

Abstract
The human glucagon receptor (GCGR) belongs to the class B G protein-coupled receptor (GPCR) family and plays a key role in glucose homeostasis and the pathophysiology of type 2 diabetes. Here we report the 3.0 Å crystal structure of full-length GCGR containing both extracellular domain (ECD) and transmembrane domain (TMD) in an inactive conformation. The two domains are connected by a 12-residue segment termed the ‘stalk’, which adopts a β-strand conformation, instead of forming an α-helix as observed in the previously solved structure of GCGR-TMD. The first extracellular loop (ECL1) exhibits a β-hairpin conformation and interacts with the stalk to form a compact β-sheet structure. Hydrogen/deuterium exchange, disulfide cross-linking and molecular dynamics studies suggest that the stalk and ECL1 play critical roles in modulating peptide ligand binding and receptor activation. These insights into the full-length GCGR structure deepen our understanding about the signaling mechanisms of class B GPCRs.
Class B GPCRs are essential components in many human physiological processes and serve as valuable drug targets for many diseases including diabetes, metabolic syndrome, osteoporosis, migraine, depression and anxiety1–4. These receptors consist of an ECD and a TMD, both of which are required for binding to their endogenous peptide ligands and regulation of cell signal transduction2,5. Previous studies suggested that tertiary interactions between the ECD and TMD play a critical role in regulating receptor activity of class B GPCRs6,7. Structures of the ECDs of several class B GPCRs have been solved2, and recently, the crystal structures of the TMDs of three class B GPCRs, the human GCGR8,9, corticotrophin-releasing factor receptor 1 (CRF1R)10 and glucagon-like peptide-1 receptor (GLP-1R)11, have been determined, providing insights into ligand recognition and selectivity of these physiologically important receptors. However, the structure of a full-length class B GPCR has remained elusive, thereby limiting our understanding of the molecular details accompanying peptide binding and signal transduction. In this study, we have solved the crystal structure of the full-length human GCGR (GCGR-FL) in an inactive conformation in complex with a negative allosteric modulator (NAM), 4-[1-(4-cyclohexylphenyl)-3-(3-methanesulfonylphenyl)ureidomethyl]-N-(2H-tetrazo-5-yl)benzamide (NNC0640), and antigen-binding fragment (Fab) of an inhibitory antibody mAb1 (Fig. 1, Extended Data Fig. 1 and Extended Data Table 1).
Figure 1. Overall structure of the GCGR-NNC0640-mAb1 complex.
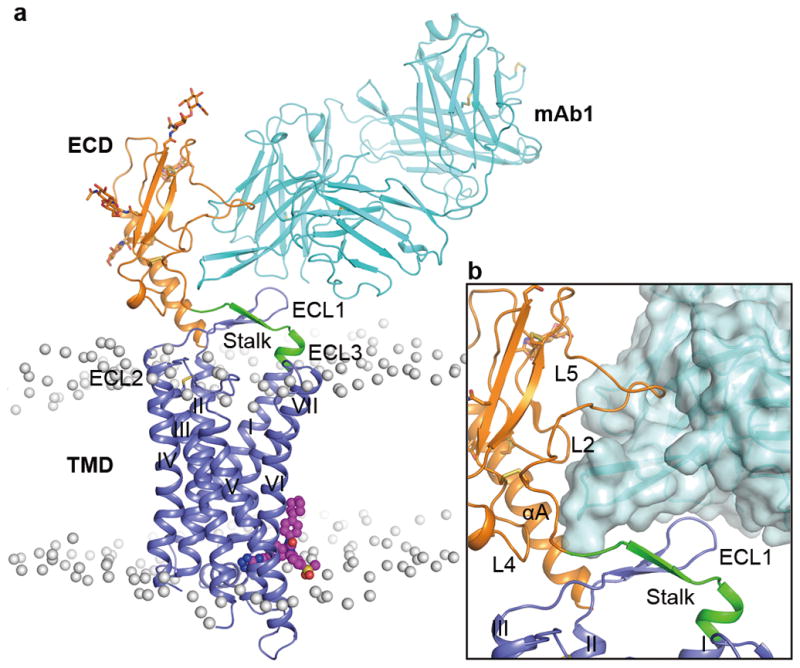
a, Structure of the GCGR-NNC0640-mAb1 complex. GCGR and mAb1 are shown in cartoon representation. The ECD (residues Q27-D124), stalk (residues G125–K136) and TMD (residues M137–W418) of the receptor and mAb1 are colored in orange, green, blue and cyan, respectively. The glycan modifications in the ECD are displayed as orange sticks. NNC0640 is shown as magenta spheres. The disulfide bonds are shown as yellow sticks. The membrane boundaries are displayed as grey spheres, which are the phosphorous atoms in each phospholipid molecule after the initial 50-ns equilibrium of the simulation system. b, Close-up view of the interface between GCGR and mAb1. The antibody mAb1 is also shown in surface representation.
Overall structure of GCGR-FL
In the GCGR-NNC0640-mAb1 complex structure, GCGR exhibits an elongated conformation with the ECD sitting on top of the TMD (Fig. 1a). The ECD comprises the common α-β-β-α fold as observed in the ECD structures of GCGR and other class B GPCRs6,12,13 with Cα RMSD of 1.3 Å compared to the same domain in the crystal structure of the GCGR-ECD bound to mAb16 (PDB ID: 4ERS). Four asparagine residues, N46, N59, N74 and N78 within the ECD are glycosylated by N-acetyl-D-glucosamines (NAGs). The TMD in the GCGR-FL structure features the canonical seven-transmembrane helical bundle (helices I–VII), which shares a similar conformation compared to the previously solved crystal structures of the GCGR-TMD8,9 with Cα root-mean-square deviation (RMSD) of 1.2 Å (PDB ID: 4L6R) and 0.8 Å (PDB ID: 5EE7). The antibody mAb1 interacts with the αA helix and loops L2, L4 and L5 of GCGR-ECD as previously reported6. Additionally, it makes close contact with ECL1 of the receptor (Fig. 1b), likely restricting conformational flexibility between the ECD and TMD. Our ligand-binding assay showed that mAb1 had little effect on the binding affinity of GCGR to NNC0640 (Extended Data Fig. 2a).
GCGR binding mode of the NAM NNC0640
The NAM NNC0640 binds to GCGR on the external surface of the TMD in a similar binding site as previously reported for another GCGR NAM MK-08939 (Fig. 2). The tetrazole ring of NNC0640 inserts into a cleft between helices VI and VII forming hydrogen bonds with S3506.41b and N4047.61b (numbers in superscript refer to the modified Ballesteros-Weinstein numbering system for class B GPCRs14,15). The benzamide group of the ligand forms an additional polar interaction with S3506.41b, while the urea carbonyl oxygen hydrogen bonds with T3536.44b. Unlike the dichlorophenylpyrazole group of MK-0893 that is oriented parallel to the membrane and makes no contact with the receptor, the cyclohexylphenyl moiety of NNC0640 forms hydrophobic contacts with helices VI and VII. Our mutagenesis studies show that the single-site mutants S3506.41bA, T3536.44bA and N4047.61bA each exhibited much lower binding affinity to [3H]-NNC0640 compared to that of the wild-type (WT) receptor and other mutants (Extended Data Fig. 2b and Extended Data Table 2), which could be explained by the importance of the multiple hydrogen bonds between the ligand and these three residues.
Figure 2. Ligand-binding mode of GCGR to NNC0640.

a, Ligand-binding site of NNC0640. GCGR is shown in grey cartoon representation. The ligand NNC0640 (magenta carbons) and GCGR residues (blue carbons) involved in ligand binding are shown in stick representation. Hydrogen bonds are displayed as green dashed lines. b, Schematic representation of interactions between GCGR and NNC0640 analyzed by LigPlot+20. The stick drawings of GCGR residues and NNC0640 are colored in blue and magenta, respectively. c, Comparison of the ligand-binding modes between NNC0640 and MK-08939. NNC0640 and MK-0893 are shown as sticks, and colored in magenta and yellow, respectively.
