

Abstract
Acute myeloid leukemia (AML) is an aggressive blood cancer that results from an abnormal expansion of uncontrollably proliferating myeloid progenitors that have lost the capacity to differentiate. AML encompasses many genetically distinct sub-types that predominantly develop de novo. However, AML can also arise from pre-malignant myeloid conditions, such as myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPNs) or develop as the result of exposure to genotoxic agents used to treat unrelated malignancies. While numerous distinct cytogenetic and molecular abnormalities associated with AML were discovered prior to the turn of the millennium, recent advances in whole genome sequencing and global genomic approaches has resulted in an explosion of newly identified molecular abnormalities. However, even with these advances, our understanding of how these mutations contribute to the etiology, pathogenesis and therapeutic responses of AMLs remains largely unknown. Recently the International Society for Experimental Hematology (ISEH) hosted a webinar entitled “Clonal Evolution of Pre-Leukemic Hematopoietic Stem Cells (HSCs) in AML” where two AML mavens, Drs. Ross Levine, MD and Ravindra Majeti, MD, PhD, discussed some of their recent, ground breaking studies that have shed light on how many of these newly identified mutations contribute to leukemogenesis and therapy resistance in AML. Here, we provide a brief overview of this webinar and discuss the basic scientific and clinical implications of the data presented.
Clonal evolution of pre-leukemic HSCs in human acute myeloid leukemia - Ravindra Majeti, MD, PhD
Recent global genome studies carried out by The Cancer Genome Atlas (TCGA) network revealed that de novo AML arises from multiple combinations of a numerous recurrent driver gene mutations [1-3]. The authors of this study grouped recurrent driver gene mutations into nine categories based on functional overlap. For example, they categorized TET2, DNMT1, DNMT3A, DMNT3B, IDH1 and IDH2 mutations together because each of these abnormalities impact global DNA methylation. Moreover, these studies revealed that individual cases of frank leukemia possess multiple functionally distinct driver mutations. As highlighted by Dr. Majeti, these data raise several intriguing questions. First, what cell type does a leukemia clone arise from? Second, is there a specific order at which mutations accumulate in a leukemia clone (i.e. are there initiating events)? Third, what basic scientific and clinical implications can be learned from the nature and genomics of leukemogenesis?
Pre-leukemic clones arise in HSCs
Given the low rate of spontaneous mutation accumulation, Dr. Majeti's group hypothesized that the leukemia cell-of-origin likely possess inherent self-renewal properties as well as an extended lifespan and therefore focused on the possibility that single leukemia clones spawn from HSCs. To test this hypothesis, Dr Majeti's lab isolated leukemic cells and residual HSCs from AML patients, based on their expression of surface markers including CD99 and TIM3. Using next generation exome sequencing, they identified a number of leukemia-associated mutations (TET2, SMC1A, NPM1, CTCF) that were also present in both frank leukemia cells as well as residual HSCs that exhibit normal stem cell activity (referred to as pre-leukemic HSCs) [4]. Going one step further, they developed custom TaqMan SNP assays specific for the recurrent mutations to interrogate purified single clones of residual HSCs. From this analysis, they found that residual HSCs from an individual patient comprise a heterogenous mixture of clones that each possesses various numbers of driver mutations. In several patients, Dr. Majeti's group found that pre-leukemic HSCs preferentially display mutations in “landscaping” genes involved in DNA methylation, chromatin modification and cohesin complex (such as IDM1, IDH2, IKZF1, DNMT3A, ASXL1) indicating that these functional mutations represent initiating events in leukemogenesis. Furthermore, they also observed that late mutations leading to frank leukemia commonly occur in genes related to proliferation and signaling activation (FLT3, KRAS/NRAS, PTPN11) [5]. From this data, Dr. Majeti's group proposed a model for preleukemic clonal evolution of leukemogenesis where normal/healthy HSCs first acquire mutations in ‘landscaping’ genes that can then also acquire subsequent mutations (secondary, tertiary, etc.). These pre-leukemic HSCs remain functionally normal until they acquire additional mutations in genes that promote proliferation at which point these clones progress into frank leukemia.
