer-related signalling pathways.
The present study provides valuable information concerning the mechanisms in NB of the miRNA-associated MYCN network that may contribute to the development of novel diagnostic biomarkers and effective therapeutic strategies for this cancer.
Materials and methods
Patient features and normal controls
Twenty-two NB primary tumour samples, equally distributed between MYCN-amplified (MNA) and MYCN-non amplified (MNnA) samples, were obtained at diagnosis before any treatment from children admitted to the Department of Oncology at Alder Hey Children's NHS Trust, Liverpool. Histopathological diagnosis was confirmed using immunohistochemistry. Details of the patients are described in Table I. Institutional written informed consent was obtained from the patients parents or legal guardians.
Table I.
Clinicopathological features of the analysed tumour cases.
| Case | Age at diagnosis | MYCN status | Primary site | Clinical stage |
|---|---|---|---|---|
| NB1a | 36 | <3 | Adrenal | 4 |
| NB2a | 65 | <3 | Adrenal | 4 |
| NB3a | 69 | <3 | Adrenal | 4 |
| NB4a | 53 | <3 | Abdomen | 4 |
| NB22 | 199 | <3 | Abdomen | 4 |
| NB45 | 3 | <3 | Adrenal | 4 |
| NB54 | 6 | <3 | Adrenal | 1 |
| NB70 | 22 | <3 | Abdomen | 4 |
| NB80 | 25 | <3 | Abdomen | 4 |
| NB96 | 111 | <3 | Adrenal | 3 |
| NB106 | 7 | <3 | Thorax | 2 |
| NB5a | 10 | >10 | Adrenal | 4 |
| NB6a | 8 | >10 | Adrenal | 4 |
| NB7a | 37 | >10 | Adrenal | 4 |
| NB8a | 16 | >10 | Abdomen | 4 |
| NB57 | 44 | >10 | Adrenal | 2 |
| NB68 | 13 | >10 | Abdomen | 4 |
| NB79 | 29 | >10 | Abdomen | 4 |
| NB81 | 37 | >10 | Adrenal | 2B |
| NB97 | 40 | >10 | Abdomen | 4 |
| NB100 | 179 | >10 | Abdomen | 4 |
| NB105 | 16 | >10 | Adrenal | 4 |
Variables were categorized as follows: age at diagnosis (months), MYCN-amplification status, primary site and clinical stage. MYCN-amplified >10; MYCN-non amplified <3.
Samples used for deep sequencing analysis.
The study underwent ethical review and approval according to the local institutional guidelines (Alder Hey Children's NHS Foundation Trust Ethics Committee, approval number 09/H1002/88).
Human DRG total RNA (Clontech Laboratories, Inc., Mountain View, CA, USA), pooled from normal tissues of 21 male/female Caucasians who had died suddenly, was used as a normal control. DRG were utilised as a normal control because of their neural crest-derived histogenesis (23) and since NB can occasionally arise from these organs (24).
RNA isolation
Tumour samples were frozen in liquid nitrogen immediately after surgical removal and stored at −80°C. Total RNA was isolated by tissues ground under liquid nitrogen using 1 ml of TRIzol LS reagent (Life Technologies, Carlsbad, CA, USA) per 50–100 mg of sample according to the manufacturers protocol. RNA concentration and purity were measured by NanoDrop 2000 (Thermo Fisher Scientific, Inc., Waltham, MA, USA), having 260/280 ratio values in the range of 1.9–2.1.
Small RNA library construction and high-throughput sequencing
Total RNA samples (5 µg) were enriched for small RNAs up to 200 bp by size selection using PureLink miRNA Isolation kit (Life Technologies) and RNA quality was assessed, before and after RNA enrichment, by the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) using a Agilent RNA 6000 Nano and a Agilent Small RNA kit, respectively. Enriched RNA samples were processed using the Small RNA Expression kit according to the manufacturers protocol (Small RNA Expression kit, rev. B; Applied Biosystems, Foster City, CA, USA). Briefly, 20 ng of RNA was first hybridized and ligated with the adapter mix ‘A’, subsequently reverse transcribed and treated with RNAse H. The cDNA libraries were then PCR amplified, purified and size-selected by PAGE, resulting in libraries containing inserted small RNA sequences of 20–40 bp length. Size, integrity and purity of the libraries were verified by the Agilent 2100 Bioanalyzer, Agilent DNA 1000 kit. The cDNA libraries were barcoded using the SOLiD RNA barcoding kit and amplified onto beads using emulsion PCR. Templated beads were deposited on slides and analysed using the Applied Biosystems SOLiD 4 sequencer.
Quantification of known microRNAs
The qualified clean reads were mapped and analysed with the ‘small RNA’ bioinformatics pipeline from the Thermo Fisher LifeScope version 2.5.1 software (https://www.thermofisher.com/it/en/home/technical-resources/software-downloads/lifescope-genomic-analysis-software.html), using as a target the human genome GRCh38/hg38 and the dataset of mature and precursor miRNA sequences (miRBase, release 21.0) (25). Any sequence match against repetitive elements of the genome (SINEs and LINEs), and against non-miRNA small RNAs (snoRNAs, piRNAs, tRNAs and rRNA fragments) were filtered out from the results.
Sequence counts were extracted and reformatted with Genomnia Perl scripts from the pipeline output. Differential expression analysis was performed with the edgeR Bioconductor statistical library version 3.1.0 on R (26) incorporated in a proprietary Genomnia pipeline. TMM-normalized sequence counts in the libraries were transformed in counts per millions (CPM) according to the formula: CPM = (transcript normalized counts/total miRNA matches) × 1,000,000. After having estimated the tagwise dispersion, genewise exact test (26) as implemented in edgeR was used to measure the significance of differential expression, using the miRNA ‘Pseudo-counts’. Sequences were filtered out to remove poorly expressed microRNAs (i.e. miRNAs with <10 CPM in more than half of the samples). miRNAs were considered significantly differentially expressed if the false discovery rate (FDR; P-value corrected for multiple comparison with the Benjamini-Hochberg procedure) obtained with this method was <0.05. The MNA vs. MNnA comparison was also performed with the edgeR generalized linear model (GLM) mode optimised for the analysis of data with sequence spikes (27), in order to confirm the obtained results with a second robust statistical approach well suited to handle outlier data.
miRNA target prediction, functional annotation and promoter motif discovery
For each differentially expressed miRNA, a list of experimentally validated targets was extracted from miRTarBase release 6 (http://mirtarbase.mbc.nctu.edu.tw/) (28). In addition, de novo target prediction was performed using the miRanda software (29) on selected downregulated and upregulated microRNAs from the comparison MNA vs. MNnA. The 3UTR sequences of all human transcripts present in Ensembl (release 78) were used as a target set for this analysis, and only predictions with total score ≥155 and total energy ≤-20 were considered as putative miRNA targets. Annotation and enrichment of functional pathways and ontology terms associated with the selected target genes were evaluated using DAVID Bioinformatics Resources 6.7 (30). The same target gene list was used as the starting dataset for the generation of a Functional Interaction network analysis and related Gene Ontology enrichment analysis with the Cytoscape Reactome FI plugin (31,32). Promoter regions of the differentially expressed miRNAs in the comparison MNA vs. MNnA were scanned for motifs by FIMO 4.10.2 software (http://meme-suite.org/tools/fimo) (33). A sequence of 5 Kb was considered as putative upstream regulatory region for each miRNA or miRNA cluster based on the histone acetylation track (H3K27Ac) reported in UCSC. Finally, the MYCN binding motifs available in JASPAR CORE database (http://jaspar.genereg.net/) were used in the promoter analysis.
Identification of isomiRs and prediction of novel microRNAs
The alignment files in BAM format corresponding to the same biological group (MNA, MNnA and Control) were merged and converted to sequence alignment/map (SAM) format with samtools (34). The files were then processed and analysed with the miRDeep2 software for miRNA prediction (35). The differential expression analysis for isomiRs was carried out with edgeR from Bioconductor, with the same analytical strategy previously described. A set of potentially novel microRNAs was also identified, by selecting among miRDeep2 predictions the hairpins with a probability >60% to be a true positive. Their expression was also evaluated in all samples. miRNA extraction, library preparation, sequencing and bioinformatics analysis were performed by Genomnia s.r.l.
Quantitative real-time PCR (qPCR)
Quantitative real-time PCR (qPCR) was performed to validate some results obtained from the high-throughput sequencing. In brief, step-loop reverse transcription and qPCR were carried out using TaqMan MicroRNA assays (Life Technologies) specific for human miR-18a-5p, miR-217, miR-491-5p and miR-628-3p, according to the manufacturers protocol. Ct values for miRNAs were normalized against U6 small nuclear RNA (RNU6) levels, used as an internal control. The relative expression of each miRNA was calculated by the comparative Ct method and expressed as fold-change. All samples were run in triplicate reactions in the StepOnePlus Real-Time system (Life Technologies) instrument. Results are expressed as means ± standard deviation (SD), and a P<0.05 was considered statistically significant.
Cell lines
The MYCN-amplified KCNR cell line was grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 1% v/v non-essential amino acid solution, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. HEK (human embryonic kidney) 293T cells were cultured in high-glucose DMEM, supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. All cell lines were maintained in a humidified incubator at 37°C and 5% CO2, and appropriately sub-cultured before confluence was reached.