Conformation of the stalk
The conformation of the stalk region (residues G125–K136) that connects the C-terminus of the ECD and the extracellular tip of helix I either defines or responds to the relative orientation between the ECD and TMD. In the previously solved structure of GCGR-TMD (PDB ID: 4L6R), the stalk region forms a 3-turn α-helical extension of helix I8. Surprisingly, the N-terminal portion of the stalk (G125–Q131) exhibits a different conformation in the inactive GCGR-FL structure by adopting a β-strand conformation that runs across the helical bundle flanked by ECL1 on one side and ECL2 and ECL3 on the other side (Fig. 3a and Extended Data Fig. 3a). With this unexpected difference in conformation of the stalk region, the relative orientation of the ECD and TMD observed in the inactive GCGR-FL crystal structure differs dramatically from what was previously predicted by modeling an active conformation of GCGR-FL in complex with its endogenous ligand glucagon7,8 (Extended Data Fig. 4a and 4b).
Figure 3. Stalk and ECL1 in the GCGR-FL structure.

a, Comparison of the stalk conformation between the GCGR-FL structure and the GCGR-TMD structure. The receptors are shown in cartoon representation. The ECD, stalk and TMD in the GCGR-FL structure are colored in orange, green and blue, respectively. The stalk and TMD in the GCGR-TMD structure (PDB ID: 4L6R) are in yellow and grey, respectively. b, Interactions between the stalk and ECL1 in the GCGR-FL structure. The stalk (residues G125-K136), ECL1 (residues R201–S217) and TMD (residues M137–T200 and D218–W418) are colored in green, magenta and blue, respectively. The residues involved in stalk-ECL1 interaction are displayed as sticks. Hydrogen bonds are shown as blue dashed lines. c, Structural superimposition between the GCGR-FL crystal structure and the model of GCGR-glucagon complex7. The ECD, stalk, ECL1 and TMD in the GCGR-FL crystal structure are colored in orange, green, magenta and blue, respectively. The receptor in the GCGR-glucagon model is in grey and glucagon is in cyan.
While it remains to be determined what role the stalk region plays in the conformation of the peptide-bound active GCGR structure, it is clear that the observed orientation of the ECD in the mAb1-bound GCGR-FL crystal structure is not compatible with the class B GPCR two-state peptide binding model where the C-terminal region of the peptide ligand targets the ECD as observed in hormone-bound ECD crystal structures of class B GPCRs, and the N-terminal region of the peptide ligand targets the TMD binding pocket2,5,16 (Extended Data Fig. 4c). A change in the relative orientation between the ECD and TMD is therefore required to enable the peptide ligand binding, raising the possibility that the stalk region that connects the ECD and TMD plays a specific role in GCGR activation. Consistent with this hypothesis, mutagenesis of the stalk region has been shown to affect peptide ligand binding8, establishing its importance either through direct interactions with the peptide or through modulation of the stalk conformation, which in turn could influence the accessible conformation of the ECD relative to the TMD. Compared to the α-helical conformation of the stalk region observed in the GCGR-TMD structure8, the extended β-strand conformation in the GCGR-FL structure allows more flexibility for the stalk to adjust its conformation to influence peptide ligand binding. Previous hydrogen/deuterium exchange (HDX) studies comparing small-molecule allosteric antagonist- and peptide antagonist-bound GCGRs revealed differential HDX behavior for the stalk region, indicating that this region is structurally dynamic upon binding of different ligands7, further supporting the important role of the stalk in peptide ligand binding.
The antibodies mAb1 and mAb23 have been studied for their effects on GCGR activation6. The investigations showed that mAb1 behaves as an antagonist by blocking ligand access through direct interactions with the residues in the ECD required for glucagon binding, while mAb23 not only blocks glucagon binding but also reduces basal receptor activity, acting as an inverse agonist. To study the structural basis for the differential effects of mAb1 and mAb23 on receptor signaling, we carried out HDX experiments with NNC0640-stabilized GCGR in complex with mAb1 or mAb23 (Extended Data Fig. 5 and Extended Data Fig. 6). The results comparing the antibody-free GCGR and the GCGR-mAb1 complex demonstrate protection to deuterium exchange in two regions within the ECD, namely the N-terminal part of αA helix (residues L32–L38) and the β4–L5 region (residues K98–Q105), both of which exhibited increased protection in the mAb1-bound GCGR (Extended Data Fig. 5b, c). This aligns well with the GCGR-NNC0640-mAb1 complex structure in which both the αA helix and L5 of the ECD interact with mAb1. For the mAb23-bound receptor, the HDX results showed protection in the same regions within αA helix and β4-L5 as observed for the GCGR-mAb1 complex (Extended Data Fig. 5e, f), indicating similar binding modes of the GCGR-ECD to mAb1 and mAb23. Additionally, the stalk region (residues I128–M137) displayed protection in the GCGR-mAb23 complex, but not in the mAb1-bound GCGR (Extended Data Fig. 5d, g), suggesting that binding of mAb23 most likely stabilizes the conformation of the stalk, which may lock GCGR in an inactive state and inhibit the basal activity of the receptor. Taken together, the GCGR-FL crystal structure combined with the HDX studies suggests that the stalk region is involved in regulating or responding to ligand binding through conformational changes and thereby may play a critical role in modulating receptor activity.
Inactive state stabilized by the stalk and ECL1
In class B GPCRs, ECL1 is highly variable in sequence and length, providing potential specificity for recognition of their cognate ligands. GCGR and some other class B GPCRs, including GLP-1R, parathyroid hormone receptor (PTHR) and gastric inhibitory polypeptide receptor (GIPR), contain a long ECL1 with 11–26 residues, as compared with 4–6 residues in other class B and most class A GPCRs. Previous studies reported that the ECL1 of GCGR might participate in binding to its endogenous ligand glucagon and regulating receptor activity17,18. However, the regulation mechanism of GCGR activation by its ECL1 remained unclear due in part to the absence of the conformational information of ECL1 in the previously solved crystal structures of GCGR-TMD. In the GCGR-FL crystal structure, ECL1 (residues R201–S217) of the receptor forms a β-hairpin with G207-D209 on the tip of the turn, which is adjacent to the extracellular tip of helix I (Fig. 3b and Extended Data Fig. 3b). The observed conformation of ECL1 is supported by the previous study using a cysteine accessibility method that suggested that this extracellular loop of GCGR is in an extended conformation, probably a β-sheet18. Furthermore, it was proposed that ECL1 forms a compact structure or interacts with other parts of the receptor18. This agrees with the GCGR-FL crystal structure in which ECL1 of the receptor runs in parallel with the stalk and its backbone from L210–T214 forms hydrogen bonds with the main chain of the residues E126–V130 on the stalk, forming a compact β-sheet structure (Fig. 3b). Additionally, the main chain nitrogen of E126 hydrogen bonds with the side chain of T214, and another hydrogen bond is formed between the side chains of E127 and S211.
