

Abstract
The hallmark of non-alcoholic fatty liver disease (NAFLD) is excessive fatty accumulation in the hepatocytes, which may be an isolated event (non-alcoholic fatty liver, NAFL) or accompanied by evidence of inflammation and cell injury with or without fibrosis (non-alcoholic steatohepatitis, NASH). NASH, the more aggressive form of NAFLD, may progress to cirrhosis and hepatocellular carcinoma. Because it has been estimated that NASH will overtake hepatitis C virus infection as the leading cause of liver transplantation in the US in the coming decade, and there are no current FDA-approved therapies for this disease, the need to find appropriate therapeutic targets is now more urgent than ever before. Diet and other life-style modifications have always been difficult to maintain and this approach alone has not slowed the rising tide of the disease. While the results of traditional therapies such as vitamin E and pioglitazone have been significant for steatosis and inflammation, they have had no effect on fibrosis, which is the strongest indicator of mortality in this condition. However, the understanding of the pathogenesis and progression of NASH has evolved and several promising novel therapies to target and possibly reverse fibrosis are being evaluated, making the future outlook of NASH therapy more optimistic.
Keywords: Nonalcoholic fatty liver disease (NAFLD), Nonalcoholic steatohepatitis (NASH), ROS (reactive oxygen species), Peroxisome proliferator-activator receptor (PPAR) agonists, Farnesoid X receptor (FXR), Glucagon-like peptide (GLP-1) agonist
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in the Western hemisphere and a leading cause of liver-related morbidity and mortality worldwide.1 An estimated 30% of Americans may be affected by this disease.2,3 NAFLD represents a clinico-pathological spectrum of disease that primarily manifests as excessive accumulation of fat in the hepatocyte (steatosis). It is considered to be the hepatic manifestation of the metabolic syndrome, whose other pathologies include obesity, insulin resistance, hypertension and hyperlipidemia. NAFLD is broadly categorized into 2 phenotypes, namely: non-alcoholic fatty liver (NAFL) which is marked by isolated steatosis, while the more aggressive subtype, non-alcoholic steatohepatitis (NASH), is characterized by cell injury, inflammatory cell infiltration and hepatocyte ballooning that may further progress to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). 4,5 NASH is expected surpass hepatitis C virus infection as the leading etiology of end-stage liver disease requiring liver transplantation in the next 5 to 15 years.6 NASH is also currently the leading etiology driving the burden of hepatocellular carcinoma.7
Despite the significant burden to the public health system, there are no FDA-approved drugs that are specifically tailored for this condition. Therefore, the need for effective treatment that manages the complex pathophysiologic processes of NASH, can no longer be ignored.8 Several novel medications targeting different stages and molecular events in the disease process are currently in the pipeline, while other previously known off-label treatments are still used. In this paper, we will review the pathophysiology and therapeutic approaches of this condition.
Pathophysiological rationale for treatment
The pathophysiology and clinical significance of the various molecular disturbances in NASH are both complex and multifactorial. However, the metabolic imbalance leading to excessive fatty accumulation in the liver appears to occur from 3 major sources: 1) increased dietary fat delivery to the liver from the gut, either due to increased intake or a dysregulated gut physiology and microbiome; 2) increased influx of free fatty acids from the non-esterified pool (mainly from white adipose tissue); and 3) increased de novo hepatic lipogenesis from excess carbohydrates (and or hyperinsulinemia from adipose tissue insulin resistance).9,10 When these processes cannot be offset by a compensatory increase in very low density lipoprotein (VLDL) secretion from the liver or use in other metabolic pathways, there is a net deposition of triglycerides and free fatty acids in hepatocytes.11
While triglyceride accumulation is believed to be relatively benign, hepatocyte lipotoxicity is thought to be chiefly caused by free fatty acids and their metabolites. These changes within the liver place extra metabolic stress on the mitochondria and endoplasmic reticulum and produce a cascade of stress-induced responses including release of reactive oxygen species (ROS) and recruitment of immune cells and other cell injury mediators. These lead to further cell injury culminating in further inflammation, programmed cell death (apoptosis) and fibrotic remodeling via collagen deposition from activated stellate cells.12,13 Alterations in gut microbiota may also lead to bacterial (product) translocation, especially the highly immune-reactive gram negative cell wall component called lipopolysaccharide (LPS), into the systemic circulation, further worsening the inflammatory process by activating macrophages and Kuppfer cells.14 (see fig. 1)
Fig 1. Pathologic processes in NAFLD and potential therapeutic targets.