Plasmid construction and luciferase assays
The 3UTR region of the human MYCN gene (3HSAR055440 derived from http://utrdb.ba.itb.cnr.it/) was amplified from genomic DNA using a proof reading Phusion High-Fidelity PCR Master Mix (Finnzymes, Espoo, Finland) with the following primers: MYCN 3UTR-F 5-GCTCTAGAACGCTTCTCAAAACTG GACAGTCAC-3 and MYCN 3UTR-R 5-GCTCTAGAAACATGAGGTATTTCAAAGTGCTATAAGATGC-3. The 880-bp amplified product was inserted into the XbaI site (underlined primer sequences) of the pRLTK vector (Promega, Madison, WI, USA) immediately downstream of the Renilla luciferase gene. The luciferase reporter construct, termed pMYCN-3UTR, was validated by specific restriction digestions as well as bi-directional sequencing by Bio-Fab Research (Rome, Italy). HEK293T cells were plated at a density of 1.5×105/well in 24-well plates and co-transfected after 24 h with 50 ng of pMYCN-3UTR Renilla luciferase construct, 10 ng of reference firefly luciferase reporter (pGL3-SV40; Promega) and 100 nM of mir-628-3p (mirVana miRNA mimic - MI0003642/MIMAT0003297 by Thermo Fisher Scientific) or miR-Ctr (miRIDIAN microRNA Mimic Negative Control #1; Dharmacon, Lafayette, CO, USA) molecules using 2 µl of Lipofectamine 2000 (Invitrogen). Cells were lysed 72 h after transfection and luciferase signals were detected by the Dual-luciferase reporter assay (Promega) using the GloMax Discover System (Promega), following the manufacturers instructions. Renilla luciferase activity was normalized to the firefly luciferase activity for each reaction. Two independent experiments were carried out, each in triplicate. All the luciferase data are expressed as the mean ± SD normalized to the negative control miRNA.
Protein extracts and western blot analysis
KCNR cells were seeded at 6.0×105/well in 12-well plates and transfected the day after with miR-628-3p or miR-Ctr (50 mM final concentration) using Lipofectamine 2000, according to the manufacturer's instructions. After 72 h, protein expression was determined by western blotting as previously described (36) with a primary antibodies against MYCN (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Tubulin (Sigma-Aldrich, St. Louis, MO, USA) was used as a normalization control for equal loading. Chemiluminescent signals were revealed by ChemiDoc XRS+ System (Bio-Rad Laboratories, Hercules, CA, USA) and densitometric analysis was performed by Image Lab 5.1 software (Bio-Rad Laboratories).
Results and Discussion
Identification and quantification of annotated miRNAs
Comprehensive miRNA, isomiR and novel miRNA identification, annotation and analysis were developed in the framework of this project and are summarized in Fig. 1. The miRNA transcriptome was profiled by sequencing of mature miRNA molecules in a panel of primary NB tumours, 4 with MYCN-amplification (MNA) and 4 without MYCN-amplification (MNnA) and from normal dorsal root ganglia (DRG). The clinical characteristics of the patients are reported in Table I. Small RNA libraries were prepared and deep sequenced by using a SOLiD 4 sequencer platform. Approximately 71 million reads mapped with high quality against the human reference genome (GRCh38/hg38, repeat masked), with an average of 8 million reads per library. The reads corresponding to annotated miRNAs were identified by mapping all reads against the human precursor and mature sequences included in miRBase v21.0. Fig. 2A shows that a total of 532 distinct known miRNAs had on average at least 10 read counts in the NB tumour samples, while 512 miRNAs were detected in the DRG sample; 382 of these were expressed in both conditions, with a 72% overlap for NB miRNAs and 75% overlap for DRG miRNAs. Thus, about a quarter of the identified known microRNAs were uniquely expressed in NB or control samples. NB-specific miRNAs might therefore play an important functional role in tumour development and progression, through the regulation of downstream targets and the biological pathways affected by these genes.
Figure 1.

Flow chart of the bioinformatic analyses performed in the present study.
Figure 2.

(A) The overlap of known expressed miRNAs (with at least 10 read counts) in a given biological condition. Neuroblastoma (NB) tumours are in dark grey colour, while DRG controls are in white. (B) Fold-change plot of miRNA levels in the NB (MNA and MNnA collectively considered) vs. DRG comparison. x-axis, average log of counts per millions; y-axis, log2 fold-change (FC). miRNAs that are differentially expressed with P<0.05 are marked as a red dot.
Differential expression analysis of miRNAs in NB vs. DRG and functional annotation analysis
Based on the high-throughput sequencing of small RNAs, we performed differential expression analysis of miRNAs comparing the two libraries from NB tumours (MNA and MNnA collectively considered) and DRG, after removing poorly expressed miRNAs. Of a total of 198 differentially expressed miRNAs in the dataset, 128 (64.6%) displayed statistically significant differential levels in NB tumours in comparison with DRG, after FDR correction. Notably, out of these 128, 47 miRNAs (36.7%) were expressed at higher levels in NBs (Table IIA), while 81 (63.3%) were expressed at lower levels (Table IIB). Fig. 2B shows the distribution of the different mature miRNA species in abundance classes comparing NB tumours (MNA and MNnA collectively considered) against DRG, allowing a survey of the entire miRNA population. Significantly dysregulated miRNAs are indicated in red. Among the differentially expressed miRNAs, some have been previously associated with NB development, such as upregulated miR-876-5p, miR-19a, as well as downregulated miR-183-5p, miR-423-3p and miR-92b-3p (18,36–39). Other miRNAs have been implicated in the pathogenesis of many types of human cancer but not in NB malignancy to date. In particular, of main interest are upregulated miR-127-5p, miR-369-3p and miR-655-3p encoded in the large 14q32 miRNA cluster and associated with oligometastases through suppression of shared target genes involved in cytoskeletal organization, cell motility and TGF-β signalling pathways (40). Also the marked downregulation of miR-328-3p in NB samples in comparison to DRG suggests its possible role as a tumour suppressor also in this malignancy as already observed in other types of cancers, such as acute myeloid leukaemia, chronic myelogenous leukaemia and glioblastoma (41,42). Therefore, future experiments will address the role of this miRNA in the proliferation, invasion and metastasis of NB tumour cells. Another new attractive result is the significant downregulation of miR-1247-5p, whose suppression has been demonstrated to play an important role in pancreatic cancer (43,44). Notably, the molecular mechanisms underlying the role of miR-1247-5p have been correlated with its capacity to directly target and silence the expression of neuropilins (44), which are transmembrane glycoproteins recently implicated in tumour functions and found as promising drug target candidates in leukaemia and lymphoma (45). Furthermore, miR-1247-5p also belongs to the 14q32 miRNA cluster (but in reverse strand in respect to the above mentioned miR-127-5p, miR-369-3p and miR-655-3p) and its aberrant expression in colorectal cancer has been linked to hypermethylation, confirming the importance of epigenetic modifications in miRNA-mediated cancer development (46). Since alterations in DNA methylation are frequent in NB and can also modulate sensitivity to cytotoxic agents (47), the analysis of the miR-1247-5p promoter region in NB samples could provide new insight into the mechanisms that correlate epigenetic processes and NB development.
Table II.
miRNA expression in NB tumours.
| A, miRNA species expressed at higher levels in NBs (MNA and MNnA are collectively considered) vs. DRG | |||||
|---|---|---|---|---|---|
| miRNA_ID | log2FC | FDR | miRNA_ID | log2FC | FDR |
| hsa-miR-876-5p | 13.31 | 0.01104 | hsa-let-7f-2-3p | 4.75 | 0.01169 |
| hsa-miR-3117-3p | 11.02 | 0.01191 | hsa-miR-495-3p | 4.62 | 0.00587 |
| hsa-miR-655-3p | 10.93 | 0.00220 | hsa-miR-450a-5p | 4.61 | 0.01676 |
| hsa-miR-215-5p | 10.83 | 0.00271 | hsa-miR-656-3p | 4.54 | 0.01232 |
| hsa-miR-25-5p | 10.19 | 0.03121 | hsa-miR-101-3p | 4.51 | 0.00866 |
