

Abstract
The lysis–lysogeny decision of bacteriophage λ has been a paradigm for a developmental genetic network, which is composed of interlocked positive and negative feedback loops. This genetic network is capable of responding to environmental signals and to the number of infecting phages. An interplay between CI and Cro functions suggested a bistable switch model for the lysis–lysogeny decision. Here, we present a real-time picture of the execution of lytic and lysogenic pathways with unprecedented temporal resolution. We monitor, in vivo, both the level and function of the CII and Q gene regulators. These activators are cotranscribed yet control opposite developmental pathways. Conditions that favor the lysogenic response show severe delay and down-regulation of Q activity, in both CII-dependent and CII-independent ways. Whereas CII activity correlates with its protein level, Q shows a pronounced threshold before its function is observed. Our quantitative analyses suggest that by regulating CII and CIII, Cro plays a key role in the ability of the λ genetic network to sense the difference between one and more than one phage particles infecting a cell. Thus, our results provide an improved framework to explain the longstanding puzzle of the decision process.
Keywords: gene regulation, lysogeny, lysis, green fluorescent protein
Bacteriophages are the most abundant species in nature and play an immense role in the turnover of bacterial ecosystems (1, 2). Yet, some bacteria and phages exist in symbiotic relationships with phage present in a dormant, lysogenic (prophage) state (3–6). Lambdoid prophages, among other types, are responsible for the expression and release of pathogenic toxins (7). λ, itself, is a temperate phage, which undergoes either lytic or lysogenic development (5, 8). A small number of phage functions are specifically required for carrying out the lysogenic response (9, 10). Studies using λ have unraveled key processes in its gene regulation and developmental pathways, suggesting the presence of a genetic switch (8, 11). The regulatory network is composed of both phage and host functions, which respond to each other, and to external factors such as the physiological conditions of the host. As examples, lysogeny is preferred upon infection of starved cells or when cells are infected at high multiplicity of infection (moi) (12, 13).
Although the interactions and structure of the λ genetic network have been extensively described, many fundamental issues still remain elusive and deserve further attention. For instance, the complex negative control of lytic functions during the lysogenic response has been generally ignored, and the relative importance of different key regulators in determining the decision is poorly understood, in particular for different values of moi. Theoretical studies have provided detailed predictions of the execution of lytic and lysogenic pathways (14–17). These predictions have not been adequately tested in an experiment.
We addressed these issues by using GFP reporter fusions that are activated after phage infection (18, 19). This approach allows for quantitative, reproducible, real-time monitoring of the activity of key regulators with high temporal resolution. The use of synchronously infected cell ensembles permits measurement of specific regulatory protein levels in parallel with monitoring their activity. We focused on monitoring the CII and Q activators, required for the lysogenic and lytic responses, respectively (see ref. 20). CII function was assayed by its activation of the pE-gfp promoter fusion (pE is also termed pRE) (20, 21). Q function was monitored by its antitermination of the pR′-tR′-gfp promoter-terminator fusion (22). Strains carrying the reporter fusions were starved and infected at high multiplicity with the tester phage to simulate a condition favoring lysogeny and were assayed for GFP in a multiwell automatic fluorimeter.