In the GCGR-FL structure, the β-sheet formed by the stalk and ECL1 stacks on top of helices I, II and III, and partially caps the orthosteric ligand-binding pocket within the TMD (Fig. 3b). Structural superimposition analysis between the GCGR-FL crystal structure and the previous GCGR-glucagon complex model7 reveals that glucagon forms spatial clashes with the stalk, suggesting that the conformation of the stalk observed in the GCGR-FL crystal structure may potentially block peptide ligand binding (Fig. 3c). The strong interactions between the stalk and ECL1 restrain movement of the stalk, and thus may inhibit receptor activation. To further validate the observed conformation of the stalk and ECL1 and their effects on receptor activation, we performed disulfide cross-linking studies to lock the interaction between these two regions (Fig. 4). The results showed that the V130stalkC/L210ECL1C GCGR mutant significantly diminished its binding ability to glucagon, while adding 1 mM dithiothreitol (DTT) rescued glucagon-binding by about 50% compared to the WT receptor (Fig. 4b). In contrast, the single-site mutations V130C and L210C retained 50–100% of glucagon-binding of the receptor, and DTT had little effect on the binding potency of these single-site mutants and the WT GCGR (Fig. 4c–e). These data suggest the formation of a disulfide-bond between V130C and L210C, and indicate that locking the stalk and ECL1 together inhibits glucagon binding. To further confirm the roles of these two regions played in receptor signaling, we measured glucagon-induced cAMP accumulation for both WT GCGR and its mutants V130C/L210C, V130C and L210C (Extended Data Fig. 2c and 2d). The data show that the engineered disulfide bond between V130C and L210C decreased the glucagon potency for GCGR by 10–100 folds compared to the WT receptor and the two single-site mutants. Adding 1 mM DTT rescued the glucagon potency of the mutant V130C/L210C to a similar level as the single-site mutant V130C. Unlike the double-cysteine mutant, cAMP accumulation of the WT receptor and the single-site mutants was not affected by DTT. In the GCGR-FL crystal structure, residues V130 on the stalk and L210 on ECL1 are in close contact with their main chains forming hydrogen bonds to stabilize the interaction between the stalk and ECL1 (Fig. 3b and 4a). Thus, the disulfide cross-linking studies support the observed conformation of the stalk and ECL1, and indicate that the interaction between the stalk and ECL1 plays an important role in modulating peptide ligand binding and receptor activation.
Figure 4. Stalk-ECL1 cross-linking assays.

a, Model of the introduced disulfide bond between the mutations V130C and L210C. GCGR is shown as cartoon. The ECD, stalk, ECL1 and TMD are colored in orange, green, magenta and blue, respectively. The disulfide bond V130C-L210C is shown as yellow sticks. b–e, Disulfide cross-linking assays of the GCGR mutant V130C/L210C (b) and the controls, WT receptor (c) and single-site mutants V130C (d) and L210C (e). Dose-response curves of [125I]-glucagon binding assay generated from three independent experiments performed in duplicate. Data are shown as means ± S.E.M. Specific [125I]-glucagon binding (% of WT) of V130C/L210C-DTT is significantly different from that of V130C/L210C (P < 0.005, two-tailed t-test). Specific [125I]-glucagon binding (% of WT) of WT-DTT, V130C-DTT and L210C-DTT is not significantly different from that of WT, V130C and L210C, respectively (P > 0.05, two-tailed t-test).
Conformational changes of the stalk and ECL1, probably the dissociation of these two fragments followed by the change of the relative orientation between the ECD and TMD, are required for GCGR binding to its cognate peptide ligand and activation of the receptor. This aligns with the HDX studies demonstrating that regions of both the stalk and ECL1 displayed increased protection in the peptide-bound GCGR compared with the small-molecule antagonist-bound receptor, suggesting that these two regions adopt variable conformations when different ligands bind to the receptor7. A previous cysteine accessibility study found that accessibility of the mutant L1982.71bC on the extracellular side of helix II was low in the absence of ligand but increased upon glucagon binding18, indicating that the residue L1982.71b, which was reported to be involved in glucagon binding8, might be buried by other residues of the receptor in the inactive state and exposed to an aqueous environment in active conformation. In the inactive GCGR-FL structure, L1982.71b is right beneath ECL1 and the stalk, which may provide a ‘shelter’ for this residue in the inactive state and change conformation to facilitate peptide ligand binding in the active state. Thus, the stalk and ECL1 most likely act as modulators between the ECD and TMD, and play critical roles in regulating signal transduction of GCGR.
The importance of the interface between the ECD, linker domain and TMD on stabilizing the inactive state of receptor was also reported in the recently published structure of the smoothened receptor (SMO)19. It was proposed that the receptor activity of SMO is controlled by ligand-regulated interactions between its ECD and TMD19. Structures of GCGR and SMO, both of which contain large ECDs, provide essential insights into the signal transduction of non class A GPCRs.
Conformational states of GCGR
To investigate the conformational flexibility of apo GCGR, we performed three independent 1-μs molecular dynamics (MD) simulations on the GCGR-FL crystal structure with mAb1 and NNC0640 removed. The MD simulations revealed significant motions of the ECD during the first 300 ns and relatively stable conformations of this region in the latter part of the simulations (Extended Data Fig. 7a). Although the ECD ended up with different conformations in the three simulations, in each simulation it underwent similar movements with the ECD shifting towards the TMD and approaching the stalk and ECL1 (Extended Data Fig. 7b–d). Throughout the duration of the three simulations, the stalk maintained its extended conformation and close contact with ECL1, and ECL1 retained the β-sheet conformation in most of the snapshots (Extended Data Fig. 7e). These simulations suggest that the basic features associated with the inactive state remain intact even in the absence of mAb1. Specifically, the ECD and stalk retain orientations that preclude binding of the glucagon peptide in a signaling competent manner.
In the MD simulations, extensive interactions between the ECD and the stalk/ECL1 region were observed (Extended Data Fig. 8a). Comparison between the predicted apo GCGR conformation and the previous model of the GCGR-glucagon complex7, as well as the crystal structure of the GCGR-NNC0640-mAb1 complex, revealed a common binding interface on the ECD of GCGR shared by the stalk/ECL1, glucagon and mAb1 (Extended Data Fig. 8b–d), further suggesting that the stalk and ECL1 may play critical roles in regulating peptide agonist binding and receptor activation. To confirm the existence of molecular contact between the ECD and stalk/ECL1 and its effect on receptor activity, we performed disulfide cross-linking studies to lock these two regions together. The results showed that the glucagon-binding ability of the receptor was reduced by the single-site mutations Q113ECDC and D209ECL1C to about 20% and 60%, respectively, while adding 1 mM DTT had little effect on the binding potency of these two single-site mutants. In contrast, the double-cysteine mutant Q113C/D209C lost its binding ability to glucagon, which was partially rescued (about 20% compared to the WT receptor) by addition of 1 mM DTT (Extended Data Fig. 2e–g). Since the recovery of 20% of the glucagon-binding ability corresponds to the effect of one of the single-site mutations Q113C, these data support the molecular contact between the ECD and ECL1 as observed in the MD simulations, and suggest that the interaction between these regions may play important roles in constraining the conformation of the apo receptor in an inactive state. Previous MD simulations and disulfide cross-linking studies indicated that the ECD of apo GCGR interacts with ECL1 and can be cross-linked to ECL37, whereas chimera studies suggested that the ECD of GCGR stabilizes the receptor in an inactive state through interactions with ECL36. These data combined with our studies suggest the existence of a dynamical interface between the ECD and TMD of GCGR.
Based on the GCGR-FL crystal structure, the results of MD simulations and disulfide cross-linking studies, together with the previous glucagon-GCGR binding studies, we postulate a hypothetical structural model of how glucagon and mAb1 are recognized by the apo GCGR (Fig. 5). In the apo state, the stalk and ECL1 may form extensive hydrophobic and polar interactions with the ECD, constraining the conformation of the apo receptor in an inactive state. With the competition of mAb1, the ECD would most likely dissociate from the stalk and ECL1, releasing its binding interface for interactions with mAb1. The antibody then interacts with the ECD to prevent the binding between glucagon’s C-terminus and the receptor, while the stalk and ECL1 form a compact β-sheet conformation to block the entrance of the N-terminus of glucagon to the binding pocket within the TMD. A hypothetical model of glucagon binding to GCGR requires the dissociation of the ECD and stalk/ECL1 to initiate the recognition between the ECD and the C-terminus of glucagon. Binding of the peptide agonist may trigger a conformational rearrangement of the ECD relative to the TMD either driven by or resulting in a conformational change of the stalk region, possibly including its dissociation with ECL1 and/or a change in secondary structure. The ECD bound to glucagon can then assume a position amenable for direct interactions between glucagon’s N-terminus and TMD resulting in activation of the receptor. Further details of the interactions between GCGR and the peptide ligand, such as the structure of GCGR-glucagon complex, are required to fully understand the activation mechanism of this receptor.