| hsa-miR-153-5p | 10.06 | 0.01678 | hsa-miR-411-5p | 4.51 | 0.02650 |
| hsa-miR-377-3p | 8.75 | 0.02109 | hsa-miR-142-3p | 4.49 | 0.04022 |
| hsa-miR-495-5p | 8.64 | 0.02699 | hsa-miR-7-1-3p | 4.44 | 0.04325 |
| hsa-miR-490-3p | 7.76 | 0.04137 | hsa-miR-98-3p | 4.42 | 0.02650 |
| hsa-miR-218-5p | 6.14 | 0.01242 | hsa-miR-1197 | 4.42 | 0.01817 |
| hsa-miR-369-3p | 6.07 | 0.00372 | hsa-miR-127-5p | 4.31 | 0.01817 |
| hsa-miR-21-5p | 6.06 | 0.01817 | hsa-miR-590-3p | 4.23 | 0.01893 |
| hsa-miR-9-3p | 5.88 | 0.03226 | hsa-miR-3613-5p | 4.23 | 0.03121 |
| hsa-miR-7-5p | 5.75 | 0.00231 | hsa-miR-136-3p | 4.22 | 0.01676 |
| hsa-miR-218-2-3p | 5.67 | 0.03121 | hsa-miR-192-5p | 4.09 | 0.03226 |
| hsa-miR-873-5p | 5.58 | 0.02699 | hsa-miR-424-5p | 4.04 | 0.02701 |
| hsa-miR-19a-3p | 5.52 | 0.03767 | hsa-miR-454-3p | 3.99 | 0.03238 |
| hsa-miR-32-5p | 5.38 | 0.01518 | hsa-miR-299-3p | 3.97 | 0.03666 |
| hsa-miR-301a-3p | 5.35 | 0.02699 | hsa-miR-590-5p | 3.83 | 0.03315 |
| hsa-miR-137 | 5.33 | 0.00499 | hsa-miR-154-3p | 3.81 | 0.03767 |
| hsa-miR-153-3p | 5.24 | 0.01676 | hsa-miR-374a-5p | 3.44 | 0.03121 |
| hsa-miR-142-5p | 5.14 | 0.03121 | hsa-miR-30e-5p | 3.32 | 0.03121 |
| hsa-miR-106b-5p | 5.10 | 0.04325 | hsa-miR-337-3p | 3.31 | 0.04571 |
| hsa-miR-380-3p | 4.95 | 0.01749 | |||
| B, miRNA species expressed at lower levels in NBs (MNA and MNnA are collectively considered) vs. DRG | |||||
| miRNA_ID | log2FC | FDR | miRNA_ID | log2FC | FDR |
| hsa-miR-6079 | −6.33 | 1.19E-20 | hsa-miR-1307-3p | −2.91 | 0.00127 |
| hsa-let-7d-3p | −6.30 | 9.76E-18 | hsa-miR-320e | −2.81 | 0.00175 |
| hsa-miR-1247-5p | −5.36 | 3.79E-06 | hsa-miR-193b-5p | −2.77 | 0.00219 |
| hsa-miR-4301 | −5.31 | 1.41E-13 | hsa-miR-1180-3p | −2.76 | 0.00512 |
| hsa-miR-328-3p | −5.19 | 1.9E-11 | hsa-miR-1306-5p | −2.75 | 0.00271 |
| hsa-miR-6074 | −4.71 | 6.15E-09 | hsa-let-7b-5p | −2.75 | 0.00200 |
| hsa-miR-193a-5p | −4.50 | 5.22E-09 | hsa-miR-204-5p | −2.74 | 0.03416 |
| hsa-miR-874-3p | −4.38 | 3.46E-09 | hsa-miR-197-3p | −2.69 | 0.00133 |
| hsa-miR-2277-3p | −4.31 | 5.28E-10 | hsa-miR-744-5p | −2.69 | 0.00763 |
| hsa-miR-760 | −4.13 | 0.00001 | hsa-miR-3605-3p | −2.66 | 0.00118 |
| hsa-miR-504-5p | −4.02 | 0.00009 | hsa-miR-99b-3p | −2.66 | 0.00286 |
| hsa-miR-532-3p | −3.89 | 0.00001 | hsa-miR-146b-3p | −2.62 | 0.00590 |
| hsa-miR-338-5p | −3.85 | 0.00053 | hsa-miR-3177-3p | −2.61 | 0.00487 |
| hsa-miR-2110 | −3.85 | 0.00000 | hsa-miR-211-5p | −2.59 | 0.03315 |
| hsa-miR-193b-3p | −3.84 | 0.00003 | hsa-miR-149-5p | −2.59 | 0.02363 |
| hsa-miR-3974 | −3.76 | 0.00001 | hsa-miR-6803-3p | −2.56 | 0.00175 |
| hsa-miR-3620-3p | −3.68 | 0.00000 | hsa-miR-133b | −2.52 | 0.00722 |
| hsa-miR-574-3p | −3.66 | 0.00014 | hsa-miR-3940-3p | −2.52 | 0.01649 |
| hsa-miR-320b | −3.61 | 0.00010 | hsa-miR-125a-5p | −2.50 | 0.00499 |
| hsa-miR-320a | −3.61 | 0.00072 | hsa-miR-6511b-3p | −2.49 | 0.00883 |
| hsa-miR-3615 | −3.60 | 0.00000 | hsa-miR-671-3p | −2.49 | 0.00512 |
| hsa-miR-23b-5p | −3.48 | 0.00009 | hsa-miR-324-3p | −2.46 | 0.01152 |
| hsa-miR-92b-3p | −3.48 | 0.00009 | hsa-miR-1270 | −2.46 | 0.00831 |
| hsa-miR-183-5p | −3.47 | 0.03136 | hsa-miR-320d | −2.43 | 0.00957 |
| hsa-miR-423-3p | −3.46 | 0.00002 | hsa-miR-6511a-3p | −2.38 | 0.00590 |
| hsa-miR-940 | −3.42 | 0.00005 | hsa-miR-92a-1-5p | −2.38 | 0.04931 |
| hsa-miR-1257 | −3.33 | 0.00118 | hsa-miR-1296-5p | −2.33 | 0.02650 |
| hsa-miR-4532 | −3.33 | 0.00002 | hsa-miR-3975 | −2.33 | 0.02850 |
| hsa-miR-589-5p | −3.19 | 0.00009 | hsa-miR-3178 | −2.31 | 0.03121 |
| hsa-miR-615-3p | −3.19 | 0.00372 | hsa-miR-1227-3p | −2.28 | 0.00936 |
| hsa-miR-210-5p | −3.19 | 0.00118 | hsa-miR-1226-3p | −2.27 | 0.02737 |
| hsa-miR-423-5p | −3.13 | 0.00015 | hsa-miR-326 | −2.27 | 0.03136 |
| hsa-miR-487a-5p | −3.11 | 0.00020 | hsa-miR-1281 | −2.25 | 0.01676 |
| hsa-miR-1343-3p | −3.07 | 0.00492 | hsa-miR-380-5p | −2.18 | 0.02911 |
| hsa-miR-133a-3p | −3.07 | 0.00092 | hsa-miR-4272 | −2.18 | 0.03136 |
| hsa-miR-4787-3p | −3.03 | 0.00219 | hsa-miR-203a-3p | −2.17 | 0.03985 |
| hsa-miR-320c | −3.02 | 0.00071 | hsa-miR-4516 | −2.10 | 0.03315 |
| hsa-miR-483-5p | −2.99 | 0.03136 | hsa-miR-431-3p | −2.03 | 0.03862 |
| hsa-miR-874-5p | −2.95 | 0.00200 | hsa-miR-4429 | −2.00 | 0.03416 |
| hsa-miR-455-3p | −2.94 | 0.00111 | hsa-miR-1229-3p | −1.94 | 0.03066 |
| hsa-miR-769-3p | −2.93 | 0.00123 | |||
FDR, false discovery rate; FC, fold-change. NB, neuroblastoma; DRG, dorsal root ganglia.
To investigate the biological implications of the dysregulated miRNAs, we assessed miRNA target genes within the regulatory network for enrichment in Gene Ontology (GO), one of the most useful methods for functional annotation and classification of gene products. GO categories of the targets of the differentially upregulated and downregulated miRNAs (NB vs. DRG) were organized into three different functional groups, i.e. Biological process, Cellular component and Molecular function (Fig. 3), among which a significant over-representation of transcription factor activity/binding, protein kinase activity, chromatin modifications, metabolic processes, cell communication, plasma membrane, cytoplasm/intracellular organelle part, cell proliferation and programmed cell death regulation were found. To further explore the biological pathways potentially affected in NB development, targets of the differentially upregulated and downregulated miRNAs (NB vs. DRG) were subjected to KEGG pathway analysis. Several cancer-related networks, including TGF-β, neurotrophin, MAPK, ErbB, Toll-like receptor and p53 signalling pathways, were significantly enriched (Tables III–IV). Indeed, the expression levels of TGFBR2, SMAD2 and SMAD4, three important components along the TGF-β cascade, were found to be downregulated in NB cells (48). VEGF, Wnt, JAK-STAT and GnRH pathways were found among the targets of the underexpressed miRNAs in accordance with their growth-inhibitory potential on NB (49–51). Moreover, some biological processes linked to cell cycle, adherents junction and focal adhesion could have also been altered. KEGG database interrogation also suggested that many of the miRNA targets contribute to the development of other human tumours, including colorectal, pancreatic cancer, melanoma and gliomas.
Figure 3.

Gene Ontology (GO) analysis of miRNA target genes according to Biological process, Cellular component and Molecular function. (A) GO categories for the predicted gene targets of the upregulated miRNAs in the NB vs. DRG comparison. (B) GO categories for the predicted gene targets of the downregulated miRNAs in the NB vs. DRG comparison. All the categories were significantly enriched (FDR <0.05). FDR, false discovery rate; DRG, dorsal root ganglia; NB, neuroblastoma.
Table III.
Enriched KEGG pathways of the target genes of the differentially upregulated miRNAs.
| KEGG pathway | No of genes | FDR |
|---|---|---|
| hsa05200:Pathways in cancer | 70 | 1.92E-29 |
| hsa05212:Pancreatic cancer | 27 | 1.58E-15 |
| hsa05222:Small cell lung cancer | 28 | 1.05E-14 |
| hsa05215:Prostate cancer | 28 | 5.60E-14 |
| hsa05210:Colorectal cancer | 26 | 1.51E-12 |
| hsa05218:Melanoma | 23 | 3.46E-11 |
| hsa05220:Chronic myeloid leukaemia | 23 | 1.24E-10 |
| hsa05214:Glioma | 20 | 3.72E-09 |
| hsa04110:Cell cycle | 27 | 4.45E-09 |
| hsa04010:MAPK signalling pathway | 39 | 9.24E-09 |
| hsa04510:Focal adhesion | 33 | 2.52E-08 |
| hsa04210:Apoptosis | 21 | 2.36E-07 |
| hsa04115:p53 signalling pathway | 18 | 1.42E-06 |
| hsa05211:Renal cell carcinoma | 18 | 2.31E-06 |
| hsa05221:Acute myeloid leukaemia | 16 | 8.40E-06 |
| hsa04620:Toll-like receptor signalling pathway | 20 | 2.42E-05 |
| hsa05223:Non-small cell lung cancer | 15 | 2.62E-05 |
| hsa05219:Bladder cancer | 13 | 8.39E-05 |
| hsa05213:Endometrial cancer | 14 | 1.36E-04 |
| hsa04722:Neurotrophin signalling pathway | 21 | 1.51E-04 |
| hsa04012:ErbB signalling pathway | 16 | 0.002529 |
| hsa04520:Adherens junction | 15 | 0.002890 |
| hsa04350:TGF-β signalling pathway | 15 | 0.012845 |
| hsa04930:Type II diabetes mellitus | 11 | 0.019442 |
| hsa04062:Chemokine signalling pathway | 22 | 0.030059 |
Comparison NB vs. DRG. Pathways significantly enriched are reported (FDR <0.05). FDR, false discovery rate; NB, neuroblastoma; DRG, dorsal root ganglia.
Table IV.