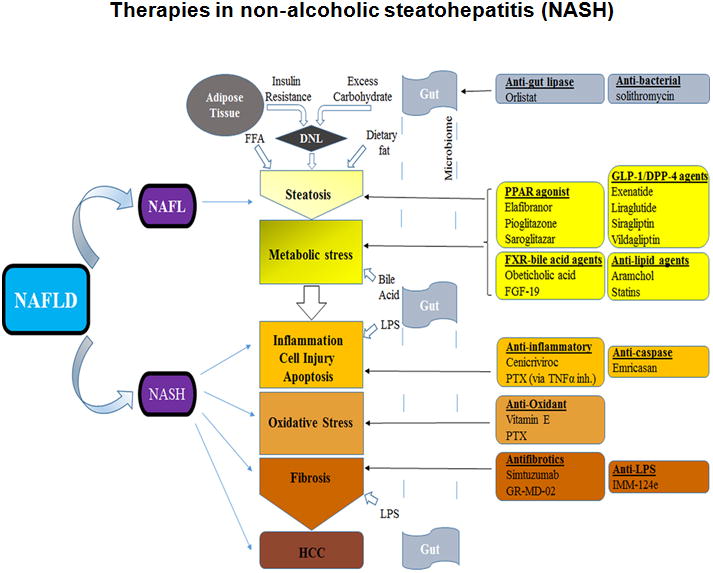
An illustration of the complex processes involved in NASH development and the expected site(s) of action of medications currently being used off-label or under investigation. DNL, de novo lipogenesis; FFA, free fatty acid; NAFL(D), non-alcoholic fatty liver (disease); NASH, non-alcoholic steatohepatitis; TNFα inh., tumor necrosis factor inhibition; PTX, pentoxifylline; HCC, hepatocellular carcinoma; black single-line arrows point to the expected site of action of the medications (but does not indicate the specific stimulatory or inhibitory process involved).
Because of the complexity of the different potential pathways involved in the pathogenesis of NASH, treatment may have to target several key elements in the process of cellular and molecular events to achieve meaningful results. Obviously, depending on the individual patient and the extent of disease activity, more than just one treatment modality may be needed to be effective.
Diet and Physical Activity
The first line of management in NASH involves lifestyle modifications, mainly sustained weight loss (through a calorie-restricted diet) and increased physical activity (exercise).15,16 While a modest weight loss of about 3% may reduce hepatic steatosis, up to 10% or more is needed to reduce inflammation and for the regression of fibrosis in NASH patients.17-19
Even in well-organized settings, only a few patients achieve and sustain a 10% weight loss.20 A more detailed review of lifestyle measures employed in NASH therapy is not the purpose of this review, but may be obtained elsewhere.21
Based of the difficulties in applying lifestyle-changing measures alone, the need to combine them with pharmacotherapy will probably remain the norm in NASH management in the foreseeable future.
Pharmaco-therapeutic Options in NASH
1) PPAR agonists
Peroxisome proliferator-activator receptors (PPARs) are a group of nuclear receptors that are expressed in the liver, adipose tissue, heart, skeletal muscle and kidney and transcriptionally regulate multiple metabolic processes including B-oxidation, lipid transport and gluconeogenesis.22 There are 3 PPAR receptors (α, β/δ and γ) which differ by tissue distribution but target the same DNA segment.23
PPARα agonists, such as fibrates, are extensively used in the treatment of hypertriglyceridemia but have been shown to have no significant benefit in NAFLD, probably because of the receptor's extensive distribution in organs outside the liver.24 PPARδ, because of its presence in macrophages, has the additional effect of decreasing macrophage and Kupffer cell activation and increasing fatty acid oxidation.25 Although a PPARβ/δ agonist (GW501516), was highly promising in initial trials, the drug may have been withdrawn due to safety concerns.26
The dual PPARα/δ agonist, elafibranor may be effective not only in improving both hepatic and peripheral insulin sensitivity in abdominally obese individuals in a liver-targeted fashion, but was also shown to resolve NASH in a phase IIb, randomized double-blind placebo controlled trial (GOLDEN-505).27 This was a multicenter, international study with NASH patients (without cirrhosis) who were randomly assigned to 3 different groups: elafibranor 80 mg/day (n = 93), elafibranor 120 mg/day (n = 91), or placebo (n = 92) for a total of 52 weeks. The primary outcome (resolution of NASH without worsening fibrosis) was not obtained when the elafibranor groups were compared to placebo in the initial analysis. However, when a modified definition was used in a post-hoc analysis, the primary outcome was obtained in patients taking elafibranor 120 mg/day compared to placebo (19% versus 12%; P = .045). In addition, the resolution of NASH was significantly higher in patients with an NAFLD activity score (NAS) ≥4 (n = 234) who took elafibranor 120 mg/day compared to placebo, both with the initial protocol definition (20% versus 11%; P = .018) and the modified criteria (19% versus 9%; P = .013). Interestingly, there was regression in the stage of fibrosis in the patients in whom NASH resolved with elafibranor 120 mg, compared to those in whom it did not (P < .001). Other metabolic parameters including liver enzymes and lipid profile were also improved in the 120mg/day group.28,29 A phase III trial (NCT02704403) is currently in the recruitment phase.
PPARγ agonists have been extensively used in diabetes as insulin sensitizers in the form of Thiazolidinediones (TZDs). They have also been shown to be effective in the treatment of NASH.30-32 In the PIVENS trial, pioglitazone was evaluated with vitamin E and placebo with improvement in NASH histology as a primary end-point. Although there was a trend towards improvement in NASH histology with pioglitazone compared to placebo (34% versus 19%; P=0.04), this did not reach the study's goal (P=0.025). On the other hand, a significant reduction in the serum alanine and aspartate aminotransferase (ALT and AST) as well as in hepatic steatosis and lobular inflammation were obtained compared to placebo (P<0.001),(P<0.001) and (P=0.004) respectively. However, in this particular trial, there was no significant improvement in fibrosis with pioglitazone compared to placebo (P=0.12). Furthermore, the known side effect of weight gain (which also occurred in the PIVENS study) and risk of congestive heart failure may limit its use in the treatment of NASH.32,33
A dual PPARα/γ agonist, saroglitazar, which has been approved to treat diabetic dyslipidemia in India has also been shown to result in a significant decrease in ALT levels in subjects with NAFLD and biopsy-proven NASH.34,35 Further clinical studies are needed in this drug to confirm this initial success.