Figure 5. Different conformational states of GCGR.

The crystal structure of GCGR-mAb1 complex, the model of apo GCGR derived from MD simulations, the hypothetical docking pose of glucagon C-terminus to GCGR and the hypothetical model of GCGR-glucagon complex are shown in two different views with the TMDs in each row in same orientation. The model of the hypothetical GCGR-glucagon complex shows a conformational change of the stalk region, possibly including its dissociation with ECL1 and/or a change in secondary structure. The structure and models are shown in cartoon and surface representations. The ECD, stalk, ECL1 and TMD of GCGR are colored in orange, green, magenta and blue, respectively. The antibody mAb1 is shown as cyan cartoon and surface. Glucagon is shown as cyan cartoon.
Methods
Cloning and insect cell expression of GCGR-FL
The codon optimized human GCGR gene (Genewiz) was cloned into a modified pFastBac1 vector with hemagglutinin (HA) signal sequence at the N-terminus and a PreScission protease site followed by a 10×His tag and a Flag tag at the C-terminus. To facilitate crystallization, T4L was inserted into ICL2 of GCGR between A256 and E260. To further improve thermostability, 45 residues (H433-F477) were truncated at the C-terminus. Our ligand-binding assay showed that protein engineering had little effect on the binding affinity of GCGR to NNC0640 (Extended Data Fig. 2a). The optimized GCGR construct was expressed in Spodoptera frugiperda (Sf9) insect cells (obtained from Invitrogen) using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Cells were infected at a density of 2 × 106 cells ml−1 with high-titer viral stock at MOI (multiplicity of infection) of 5.0. Cells were collected after 48 h post infection and stored at −80 ºC until use.
Purification of GCGR-NNC0640-mAb1 complex
The cells expressing the GCGR-T4L protein were lysed in a buffer containing 10 mM HEPES, pH 7.5, 20 mM KCl, 10 mM MgCl2 and EDTA-free protease inhibitor cocktail tablets (Roche), then prepared with 3 times wash of high salt buffer containing 10 mM HEPES, pH 7.5, 1 M NaCl, 20 mM KCl and 10 mM MgCl2. Purified membranes were resuspended in 10 ml lysis buffer with 40% glycerol and stored at −80 ºC.
Purified membranes were thawed in 30 ml buffer containing 10 mM HEPES, pH 7.5, 20 mM KCl, 10 mM MgCl2, 13% glycerol, 200 μM NNC0640 and EDTA-free protease inhibitor cocktail (Roche) at 4 ºC for 1 h. The GCGR protein was extracted by adding 10 ml solubilization buffer containing 4% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace) and 0.8% (w/v) cholesteryl hemisuccinate (CHS, Sigma) at 4 ºC for 3 h. The supernatant was isolated by ultracentrifugation at 160,000 g for 30 min, and the final concentrations of NaCl and DDM were adjusted to 0.8 M and 0.5% by adding 40 ml buffer containing 50 mM HEPES, pH 7.5 and 1.6 M NaCl. The supernatant was incubated with TALON resin (Clontech) overnight at 4 ºC.
The TALON resin was washed with 25 column volumes of wash buffer 1 containing 25 mM HEPES, pH 7.5, 500 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% glycerol, 30 μM NNC0640 and 30 mM imidazole, and then followed by incubation with mAb1 at a molar ratio of 1:1.2 in 2 ml of wash buffer 2 containing 25 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% glycerol, 30 μM NNC0640 and 10 mM imidazole at 4 ºC for 2 h. The unbound mAb1 was removed by washing the resin with 13 column volumes of wash buffer 2. The GCGR-NNC0640-mAb1 complex was eluted with 5 column volumes of 25 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% glycerol, 30 μM NNC0640 and 300 mM imidazole. The PD MiniTrap G-25 column (GE Healthcare) was used to remove imidazole. The sample was treated overnight with His-tagged PreScission protease (home-made) to remove the C-terminal His- and FLAG-tags, and His-tagged PNGase F (home-made) was also added to deglycosylate the receptor. The Ni-NTA resin (Qiagen) was incubated with the sample at 4 ºC for 1 h to remove the cleaved His-tag and PreScission protease. The purified GCGR-NNC0640-mAb1 complex was concentrated to 20–30 mg/ml with a 100 kDa molecular weight cut-off concentrator (Millipore).
Crystallization in lipidic cubic phase and data collection
Crystallization was performed using the lipidic cubic phase (LCP) method21. The GCGR-NNC0640-mAb1 protein (20–30 mg/ml) was mixed with lipid (monoolein/cholesterol 10:1 by mass) at weight ratio of 2:3 using a syringe mixer. The LCP mixture was dispensed onto 96-well glass sandwich plates (Shanghai FAstal BioTech) in 35 nl drop and overlaid with 800 nl precipitant solution using a Mosquito LCP robot (TTP Labtech). Protein reconstitution in LCP and crystallization trials were performed at room temperature (19–22 ºC). Plates were incubated and imaged at 20 ºC using an automated incubator/imager (RockImager, Formulatrix). Crystals grew in 100 mM HEPES, pH 7.0, 200–300 mM potassium phosphate monobasic, 20–30% (v/v) PEG 500DME and 10–60 mM gly-gly-glycine, and reached to a full size of 60–100 μm after 15 d. Crystals were harvested using 75–100 μm MiTeGen micromounts (M2-L19–50/150, MiTeGen) and immediately flash frozen in liquid nitrogen. Data collection was performed at the SPring-8 beam line 41XU, Hyogo, Japan, using a Pilatus3 6M detector (X-ray wavelength 1.0000 Å). The crystals were exposed with a 10 μm × 8 μm mini-beam for 0.2 s and 0.2° oscillation per frame. Due to radiation damage, data collection was limited to 10–15° per crystal. Diffraction data from 26 crystals were integrated and scaled using XDS22.
The crystals for X-ray free-electron laser (XFEL) data collection were obtained23 by injecting 6–8 μl of LCP sample as a continuous column into a syringe filled with 80 μl precipitant solution comprised of 100 mM HEPES, pH 7.0, 300 mM potassium phosphate monobasic, 25% (v/v) PEG 500DME and 100 mM gly-gly-glycine. The syringe was sealed up and incubated at 20 ºC. The excess precipitant was removed after the crystals appeared. The 7.9 MAG was added to absorb the residual precipitant solution and avoid the problem of lipid freezing upon injecting LCP in vacuum24. LCP-SFX experiments were carried out at the Coherent X-ray Imaging (CXI) instrument25 at the Linac Coherent Light Source (LCLS) in the SLAC National Accelerator Laboratory (Menlo Park, California, USA). X-ray pulses of 40 fs duration at a wavelength of 1.3 Å (9.5 keV) were attenuated to ~6% (9×1010 photons/pulse) and focused to ~1.5 μm diameter at the interaction point using Kirkpatrick-Baez mirrors26. GCGR-NNC0640-mAb1 complex crystals in LCP were injected across the XFEL beam using an LCP injector24 with a 50 μm diameter nozzle at a flow rate of ~0.2 μl/min. Diffraction patterns were collected at 120 Hz using the Cornell-SLAC Pixel Array Detector (CSPAD). Over 1 million data frames were collected corresponding to ~2.3 h of data acquisition time. Of these frames, ~6.5% contained potential crystal hits as identified using Cheetah27 (more than 15 Bragg peaks of minimum 2 pixels in size and a signal to noise ratio better than 7 after local background subtraction). Of the 91,626 potential crystal hits, 57,573 diffraction patterns could be auto-indexed by CrystFEL28 (indexing rate of 63%) using a combination of MOSFLM29, asdf28 and DirAx30. Reflections from different crystals in random orientations were merged using a Monte Carlo integration of each reflection by CrystFEL28. The data used for the structure refinement were truncated at 3.0 Å based on the criteria of data correlation coefficient (CC*) cutoff of 0.5. The statistics of the final data used in structure refinement are shown in Extended Data Table 1.