Materials and Methods
Plasmid, Bacterial, and Phage Strains. Promoter-GFP fusion plasmids were constructed by inserting the PCR products of the λ promoters into pSA11 (23), between the BamHI and XbaI sites (replacing the ptac promoter). The pSA11 plasmid, that carries a pBR322 origin of replication, is a medium copy number plasmid with an average of ≈20 copies per cell. CII activity was reported by the pE-gfp plasmid carrying DNA between λ coordinates 5′-38635 and 38336–3′, whereas Q activity was reported by the pR′-tR′-gfp plasmid carrying DNA between λ coordinates 5′-44300 and 44831–3′. A reference pNull-gfp plasmid carrying λ coordinates 5′-35101–35361–3′ is a control without a promoter. All constructs were verified by sequence and introduced into strain W3110. Recombineering with oligonucleotides (24) was used to construct a λ mutant paQ377 (TTGC-GAGCAC-TTGC changed to TGGC-GAGCAC-CTGC) by modifying the TTGC CII-binding sites of the paQ promoter. These mutations do not change the amino acid sequence of Q, and cause a clear plaque phenotype. The conditional amber λ cro071 mutant phage was constructed by replacing the Tyr-10 codon of the cro gene by a suppressible UAG chain termination codon (24) in a λcI+ background. The λcI60cro071, λcII68cro071, and λcI60cII68cro071 were constructed by classical recombination with λcro071 (24). All other phages were taken from our collections.
Fluorescence Assays. Strains carrying the promoter-gfp plasmids were grown overnight in LB medium supplemented with 0.2% maltose and 50 μg/ml ampicillin. Cells were concentrated by centrifugation to 6 × 109 cells per ml, and 25-μl samples then were infected with different amounts of freshly prepared phage lysates (at the specified moi) on ice. After incubation for 30 min on ice, the infected cells were diluted to 0.5 ml in M9 supplemented with 0.5% glycerol, 0.2% maltose, 0.01% B1, and 50 μg/ml ampicillin, and 200 μl (6 × 107 cells) were assayed in duplicates in 96-well plates for GFP activity. Plates were incubated at 37°C and assayed at 2-min intervals in a SPECTRAFluor Plus fluorimeter (Tecan, Maennedorf, Switzerland) with a short shaking interval between assays. Both fluorescence and optical density were measured. We found a full correlation between the drop in the optical density and the drop in the rate of GFP expression due to lysis. The error between duplicate wells was ≈10% (Fig. 1O). The pR′-gfp fusion, lacking the terminator, was found to be constitutively expressed at high levels in both lysogenic and nonlysogenic cells and was not studied further (data not shown). We checked the relationship between GFP synthesis and fluorescence levels in our system by using a pSA11 plasmid (pTaq-gfp) and found linearity between fluorescence values and isopropyl β-d-thiogalactoside concentrations. Furthermore, we tested the effect of WT λ infection on GFP expression from the pSA11 plasmid and did not find any change in GFP maturation, folding, and stability due to phage infection.
Fig. 1.

The effect of phage mutants on the kinetics of reporter fusions activation by CII and Q. CII activity is reported by pE-gfp fusions (blue diamonds), whereas Q activity is reported by pR′-tR′-gfp fusions (red circles). (A–C) Total fluorescence as function of time of infected cultures with λc+ (A), λcI– (B), and λcII– (C) mutants. (D–N) Promoter activity of pE (blue diamonds) and Q (red circles) as a function of time after infection with λc+ (D), λcI– (E), λcII– (F), λcro– (G), λcro–cI– (H), λcro–cII– (I), λpaQ– (J), λpE– (K), λcIII– (L), λQ– (M), and λcII–Q– (N) mutants. The increase in cell mass, measured by following the optical density at 595 nM, was similar for the first hour in all experiments. All measurements were carried out at a moi of 6. For additional information, see Materials and Methods.(O) Data reproducibility: Normalized fluorescence measured on days 2–4 as function of the normalized fluorescence on day 1. Yellow lines denote 10% deviation limits.
Data Analysis. For a given promoter (pE, pR′-tR′, or pNull), the average of the total output fluorescence values from duplicate wells was determined for each time point. Then, the average value obtained at each time point from uninfected cells (also in duplicate wells) was subtracted, to cancel out the intrinsic background fluorescence from the bacterial cells. Next, the GFP fluorescence levels expressed from the pNull-gfp negative control plasmid (between 0.1 and 0.4 fluorescent units) were subtracted from those of pE and pR′-tR′, yielding the specific activities of these promoters. The time scale was corrected in all experiments by subtracting 16 min, a time lapse that we estimated to be required for heating the multiwell plate to 37°C and for expression and maturation of GFP by using a lac promoter GFP fusion as a standard.