Clinical Implications from the Clonal Evolution of Pre-leukemic HSCs
While many AML patients initially respond to chemotherapy, the majority of patients relapse with a chemotherapy-resistant disease. To determine the clonal basis of remission and relapse, Dr. Majeti's group extended their methods for determining clonal evolution to AML patients both before and after treatment. From these analyses, Dr. Majeti's group observed that patients relapse with one of four distinct models of clonal relapse. In the first model, referred to as the dominant presenting clone model, relapse patients present with the same dominant clone found at diagnosis, which was already resistant to chemotherapy. Second, in the minor presenting clone model, the relapse leukemia is largely comprised of a clone that was under-represented prior to treatment. Third, referred to as the further leukemic clonal evolution model of relapse, in which a leukemic clone acquires additional mutations that make it chemo-resistant. In the fourth model of clonal relapse, referred to as further pre-leukemic clonal evolution, patients relapse with a clone that was initially pre-leukemic prior to treatment but then acquired additional mutations. Dr. Majeti also emphasized that these models may not be mutually exclusive and it could be possible that multiple models of relapse could occur within the same patient.
Epigenetic Mutations in Pre-Malignant Hematopoiesis and Leukemic Transformation - Ross L. Levine, MD
While Dr. Majeti's seminar focused on deciphering the order of mutation accumulation during the malignant clonal outgrowth of HSCs, Dr. Levine's centered on functionally characterizing how mutations in epigenetic regulators initiate leukemogenesis and cooperate with other leukemogenic mutations to drive the development of frank leukemia. Dr. Levine began by making reference to the phenomenon, as also described by Dr. Majeti, that the acquisition of mutations in epigenetic regulators confers a competitive self-renewal advantage to multipotent HSCs in vivo and sets the stage for the clonal expansion of pre-malignant HSCs [6-10]. It was also noted that these mutations frequently occur in genes that regulate DNA methylation and chromatin state and that these mutations are associated with a worse outcome in response to therapy. These observations led Dr. Levine to pose several key and clinically relevant questions. First, do mutations in epigenetic modifiers have a role in leukemia maintenance or do they merely serve to increase stem cell fitness and initiate leukemogenesis? Second, if these genes do play a role in leukemia maintenance then will chemicals that revert the pre-malignant phenotypes induced by these enzymes (i.e. cause reversion of DNA methylation patterns or chromatin state) have therapeutic efficacy? Third, given that mutations in epigenetic regulators are associated with differential patient outcome, do they also promote resistance to standard anti-AML therapies?
Combined deletion of Tet2 with Flt3ITD expression induces a therapy-resistant AML in mice
In an effort to address these questions, Dr. Levine's group generated a genetically engineered mouse model (GEMM) of AML that harbours a conditional bi-allelic knockout of Tet2 in combination with a knock-in of the mutant Flt3 internal tandem duplication (ITD). As previously described by Dr. Levine's group, these mice develop a fully penetrant AML disease phenotype that is resistant to treatment with the front line combination therapy of Doxorubicin and Cytarabine [11]. By prospectively purifying different phenotypic compartments of the AML, they identified a leukemic stem cell (LSC)-like population within the lineage negative, c-Kit positive, Sca-1 positive (LSK) compartment capable of propagating the disease following transplantation into secondary recipients, whereas malignant cells displaying a mature granulocyte-macrophage progenitor (GMP) immunophenotype were incapable of doing so. Consistent with these observations, gene expression profiling revealed that leukemic LSKs, rather than differentiated leukemic GMP progeny, more closely resemble normal LSK cells. Interestingly, treatment of leukemic mice with a FLT3 inhibitor (AC220) largely decreased the disease burden of leukemic blasts but was unable to eliminate LSCs, suggesting that specific targeting of the second transforming hit does not prevent disease propagation in this model.
Tet2 deletion and Flt3ITD cooperate to synergistically alter gene expression
To identify the downstream molecular mechanisms that contribute to AML in the Tet2-/- Flt3ITD mice, Dr. Levine's group performed global gene expression analysis (RNA-seq) on LSKs purified from wild type (wt), each single mutant and double mutant mice. Gene set enrichment analysis comparing wt and Tet2-/- Flt3ITD LSKs revealed that an expression module associated with the Gata transcription factor family was significantly repressed in leukemic LSCs. A more refined gene expression array analysis revealed that Gata2 expression was significantly down-regulated in Tet2-/- Flt3ITD LSKs compared to their wt counterparts. Subsequent analysis of DNA methylation patterns showed that the Gata2 promoter was hypermethylated in leukemic LSCs compared to wt LSKs. Perhaps of most significance, a comparison of DNA methylation patterns among LSKs purified from wt, each single mutant or double mutant mice displayed that the combined effects of these two mutations resulted in a synergistic increase in the number of differentially methylated regions and in altered gene expression changes compared to each mutation alone. Confirming that Gata2 is a synergistic target of combined Tet2 loss and Flt3ITD expression, they showed that re-introduction of Gata2 expression in Tet2-/- Flt3ITD LSKs resolves the AML phenotype by enforcing differentiation of mutant LSCs.