Structure determination
Both the synchrotron data and XFEL data were initially merged according to the apparent Laue group of mmm. Molecular replacement searches were performed in all possible space groups of mmm, but no satisfying structure solution was found. The data were then reprocessed with the Laue group of 2/m, and the axis with length of 245.3 Å was selected as the 21 screw axis based on systematic absences of the merged synchrotron data, with β angle very close to 90 degree (90.01º).
Both the large crystals for synchrotron data collection and the small crystals for XFEL data collection appeared to be pseudo-merohedrally twinned based on the L-test analysis by Phenix Xtriage31 with the multivariate score of 7.8. Despite the challenge of twinned data, the GCGR-NNC0640-mAb1 complex structure was solved by molecular replacement (MR) implemented in Phaser32 using the models of GCGR-TMD, mAb1 bound ECD of GCGR, and T4L (PDB IDs: 4L6R, 4LF3, and 2RH1, respectively). Two molecules of GCGR-TMD, two molecules of mAb1-bound GCGR-ECD and one molecule of T4L were found sequentially by MR search. The second T4L was partially resolved based on the electron density.
The structure was initially solved and refined against the synchrotron data without using a twin law to an Rfree of ~ 33% with REFMAC33 and BUSTER34. The model maps from the data were of sufficient quality to interpret the overall structure of the GCGR-NNC0640-mAb1 complex, and both the stalk and ECL1 were built based on the electron map. The model then underwent iterated cycles of manual building into |2Fo| - |Fc| maps with Coot35 and refinement with REFMAC33, where rigid body, individual positions and TLS refinements were used along with NCS restraints and a twin law (h, -k, -l). The final structure refined by synchrotron data was then used as a search model for the XFEL data, and the XFEL structure was refined in a similar strategy as described above. Both structures have been carefully refined and the ramachandran plot analysis indicates that 100% of the residues are in favorable or allowed regions (no outliers).
The structures of the GCGR-NNC0640-mAb1 complex were determined to 3.0 and 3.2 Å resolution using the XFEL data and synchrotron data, respectively (Extended Data Table 1). The two structures are similar with Cα RMSD of 0.6 Å. Structure analysis and discussion are based on the structure solved using the XFEL data at higher resolution.
Expression and purification of the Fab fragment of mAb1
The genes of light chain and heavy chain of mAb1 Fab fragment were synthesized with CD33 signal peptide and cloned into the vector pJSV002 for mammalian cell expression. The plasmids were then transfected into HEK293-6E cells (obtained from Invitrogen) at density of 1.0 × 106 cells ml−1 with DNA molar ratio of 1:1. The transfection was performed following Invitrogen’s Freestyle_293 expression manual. The cell culture supernatant was filtered after 120 h post transfection and applied to a protein G affinity column (GE Healthcare) that was pre-equilibrated in phosphate buffered saline (PBS). The bound Fab was eluted with 100 mM glycine-HCl, pH 2.8. Fractions were collected and neutralized immediately with 1/20 volume of 2 M Tris-HCl, pH 9.0. The pooled fraction was then diluted into 20 mM Na acetate, pH 5.5 and applied to a SP HP column (GE Healthcare). The bound Fab was eluted with a 100–300 mM linear gradient of NaCl in 20 mM Na acetate, pH 5.5 and buffer-exchanged to PBS on a G25 desalting column (GE Healthcare). Purified protein was sterilized by filtration through a 0.2 mm filter unit (Sartorius). The purity of the protein sample was analyzed by SDS-PAGE and size-exclusion chromatography. The Fab identity was confirmed by mass spectrometry.
Construction of GCGR mutants and cell transfection
The complementary DNA (cDNA) encoding the human GCGR was originally obtained from GeneCopoeia and cloned into the expression vector pcDNA3.1/V5-His-TOPO (Invitrogen) at the HindIII and EcoRI sites. The double cysteine mutants were constructed by PCR-based site-directed mutagenesis. CHO-K1 cells (obtained from ATCC) were seeded onto 96-well poly-D-lysine treated cell culture plates (PerkinElmer) at a density of 3 × 104 cells per well. After overnight culture, the cells were transiently transfected with WT or mutant GCGR DNA using Lipofectamine 2000 transfection reagent (Invitrogen).
Whole-cell glucagon binding assay
CHO-K1 cells were cultured in F-12 medium with 10% (v/v) fetal bovine serum and harvested after 24 h post transfection, washed twice, and incubated with blocking buffer (F12 supplemented with 33 mM HEPES, pH 7.4, and 0.1% bovine serum albumin (BSA)) for 2 h at 37 °C. Cells were treated with PBS or 1mM DTT for 10 min before homogeneous binding. They were then washed twice with PBS and incubated in binding buffer (PBS supplemented with 10% BSA, pH 7.4) with constant concentration of [125I]-glucagon (40 pM) and different concentrations of unlabeled glucagon (3.57 pM to 1 μM) at room temperature for 3 h. Cells were washed three times with ice-cold PBS and lysed by 50 μl lysis buffer (PBS supplemented with 20 mM Tris-HCl, 1% Triton X-100, pH 7.4). The plates were subsequently counted for radioactivity (counts per minute, CPM) in a scintillation counter (MicroBeta2™ Plate Counter, PerkinElmer) using a scintillation cocktail (OptiPhase SuperMix, PerkinElmer).
cAMP assay
HEK-293T cells (obtained from and certified by the Cell Bank at the Chinese Academy of Science) were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% (v/v) fetal bovine serum, 50 IU/ml penicillin and 50 ug/ml streptomycin. Cells were maintained at 37 °C in 5% CO2 incubator and seeded onto 6-well cell culture plates before transfection. After overnight culture, the cells were transiently transfected with WT or mutant GCGR DNA using Lipofectamine 2000 transfection reagent (Invitrogen). The transfected cells were seeded onto 384-well plates (8,000 cells per well) after 24 h post transfection. cAMP accumulation was measured using the LANCE cAMP kit (PerkinElmer) according to the manufacturer’s instructions. Briefly, transfected cells were incubated for 30 min in assay buffer (DMEM, 1 mM 3-isobutyl-1-methylxanthine) with different concentrations of glucagon (0.001 pM to 10 nM) at 37 °C. The reactions were stopped by adding lysis buffer containing LANCE reagents. Plates were then incubated for 60 min at room temperature and time-resolved FRET signals were measured at 620 nm and 650 nm by an EnVision multilabel plate reader (PerkinElmer).
NNC0640 binding assay
NNC0640 binding was analyzed using plasma membranes prepared from HEK293T cells transiently expressing WT and mutant GCGRs. Approximately 1.2 × 108 transfected HEK293T cells were harvested, suspended in 10 ml ice-cold membrane buffer (20 mM HEPES-NaOH and 10 mM EDTA, pH 7.4) and centrifuged for 5 min at 200 g. The resulting pellet was resuspended in cold membrane buffer then homogenized and centrifuged for 15 min at 40,000 g. The pellet was resuspended, homogenized and centrifuged again, and the precipitate containing the plasma membranes was suspended in the membrane buffer containing protease inhibitor (Sigma-Aldrich) and stored at −80 °C. Protein concentration was determined using a protein BCA assay kit (Pierce Biotechnology).