2) FXR-bile acid axis
The bile acid intracellular receptor, farnesoid X receptor (FXR), negatively regulates bile acid synthesis and decreases hepatic gluconeogenesis, lipogenesis and steatosis.36,37 Obeticholic acid, or OCA, is a synthetic bile acid derivative and FXR agonist that was recently studied in biopsy-proven NASH patients without cirrhosis in the FLINT trial (a multicenter, randomized, double-blind, placebo controlled study). In this study, 283 patients were randomly assigned to receive either a daily dose of 25mg OCA (n=141) or placebo (n=142) for 72 weeks. The findings showed a significant histological improvement in the OCA group (45% versus 21% of control; P=0.0002) as well as an improved fibrosis score (35% versus 19% of controls; p=0.004). The development of the adverse effect, pruritus, was noted in OCA-treated patients (23%) leading to discontinuation of the medication in some cases and raising questions about drug tolerability. Even though it is reversible, there was also worsening of the lipid profile in subjects treated with OCA, which may require closer monitoring for possible cardiovascular consequences.38 Otherwise, thus far, OCA appears to be highly promising for the treatment of NASH and its upcoming phase III trial (NCT02548351), with more ambitious end-points (including mortality and liver-related morbidity), will hopefully confirm efficacy of this treatment in these patients.
FGF-19 is a hormone that is transcriptionally regulated via FXR activation in response to the postprandial influx of bile acids in the terminal ileum. FGF-19 then binds to the FGFR4/β-klotho receptor complex on the hepatocyte cell membrane, thus suppressing gluconeogenesis while promoting glycogen synthesis.39,40 It has been suggested that FGFR4 activation could have a cancer-promoting effect on hepatocytes. Thus non-tumorigenic variants have been developed with some success in initial studies.41,42 NGM-282 is considered to be one non-cancer-promoting agent and it is currently in a phase II trial (NCT02443116) to evaluate its effect in NASH patients over a 12-week treatment period.There are ongoing studies evaluating other FXR-modulating, as well as bile acid-sequestrating agents, in either animal testing phases or in preliminary clinical trials (see fig.1).
3) Lipid-altering agents
Stearoyl-CoA desaturase (SCD) is an enzyme that catalyzes the rate-limiting step in the synthesis of monounsaturated fatty acids such as oleic acid.43 Located in the endoplasmic reticulum, animal studies have shown that a deficiency (or inhibition) of the SCD-1 isoform is associated with decreased liver steatosis as well as improved insulin sensitivity.44-46 Obese subjects with NASH appear to have greater stearoyl-CoA desaturase 1 activity (SCD1, p < 0.002) than those with a normal liver, which is also reflected at the level of gene expression.47
Aramchol, an SCD-1 inhibitor, is a cholic acid-arachidic acid conjugate that was evaluated in a double-blind, placebo-controlled trial of 60 patients with biopsy-proven NAFLD (only 6 had NASH) who were randomized assigned to daily 100mg or 300mg Aramchol versus placebo (n=20 per group). The drug was associated with reduced hepatic fat content when administered as a 300mg/day regimen versus placebo for 3 months.48 Aramchol is currently being evaluated in a multicenter phase IIb trial to determine its effectiveness in NASH patients.
Statins are HMG-CoA reductase inhibitors that are extensively used in both primary and secondary prevention of cardiovascular disease due to their effective plasma lipid-lowering abilities.49 Dyslipidemia is a common feature of both the metabolic syndrome and NAFLD, placing patients at increased risk of cardiovascular events.50,51 Moreover, NAFLD alone is an independent risk factor for cardiovascular disease. However, statins have been found to be underused for this condition, even though they are considered to be generally safe at moderate doses in patients with chronic liver disease.52,53 In fact, even though there are only a few studies on the use of statins in NASH, in a cohort of 1201 Europeans who underwent liver biopsy for suspected NASH, these agents were found to provide protection from steatosis (p=0.004), steatohepatitis (p<0.001), and stage F2-F4 fibrosis (p=0.017).54 A small, prospective but uncontrolled study also showed resolution of NASH in 19/20 patients with a 10mg/day dose of rosuvastatin for 12 months.55
4) Incretin-based Therapies
Glucagon-like peptide 1 (GLP-1) is a hormone that belongs to the incretin group of proteins that is secreted in the distal ileum and proximal colon by L cells.56 Besides stimulating the pancreas to cause beta cell proliferation and enhance insulin biosynthesis, GLP-1 also interacts with receptors in other parts of the GI tract and in the lung, kidney and CNS. Thus, GLP-1 has several metabolic functions including delayed gastric emptying, appetite suppression, enhanced liver glucose uptake and peripheral insulin sensitivity, as well as glucose-dependent insulin secretion while inhibiting the release of glucagon from α-cells.57,58 GLP-1 undergoes rapid degradation by the dipeptidyl peptidase 4 (DPP-4) enzyme; therefore, these medications are made to resist this immediate cleavage by DDP-4.56
While GLP-1 receptor agonists such as exenatide and liraglutide have mainly been approved for diabetes mellitus type 2, a meta-analysis of several studies has shown significant results in patients with NASH. These beneficial effects include decreased serum ALT levels, as well as an improvement in hepatic fat content and fibrosis. The associated weight loss with these medications make them potentially attractive for use in patients with NASH and the metabolic syndrome.59-61
The LEAN study evaluated the safety and efficacy of liraglutide in NASH in 52 patients randomized to receive either 1.8mg/day of liraglutide or placebo for 48 weeks (n=26 in each group). The study found that 39% of patients in the liraglutide group met the primary end-point (resolution of NASH without worsening of fibrosis) compared to 9% in the placebo group (p=0.019). While fibrosis only progressed in 9% of patients in the liraglutide group, it occurred in 36% (p=0.019) of the placebo group. 62 This makes liraglutide one of the most attractive potential therapies available for NASH.