Collectively, this molecular data strongly suggest that the process of leukemic transformation is not simply the step-wise addition of two different mutagenic events, which each impact upon separate biological functions of the cell. Rather, it would appear that the presence of the Flt3ITD mutation synergistically facilitates additional epigenetic reprogramming of the LSK compartment in the context of the Tet2 loss of function, resulting in gene expression changes that are not simply the sum of the two individual mutant phenotypes (Figure 1).
Figure 1. Schematic representation of mutational cooperativity during leukemogenesis.
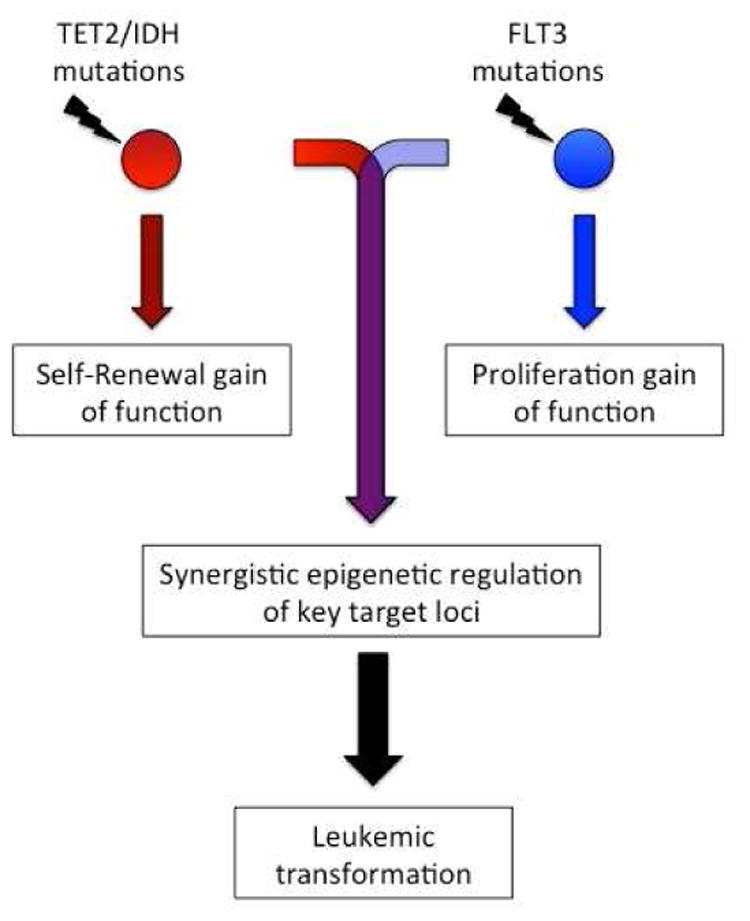
Individual mutations in genes that impact on DNA methylation such as TET2 or IDH1/2, remodel the DNA methylome of normal HSCs resulting in enhanced self-renewal, but do not provide the necessary proliferation stimulus to drive leukemic transformation. In contrast, mutations in the tyrosine kinase receptor FLT3 drive proliferation, leading to the onset of a myeloproliferative neoplasm but lacking the ability to directly precipitate acute leukemia. Combined mutation of TET2/IDH and FLT3 facilitates a synergistic reprogramming of key genetic loci which is required for leukemic transformation and is likely required the maintenance of leukemia propagating cells.
Reversing the DNA methylation patterns induced by Tet2 loss has therapeutic efficacy
To explore the therapeutic potential of targeting of aberrant DNA methylation caused by Tet2 loss in AML, Dr. Levine's group treated Tet2-/- Flt3ITD mice with a pharmacological inhibitor of DNA methylation, 5-azacytidine (Aza). Chronic administration of Aza resulted in a normalization of total white blood cells and blasts in the peripheral blood of diseased Tet2-/- Flt3ITD mice as well as a reversal of the splenomegaly, anemia and thrombocytopenia phenotypes. However, this effect was not achieved by the elimination of mutant cells, since these cells were still present, but rather by the reversal of the DNA methylation signature associated with AML. Presumably, this lack of molecular remission could mean that the AML might relapse with time or upon cessation of therapy. Preliminary data describing the application of combined therapy against both synergistic mutations, using Aza and AC220, suggested that the burden of mutant cells could be reduced using this approach.