Enriched KEGG pathways of the target genes of the differentially downregulated miRNAs.
| KEGG pathway | No. of genes | FDR |
|---|---|---|
| hsa05200:Pathways in cancer | 66 | 4.11E-30 |
| hsa05215:Prostate cancer | 31 | 3.56E-19 |
| hsa05210:Colorectal cancer | 29 | 1.40E-17 |
| hsa05212:Pancreatic cancer | 26 | 6.02E-16 |
| hsa05220:Chronic myeloid leukaemia | 26 | 1,88E-15 |
| hsa05218:Melanoma | 24 | 1.33E-13 |
| hsa04520:Adherens junction | 24 | 7.66E-13 |
| hsa05219:Bladder cancer | 18 | 1.82E-11 |
| hsa05214:Glioma | 21 | 1.91E-11 |
| hsa05213:Endometrial cancer | 19 | 7.55E-11 |
| hsa05223:Non-small cell lung cancer | 19 | 1.61E-10 |
| hsa05211:Renal cell carcinoma | 20 | 2.15E-09 |
| hsa04722:Neurotrophin signalling pathway | 25 | 6.68E-09 |
| hsa04510:Focal adhesion | 31 | 1.37E-08 |
| hsa04110:Cell cycle | 24 | 5.71E-08 |
| hsa05222:Small cell lung cancer | 20 | 7.21E-08 |
| hsa04370:VEGF signalling pathway | 18 | 7.50E-07 |
| hsa04012:ErbB signalling pathway | 19 | 1.13E-06 |
| hsa04310:Wnt signalling pathway | 24 | 2.90E-06 |
| hsa04010:MAPK signalling pathway | 31 | 1.62E-05 |
| hsa05216:Thyroid cancer | 11 | 3.64E-05 |
| hsa04115:p53 signalling pathway | 15 | 9.64E-05 |
| hsa05221:Acute myeloid leukaemia | 14 | 9.79E-05 |
| hsa04660:T cell receptor signalling pathway | 18 | 2.45E-04 |
| hsa04662:B cell receptor signalling pathway | 15 | 3.51E-04 |
| hsa04350:TGF-β signalling pathway | 16 | 3.89E-04 |
| hsa04360:Axon guidance | 19 | 6.88E-04 |
| hsa04320:Dorso-ventral axis formation | 9 | 0.001784 |
| hsa04914:Progesterone oocyte maturation | 15 | 0.002008 |
| hsa04916:Melanogenesis | 15 | 0.011147 |
| hsa04620:Toll-like receptor signalling pathway | 15 | 0.014125 |
| hsa04630:Jak-STAT signalling pathway | 18 | 0.039146 |
| hsa04912:GnRH (gonadotropin-releasing hormone) signalling pathway | 14 | 0.047092 |
Comparison NB vs. DRG. Pathways significantly enriched are reported (FDR <0.05). FDR, false discovery rate; NB, neuroblastoma; DRG, dorsal root ganglia.
Differential expression analysis of miRNAs in NB with or without MYCN amplification
The miRNA expression profiles of MNA and MNnA tumours largely overlapped as shown by a Venn diagram (Fig. 4A), suggesting that only a small number of miRNAs are specific for each subgroup. After removing poorly expressed miRNAs, 70 molecules were found to be differentially expressed between MNA and MNnA tumours. Of these, 23 had a P<0.05 after FDR correction: 17 miRNAs were significantly upregulated in MNA samples, while 6 miRNAs were significantly downregulated (Table V). The most homogeneously expressed molecules among different tumours inside each subgroup were miR-16-2-3p, miR-18a-5p, miR-217, miR-338-5p, miR-489-3p, miR-491-5p, miR-628-3p, miR-628-5p and miR-3065-3p (data not shown). Among these, miR-18a-5p, miR-217, miR-491-5p and miR-628-3p were selected as candidates to validate the miRNA expression data by qPCR experiments in the 8 deep-sequencing-analysed NB tumours and in 14 additional NB samples as well as in 4 human NB cell lines (MNA LAN5 and KCNR and MNnA SH-SY5Y and SK-N-SH). qPCR demonstrated that the expression levels of the selected dysregulated miRNAs showed strong overlap with the deep-sequencing findings, thus confirming the overexpression of miR-18a-5p and miR-217 as well as the downregulation of miR-491-5p and miR-628-3p in the MNA (Fig. 4B) in comparison with the MNnA tumours. The same trend was found in MNA vs. MNnA NB cell lines (data not shown). The differential expression of a substantial number of miRNAs between favourable and unfavourable NBs was consistent with previous reports (Table V), reinforcing their fundamental role in NB development (18,48,52,53,56,62,63,74,76). In particular, the present study confirms how the majority of the differentially expressed miRNAs positively correlate with MYCN amplification, suggesting that miRNA induction might be a general mechanism caused by MYCN oncogene in high-risk NB tumours. Additionally, bioinformatic analysis of the promoter regions of the overexpressed miRNAs identified MYCN-binding sites (38,54) in 5 out of 17 miRNAs (Table V), and this is consistent with a direct transcriptional regulation of these molecules by MYCN. Overexpression of miR-17-5p, miR-18a-5p, miR-20a-5p and miR-92a-3p, belonging to the polycistronic miR-17-92 cluster at 13q31.3, was consistently higher in MNA cell lines compared to the MNnA tumours as previously reported (55,56) and in line with the evidence that MYCN directly activates miR-17-92 expression in NB cell lines (55). Indeed, the miR-17-92 cluster has a unique promoter control region and encodes for a precursor transcript that contains six tandem-loop hairpin structures, which in turn produce the six mature molecules miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92a-1 (57,58). These miRNAs may act independently or synergistically to target multiple mRNAs encoding for proteins implicated in cancer-related pathways (57,58). Given the miR-17-92 oncogenic role, inhibition of the clustered oncomiRs by delivering single-stranded antisense oligonucleotides (59) may represent a novel and attractive treatment approach for NB therapy. Regarding this aspect, Fontana et al (60) demonstrated that in vitro and in vivo treatment of MNA NB cells with a specific antagomiR against miR-17-5p was able to dramatically reduce proliferation and to activate massive apoptosis through p21 and BIM over-expression, respectively. Concerning some downregulated miRNAs in MNA tumours, our data are in agreement with recently published studies. In particular, miR-491-5p was found to be significantly downregulated at NB metastatic sites compared with primary tumours by Guo et al (61), and miR-628 was confirmed to be moderately expressed in favourable NBs and nearly absent in unfavourable cases, thus, supporting its tumour-suppressive function and its potential as a good candidate in discriminating between these two NB subsets (38,62,63). Indeed, among the downregulated miRNAs, miR-628-3p has one pairing region at position 718–724 of the MYCN 3 untranslated region (3UTR; Fig. 5A), as predicted by Miranda software (best score ≥120). To verify whether miR-628-3p was able to directly target MYCN mRNA, HEK293T cells were co-transfected either with a reporter construct containing 880 base pairs of the human MYCN 3UTR downstream of the Renilla luciferase open reading frame (Fig. 5A) with either miR-628-3p mimic or negative control miRNA (miR-Ctr). Overexpression of miR-628-3p upon transfection was confirmed by qPCR assays (data not shown). Luciferase activity was specifically reduced to ~35% (P<0.01) in cells transfected with miR-628-3p compared to the control group (Fig. 5B), thus indicating that miR-628-3p functionally interacts with the MYCN 3′UTR and decreases the expression of the corresponding protein. Indeed, miR-628-3p mimics transiently introduced into KCNR cells, an in vitro model of MNA NB, were able to slightly reduce endogenous MYCN protein levels as compared with miR-Ctr positive samples (Fig. 5C), suggesting a functional role of miR-628-3p in MYCN gene expression. Finally, our analysis identified a set of differentially expressed miRNAs, such as miR-130b-5p, miR-217, miR-3065-3p and miR-3182, known as oncomiRs in several other tumours (64–67), but whose role in NB has not yet been investigated (Table V). Notably, miR-130b and miR-301b belong to the same genomic cluster located on chromosome 22 and are significantly upregulated in triple-negative (lacking the expression of the oestrogen receptor, the progesterone receptor and the human epidermal growth factor receptor 2) breast cancer (64), having a direct role in cyclin G2 regulation. miR-338 and miR-3065 belong to the brain-specific genomic cluster located on chromosome 17 in an intronic region within the apoptosis-associated tyrosine kinase (AATK) gene, thus having a tumour-suppressor function (68). Accordingly, it was described that miR-338-3p strand has the potential to inhibit cell proliferation, cell migration and invasion in human NB cells by affecting the PTEN/Akt pathway (69). miR-3065 was recently mapped to the antisense strand of miR-338 (70) and regarded as a novel promising candidate contributing to clear cell renal cell development (71). Furthermore, our miRNA promoter in silico analysis showed that downregulated miR-338-5p, miR-489-3p, miR-628-3p and miR-3065 harbour putative MYCN binding motifs in their regulatory regions (Table V), suggesting an inverse correlation and possible feedback regulatory circuits between MYCN amplification and the reduced levels of these miRNAs. To this concern, recent studies have supported the dual role of MYCN in controlling gene expression, being not only a transcriptional activator but also a mediator of transcriptional silencing by facilitating DNA methylation (54,72,73). Thus, it will be interesting to characterise the molecular mechanisms that regulate miRNA expression.
Figure 4.

(A) Distribution of different miRNAs with at least 10 read counts in a given biological condition. MYCN-non amplified tumours are in dark grey, while MYCN-amplified tumours are in white. (B) Validation of miRNA array data by quantitative real-time polymerase chain reaction (qPCR). Relative expression levels for miR-18a-5p, miR-217, miR-491-5p and miR-628-3p in MNA (n=11) in comparison to MNnA (n=11) NB cases. Histograms indicate the mean value ± SD of independent tumours (*P<0.05). MNA, MYCN-amplified; MNnA, MYCN-non amplified.
Table V.