Dipeptidyl peptidase 4 (DPP-4) inhibitors, such as sitagliptin and vildapliptin, act on the enzyme DPP-4, which is known to rapidly degrade GLP-1. Thus, these medications are expected to prolong the action of GLP-1. Thus far, results in sitagliptin have been mixed for liver enzyme levels, fat content and the measurement of fibrosis and there is no convincing consensus on the effectiveness of this group of medications in NASH at this time.35,63-66 Further studies are needed to fully confirm their clinical efficacy.
5) Agents targeting inflammation, cell injury or death (apoptosis) and oxidative stress
At present vitamin E is considered to be a first line pharmacological treatment in the management of NASH especially when diet and other lifestyle changes are insufficient. Vitamin E, with its known antioxidant effects, was initially shown to be effective in NASH in a successful pilot study.67 This was followed by the PIVENS trial comparing non-diabetic, biopsy-proven NASH patients who received 800 IU/day of vitamin E (800mg/day; n=84) to patients receiving pioglitazone (30mg/day; n=80) and placebo (n=83). This study showed that vitamin E was better than placebo in reducing ALT levels (P<0.001), liver steatosis (P0.005) and inflammation (P=0.02) and also showed that vitamin E effectively promoted resolution of NASH (43 % versus 19% in placebo; P=0.001). However, there was no improvement in the liver fibrosis score (P=0.24).32 Similar findings were recorded in the TONIC trial, which was performed in children and adolescents.68
Although vitamin E was not specifically studied in diabetic NASH patients, indirect evidence from pooled data from the PIVENS trial and the placebo arm of the FLINT trial who received vitamin E supports the efficacy of vitamin E in diabetics as well. In that analysis, whatever the status of diabetes, the histological features of the liver were markedly higher in patients receiving vitamin E than in those who did not receive the medication.69
Even though no significant major adverse events have been reported with vitamin E in the above mentioned studies, prolonged use of this medication, especially at higher doses, may influence mortality. Although the effect on all-cause mortality was not supported by subsequent studies on the subject, 70,71 some studies have linked long-term use of vitamin E to increased risks of prostate cancer and hemorrhagic stroke.72,73 Therefore, these potential risks need to be considered in relation to the extent of disease activity (NASH) and the patient's health when deciding on treatment. A dose of 400 IU daily could be a solution in certain circumstances.74
Tumour necrosis factor (TNF)α signaling stands at the crossroads of the major pathways mediating hepatocyte cell injury and caspase-regulated programmed cell death (apoptosis) in NASH.75,76 Emricasan is a pan-caspase inhibitor that was initially studied in patients with chronic liver diseases of diverse etiologies, and was found to lower ALT levels, in particular in patients with hepatitis C and with NASH.77 The efficacy of Emricasan is being evaluated in a phase IIb trial (ENCORE-NF; NCT02686762) in patients with NASH and fibrosis.
Pentoxifylline (PTX) is a methylxanthine derivative that has an inhibitory effect on phosphodiesterase as well as on TNFα, and could therefore modulate the functions of other inflammatory cytokines.78,79 Although it was previously used in a subset of patients with alcoholic hepatitis, the improved survival reported in these patients is currently in serious question.80,81 Nevertheless, significant histologic improvement was found with PTX in patients with biopsy-proven NASH (400mg three times daily for a year), based on an NAS of ≥2 in 38.5% (n=26) of the treatment group compared to 13.8% (n=29) with placebo (P=0.036). Although fibrosis decreased in more patients in the PTX group (35%) than in the placebo group (15%), this was not statistically significant. Another study suggests that the mechanism of PTX may be due, at least in part, to a reduction in lipid peroxidation, through this agents oxygen radical scavenging properties and glutathione-replenishing ability.82-84 Larger, robust studies are needed to confirm the efficacy of PTX in the treatment of NASH.