For homogeneous binding, cell membrane homogenates (10 μg protein per well) were incubated in membrane binding buffer with constant concentration of [3H]-NNC0640 (50 nM, labeled by PerkinElmer) and serial dilutions of unlabeled NNC0640 (0.26 nM to 100 μM) at room temperature for 3 h. Nonspecific binding was determined in the presence of 100 μM NNC0640. Following incubation, the samples were filtered rapidly in vacuum through glass fiber filter plates (PerkinElmer). After soaking and rinsing 4 times with ice-cold PBS, the filters were dried and counted for radioactivity in a scintillation counter (PerkinElmer).
Hydrogen/Deuterium Exchange Mass spectrometry (HDX-MS)
HDX experiments on the antibody-bound and antibody-free GCGRs were carried out at 4 °C using a system as previously described7. The GCGR construct used in the HDX studies lacks the T4L fusion protein. The ligand NNC0640 was added during protein purification to improve protein stability. Briefly, 15 μM of the receptor protein was incubated in a D2O containing buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% glycerol) for a range of exchange times from 10 s to 1 h before quenching the deuterium exchange reaction with an acidic quench solution (pH 2.4). All mixing and digestions were carried out on a LEAP Technologies Twin HTS PAL liquid handling robot housed inside a temperature-controlled cabinet. Digestion was performed in-line with chromatography using an immobilized pepsin column. Mass spectra were acquired on a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific) and peptide identification from the MSMS data was done using Mascot. HDX experiments for each pairwise comparison (antibody-free vs. mAb1-bound GCGR or antibody-free vs. mAb23-bound GCGR) were run separately under the same conditions and percent deuterium exchange values for peptide isotopic envelopes at each time point were calculated and processed using the Workbench software36.
MD simulation
We performed three 1-μs all-atom MD simulations on the full-length GCGR extracted from the GCGR-NNC0640-mAb1 complex structure to investigate the conformational dynamics of the apo receptor. The apo GCGR structure without the ligand NNC0640 and mAb1 was used as the starting model for the MD simulation, which was embedded in a 90 Å × 90 Å palmitoyl oleoyl phosphatidyl choline (POPC) bilayer and the lipids located within 1 Å of the receptor were removed. The system was solvated in a box (90×90×160 Å3) with TIP3P waters and 0.15 M NaCl, including 124,478 atoms. Three parallel MD simulations were performed using the GROMACS 5.0.4 package37 with isothermal–isobaric (NPT) ensemble and periodic boundary condition. The CHARMM36-CAMP force field38 was used for the protein, the POPC phospholipids, ions and water molecules. Energy minimizations were first performed to relieve unfavorable contacts in the system, followed by equilibration steps of 50 ns in total to equilibrate the lipid bilayer and the solvent with restraints on the main chain of GCGR. Subsequently, three 1-μs production runs were performed. The temperature of the systems was maintained at 310 K using the v-rescale method39 with a coupling time of 0.1 ps. The pressure was kept at 1 bar using the Parrinello-Rahman40 with τp = 1.0 ps and a compressibility of 4.5 × 10−5 bar−1. SETTLE41 constraints and LINCS42 constraints were applied to the hydrogen-involved covalent bonds in water molecules and in other molecules, respectively, and the time step was set to 2 fs. Electrostatic interactions were calculated with the Particle-Mesh Ewald (PME) algorithm43 with a real-space cutoff of 1.2 nm.
Data availability
Atomic coordinates and structure factor files for the GCGR-NNC0640-mAb1 complex structures solved using the XFEL data and synchrotron data have been deposited in the Protein Data Bank with indentification codes 5XEZ and 5XF1, respectively.
Extended Data
Extended Data Figure 1. Snake plot of the GCGR construct used for crystallization and crystal packing of the GCGR-NNC0640-mAb1 complex structure.

a, Snake plot of the GCGR-T4L fusion construct used for crystallization. The eight cysteine residues forming disulfide bonds are shown in yellow with four yellow lines illustrating the disulfide bonds. b, Crystal packing of the GCGR-NNC0640-mAb1 complex structure. GCGR and mAb1 are shown in cartoon representation. The ECD, stalk and TMD of the receptor are colored in orange, green and blue, respectively. The T4L fusion is in grey and mAb1 is in cyan. NNC0640 is displayed as magenta spheres. The unit cell is shown as a red box.
Extended Data Figure 2. Binding of [3H]-NNC0640 and [125I]-glucagon to WT and mutant GCGRs and glucagon-induced cAMP assays.

a, Binding of [3H]-NNC0640 to membrane preparations from Sf9 cells expressing WT and the engineered GCGR for crystallization. Data are shown as means ± S.E.M. from three independent experiments performed in duplicate. “Construct” indicates the construct used for crystallization, containing the T4L fusion at ICL2 and 45 residues truncated at the C-terminus of the receptor. b, Binding of [3H]-NNC0640 to membrane preparations from HEK293T cells expressing WT and mutant GCGRs. Data are shown as means ± S.E.M. from three independent experiments performed in duplicate. The IC50 values for the WT and mutant GCGRs from at least three independent experiments are listed in Extended Data Table 2. c, Glucagon-induced cAMP accumulation measurement of the mutants V130C/L210C, V130C and L210C and the WT GCGR. d, Glucagon-induced cAMP accumulation measurement of the mutants V130C/L210C, V130C and L210C and the WT GCGR in presence of 1 mM DTT. Dose-response curves of cAMP accumulation assays generated from three independent experiments performed in duplicate. Data are shown as means ± S.E.M. e–g, Disulfide cross-linking assays of the GCGR mutant Q113C/D209C (e) and the controls, single-site mutants Q113C (f) and D209C (g). Dose-response curves of [125I]-glucagon binding assay generated from three independent experiments performed in duplicate. Data are shown as means ± S.E.M.
Extended Data Figure 3. Electron densities for the stalk and ECL1 in the GCGR-FL crystal structure.

a, Electron densities for the stalk. The stalk region is shown as sticks and colored in green. The rest of the receptor is shown in cartoon representation and colored in orange (ECD), magenta (ECL1) and blue (TMD). Electron densities are contoured at 1.0 σ from a composite omit map and colored in blue. b, Electron densities for ECL1. ECL1 is shown as sticks and colored in magenta. The rest of the receptor is shown in cartoon representation and colored in orange (ECD), green (stalk) and blue (TMD). Electron densities are contoured at 1.0 σ from a composite omit map and colored in blue.
Extended Data Figure 4. Comparison between the GCGR-FL crystal structure and previously published structure and models.

a and b, Comparison between the GCGR-mAb1 complex structure and the previous model of GCGR-glucagon complex7. Only the receptors in the GCGR-mAb1 complex structure and the model are shown in cartoon representation and colored in blue and yellow, respectively. The ECDs are also shown in surface representation. The stalk in the GCGR-FL structure is in green, and the stalk in the model is in magenta. (b) Side view; (c) Top view. c, Comparison between the GCGR-FL structure and crystal structure of GLP-1R’s ECD bound to its endogenous ligand GLP-1. Structural superimposition shows spatial clashes between ECL1 and helix II of GCGR and GLP-1. The receptor in the GCGR-FL structure is shown as blue cartoon, and the ligand NNC0640 is displayed as magenta spheres. The complex structure of GLP-1R’s ECD bound to GLP-1 (PDB ID: 3IOL) is shown in cartoon representation. The ECD of GLP-1R is colored in green, and GLP-1 is in red.
Extended Data Figure 5. HDX studies for the NNC0640-stabilized GCGR in complex with mAb1 or mAb23.