The promoter activity is given by the first derivative of the fluorescence data. To determine the instantaneous promoter activity, we first smoothed the data by averaging each time point with the previous two and next two points. Next, we calculated the first derivative of the fluorescence readings by subtracting from each time point the previous one and dividing by the time interval between measurements. Last, the resulting derivative was again smoothed by averaging each time point with both the previous and subsequent ones. Fits of the data of promoter activity (PA) vs. regulatory protein levels (x) (see Fig. 2) were carried out by using a Hill function (origin software, Microcal Software, Northampton, MA):
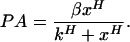 |
Fig. 2.

Promoter activity vs. regulatory protein levels. (A) Western blots for CII after infection with λc+ and Q after infection with λcII– carried out on samples taken from multiwell plate experiments at the indicated times and resolved on NuPAGE 4–12% Bis-Tris precast gels (Invitrogen). (B) CII (blue) and Q (red) levels as a function of time. Levels and their SD were determined with the National Institutes of Health image program, from two different experiments. The level of Q protein after infection with λc+ reached ≈20% of that observed in λcII– infection (data not shown). (C and D) Promoter activities of pE after infection with λc+ (C) and pR′-tR′ after infection with λcII– (D), as calculated from fluorescence measurements, vs. CII and Q concentrations, respectively. Cultures were infected at a moi of 6. Fluorescence measurements and protein levels were measured from the same infections under identical conditions. The lines represent a fit to the data by using a Hill function, with a fitted Hill coefficient H = 0.98 ± 0.15 (C) and H = 6.4 ± 3.3 (D) (see Materials and Methods).
Results and Discussion
Infections with the WT phage λc+ show activation of the pE-gfp, fusion, starting ≈7–8 min after infection (Fig. 1A). The expression from the lytic pR′-tR′-gfp fusion was low under these conditions. Low levels of Q protein were observed with Q-specific Abs (data not shown). We found that the three CII-activated promoters pE, pI, and paQ (20, 21, 25) exhibited a similar pattern of expression, although the level of expression from the latter two promoters was lower (O.K., A.R., D.L.C., J.S., and A.B.O., unpublished data). The derivative of the fluorescence values as a function of time (19) (see Materials and Methods) provides a temporal profile of the rate of synthesis of the GFP reporter (Fig. 1D) (19). The activation kinetics of the CII-dependent promoter (pE) is bell-shaped, indicating initial expression of CII followed by a rapid decrease (Fig. 1D) caused by turnoff of cII transcription and degradation of CII by host protease (Fig. 2 A and B) (19, 26).
We perturbed the λ genetic network by using cI and cII mutants that form clear plaques and have a reduced lysogenic response. Neither mutant expresses functional CI repressor (10). Infection with a λcII68 mutant yielded no expression from pE, confirming pE dependence on a functional CII, but allowed Q-dependent (lytic) expression from pR′-tR′, starting ≈28 min after infection (Fig. 1 C and F; see also Fig. 1O for reproducibility of the experimental system). This expression increased dramatically before lysis began at ≈80 min after infection. No increase in expression from pR′-tR′ was found after infections with λc+Q152 and λcII68Q81 carrying a conditional amber mutation in the Q gene (Fig. 1 M and N). Infection with a λcI60 mutant yielded expression from pE, because CII protein was made during this infection (Fig. 1 B and E). This infection also yielded Q function starting at ≈40 min, an even longer delay relative to infection by λcII68. This result suggests that CII delays the function of Q.
The CII-dependent delay in Q function might be caused by CII activation of pE and/or paQ; both are antisense promoters to the pR transcript. We tested two promoter mutants, one in pE and the other in paQ. The pE mutant, λcy42, had the same delayed kinetics as λcI60. The λpaQ377 mutant, conversely, expressed Q function much earlier, like the λcII68 mutant (Fig. 1 J and K). Thus, the CII-dependent delay in expression of Q requires a functional paQ promoter and is independent of pE and of CI.