During liver injury, hepatocytes and other liver cell types release inflammatory chemokines such as CCL2 (MCP1) and CCL5 (RANTES) that help recruit macrophages and other inflammatory cells to the site.85,86 CCL2 and CCL5 act via their respective receptors, CCR2 and CCR5, located on a host of inflammatory cell types to elicit migration. Interestingly, CCL2 and CCL5 are two of 17 up-regulated genes in NASH patients.87 Cenicriviroc is a CCR2/CCR5 antagonist and the results of a phase IIb trial (CENTAUR) with this drug are awaited in NASH patients.
Other agents that may affect inflammation, cell injury or apoptosis as well immune related factors are currently in ongoing clinical trials. For example, GS-4997, an inhibitor of apoptosis signal-regulating kinase 1 (ASK1), a MAP3 kinase that may play a role in hepatic steatosis and fibrosis is being studied for this indication.35
6) Gut and Microbiome related therapies
Disturbed gut-liver barrier integrity has been reported to be essential in the pathogenesis of NAFLD and NASH since bacteria and their products (especially lipopolysaccharide or LPS) can escape into the systemic circulation causing a massive inflammatory response from the liver.88 Thus, any therapies that prevent this bacterial or LPS ‘translocation’ to the systemic circulation and liver are of interest.
IMM-124e is an IgG-enhanced bovine-derived colostrum with favorable results in preliminary clinical studies including improved glycemic control, insulin resistance and lipid profile.89 Further studies are ongoing.
Orlistat is an FDA-approved lipase inhibitor to treat obesity whose action is located in the gut to reduce dietary fat absorption (8626884). Although it seems to improve liver enzyme levels and liver content, its efficacy in NASH has not yet been clearly evaluated.90
Other agents targeting the microbiome include a macrolide antibiotic, solithromycin, which is currently in a phase II trial (NCT02510599).
7) Antifibrotic Therapies
Recent work has shown the stage of liver fibrosis to be the strongest predictor of mortality in patients with NAFLD. Hence, there is great interest in developing appropriate therapeutics to target various elements in fibrogenesis.91
Simtuzumab is a monoclonal antibody targeting lysyl oxidase-like 2 (LOXL2), which is a key matrix enzyme in collagen formation and highly expressed in the liver.92 The drug is currently being studied in a phase II trial in both cirrhotic and non-cirrhotic subjects with NASH (NCT01672866).
Galectin-3 is a protein expressed predominantly in immune cells that recognizes and binds to galactose residues. Galectin-3 is an essential protein in liver fibrogenesis and thus a good target of the inhibitor, GR-MD-02. This drug is currently in two phase II clinical trials in NASH patients with fibrosis/cirrhosis (NCT02462967).
Summary
In the absence of any existing FDA-approved medications for the treatment of NAFLD/NASH, the mainstay of management continues to be dietary and other lifestyle changes tailored to the individual patient. Since the acceptable threshold for weight loss needed to resolve NASH and for the regression of fibrosis (more than 10% of body weight) is difficult to obtain even in highly motivated individuals, pharmacotherapies are urgently needed.
Numerous medications have been added to the pipeline of novel therapies, increasing the promise of successful treatment of NASH in the future. In the meantime, first line drugs such as vitamin E and pioglitazone continue to serve their purpose in carefully selected patients, with or without diabetes.
Key Points.
NASH is the more aggressive subtype of NAFLD and may replace hepatitis C as the leading cause of liver transplantation by 2020
The presence of fibrosis is the strongest predictor of mortality in patients with this disease.
Beside life-style changes, there are no FDA-approved medications for patients with NASH
Common medications that have showed good results in reducing disease activity are vitamin E and pioglitazone, but these have no consistently reliable effect on fibrosis
There are multiple other promising drugs targeting fibrosis and other elements involved in the pathogenesis of NASH that are currently in various stages of clinicial study
Acknowledgments
Funding source: This manuscript was supported by a training grant T32 DK 7150-40 and RO1 DK 081450 from NIDDK to Dr. Arun Sanyal.
Footnotes
Conflicts of Interest: Dr. Oseini: None to report
Dr. Sanyal: ICMJE COI form submitted separately
Contributor Information
Abdul Oseini, MCV Box 980341, Richmond, VA 23298-0341, (804) 828 6314, (804) 828 2992.
Arun J. Sanyal, MCV Box 980341, Richmond, VA 23298-0341, (804) 828 2992, (804) 828 6314.