Differentially expressed miRNAs in MNA vs. MNnA NB tumours.
| miRNA | log2FC | FDR | Status | Refs. | MYCN-binding motif |
|---|---|---|---|---|---|
| miR-517b-3p | 7.40 | 0.01263 | Upregulated | Novel in NB | 0 |
| miR-183-5p | 4.07 | 0.01311 | Upregulated | (52) | 0 |
| miR-182-5p | 3.93 | 0.01263 | Upregulated | (53) | 0 |
| miR-451a | 3.45 | 0.01851 | Upregulated | (61) | 0 |
| miR-217 | 3.29 | 0.00962 | Upregulated | Novel in NB | 0 |
| miR-18b-5p | 3.18 | 0.01137 | Upregulated | (18) | 2 |
| miR-3182 | 2.85 | 0.02742 | Upregulated | Novel in NB | 0 |
| miR-105-5p | 2.84 | 0.03751 | Upregulated | (62) | 0 |
| miR-20a-5p | 2.80 | 0.01263 | Upregulated | (18,48,62,63,74) | 1 |
| miR-130b-3p | 2.79 | 0.01263 | Upregulated | (61) | 0 |
| miR-935 | 2.69 | 0.02807 | Upregulated | (62) | 0 |
| miR-18a-5p | 2.64 | 0.01263 | Upregulated | (56,62,63,74) | 1 |
| miR-130b-5p | 2.64 | 0.01440 | Upregulated | Novel in NB | 0 |
| miR-17-5p | 2.57 | 0.01851 | Upregulated | (18,48,55,62) | 1 |
| miR-301b-3p | 2.44 | 0.03073 | Upregulated | Novel in NB | 0 |
| miR-16-2-3p | 2.38 | 0.01263 | Upregulated | Novel in NB | 0 |
| miR-92a-3p | 2.12 | 0.04003 | Upregulated | (18,55,62,63,76) | 1 |
| miR-489-3p | −3.25 | 0.01263 | Downregulated | Novel in NB | 2 |
| miR-628-5p | −2.53 | 0.01137 | Downregulated | (62,63) | 0 |
| miR-338-5p | −2.40 | 0.01263 | Downregulated | Novel in NB | 1 |
| miR-491-5p | −2.23 | 0.01263 | Downregulated | (76) | 0 |
| miR-628-3p | −2.16 | 0.01851 | Downregulated | (18,48,62,63) | 1 |
| miR-3065-3p | −2.10 | 0.03073 | Downregulated | Novel in NB | 1 |
FDR, false discovery rate; FC, fold-change; NB, neuroblastoma.
Figure 5.

(A) Schematic representation of the luciferase construct with a fragment of 880 bp of the MYCN 3UTR, including the predicted target sequence for miR-628-3p. (B) Luciferase activity in HEK293T cells overexpressing miR-628-3p and the MYCN-3UTR vector relative to cells transfected with miR-Ctr. Bars represent the mean of two independent experiments ± SD, each carried out in triplicate. (**P<0.01). (C) Reduction of MYCN protein levels in KCNR cells transiently transfected with miR-628-3p mimics in comparison to miR-Ctr samples. Tubulin was used as loading control. Representative image from three independent experiments. The intensity of the MYCN bands was evaluated by densitometric analysis, normalized to the tubulin signal, and reported with respect to untreated cells (miR-Ctr).
miRNA target prediction and functional annotation in NBs with or without MYCN amplification
Using the miRTarBase web tool, we found a relevant number of cancer-related genes, such as PTEN, KRAS and EZH2 among the validated target genes for the differentially expressed miRNAs. Indeed, since the availability of experimentally validated targets in miRTarBase was variable and possibly biased towards the more extensively investigated miRNAs, we decided to extend our analysis and perform a de novo target prediction using the miRanda software. In particular, we considered miRNAs with a homogeneous expression level between tumours inside each group: upregulated miR-16-2-3p, miR-18a-5p, miR-217 and downregulated miR-338-5p, miR-489-3p, miR-491-5p, miR-628-3p, miR-628-5p and miR-3065-3p. The 3UTR sequences of all human transcripts present in Ensembl were screened for the identification of putative microRNA binding sites: a total of 867 putative target genes were identified for the upregulated miRNAs, while 2148 target genes were identified as potentially modulated by the downregulated miRNAs. The predicted targets were grouped into functional modules on the basis of their functional interactions, and for each module an enrichment analysis was performed with the Cytoscape Reactome FI plugin. Fig. 6 shows the main functional module putatively regulated by miR-16-2-3p, miR-18a-5p and miR-217, which are overexpressed in MNA tumours. The different interaction networks are enriched in gene products implicated in cancer-related biological processes, such as DNA-repair and apoptosis, as well as axon guidance, adenylate cyclase activity and the FGFR signalling pathway. Target genes associated to each specific overexpressed miRNA are reported in Table VI. The GO categories associated with the predicted miRNA targets showed significant overlap with biological processes that are over-represented in the MYCN target genes (38), confirming the existence in NB of a miRNA:MYCN related network able to broadly perturb gene expression (38,74,75). Fig. 7 shows the main functional module putatively regulated by miR-338-5p, miR-489-3p, miR-491-5p, miR-628-3p, miR-628-5p and miR-3065-3p which are underexpressed in MNA samples. The enriched interactions consist of proteins involved in synaptic transmission as well as TLR and EGFR signalling pathways. Target genes associated to each specific downregulated miRNA are reported in Table VII. The complete list of network modules and GO BP - pathway enrichment is included in the Table VIII. Our results suggest that the selected differentially expressed miRNAs are relevant in NB biology and could be of potential importance to stratify high-risk and low-risk tumours. Further investigations will be necessary to validate the specific target genes and to assess the downstream effects.
Figure 6.
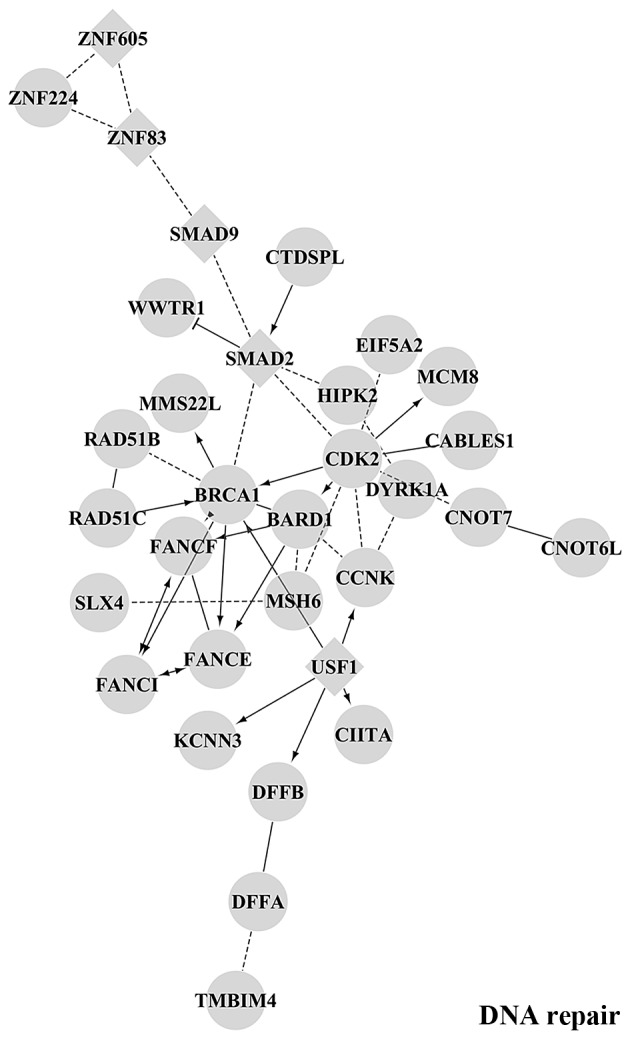
Main functional module by GO Biological Process Enrichment Analysis of Functional Interaction Networks from target genes of upregulated microRNAs (miR-16-2-3p, miR-18a-5p and miR-217) in the MNA vs. MNnA comparison. Diamond-shaped nodes represent transcription factors; round-shaped nodes represent genes; solid lines indicate direct interactions; dashed lines represent indirect relationship.
Table VI.
Predicted targets of overexpressed miRNAs, involved in DNA repair, apoptotic signalling pathway, FGF receptor signalling pathway, axon guidance and negative regulation of adenylate cyclase activity.
| miRNA | Target genes |
|---|---|
| miR-16-2-3p | AATK, ATP2A2, BCL2, BNIP2, BRCA1, CASP10, CNOT7, FANCE, FANCF, FRS2, GALR1, GNAI3, IL16, KCNN3, KLRD1, MAPK8, MDM2, PRKX, PTGER3, RAD51C, RICTOR, RPS6KA5, SCN7A, SLC8A1, SMAD2, SOS1, SRGAP1, STX7, WASF3, ZNF83 |
| miR-18a-5p | ACAP2, ACTR3, ADRBK2, ANGPT2, ATP2A2, BNIP2, CABLES1, CACNA2D2, CACNB3, CASP10, CASP2, CCNK, CCR2, CD47, CD59, CDK2, CDK5R1, CHRM2, CIITA, CNOT7, CTDSPL, DAB1, DFFA, DFFB, EIF5A2, EPHA5, ESR1, FANCI, FRS2, FZD4, GNAI3, GNB5, GRM1, GRM4, HEBP1, HIPK2, HOMER2, INSR, ITGA2, KCNN3, KIF3B, KIF5A, KLRD1, LCK, MAPK8, MAVS, MDM2, MMS22L, MYLK, NEO1, NEURL1B, NOTCH2, NTRK2, OPRL1, OTUD7B, PCSK2, PDGFRB, PRKX, RAD51B, RAD51C, RAP1A, RPS6KA5, SH3KBP1, SLC6A2, SMAD2, SOS1, SRGAP1, TNFSF12, TUBB3, WASF3, WHSC1, WNT9A, WWTR1 |
| miR-217 | ACAP2, ADAM12, ADRBK2, ARHGEF7, BARD1, BCL2, BNIP2, BTC, CD59, CFLAR, CIITA, CNOT6L, CNOT7, COL21A1, CREB5, DAB1, DMRTB1, DOCK4, DYNLL2, DYRK1A, EPHA5, ERC1, FGF16, FZD4, GALR1, GFRA1, GNAI3, HOMER2, HOXD8, HSD17B2, IL16, ITGA2, ITGA9, KCNN3, MAP1B, MAP4K4, MAVS, MCM8, MDM2, MMS22L, MSH6, MYLK, NOTCH2, NTRK2, OSBPL1A, OTUD7B, PDCD1, PFDN1, PRKCA, PRKX, RASGRP3, RGS6, RICTOR, ROBO2, RPS6KA5, SCN7A, SIPA1L2, SLC8A1, SLX4, SMAD2, SMAD9, SOS1, STX1A, STX7, STXBP5, TG, TMBIM4, TNFSF10, USF1, WHSC1, ZNF224, ZNF605 |
Figure 7.