Summary of the Basic Scientific and Clinical Implications
The data presented here by Drs. Majeti and Levine has both significant clinical implications and also lead towards an improved understanding of how driver mutations contribute to AML pathogenesis. Dr. Majeti's single clone sequencing studies of human AML patient samples revealed that mutations in epigenetic regulators, chromatin modifiers and members of the cohesin complex in otherwise healthy HSCs are key initiating events in the development of AML. As put forth by Dr. Levine, these observations raise two extremely important and related questions: Are these initiator mutations also required for leukemia maintenance and, if so, will targeting these mutations have therapeutic efficacy? By sequencing the leukemia clones of relapsed AML patients both prior- and post-therapy, Dr. Majeti's group found that some patients may relapse with a disease that arises from pre-leukemic clones that acquire additional mutations, indicating that eliminating pre-leukemic HSCs may be key to preventing relapse in certain cases of AML. Recent data from Nancy Speck's group provide a possible novel insight into such mechanism, showing that Runx1 deficiency can lead to the generation of stress-resistant pre-leukemic HSCs (via reduced ribosome biogenesis and p53/apoptosis levels) offering them a competitive advantage over wild type HSCs[12]. Moreover, Dr. Levine provided data showing that initiator mutations, such as in TET2 or IDH1/2, synergistically cooperate with late-stage driver mutations like Flt3ITD to initiate and maintain frank leukemia. Furthermore, using a GEMM of AML driven by the combination of Tet2 loss with Flt3ITD expression, Dr. Levine also showed that pharmacologically correcting the molecular defects associated with initiator mutations reduces disease burden and partly restores normal HSC function to pre-leukemic HSCs. Collectively, these data suggest that initiator mutations do support leukemia maintenance and that pharmacologically targeting the molecular consequences of these mutations will have therapeutic efficacy.
Moving therapeutic strategies forward: Combination Therapy
Although Dr. Levine's group observed that 5-Azacytidine (Aza) alone reduced disease burden and partly restored HSC function, the treatment was unable to eliminate mutant cells, raising the possibility that these cells could later promote disease relapse and further emphasizing the importance of eliminating pre-leukemic HSCs. This observation combined with Dr. Levine's data showing that Tet2 loss and Flt3 mutations synergistically cooperate to promote leukemogenesis suggested that the combination of Aza and a Flt3 inhibitor, AC220 may cooperate to block AML progression and eliminate mutant cells. In fact, preliminary data presented by Dr. Levine indicate that this therapeutic combination is able to eliminate mutant cells and reduce disease burden more effectively than each agent alone. Dr. Levine also mentioned that his group has obtained similar results with this treatment regimen in a GEMM where AML is driven by the combined expression of an IDH1 mutant with and Flt3ITD mutation. Interestingly, Dr. Majeti's group recently published a study, not discussed in this webinar, where they discovered that chemical inhibitors of BCL-2 selectively kill AML cells carrying IDH1 or IDH2 mutations raising the possibility that BCL-2 inhibitors may synergistically cooperate with FLT3 inhibitors in AML patients that bear both IDH and FLT3 mutations[13].
Collectively, the studies discussed by Drs. Levine and Majeti have established that pre-leukemic mutations that are essential for the initial development of hematological malignancies, such as AML, are not only critical for the early stages of leukemogenesis but also play a key role in disease maintenance and therapy resistance. These findings indicate that targeting/correcting the molecular aberrations induced by initiating mutations will need to be considered when developing novel therapeutic strategies for eliminating leukemia propagating cells in AML.
The full webinar can be viewed via the ISEH website at http://iseh.site-ym.com/?ISEHWebinars
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walter MJ, Payton JE, Ries RE, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12950–12955. doi: 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. The New England journal of medicine. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science translational medicine. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2548–2553. doi: 10.1073/pnas.1324297111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quivoron C, Couronne L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nature genetics. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Challen GA, Sun D, Mayle A, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell stem cell. 2014;15:350–364. doi: 10.1016/j.stem.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasaki M, Knobbe CB, Munger JC, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shih AH, Jiang Y, Meydan C, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer cell. 2015;27:502–515. doi: 10.1016/j.ccell.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai X, Gao L, Teng L, Ge J, Oo ZM, Kumar AR, Gilliland DG, Mason PJ, Tan K, Speck NA. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell. 2015 Aug 6;17(2):165–77. doi: 10.1016/j.stem.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nature medicine. 2015;21:178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]