References
- 1.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nature reviews Gastroenterology & hepatology. 2013 Nov;10(11):686–690. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- 2.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004 Dec;40(6):1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 3.Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011 Jan;140(1):124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 4.Karlas T, Wiegand J, Berg T. Gastrointestinal complications of obesity: non-alcoholic fatty liver disease (NAFLD) and its sequelae. Best practice & research Clinical endocrinology & metabolism. 2013 Apr;27(2):195–208. doi: 10.1016/j.beem.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. Journal of hepatology. 2010 Aug;53(2):372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011 Oct;141(4):1249–1253. doi: 10.1053/j.gastro.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 7.Sanyal A, Poklepovic A, Moyneur E, Barghout V. Population-based risk factors and resource utilization for HCC: US perspective. Current medical research and opinion. 2010 Sep;26(9):2183–2191. doi: 10.1185/03007995.2010.506375. [DOI] [PubMed] [Google Scholar]
- 8.Sanyal AJ, Friedman SL, McCullough AJ, Dimick-Santos L. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases-U.S. Food and Drug Administration Joint Workshop. Hepatology. 2015 Apr;61(4):1392–1405. doi: 10.1002/hep.27678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asrih M, Jornayvaz FR. Diets and nonalcoholic fatty liver disease: the good and the bad. Clin Nutr. 2014 Apr;33(2):186–190. doi: 10.1016/j.clnu.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014 Mar;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008 Feb;134(2):424–431. doi: 10.1053/j.gastro.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010 Aug;52(2):774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 13.Cheung O, Sanyal AJ. Abnormalities of lipid metabolism in nonalcoholic fatty liver disease. Seminars in liver disease. 2008 Nov;28(4):351–359. doi: 10.1055/s-0028-1091979. [DOI] [PubMed] [Google Scholar]
- 14.Betrapally NS, Gillevet PM, Bajaj JS. Gut microbiome and liver disease. Translational research : the journal of laboratory and clinical medicine. 2016 Jul 15; doi: 10.1016/j.trsl.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. The American journal of gastroenterology. 2012 Jun;107(6):811–826. doi: 10.1038/ajg.2012.128. [DOI] [PubMed] [Google Scholar]
- 16.Huang MA, Greenson JK, Chao C, et al. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. The American journal of gastroenterology. 2005 May;100(5):1072–1081. doi: 10.1111/j.1572-0241.2005.41334.x. [DOI] [PubMed] [Google Scholar]
- 17.Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology. 2009 Jan;49(1):80–86. doi: 10.1002/hep.22575. [DOI] [PubMed] [Google Scholar]
- 18.Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010 Jan;51(1):121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology. 2015 Aug;149(2):367–378 e365. doi: 10.1053/j.gastro.2015.04.005. quiz e314-365. [DOI] [PubMed] [Google Scholar]
- 20.Wadden TA, Volger S, Sarwer DB, et al. A two-year randomized trial of obesity treatment in primary care practice. The New England journal of medicine. 2011 Nov 24;365(21):1969–1979. doi: 10.1056/NEJMoa1109220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lassailly G, Caiazzo R, Pattou F, Mathurin P. Perspectives on Treatment for Nonalcoholic Steatohepatitis. Gastroenterology. 2016 Jun;150(8):1835–1848. doi: 10.1053/j.gastro.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007 Jan 30;115(4):518–533. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
- 23.Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Seminars in cell & developmental biology. 2012 Aug;23(6):631–639. doi: 10.1016/j.semcdb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Musso G, Gambino R, Cassader M, Pagano G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology. 2010 Jul;52(1):79–104. doi: 10.1002/hep.23623. [DOI] [PubMed] [Google Scholar]
- 25.Bojic LA, Huff MW. Peroxisome proliferator-activated receptor delta: a multifaceted metabolic player. Current opinion in lipidology. 2013 Apr;24(2):171–177. doi: 10.1097/MOL.0b013e32835cc949. [DOI] [PubMed] [Google Scholar]
- 26.Riserus U, Sprecher D, Johnson T, et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008 Feb;57(2):332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- 27.Cariou B, Hanf R, Lambert-Porcheron S, et al. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes care. 2013 Oct;36(10):2923–2930. doi: 10.2337/dc12-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology. 2016 May;150(5):1147–1159 e1145. doi: 10.1053/j.gastro.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 29.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005 Jun;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 30.Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. The New England journal of medicine. 2006 Nov 30;355(22):2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 31.Promrat K, Lutchman G, Uwaifo GI, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004 Jan;39(1):188–196. doi: 10.1002/hep.20012. [DOI] [PubMed] [Google Scholar]