Main functional module by GO Biological Process Enrichment Analysis of Functional Interaction Networks from target genes of downregulated microRNAs (miR-338-5p, miR-489-3p,miR-491-5p, miR-628-3p, miR-628-5p and miR-3065-3p) in the MNA vs. MNnA comparison. Diamond-shaped nodes represent transcription factors; round-shaped nodes represent genes; solid lines indicate direct interactions; dashed lines represent indirect relationship.
Table VII.
Predicted targets of underexpressed miRNAs, involved in TLR signalling pathway, EGFR signalling pathway and synaptic transmission.
| miRNA | Target genes |
|---|---|
| miR-338-5p | BNIP2, C5AR1, CD28, CLOCK, CNR1, CREB1, CX3CR1, DGKI, DSC2, ELK4, ENAH, EPN1, GNB5, GRIN2B, IMPAD1, INPP4B, INSR, KCNJ15, KCNJ6, KLRD1, KRAS, LEPR, MAFB, MAP3K2, MBP, MYLK, NFAT5, NPFFR1, NTRK3, PAG1, PEAK1, PGR, PKN2, PPARGC1A, PRKCB, RPS6KA5, SNTB1, SSTR2, UGT8, UNC5C |
| miR-489-3p | ABI2, ABLIM3, ADCY6, ATP6AP1, CACNA1B, CACNA1D, CACNG8, CDS2, CFLAR, CLOCK, CREB1, CREB5, CRKL, DBNL, DGKI, DUSP4, ELK4, EPN1, GNA11, GNAI1, GNB5, GRB10, GRIA4, GRIK2, GRIN2B, HNF4G, IGF1R, KCNJ15, KCNJ6, KLRD1, MAP2K1, MAP3K2, MAPK1, MAPK14, MBP, MEF2A, MYLK, MYO10, NFAT5, NFKBIA, NTRK3, PAK3, PAQR5, PDE3A, PHLPP2, PIK3C3, PKP1, PLXNA4, PPP2R2C, PTGFR, PTPN23, RALGAPA2, RAPGEF3, RASGRF1, RPS6KA5, SCN4B, SEMA6A, SHC4, SIRPA, SNX17, SSTR2, TANK, TNK1, UNC5C |
| miR-491-5p | ADCY1, ADCY5, AKT2, BATF2, CACNA1C, CACNA2D1, CAMK2B, CCNK, CDS2, CFLAR, CREB1, CREB3L2, CRKL, CRTC2, CX3CR1, DBNL, DEDD, DGKG, DISC1, DOCK5, DSC2, DUSP4, DUSP7, DUSP8, EFS, ELMSAN1, EPN1, EPS15L1, FGFR3, FOSB, GAB3, GABBR1, GABRR1, GALR1, GLP2R, GNA11, GNAO1, GNG7, GPR132, GRIK4, HLA-A, HNF1A, IL16, INPP4B, INPP5E, IQGAP1, KCNJ12, KRAS, MAPK1, MBP, MED15, MED17, MED24, MEF2D, MYLK, NMT1, NTRK3, PAG1, PAQR5, PEAK1, PGR, PI4K2A, PLXNA4, POU2F2, PRKCB, PTPN23, RALGAPA2, RAPGEF3, RASD1, RGS11, RGS19, RIN3, RORC, RPS6KA2, RPS6KA5, RUSC1, S1PR4, SARM1, SEMA6A, SH3GL1, SIRPA, SNX17, SPIB, SQSTM1, SSTR2, SSTR5, STAT6, TRAF3, TSHR, VIPR1, ZNF76 |
| miR-628-3p | ABI2, ABRA, CACNA2D1, CHRNA7, CREB3L2, CXCL12, CXCR2, DGKI, DSC2, DSP, DUSP16, ENAH, EPS15L1, GRIA3, GRIK4, HLF, IGF1R, IMPAD1, IRS1, KCNJ15, KCNJ6, KLRD1, KRAS, MCHR2, PAK3, PEAK1, PGR, PIAS2, PLA2G16, PRKCB, PTEN, RET, RPS6KA5, SARM1, SH3BP5 |
| miR-628-5p | ABI2, ATP6AP1, CACNA1C, CALM2, CAMK2G, CD72, CFLAR, CHRNA7, CREB1, DGKZ, DOCK5, DSC2, EDN1, ENAH, FSHB, GMFB, HNF4G, IGF1R, IL16, IL1R1, IL4R, INPP4A, IQGAP1, KCNJ6, MAPK1, MED24, MYO18Av NTRK3, PAG1, PAK3, PCCB, PEAK1, PGR, PPP1R3D, RALGAPA2, RET, SHC4, SLC23A1, TCF4, TRAF3, VLDLR, WNT2B, YY1AP1 |
| miR-3065-3p | ABI2, AHNAK, ARC, ATG9A, BAIAP2, BATF2, BDNF, BID, CD40, CD59, CHD2, CHRNA7, CIITA, CLOCK, CREB1, CREB5, CRMP1, CRTC3, DAPK1, ABI2, AHNAK, ARC, ATG9A, BAIAP2, BATF2, BDNF, BID, CD40, CD59, CHD2, CHRNA7, CIITA, CLOCK, CREB1, CREB5, CRMP1, CRTC3, DAPK1, DMRT2, DSC2, DUSP22, EAF2, ELK4, ELMSAN1, ENAH, FADD, FOS, FOSB, GABRR1, GCDH, GSDMA, HLA-DOA, HLF, HNF1A, IL1R1, KRT8, LDHA, MAF, MAFB, MAP4K4, MAPK10, MAPK11, MAPKAPK2, MAPKAPK3, MBP, MED15, MED24, MEF2D, NFATC2, NFATC3, NFKBIA, PER1, PKP1, PKP2, PLXNA4, POU2AF1, POU2F2, PTPRH, RPS6KA4, RPS6KA5, SARM1, SMPD3, TAB1, TAL1, TBX21, TNFRSF8, TNIP2, TRADD, TRAF3, TRAFD1, WASF2, ZNF142 |
Table VIII.
Main modules involved in the functional interaction network built with Cytoscape from the predicted target genes of the selected microRNAs.
| Enriched term | DNA repair | FGF signalling pathway | Apoptotic signalling pathway | Axon guidance | Negative regulation of adenylate cyclase activity |
|---|---|---|---|---|---|
| Number of target genes annotated with this term | 10 | 5 | 5 | 6 | 4 |
| P-value | 0 | 0 | 0 | 0 | 0 |
| FDR | <1.000e-03 | 1.90E+01 | 1.00E+00 | 1.30E+01 | <1.000e-03 |
| Gene list | RAD51C | BTC | CFLAR | ITGA2 | PRKCA |
| RAD51B | FGF16 | TNFSF12 | CACNB3 | GNAI3 | |
| SLX4 | FRS2 | CASP10 | NEO1 | GALR1 | |
| FANCI | LCK | TNFSF10 | RPS6KA5 | CCR2 | |
| FANCE | PDGFRB | CASP2 | ROBO2 | ||
| FANCF | SRGAP1 | ||||
| MSH6 | |||||
| CDK2 | |||||
| BRCA1 | |||||
| BARD1 | |||||
| Enriched term | TLR signalling pathway | EGFR signalling pathway | Synaptic transmission | ||
| Number of target genes annotated with this term | 18 | 13 | 27 | ||
| P-value | 0 | 0 | 0 | ||
| FDR | <2.000e-04 | <1.000e-03 | <1.000e-03 | ||
| Gene list | CREB1 | BCAR1 | ADCY1 | ||
| DUSP4 | CD28 | ADCY6 | |||
| DUSP7 | EPN1 | ADCY7 | |||
| FADD | EPS15L1 | CACNA1B | |||
| FOS | FGFR3 | CACNA1C | |||
| MAPK1 | IRS1 | CACNA1E | |||
| MAPK10 | KRAS | CACNG8 | |||
| MAPK11 | MAP2K1 | CAMK2B | |||
| MAPK14 | PAG1 | CAMK2G | |||
| MAPKAPK2 | PHLPP2 | GABBR1 | |||
| MAPKAPK3 | PLCG1 | GJD2 | |||
| MEF2A | PTEN | GNAI1 | |||
| NFKBIA | TRIB3 | GNG7 | |||
| RPS6KA2 | GRIA3 | ||||
| RPS6KA5 | GRIA4 | ||||
| SARM1 | GRIK2 | ||||
| TAB1 | GRIK4 | ||||
| TRAF3 | GRIN1 | ||||
| GRIN2B | |||||
| GRM4 | |||||
| KCNJ12 | |||||
| KCNJ15 | |||||
| KCNJ6 | |||||
| LRP6 | |||||
| MTNR1B | |||||
| PRKCB | |||||
| VIRP1 | |||||
The most highly enriched Gene Ontology Biological Process (GO BP) in each module with the corresponding P-value and FDR for the target genes of upregulated (A) and downregulated (B) miRNAs in the MNA vs. MNnA comparison. FDR, false discovery rate.