a, Interaction between GCGR and mAb1. The receptor is shown in grey cartoon representation. The regions of αA helix (residues L32–L38) and β4-L5 (residues K98–Q105) in ECD and the stalk (residues I128–M137), which showed increased protection in the antibody-bound GCGRs, are colored in red, blue and green, respectively. The antibody mAb1 is shown as cyan surface and cartoon. b–d, HDX plots for the regions of ECD αA helix (b), ECD β4-L5 (c) and the stalk (d) in the antibody-free and mAb1-bound GCGRs. e–g, HDX plots for the regions of ECD αA helix (e), ECD β4-L5 (f) and the stalk (g) in the antibody-free and mAb23-bound GCGRs. HDX data are plotted as means ± S.D. of three independent experiments.
Extended Data Figure 6. Differential perturbation heat map view of the HDX studies.

a, Heat map view of the GCGR-NNC0640-mAb1 complex colored according to the heat map coloring scheme used by the software HDX Workbench36. Each bar represents a peptide showing the average difference (across 6 time points) in D2O uptake between the receptor-antibody complex and the antibody-free receptor with the standard deviation between replicates and the peptide charge states shown in parentheses. The regions that revealed statistically significant reduction in deuterium uptake in the receptor-antibody complex compared to the antibody-free receptor are colored in green and boxed in red. The D2O difference between the antibody-bound and antibody-free GCGRs at two consecutive time points has a P-value < 0.05 or a single time point has a P-value < 0.01. The regions with no significant change are in grey and the regions that have no peptides covering the sequence in MSMS and HDX runs are shown as gaps. b, Heat map view of the GCGR-NNC0640-mAb23 complex.
Extended Data Figure 7. MD simulations of the apo GCGR.

a, Main chain RMSD values of the ECD versus simulation time in the three 1-μs MD simulations. The values were calculated from snapshots at 100 ps intervals. All the structures were superimposed to the crystal structure of GCGR-FL using the main chain atoms of residues S150-L160 (helix I), I176–V193 (helix II), A227–G246 (helix III), G271–P275 (helix IV), V311-I321 (helix V), T351-L358 (helix VI) and Q392–Y400 (helix VII). b–d, Comparison between the results of simulations and the GCGR-FL crystal structure. The GCGR-FL crystal structure is shown as grey cartoon. The results of the three simulations are shown in cartoon representation, and colored in yellow, cyan and orange, respectively. The ECDs of the receptors are also shown in surface representation. The N-terminal portion of stalk (residues G125–Q131) and the ECL1 region (residues T200-D218), which are analyzed in panel e, are colored in green and magenta, respectively. The red arrows indicate the movements of the ECDs and ECL1 (d) in the simulations. e, Secondary structure as a function of time for the stalk (residues G125–Q131) and ECL1 (residues T200-D218) regions in the crystal structure and simulations.
Extended Data Figure 8. Interactions between the ECD and stalk/ECL1 in MD simulations and comparison with the binding modes of glucagon and mAb1.

a, Interactions between the ECD and stalk/ECL1 in one typical MD simulation snapshot. The residues Y202, K205 and I206 on the N-terminal half of ECL1 make hydrophobic contacts with a hydrophobic core formed by residues L32, F33, W36, Y65, Y84, L85, P86 and W87 on the αA helix, L2 and L5 of the ECD. Additionally, the negatively charged residues E127 and E129 on the stalk and D208 and D209 on the tip of ECL1 tend to form salt bridges with the basic residues R111 and R116 on the L5 of the ECD. The receptor is shown in cartoon representation. The ECD, stalk, ECL1 and TMD are colored in orange, green, magenta and blue, respectively. The residues involved in the interaction are shown as sticks. b–d, Interaction interfaces on the ECD for the stalk/ECL1 in the MD simulations (b), glucagon in the GCGR-glucagon complex model7 (c) and mAb1 in the GCGR-NNC0640-mAb1 complex structure (d). The ECDs of the receptor are shown as grey cartoon and surface. The residues involved in the interactions are shown as sticks and colored in orange.
Extended Data Table 1.
Data collection and structure refinement statistics.
| Data Collection | ||||
|---|---|---|---|---|
| XFEL | Synchrotron | |||
| Number of crystals used | 57,573 | 26 | ||
| Space group | P21 | P21 | ||
| Cell dimensions | ||||
| a, b, c (Å) | 72.6, 245.3, 96.3 | 71.7, 248.8, 93.5 | ||
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | ||
| Number of reflections processed | 13, 392, 502 | 137, 681 | ||
| Number of unique reflections | 67,598 | 49,677 | ||
| Resolution (Å) | 31.0-3.00 (3.10-3.00)* | 50.0-3.20 (3.35-3.20) | ||
| Rmerge (%) | 22.8 (184)† | 23.6 (87.2) | ||
| CC*‡ | 0.99 (0.51) | 0.99 (0.72) | ||
| SNR or l/σ | 4.8 (0.68) | 3.1 (0.8) | ||
| Completeness (%) | 100.0 (100.0) | 92.8 (93.1) | ||
| Redundancy | 198 (17.6) | 2.8 (2.7) | ||
| Refinement | ||||
| Resolution (Å) | 31.0-3.00 | 50.0-3.20 | ||
| Number of reflections (test set) | 63,632 (3,311) | 48,212(2,547) | ||
| Rwork / Rfree (%) | 21.0/24.5 | 20.9/22.1 | ||
| Number of atoms | ||||
| Protein | 14,808 | 14,741 | ||
| Ligand | 82 | 82 | ||
| Others | 182 | 225 | ||
| Overall B values (Å2) | A | B | A | B |
| GCGR | 95.9 | 110.8 | 124.1 | 135.4 |
| ECD | 60.7 | 65.2 | 94.0 | 102.4 |
| Stalk | 79.1 | 87.5 | 109.5 | 133.9 |
| ECL1 | 63.4 | 69.5 | 104.6 | 112.9 |
| TMD | 94.8 | 113.9 | 136.4 | 148.9 |
| T4L | 98.0 | 96.3 | 118.6 | 118.3 |
| mAb1 (heavy chain) | 61.2 | 58.4 | 83.0 | 79.1 |
| mAb1 (light chain) | 58.5 | 51.4 | 79.8 | 74.4 |
| Ligand | 71.6 | 142.2 | 165.5 | 161.0 |
| NAG | 106.0 | 78.5 | 96.1 | 87.2 |
| RMSD | ||||
| Bond lengths (Å) | 0.009 | 0.014 | ||
| Bond angles (°) | 1.4 | 1.8 | ||
| Ramachandran plot statistics (%)§ | ||||
| Favored regions | 94.2 | 89.5 | ||
| Allowed regions | 5.8 | 10.5 | ||
| Disallowed regions | 0.0 | 0.0 | ||
Extended Data Table 2.
Binding of [3H]-NNC0640 to membrane preparations from HEK293T cells expressing WT and mutant GCGRs.
| GCGR constructs | IC50* (nM) | Span† (% of WT) | Expression‡ (% of WT) |
|---|---|---|---|
| WT | 69.2±4.4 | 100 | 100 |
| R346A | 203.8±54.6 | 85.8±13.5 | 98.7±2.0 |
| K349A | 148.7±41.1 | 18.2±3.6 | 89.6±5.5 |
| S350A | 658.9±89.7 | 56.4±15.2 | 130.9±15.6 |
| T353A | NB§ | NB | 75.8±5.3 |
| L357A | 83.3±17.9 | 95.8±6.1 | 73.7±16.1 |
| L395A | 109.8±16.0 | 95.1±24.6 | 144.9±56.9 |
| L403A | 152.7±34.1 | 72.1±8.4 | 60.3±26.3 |
| N404A | 612.6±138.1 | 21.6±3.0 | 34.0±14.1 |
Values shown are means ± S.E.M. of at least three independent experiments.
Specific [3H]-NNC0640 binding (span) is defined as the window between the highest (no unlabeled ligand) and the lowest binding (unlabeled ligand at the highest concentration examined).
Protein expression levels of GCGR constructs in radioligand displacement studies are determined by flow cytometry with an anti-GCGR antibody and reported as percent compared to WT GCGR.