Even in the absence of CII, it takes >20 min for Q-dependent activity to appear. This result is not explained by a delay in Q transcription. The 6-kb distance between Q and the pR promoter could be transcribed by RNA polymerase in <3 min (27). Furthermore, the removal of a dispensable 2.5-kb segment containing several transcription terminators (nin5 deletion; ref. 28) had little measurable effect on this CII-independent delay (data not shown). To explain the CII-independent delay, we hypothesized that Q activation of late gene expression may be particularly sensitive to functional threshold effects (29, 30). To test this hypothesis, we compared Q protein levels with Q function (Fig. 2D). The data show that Q function could be detected only when Q protein levels reached 40–50% of maximal concentration, demonstrating a strong threshold effect on Q function. By using a two-plasmid system with one plasmid expressing Q and the second carrying a Q reporter fusion, a similar nonlinear response for Q activity was observed. Furthermore, to reach maximal activity, 25-fold higher levels of Q protein were required in vivo than in vitro (30). It was suggested that this threshold effect might be due to differences in pause time of RNA polymerase at the site where Q acts or efficiency of Q dimerization (30). Alternatively, it is possible that high cooperativity of Q is required in vivo or that a host factor limits Q activity when Q concentration is low.
Thus, our results indicate that although CII and Q are both transcribed from the pR promoter, CII functions relatively rapidly (Fig. 2C), whereas Q activity is delayed both by threshold control and by CII-dependent inhibition via paQ (Fig. 2D). We reason that in the lysogenic pathway, these delays prevent Q function before the establishment of CI repression. In the lytic pathway, where CII inhibition of Q does not occur, the threshold control may coordinate lytic gene expression with phage DNA replication.
To test for the role of Cro in the decision process, we constructed a cro suppressible phage mutant, λcro071 (see Materials and Methods), in a λcI+ background. Phage-carrying mutations in the cro gene followed exclusively the lysogenic pathway and formed plaques only on a suppressor host (20). Infection with λcro071 prolonged CII-dependent pE expression and led to low Q activity (compare Fig. 1 D to G). The double mutant λcI60cro071, being constitutive for CII, directed high CII activity and very low Q expression, resulting in an abortive infection where neither lysogeny nor lysis can occur (Fig. 1H). Indeed, this phage is unable to form plaques on the nonsuppressible host. The lytic phenotype is compensated by a cII mutation as seen by the functional assay for Q (Fig. 1I) and by the ability of λcII68cro071 and λcI60cII68cro071 mutants to form clear plaques (data not shown). We conclude that in the absence of Cro, the persistent high level of CII prevents Q activity.
The CIII protein stabilizes CII against rapid degradation by FtsH (31). Infection with a λcIII– phage, λcIII67 retains some ability to follow the lysogenic response at high mois (9, 32). After infection with λcIII67, only low levels of pE activity were observed, and the CII-dependent delay in Q activity was not seen, although the levels of Q were low (Fig. 1L). One could speculate that, in the absence of CIII, CII levels are too low to activate paQ, and therefore no CII-dependent delay in Q activity takes place. We have no simple explanation for the low levels of Q activity in infections with λcIII–.
It was shown that lysogenization frequency depends on the number of phages infecting a cell, i.e., at moi = 1, most cells undergo lysis, whereas infection at moi ≥ 2 leads to a high frequency of lysogeny (12). The teleological rationale is that the lysogenic response is favored once the bacterial population becomes limiting, and phage progeny may be unable to find new bacterial cells for infection. We questioned whether moi affects CII and Q functions as it affected lysogeny. After infections with a λcI+ phage, expression from the pE reporter fusion increased with phage multiplicity, whereas Q function decreased (Fig. 3 A and B, respectively). The expression behavior is affected not only by CII levels but also by the presence of CI. Infection by a λcI– mutant clearly shows a dissociation between a clear moi response in pE expression, compared with almost no moi response on the level of expression from the pR′-tR′ reporter fusion, although the CII-dependent delay in Q activity does increase with moi (Fig. 3 C and D). These results further emphasize the participation of CI repression in regulating Q activity.