IsomiRs and novel miRNAs
Recent findings identified a large number of sequences with different 5 or 3 flanking regions, called isomiRs (76). From a biological point of view, the most interesting isomiRs are the 5′-isomiRs (having different 5-ends compared to their corresponding canonical miRNAs) as they are potentially able to recognize different targets with respect to the main isoform. A list of isomiRs (length and sequence variants) was generated from the miRDeep2 output with proprietary scripts, and their expression was evaluated in all samples. As expected, compared to the total number of isomiRs detected in our dataset, 5-isomiRs were less frequent: out of 18,809 isomiRs with at least 10 read counts in at least one library, 3,769 were 5-isomiRs (20%), as described in Fig. 8A. The presence of miRNA length variants suggests that these isomiRs might have different regulatory functions or efficiencies, this allowing a finely tuned control of global gene expression. Furthermore, in silico data analysis identified 18 putative novel miRNAs expressed in the tumour samples. Two of these were differentially expressed in MNA vs. MNnA tumours. In particular, a hairpin with genomic coordinates chr4:180531388-180531451 was detected as significantly overexpressed in the MNA tumours (P=0.001; FDR=0.02) using the ‘GLM’ analysis, and it demonstrated a uniform expression within each biological subgroup. The secondary stem-loop structure of this putative novel miRNA is shown in Fig. 8B. According to the miRanda predictions, this candidate miRNA has ~900 putative targets, which are enriched in transcriptional regulation and protein phosphorylation ontology terms. Interestingly, a significant number of putative target genes, highlighted in red in Fig. 8C, are correlated with the mTOR signalling cascade, outlining the evidence for the aberrant activation of this pathway in NB (77–79). For this reason, the putative new miRNA may be an interesting candidate for future functional studies in order to determine its biological relevance in mTOR signalling, which in turn might lead to the identification of further molecular targets in NB tumour.
Figure 8.

(A) Distribution of isomiRs and sequence variants in all examined tumours. (B) Secondary stem-looped structure of a putative novel miRNA significantly overexpressed in MNA cases. (C) Schematic representation of mTOR signalling pathway. Red characters represent key genes regulated by the predicted novel miRNA. MNA, MYCN-amplified.
In conclusion, in the present study, NGS technology and bioinformatics analysis allowed the identification of a specific set of miRNAs that are differentially expressed in NB tumours compared to DRG, or in NB tumours with or without MYCN amplification. A relevant percentage of these microRNAs has been already found to be involved in NB, another percentage has been shown to be involved in the pathogenesis/progression of other tumours, while other miRNAs are new. Target prediction and functional annotation highlighted the possible roles of the identified microRNAs, and in vitro assays provide initial evidence of the contribution of miR-628-3p to MYCN regulation. New potential miRNA candidates not previously reported were also found, but further studies are needed to clarify their role in NB development.
Altogether, these findings may provide further insight concerning NB development and may also contribute to the identification of novel potential therapeutic targets in this highly lethal childhood tumour.
Acknowledgements
The present study was supported by the ‘Associazione Onlus Fabrizio Procaccini’, ‘IO, domani…Associazione Onlus per la Lotta contro i Tumori Infantili’ (ALTI), ‘Isle of Man Anti-Cancer Association’, Italian ‘Ministero dell'Istruzione, dell'Università e della Ricerca’ (MIUR) and Genomnia Srl R&D funds.
References
- 1.Irwin MS, Park JR. Neuroblastoma: Paradigm for precision medicine. Pediatr Clin North Am. 2015;62:225–256. doi: 10.1016/j.pcl.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, et al. INRG Task Force: The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J Clin Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwab M, Varmus HE, Bishop JM, Grzeschik KH, Naylor SL, Sakaguchi AY, Brodeur G, Trent J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature. 1984;308:288–291. doi: 10.1038/308288a0. [DOI] [PubMed] [Google Scholar]
- 5.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshimoto M, De Toledo SR Caminada, Caran EM Monteiro, de Seixas MT, de Martino Lee ML, de Campos Vieira Abib S, Vianna SM, Schettini ST, Andrade J Anderson Duffles. MYCN gene amplification. Identification of cell populations containing double minutes and homogeneously staining regions in neuroblastoma tumors. Am J Pathol. 1999;155:1439–1443. doi: 10.1016/S0002-9440(10)65457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guglielmi L, Cinnella C, Nardella M, Maresca G, Valentini A, Mercanti D, Felsani A, DAgnano I. MYCN gene expression is required for the onset of the differentiation programme in neuroblastoma cells. Cell Death Dis. 2014;5:e1081. doi: 10.1038/cddis.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaneko Y, Suenaga Y, Islam SM, Matsumoto D, Nakamura Y, Ohira M, Yokoi S, Nakagawara A. Functional interplay between MYCN, NCYM, and OCT4 promotes aggressiveness of human neuroblastomas. Cancer Sci. 2015;106:840–847. doi: 10.1111/cas.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau DT, Flemming CL, Gherardi S, Perini G, Oberthuer A, Fischer M, Juraeva D, Brors B, Xue C, Norris MD, et al. MYCN amplification confers enhanced folate dependence and methotrexate sensitivity in neuroblastoma. Oncotarget. 2015;6:15510–15523. doi: 10.18632/oncotarget.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beltran H. The N-myc Oncogene: Maximizing its targets, regulation, and therapeutic potential. Mol Cancer Res. 2014;12:815–822. doi: 10.1158/1541-7786.MCR-13-0536. [DOI] [PubMed] [Google Scholar]
- 11.Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA. 2003;100:9779–9784. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 13.He L, Hannon GJ. MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1415. [DOI] [PubMed] [Google Scholar]
- 14.Kozomara A, Griffiths-Jones S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database):D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domingo-Fernandez R, Watters K, Piskareva O, Stallings RL, Bray I. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr Surg Int. 2013;29:101–119. doi: 10.1007/s00383-012-3239-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leichter AL, Sullivan MJ, Eccles MR, Chatterjee A. MicroRNA expression patterns and signalling pathways in the development and progression of childhood solid tumours. Mol Cancer. 2017;16:15. doi: 10.1186/s12943-017-0584-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mei H, Lin ZY, Tong QS. The roles of microRNAs in neuroblastoma. World J Pediatr. 2014;10:10–16. doi: 10.1007/s12519-014-0448-2. [DOI] [PubMed] [Google Scholar]
- 19.Bray I, Tivnan A, Bryan K, Foley NH, Watters KM, Tracey L, Davidoff AM, Stallings RL. MicroRNA-542-5p as a novel tumor suppressor in neuroblastoma. Cancer Lett. 2011;303:56–64. doi: 10.1016/j.canlet.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tivnan A, Tracey L, Buckley PG, Alcock LC, Davidoff AM, Stallings RL. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer. 2011;11:33. doi: 10.1186/1471-2407-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Tsai YH, Fang Y, Tseng SH. Micro-RNA-21 regulates the sensitivity to cisplatin in human neuroblastoma cells. J Pediatr Surg. 2012;47:1797–1805. doi: 10.1016/j.jpedsurg.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Ryan J, Tivnan A, Fay J, Bryan K, Meehan M, Creevey L, Lynch J, Bray IM, O'Meara A, Tracey L, et al. MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br J Cancer. 2012;107:967–976. doi: 10.1038/bjc.2012.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teillet MA, Kalcheim C, Le Douarin NM. Formation of the dorsal root ganglia in the avian embryo: Segmental origin and migratory behavior of neural crest progenitor cells. Dev Biol. 1987;120:329–347. doi: 10.1016/0012-1606(87)90236-3. [DOI] [PubMed] [Google Scholar]
- 24.Beckwith JB, Martin RF. Observations on the histopathology of neuroblastomas. J Pediatr Surg. 1968;3:106–110. doi: 10.1016/0022-3468(68)90989-5. [DOI] [PubMed] [Google Scholar]
- 25.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database):D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, et al. miRTarBase update 2014: An information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42(D1):D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 31.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Huang Q, Liu ZP, Wang Y, Wu LY, Chen L, Zhang XS. NOA: A novel network ontology analysis method. Nucleic Acids Res. 2011;39:e87. doi: 10.1093/nar/gkr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant CE, Bailey TL, Noble WS. FIMO: Scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedländer MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 36.Megiorni F, Camero S, Ceccarelli S, McDowell HP, Mannarino O, Marampon F, Pizer B, Shukla R, Pizzuti A, Marchese C, et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget. 2016;7:79342–79356. doi: 10.18632/oncotarget.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Afanasyeva EA, Hotz-Wagenblatt A, Glatting KH, Westermann F. New miRNAs cloned from neuroblastoma. BMC Genomics. 2008;9:52. doi: 10.1186/1471-2164-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mestdagh P, Fredlund E, Pattyn F, Schulte JH, Muth D, Vermeulen J, Kumps C, Schlierf S, De Preter K, Van Roy N, et al. MYCN/c-MYC-induced microRNAs repress coding gene networks associated with poor outcome in MYCN/c-MYC-activated tumors. Oncogene. 2010;29:1394–1404. doi: 10.1038/onc.2009.429. [DOI] [PubMed] [Google Scholar]
- 39.Bienertova-Vasku J, Mazanek P, Hezova R, Curdova A, Nekvindova J, Kren L, Sterba J, Slaby O. Extension of microRNA expression pattern associated with high-risk neuroblastoma. Tumour Biol. 2013;34:2315–2319. doi: 10.1007/s13277-013-0777-0. [DOI] [PubMed] [Google Scholar]