If the span was < 10% of the WT GCGR, IC50 is reported as no binding (NB).
Acknowledgments
This work was supported by the “National Basic Research Program of China” grants 2014CB910400 (B.W.) and 2015CB910304 (H.J.), CAS Strategic Priority Research Program XDB08020000 and CAS grant QYZDB-SSW-SMC024 (B.W.), the National Science Foundation of China grants 31422017 (B.W.), 81525024 (Q.Z.), 81230076 (H.J.) and 81573479 (D.Y.), the National Health and Family Planning Commission grants 2012ZX09304-011, 2013ZX09401003-005, 2013ZX09507001 and 2013ZX09507-002 (M.-W.W.), Shanghai Science and Technology Development Fund 15DZ2291600 (M.-W.W.), the National Institutes of Health grant R01 GM108635 (V.C.), and the National Science Foundation STC award 1231306 (G.N., U.W., T.D.G., V.C.). The authors thank A. Walker for assistance with manuscript preparation, Y. Feng and C. Ji for technical assistance, M. Hunter, A. Batyuk, A. Ishchenko, L. Johansson, B. Stauch, M. Audet, M. Liang, M. Seaberg and P. Walter for their help with XFEL data collection, B. Yu for help with mAb1 protein sample preperation, and the special program for applied research on super computation of the NSFC-Guangdong Joint Fund (the second phase). The synchrotron radiation experiments were performed at the BL41XU of SPring-8 with approval of the Japan Synchrotron Radiation Research Institute (proposal no. 2016A2517, 2016A2518, 2016B2517 and 2016B2518). We thank the beamline staff members K. Hasegawa, H. Okumura, and H. Murakami of the BL41XU for help on X-ray data collection. Use of the LCLS, SLAC National Accelerator Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Parts of the sample injector used at LCLS for this research was funded by the National Institutes of Health, P41GM103393, formerly P41RR001209.
Footnotes
S.R.-R., X.W. and J.L. are employees of Novo Nordisk, a pharmaceutical company focused on class B GPCRs for type 2 diabetes. R.C.S. is a founder and board member of Bird Rock Bio, a company focused on GPCR therapeutic antibodies. The remaining authors declare no competing financial interests
Readers are welcome to comment on the online version of the paper
Author Contributions Ha.Z. and A.Q. optimized the construct, developed the purification procedure and purified the GCGR proteins for crystallization, performed crystallization trials and optimized crystallization conditions. D.Y. designed, performed and analysed disulfide cross-linking, ligand binding and cAMP assays of WT and mutant GCGRs. L.Y. performed and analysed MD simulations. V.D. performed and analysed HDX data. Hu.Z. collected the X-ray diffraction data at the synchrotron. G.W.H. helped with structure determination and refinement. T.D.G. processed the XFEL data. R.G.S., U.W., G.N. and W.L. helped with XFEL sample preperation and data collection. Y.W. helped with construct and crystal opimization. L.M. expressed the GCGR proteins. A.D., X.C. and G.L. helped with disulfide cross-linking and ligand binding assays. X.W. prepared mAb1 protein sample. Z.G. and Y.D. helped with data analysis. C.d.G., S.R.-R., G.S., J.L., V.C. and M.A.H. helped with structure analysis/interpretation and commented on the manuscript. P.R.G. oversaw HDX assay. V.C. oversaw collection of the XFEL data. H.Y. and H.J. oversaw MD simulations. M.-W.W. oversaw the disulfide cross-linking, ligand binding and cAMP assays. R.C.S, H.J. and M.-W.W. oversaw structure analysis/interpretation and edited the manuscript. B.W. and Q.Z. initiated the project, planned and analysed experiments, solved the structures, supervised the research and wrote the manuscript with input from all co-authors.
References
- 1.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Hollenstein K, et al. Insights into the structure of class B GPCRs. Trends Pharmacol Sci. 2014;35:12–22. doi: 10.1016/j.tips.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finan B, et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell. 2016;167:843–857. doi: 10.1016/j.cell.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 4.Longuet C, et al. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008;8:359–371. doi: 10.1016/j.cmet.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parthier C, Reedtz-Runge S, Rudolph R, Stubbs MT. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Koth CM, et al. Molecular basis for negative regulation of the glucagon receptor. Proc Natl Acad Sci USA. 2012;109:14393–14398. doi: 10.1073/pnas.1206734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, et al. Conformational states of the full-length glucagon receptor. Nat Commun. 2015;6:7859. doi: 10.1038/ncomms8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siu FY, et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jazayeri A, et al. Extra-helical binding site of a glucagon receptor antagonist. Nature. 2016;533:274–277. doi: 10.1038/nature17414. [DOI] [PubMed] [Google Scholar]
- 10.Hollenstein K, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- 11.Song G, et al. Structure of the human GLP-1 receptor complex with allosteric modulators. Nature. 2017 doi: 10.1038/nature22378. companion article. [DOI] [PubMed] [Google Scholar]
- 12.Grace CR, et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc Natl Acad Sci USA. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of strcture-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 15.Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc Natl Acad Sci USA. 2013;110:5211–5216. doi: 10.1073/pnas.1221585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mann R, Wigglesworth MJ, Donnelly D. Ligand-receptor interactions at the parathyroid hormone receptors: subtype binding selectivity is mediated via an interaction between residue 23 on the ligand and residue 41 on the receptor. Mol Pharmacol. 2008;74:605–613. doi: 10.1124/mol.108.048017. [DOI] [PubMed] [Google Scholar]
- 17.Unson CG, et al. Roles of specific extracellular domains of the glucagon receptor in ligand binding and signaling. Biochemistry. 2002;41:11795–11803. doi: 10.1021/bi025711j. [DOI] [PubMed] [Google Scholar]
- 18.Roberts DJ, Vertongen P, Waelbroeck M. Analysis of the glucagon receptor first extracellular loop by the substituted cysteine accessibility method. Peptides. 2011;32:1593–1599. doi: 10.1016/j.peptides.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Byrne EF, et al. Structural basis of Smoothened regulation by its extracellular domains. Nature. 2016;535:517–522. doi: 10.1038/nature18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 21.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu W, Ishchenko A, Cherezov V. Preparation of microcrystals in lipidic cubic phase for serial femtosecond crystallography. Nat Protoc. 2014;9:2123–2134. doi: 10.1038/nprot.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weierstall U, et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun. 2014;5:3309. doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang MN, et al. The Coherent X-ray Imaging instrument at the Linac Coherent Light Source. J Synchrotron Radiat. 2015;22:514–519. doi: 10.1107/S160057751500449X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siewert F, et al. Ultra-precise characterization of LCLS hard X-ray focusing mirrors by high resolution slope measuring deflectometry. Optics Express. 2012;20:4525–4536. doi: 10.1364/OE.20.004525. [DOI] [PubMed] [Google Scholar]
- 27.White TA, et al. Crystallographic data processing for free-electron laser sources. Acta Crystallogr D Biol Crystallogr. 2013;69:1231–1240. doi: 10.1107/S0907444913013620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White TA, et al. CrystFEL: a software suite for snapshot serial crystallography. J Appl Crystallogr. 2012;45:335–341. [Google Scholar]
- 29.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duisenberg AJM. Indexing in Single-Crystal Diffractometry with an Obstinate List of Reflections. J Appl Crystallogr. 1992;25:92–96. [Google Scholar]
- 31.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Smart OS, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pascal BD, et al. HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klauda JB, et al. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 40.Parrinello M, Rahman A. Polymorphic transitions in single-crystals - A new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 41.Miyamoto S, Kollman PA. Settle - an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 42.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 43.Essmann U, et al. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 44.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Atomic coordinates and structure factor files for the GCGR-NNC0640-mAb1 complex structures solved using the XFEL data and synchrotron data have been deposited in the Protein Data Bank with indentification codes 5XEZ and 5XF1, respectively.