- 32.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England journal of medicine. 2010 May 6;362(18):1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. Jama. 2007 Sep 12;298(10):1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 34.Pai V, Paneerselvam A, Mukhopadhyay S, et al. A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V) Journal of diabetes science and technology. 2014 Jan 16;8(1):132–141. doi: 10.1177/1932296813518680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. 2016 Sep 19; doi: 10.1136/gutjnl-2016-312431. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. The Journal of clinical investigation. 2004 May;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. Journal of lipid research. 2012 Sep;53(9):1723–1737. doi: 10.1194/jlr.R024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015 Mar 14;385(9972):956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011 Mar 25;331(6024):1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nies VJ, Sancar G, Liu W, et al. Fibroblast Growth Factor Signaling in Metabolic Regulation. Frontiers in endocrinology. 2015;6:193. doi: 10.3389/fendo.2015.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo J, Ko B, Elliott M, et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Science translational medicine. 2014 Jul 30;6(247):247ra100. doi: 10.1126/scitranslmed.3009098. [DOI] [PubMed] [Google Scholar]
- 42.Luo J, Ko B, To C, et al. Treatment with Ngm282 Significantly improves liver histopathology in a mouse model of non-alcoholic steatohepatitis (Nash) Journal of hepatology. 2015;62:S694–S. [Google Scholar]
- 43.Hodson L, Fielding BA. Stearoyl-CoA desaturase: rogue or innocent bystander? Progress in lipid research. 2013 Jan;52(1):15–42. doi: 10.1016/j.plipres.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Ntambi JM. The regulation of stearoyl-CoA desaturase (SCD) Progress in lipid research. 1995;34(2):139–150. doi: 10.1016/0163-7827(94)00010-j. [DOI] [PubMed] [Google Scholar]
- 45.Miyazaki M, Flowers MT, Sampath H, et al. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell metabolism. 2007 Dec;6(6):484–496. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 46.Issandou M, Bouillot A, Brusq JM, et al. Pharmacological inhibition of stearoyl-CoA desaturase 1 improves insulin sensitivity in insulin-resistant rat models. European journal of pharmacology. 2009 Sep 15;618(1-3):28–36. doi: 10.1016/j.ejphar.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Walle P, Takkunen M, Mannisto V, et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism: clinical and experimental. 2016 May;65(5):655–666. doi: 10.1016/j.metabol.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 48.Safadi R, Konikoff FM, Mahamid M, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2014 Dec;12(12):2085–2091 e2081. doi: 10.1016/j.cgh.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 49.Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014 Jun 24;129(25 Suppl 2):S1–45. doi: 10.1161/01.cir.0000437738.63853.7a. [DOI] [PubMed] [Google Scholar]
- 50.Srikanth S, Deedwania P. Management of Dyslipidemia in Patients with Hypertension, Diabetes, and Metabolic Syndrome. Current hypertension reports. 2016 Oct;18(10):76. doi: 10.1007/s11906-016-0683-0. [DOI] [PubMed] [Google Scholar]
- 51.Katsiki N, Mikhailidis DP, Mantzoros CS. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism: clinical and experimental. 2016 Aug;65(8):1109–1123. doi: 10.1016/j.metabol.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Pastori D, Polimeni L, Baratta F, Pani A, Del Ben M, Angelico F. The efficacy and safety of statins for the treatment of non-alcoholic fatty liver disease. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2015 Jan;47(1):4–11. doi: 10.1016/j.dld.2014.07.170. [DOI] [PubMed] [Google Scholar]
- 53.Blais P, Lin M, Kramer JR, El-Serag HB, Kanwal F. Statins Are Underutilized in Patients with Nonalcoholic Fatty Liver Disease and Dyslipidemia. Digestive diseases and sciences. 2016 Jun;61(6):1714–1720. doi: 10.1007/s10620-015-4000-6. [DOI] [PubMed] [Google Scholar]
- 54.Dongiovanni P, Petta S, Mannisto V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. Journal of hepatology. 2015 Sep;63(3):705–712. doi: 10.1016/j.jhep.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 55.Kargiotis K, Athyros VG, Giouleme O, et al. Resolution of non-alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World journal of gastroenterology. 2015 Jul 7;21(25):7860–7868. doi: 10.3748/wjg.v21.i25.7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006 Nov 11;368(9548):1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 57.Drucker DJ. The biology of incretin hormones. Cell metabolism. 2006 Mar;3(3):153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 58.Abu-Hamdah R, Rabiee A, Meneilly GS, Shannon RP, Andersen DK, Elahi D. Clinical review: The extrapancreatic effects of glucagon-like peptide-1 and related peptides. The Journal of clinical endocrinology and metabolism. 2009 Jun;94(6):1843–1852. doi: 10.1210/jc.2008-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carbone LJ, Angus PW, Yeomans ND. Incretin-based therapies for the treatment of non-alcoholic fatty liver disease: A systematic review and meta-analysis. Journal of gastroenterology and hepatology. 2016 Jan;31(1):23–31. doi: 10.1111/jgh.13026. [DOI] [PubMed] [Google Scholar]
- 60.Buse JB, Klonoff DC, Nielsen LL, et al. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: an interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clinical therapeutics. 2007 Jan;29(1):139–153. doi: 10.1016/j.clinthera.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 61.Armstrong MJ, Houlihan DD, Rowe IA, et al. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: individual patient data meta-analysis of the LEAD program. Alimentary pharmacology & therapeutics. 2013 Jan;37(2):234–242. doi: 10.1111/apt.12149. [DOI] [PubMed] [Google Scholar]
- 62.Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016 Feb 13;387(10019):679–690. doi: 10.1016/S0140-6736(15)00803-X. [DOI] [PubMed] [Google Scholar]
- 63.Yilmaz Y, Yonal O, Deyneli O, Celikel CA, Kalayci C, Duman DG. Effects of sitagliptin in diabetic patients with nonalcoholic steatohepatitis. Acta gastro-enterologica Belgica. 2012 Jun;75(2):240–244. [PubMed] [Google Scholar]
- 64.Fukuhara T, Hyogo H, Ochi H, et al. Efficacy and safety of sitagliptin for the treatment of nonalcoholic fatty liver disease with type 2 diabetes mellitus. Hepato-gastroenterology. 2014 Mar-Apr;61(130):323–328. [PubMed] [Google Scholar]