Fig. 3.

Effect of moi on regulator activities. Total fluorescence as a function of time from pE-gfp (blue, A, E, I, C, G, and K) and pR′-tR′-gfp fusions (red, B, F, J, D, H, and L) after cell infection with λc+ (A and B), λcI– (C and D), λcro–cI– (E and F), λcro– (G and H), λcII– (I and J), and λcIII– (K and L) mutants, at moi 1, 2, 4, 6, and 10, shown as symbols with increasing color density (brightest represents lowest moi).
We compared the results presented in Fig. 3 with the calculated fraction of cells in the bacterial population infected with n or more phages (n = 1–6) as determined by the Poisson distribution (12). This analysis (details not shown) suggests that maximal CII activity for cells infected with λcI– mutant is at n ≥ 2, whereas very low CII activity for n = 1 (for moi of 1, 2, 4, and possibly 6). Infections with λcI+ phage yielded a more complex response. Maximal CII activity was found for cells infected with n ≥ 2, but the increase in CII activity per infected phage was diminished as the moi increased. These results suggest that a functional threshold of CII must be reached for lysogeny to be established. Thus, the λ genetic network is able to “measure” the number of infecting phage particles through the CII level. This measurement, we find, is highly dependent on the presence of Cro. The double-mutant λcI–cro– shows very little response to moi for both CII and Q activity (Fig. 3 E and F, respectively), and maximal CII activity is achieved at n = 1. Infection with λcro– exhibits CII activity that depends on moi with high CII activity observed even at n = 1. However, we observed shutoff of CII expression, presumably by CI repressor (Fig. 3G). Q function, conversely, is very low for all moi (Fig. 3H). This result suggests that the levels of CII in λcro– infection at moi of 1 are sufficient to cause the infected complex to follow exclusively the lysogenic pathway. Note that in the absence of early repression of pR by Cro, CII levels are much higher than in its presence. Based on our results, we are led to conclude that in cro mutants, high levels of CII are able to down-regulate Q expression dramatically, independent of the activity of CI. Infections by a λcII– mutant, similar to infection by a λcI– mutant, lead to maximal Q activity but do not show a delay in Q activity (Fig. 3 I and J). Infections by a λcIII– phage demonstrate the importance of CIII in allowing for maximal manifestation of CII activity (Fig. 3 K and L). Interestingly, although pE activity increases and Q activity decreases as a function of moi, no moi effect on the onset of Q was observed, suggesting a possible unique role of CIII in Q activity.
Our analysis of the λ genetic network supports many of the conclusions reached from classical approaches, provides a detailed kinetic description of CII and Q functions, and leads to the following view of the lysis–lysogeny decision (Fig. 4 A and B). Under conditions that favor lysogenization, when two or more phage particles infect a cell, CII levels are sufficient for the lysogenic response as shown in the schematic kinetic diagram (Fig. 4C). Under such conditions, Cro probably plays a very limited role. Thus, during the lysogenic pathway, CII and CI activities are kinetically coordinated such that CI can replace CII in inhibiting lytic gene expression as CII is being turned off (Fig. 4C). CI repressor executes repression at the commitment stage but appears not to participate in the decision process. It seems that the synthesis of CII is programmed for rapid overshoot of CI expression. This overshoot was noted earlier, based on measurements of CI expression after infection (10); it may be required for the high cooperativity of the CI repressor. Our kinetic assays of CII activity show that after infection by WT phages, CII function is sufficient to activate the CII-activated promoters carried by the infecting phages, as well as the ≈20 pE-gfp reporter fusions carried by the plasmids.