- 40.Uppal A, Wightman SC, Mallon S, Oshima G, Pitroda SP, Zhang Q, Huang X, Darga TE, Huang L, Andrade J, et al. 14q32-encoded microRNAs mediate an oligometastatic phenotype. Oncotarget. 2015;6:3540–3552. doi: 10.18632/oncotarget.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu L, Chen R, Zhang Y, Fan W, Xiao F, Yan X. Low expression of circulating microRNA-328 is associated with poor prognosis in patients with acute myeloid leukemia. Diagn Pathol. 2015;10:109. doi: 10.1186/s13000-015-0345-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan J, Zheng Z, Zheng Y, Lu X, Xu L, Lin L. microRNA-328 is a favorable prognostic marker in human glioma via suppressing invasive and proliferative phenotypes of malignant cells. Int J Neurosci. 2015;126:145–153. doi: 10.3109/00207454.2014.1002610. [DOI] [PubMed] [Google Scholar]
- 43.Anwar SL, Albat C, Krech T, Hasemeier B, Schipper E, Schweitzer N, Vogel A, Kreipe H, Lehmann U. Concordant hypermethylation of intergenic microRNA genes in human hepatocellular carcinoma as new diagnostic and prognostic marker. Int J Cancer. 2013;133:660–670. doi: 10.1002/ijc.28068. [DOI] [PubMed] [Google Scholar]
- 44.Shi S, Lu Y, Qin Y, Li W, Cheng H, Xu Y, Xu J, Long J, Liu L, Liu C, et al. miR-1247 is correlated with prognosis of pancreatic cancer and inhibits cell proliferation by targeting neuropilins. Curr Mol Med. 2014;14:316–327. doi: 10.2174/1566524014666140228120014. [DOI] [PubMed] [Google Scholar]
- 45.Karjalainen K, Jaalouk DE, Bueso-Ramos CE, Zurita AJ, Kuniyasu A, Eckhardt BL, Marini FC, Lichtiger B, OBrien S, Kantarjian HM, et al. Targeting neuropilin-1 in human leukemia and lymphoma. Blood. 2011;117:920–927. doi: 10.1182/blood-2010-05-282921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan H, Choi AJ, Lee BH, Ting AH. Identification and functional analysis of epigenetically silenced microRNAs in colorectal cancer cells. PLoS One. 2011;6:e20628. doi: 10.1371/journal.pone.0020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charlet J, Schnekenburger M, Brown KW, Diederich M. DNA demethylation increases sensitivity of neuroblastoma cells to chemotherapeutic drugs. Biochem Pharmacol. 2012;83:858–865. doi: 10.1016/j.bcp.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Mestdagh P, Boström AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, von Stedingk K, Ghesquière B, Schulte S, Dews M, et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol Cell. 2010;40:762–773. doi: 10.1016/j.molcel.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miao T, Wu D, Zhang Y, Bo X, Xiao F, Zhang X, Magoulas C, Subang MC, Wang P, Richardson PM. SOCS3 suppresses AP-1 transcriptional activity in neuroblastoma cells through inhibition of c-Jun N-terminal kinase. Mol Cell Neurosci. 2008;37:367–375. doi: 10.1016/j.mcn.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Morgan K, Stewart AJ, Miller N, Mullen P, Muir M, Dodds M, Medda F, Harrison D, Langdon S, Millar RP. Gonadotropin-releasing hormone receptor levels and cell context affect tumor cell responses to agonist in vitro and in vivo. Cancer Res. 2008;68:6331–6340. doi: 10.1158/0008-5472.CAN-08-0197. [DOI] [PubMed] [Google Scholar]
- 51.Weng WC, Lin KH, Wu PY, Lu YC, Weng YC, Wang BJ, Liao YF, Hsu WM, Lee WT, Lee H. Calreticulin regulates VEGF-A in neuroblastoma cells. Mol Neurobiol. 2015;52:758–770. doi: 10.1007/s12035-014-8901-8. [DOI] [PubMed] [Google Scholar]
- 52.Lodrini M, Oehme I, Schroeder C, Milde T, Schier MC, Kopp-Schneider A, Schulte JH, Fischer M, De Preter K, Pattyn F, et al. MYCN and HDAC2 cooperate to repress miR-183 signaling in neuroblastoma. Nucleic Acids Res. 2013;41:6018–6033. doi: 10.1093/nar/gkt346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rihani A, Van Goethem A, Ongenaert M, De Brouwer S, Volders PJ, Agarwal S, De Preter K, Mestdagh P, Shohet J, Speleman F, et al. Genome wide expression profiling of p53 regulated miRNAs in neuroblastoma. Sci Rep. 2015;5:9027. doi: 10.1038/srep09027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy DM, Buckley PG, Bryan K, Das S, Alcock L, Foley NH, Prenter S, Bray I, Watters KM, Higgins D, et al. Global MYCN transcription factor binding analysis in neuroblastoma reveals association with distinct E-box motifs and regions of DNA hypermethylation. PLoS One. 2009;4:e8154. doi: 10.1371/journal.pone.0008154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 2008;122:699–704. doi: 10.1002/ijc.23153. [DOI] [PubMed] [Google Scholar]
- 56.Lovén J, Zinin N, Wahlström T, Müller I, Brodin P, Fredlund E, Ribacke U, Pivarcsi A, Påhlman S, Henriksson M. MYCN-regulated microRNAs repress estrogen receptor-alpha (ESR1) expression and neuronal differentiation in human neuroblastoma. Proc Natl Acad Sci USA. 2010;107:1553–1558. doi: 10.1073/pnas.0913517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20:1603–1614. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorsen SB, Obad S, Jensen NF, Stenvang J, Kauppinen S. The therapeutic potential of microRNAs in cancer. Cancer J. 2012;18:275–284. doi: 10.1097/PPO.0b013e318258b5d6. [DOI] [PubMed] [Google Scholar]
- 60.Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, Boldrini R, Donfrancesco A, Federici V, Giacomini P, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One. 2008;3:e2236. doi: 10.1371/journal.pone.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo J, Dong Q, Fang Z, Chen X, Lu H, Wang K, Yin Y, Cai X, Zhao N, Chen J, et al. Identification of miRNAs that are associated with tumor metastasis in neuroblastoma. Cancer Biol Ther. 2010;9:446–452. doi: 10.4161/cbt.9.6.10894. [DOI] [PubMed] [Google Scholar]
- 62.Schulte JH, Marschall T, Martin M, Rosenstiel P, Mestdagh P, Schlierf S, Thor T, Vandesompele J, Eggert A, Schreiber S, et al. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res. 2010;38:5919–5928. doi: 10.1093/nar/gkq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rahmann S, Martin M, Schulte JH, Köster J, Marschall T, Schramm A. Identifying transcriptional miRNA biomarkers by integrating high-throughput sequencing and real-time PCR data. Methods. 2013;59:154–163. doi: 10.1016/j.ymeth.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 64.Chang YY, Kuo WH, Hung JH, Lee CY, Lee YH, Chang YC, Lin WC, Shen CY, Huang CS, Hsieh FJ, et al. Deregulated microRNAs in triple-negative breast cancer revealed by deep sequencing. Mol Cancer. 2015;14:36. doi: 10.1186/s12943-015-0301-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Hu Y, Fang JY, Xu J. Gain-of-function miRNA signature by mutant p53 associates with poor cancer outcome. Oncotarget. 2016;7:11056–11066. doi: 10.18632/oncotarget.7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pehserl AM, Ress AL, Stanzer S, Resel M, Karbiener M, Stadelmeyer E, Stiegelbauer V, Gerger A, Mayr C, Scheideler M, et al. Comprehensive analysis of miRNome alterations in response to sorafenib treatment in colorectal cancer cells. Int J Mol Sci. 2016;17:E2011. doi: 10.3390/ijms17122011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang H, Zhi H, Ma D, Li T. MiR-217 promoted the proliferation and invasion of glioblastoma by repressing YWHAG. Cytokine. 2017;92:93–102. doi: 10.1016/j.cyto.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 68.Kos A, Loohuis NF Olde, Wieczorek ML, Glennon JC, Martens GJ, Kolk SM, Aschrafi A. A potential regulatory role for intronic microRNA-338-3p for its host gene encoding apoptosis-associated tyrosine kinase. PLoS One. 2012;7:e31022. doi: 10.1371/journal.pone.0031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X, Pan M, Han L, Lu H, Hao X, Dong Q. miR-338-3p suppresses neuroblastoma proliferation, invasion and migration through targeting PREX2a. FEBS Lett. 2013;587:3729–3737. doi: 10.1016/j.febslet.2013.09.044. [DOI] [PubMed] [Google Scholar]
- 70.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Müller S, Nowak K. Exploring the miRNA-mRNA regulatory network in clear cell renal cell carcinomas by next-generation sequencing expression profiles. BioMed Res Int. 2014;2014:948408. doi: 10.1155/2014/948408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murphy DM, Buckley PG, Das S, Watters KM, Bryan K, Stallings RL. Co-localization of the oncogenic transcription factor MYCN and the DNA methyl binding protein MeCP2 at genomic sites in neuroblastoma. PLoS One. 2011;6:e21436. doi: 10.1371/journal.pone.0021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Corvetta D, Chayka O, Gherardi S, DAcunto CW, Cantilena S, Valli E, Piotrowska I, Perini G, Sala A. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. J Biol Chem. 2013;288:8332–8341. doi: 10.1074/jbc.M113.454280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buechner J, Einvik C. N-myc and noncoding RNAs in neuroblastoma. Mol Cancer Res. 2012;10:1243–1253. doi: 10.1158/1541-7786.MCR-12-0244. [DOI] [PubMed] [Google Scholar]
- 75.Jackstadt R, Hermeking H. MicroRNAs as regulators and mediators of c-MYC function. Biochim Biophys Acta. 2015;1849:544–553. doi: 10.1016/j.bbagrm.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 76.Guo L, Chen F. A challenge for miRNA: Multiple isomiRs in miRNAomics. Gene. 2014;544:1–7. doi: 10.1016/j.gene.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 77.Iżycka-Świeszewska E, Drożyńska E, Rzepko R, Kobierska-Gulida G, Grajkowska W, Perek D, Balcerska A. Analysis of PI3K/AKT/mTOR signalling pathway in high risk neuroblastic tumours. Pol J Pathol. 2010;61:192–198. [PubMed] [Google Scholar]
- 78.King D, Yeomanson D, Bryant HE. PI3King the lock: Targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol. 2015;37:245–251. doi: 10.1097/MPH.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 79.Vaughan L, Clarke PA, Barker K, Chanthery Y, Gustafson CW, Tucker E, Renshaw J, Raynaud F, Li X, Burke R, et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget. 2016;7:57525–57544. doi: 10.18632/oncotarget.10544. [DOI] [PMC free article] [PubMed] [Google Scholar]