- 65.Kato H, Nagai Y, Ohta A, et al. Effect of sitagliptin on intrahepatic lipid content and body fat in patients with type 2 diabetes. Diabetes research and clinical practice. 2015 Jul;109(1):199–205. doi: 10.1016/j.diabres.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 66.Cui J, Philo L, Nguyen P, et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. Journal of hepatology. 2016 Aug;65(2):369–376. doi: 10.1016/j.jhep.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanyal AJ, Mofrad PS, Contos MJ, et al. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2004 Dec;2(12):1107–1115. doi: 10.1016/s1542-3565(04)00457-4. [DOI] [PubMed] [Google Scholar]
- 68.Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. Jama. 2011 Apr 27;305(16):1659–1668. doi: 10.1001/jama.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kowdley K, Wilson L, Van Natta M, et al. Efficacy and Safety of Vitamin E in Nonalcoholic Steatohepatitis Patients With and Without diabetes: Pooled analysis from the PIVENS and FLINT NIDDK NASH CRN Trials. Hepatology. 2015;62(264A–A) [Google Scholar]
- 70.Abner EL, Schmitt FA, Mendiondo MS, Marcum JL, Kryscio RJ. Vitamin E and all-cause mortality: a meta-analysis. Current aging science. 2011 Jul;4(2):158–170. doi: 10.2174/1874609811104020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama. 2007 Feb 28;297(8):842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 72.Klein EA, Thompson IM, Jr, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) Jama. 2011 Oct 12;306(14):1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010 Nov 04;341:c5702. doi: 10.1136/bmj.c5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rinella ME, Sanyal AJ. Management of NAFLD: a stage-based approach. Nature reviews Gastroenterology & hepatology. 2016 Apr;13(4):196–205. doi: 10.1038/nrgastro.2016.3. [DOI] [PubMed] [Google Scholar]
- 75.Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clinics in liver disease. 2009 Nov;13(4):565–580. doi: 10.1016/j.cld.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Frontiers in bioscience : a journal and virtual library. 2005 Sep 01;10:3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- 77.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007 Aug;46(2):324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 78.D'Hellencourt CL, Diaw L, Cornillet P, Guenounou M. Differential regulation of TNF alpha, IL-1 beta, IL-6, IL-8, TNF beta, and IL-10 by pentoxifylline. International journal of immunopharmacology. 1996 Dec;18(12):739–748. doi: 10.1016/s0192-0561(97)85556-7. [DOI] [PubMed] [Google Scholar]
- 79.Genoves P, Garcia D, Cejalvo D, et al. Pentoxifylline in liver ischemia and reperfusion. Journal of investigative surgery : the official journal of the Academy of Surgical Research. 2014 Apr;27(2):114–124. doi: 10.3109/08941939.2013.835454. [DOI] [PubMed] [Google Scholar]
- 80.Thursz MR, Richardson P, Allison M, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. The New England journal of medicine. 2015 Apr 23;372(17):1619–1628. doi: 10.1056/NEJMoa1412278. [DOI] [PubMed] [Google Scholar]
- 81.Mathurin P, Louvet A, Duhamel A, et al. Prednisolone with vs without pentoxifylline and survival of patients with severe alcoholic hepatitis: a randomized clinical trial. Jama. 2013 Sep 11;310(10):1033–1041. doi: 10.1001/jama.2013.276300. [DOI] [PubMed] [Google Scholar]
- 82.Zein CO, Lopez R, Fu X, et al. Pentoxifylline decreases oxidized lipid products in nonalcoholic steatohepatitis: new evidence on the potential therapeutic mechanism. Hepatology. 2012 Oct;56(4):1291–1299. doi: 10.1002/hep.25778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Freitas JP, Filipe PM. Pentoxifylline. A hydroxyl radical scavenger. Biological trace element research. 1995 Jan-Mar;47(1-3):307–311. doi: 10.1007/BF02790131. [DOI] [PubMed] [Google Scholar]
- 84.Koppe SW, Sahai A, Malladi P, Whitington PF, Green RM. Pentoxifylline attenuates steatohepatitis induced by the methionine choline deficient diet. Journal of hepatology. 2004 Oct;41(4):592–598. doi: 10.1016/j.jhep.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 85.Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014 Sep;147(3):577–594 e571. doi: 10.1053/j.gastro.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 86.Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Frontiers in physiology. 2012;3:213. doi: 10.3389/fphys.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bertola A, Bonnafous S, Anty R, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PloS one. 2010 Oct 22;5(10):e13577. doi: 10.1371/journal.pone.0013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Compare D, Coccoli P, Rocco A, et al. Gut--liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutrition, metabolism, and cardiovascular diseases : NMCD. 2012 Jun;22(6):471–476. doi: 10.1016/j.numecd.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 89.Mizrahi M, Shabat Y, Ben Ya'acov A, et al. Alleviation of insulin resistance and liver damage by oral administration of Imm124-E is mediated by increased Tregs and associated with increased serum GLP-1 and adiponectin: results of a phase I/II clinical trial in NASH. Journal of inflammation research. 2012;5:141–150. doi: 10.2147/JIR.S35227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zelber-Sagi S, Kessler A, Brazowsky E, et al. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2006 May;4(5):639–644. doi: 10.1016/j.cgh.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 91.Ekstedt M, Hagstrom H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015 May;61(5):1547–1554. doi: 10.1002/hep.27368. [DOI] [PubMed] [Google Scholar]
- 92.Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nature medicine. 2010 Sep;16(9):1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]