Fig. 4.

A schematic model for the lysis–lysogeny decision process. (A) Elements of the λ genetic network participating in the lysogenic response. (B) Elements of the λ genetic network active during the lytic pathway. Arrows denote positive actions, and bars denote negative actions. A bar also denotes the threshold or cooperativity effect delaying Q activity. Both schemes focus on functions studied in this work. (Promoters are shown in green, and proteins are shown in orange.) Black connecting lines represent parts of the network operating in both the lytic and the lysogenic response. (C) Simplified kinetic model suggested by our experiments in which CII (blue line) activates CI synthesis (green line) and represses Q activity (red line) during a lysogenic response. When insufficient levels of CII are accumulated, the lytic response is the default of the genetic network.
The delays placed on Q function permit the extension of the decision period and ensure the viability of the newly established lysogenic cells. When only a single phage infects a cell, Cro function plays a vital role in reducing transcription from pL and pR promoters, thereby limiting CII function (Fig. 4 B and C). This action and the instability of CII ensure lytic development, because there is not sufficient CII both to promote CI synthesis from the pE promoter and to carry out the CII-dependent delay of Q activity. The threshold control of Q function, however, could play an important role in both lytic and lysogenic pathways. We suspect a similar role for Cro after prophage induction. Lysogeny, however, does occur at low frequency when one phage infects. This rare event may reflect a unique cell physiology at the time of infection or a stochastic process.
Our study, revealing the CII and Q dynamic controls, could serve as a benchmark to test models and computer simulations of the λ genetic network such as those proposed recently (14–17). A more complete understanding of the operation of the λ genetic network will require further detailed study of individual steps, the identification and characterization of other modules, known or unknown, by using the current methods as well as genetics, theoretical modeling, and mathematical analyses.
Acknowledgments
We thank Simi Koby for her help at the early stages of the work; Shoshy Altuvia, Ariella Oppenheim, and Ilan Rosenshine for fruitful discussions; Jeff Roberts (Cornell University, Ithaca, NY) for Q Abs; and Ariella Oppenheim for critical reading of the manuscript. This research was supported in part by Israel Science Foundation Grants 489/01-1 and 340/04.
Author contributions: O.K., D.L.C., J.S., and A.B.O. designed research; O.K., A.R., and A.B.O. performed research; O.K., A.R., N.F., J.S., and A.B.O. contributed new reagents/analytic tools; O.K., A.R., N.F., D.L.C., J.S., and A.B.O. analyzed data; and O.K., N.F., D.L.C., J.S., and A.B.O. wrote the paper.
Abbreviation: moi, multiplicity of infection.
References
- 1.Wommack, K. E. & Colwell, R. R. (2000) Microbiol. Mol. Biol. Rev. 64, 69–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hendrix, R. W. (2003) Curr. Opin. Microbiol. 6, 506–511. [DOI] [PubMed] [Google Scholar]
- 3.Friedman, D. I. & Court, D. L. (2001) Curr. Opin. Microbiol. 4, 201–207. [DOI] [PubMed] [Google Scholar]
- 4.Stopar, D., Cerne, A., Zigman, M., Poljsak-Prijatelj, M. & Turk, V. (2003) Microb. Ecol. 46, 249–256. [DOI] [PubMed] [Google Scholar]
- 5.Campbell, A. (2003) Nat. Rev. Genet. 4, 471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedulla, M. L., Ford, M. E., Houtz, J. M., Karthikeyan, T., Wadsworth, C., Lewis, J. A., Jacobs-Sera, D., Falbo, J., Gross, J., Pannunzio, N. R., et al. (2003) Cell 113, 171–182. [DOI] [PubMed] [Google Scholar]
- 7.Wagner, P. L., Livny, J., Neely, M. N., Acheson, D. W., Friedman, D. I. & Waldor, M. K. (2002) Mol. Microbiol. 44, 957–970. [DOI] [PubMed] [Google Scholar]
- 8.Ptashne, M. (2004) Genetic Switch: Phage Lambda Revisited (Cold Spring Harbor Lab. Press, Woodbury, NY).
- 9.Kaiser, A. D. (1957) Virology 3, 42–61. [DOI] [PubMed] [Google Scholar]
- 10.Reichardt, L. & Kaiser, A. D. (1971) Proc. Natl. Acad. Sci. USA 68, 2185–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisen, H., Brachet, P., Pereira da Silva, L. & Jacob, F. (1970) Proc. Natl. Acad. Sci. USA 66, 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kourilsky, P. & Knapp, A. (1974) Biochimie 56, 1517–1523. [PubMed] [Google Scholar]
- 13.Reichardt, L. F. (1975) J. Mol. Biol. 93, 267–288. [DOI] [PubMed] [Google Scholar]
- 14.McAdams, H. H. & Shapiro, L. (1995) Science 269, 650–656. [DOI] [PubMed] [Google Scholar]
- 15.Arkin, A., Ross, J. & McAdams, H. H. (1998) Genetics 149, 1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aurell, E., Brown, S., Johanson, J. & Sneppen, K. (2002) Phys. Rev. E 65, 051914-1–051914-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhu, X. M., Yin, L., Hood, L. & Ao, P. (2004) Funct. Integr. Genomics 4, 188–195. [DOI] [PubMed] [Google Scholar]
- 18.Cormack, B. P., Valdivia, R. H. & Falkow, S. (1996) Gene 173, 33–38. [DOI] [PubMed] [Google Scholar]
- 19.Ronen, M., Rosenberg, R., Shraiman, B. I. & Alon, U. (2002) Proc. Natl. Acad. Sci. USA 99, 10555–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendrix, R. W., Roberts, J. W., Stahl, F. W. & Weisberg, R. A. (1983) Lambda II (Cold Spring Harbor Lab. Press, Woodbury, NY).
- 21.Hoopes, B. C. & McClure, W. R. (1985) Proc. Natl. Acad. Sci. USA 82, 3134–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marr, M. T., Datwyler, S. A., Meares, C. F. & Roberts, J. W. (2001) Proc. Natl. Acad. Sci. USA 98, 8972–8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlosser-Silverman, E., Elgrably-Weiss, M., Rosenshine, I., Kohen, R. & Altuvia, S. (2000) J. Bacteriol. 182, 5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oppenheim, A. B., Rattray, A. J., Bubunenko, M., Thomason, L. C. & Court, D. L. (2004) Virology 319, 185–189. [DOI] [PubMed] [Google Scholar]
- 25.Ho, Y. S. & Rosenberg, M. (1985) J. Biol. Chem. 260, 11838–11844. [PubMed] [Google Scholar]
- 26.Kobiler, O., Oppenheim, A. B. & Herman, C. (2004) J. Struct. Biol. 146, 72–78. [DOI] [PubMed] [Google Scholar]
- 27.Gotta, S. L., Miller, O. L. & French, S. L. (1991) J. Bacteriol. 173, 6647–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costantino, N., Zuber, M. & Court, D. (1990) J. Bacteriol. 172, 4610–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Echols, H., Court, D. & Green, L. (1976) Genetics 83, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang, X. J., Hart, C. M., Grayhack, E. J. & Roberts, J. W. (1987) Genes Dev. 1, 217–226. [DOI] [PubMed] [Google Scholar]
- 31.Kobiler, O., Koby, S., Teff, D., Court, D. & Oppenheim, A. B. (2002) Proc. Natl. Acad. Sci. USA 99, 14964–14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kornitzer, D., Altuvia, S. & Oppenheim, A. B. (1991) Proc. Natl. Acad. Sci. USA 88, 5217–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]