

Abstract
Background
Diabetic retinopathy (DR) is a microvascular disease that results from retinal vascular degeneration and defective repair due to diabetes induced endothelial progenitor dysfunction.
Objective
Understanding key molecular factors involved in vascular degeneration and repair is paramount for developing effective DR treatment strategies. We propose that diabetes-induced activation of acid sphingomyelinase (ASM) plays essential role in retinal endothelial and CD34+ circulating angiogenic cell (CAC) dysfunction in diabetes.
Methods
Human retinal endothelial cells (HRECs) isolated from control and diabetic donor tissue and human CD34+ CACs from control and diabetic patients were used in this study. ASM mRNA and protein expression was assessed by quantitative PCR and ELISA, respectively. To evaluate the effect of diabetes-induced ASM on HRECs and CD34+ CACs function, tube formation, CAC incorporation into endothelial tubes, and diurnal release of CD34+ CACs in diabetic individuals was determined.
Results
ASM expression level was significantly increased in HRECs isolated from diabetic compared to control donor tissue, as well as CD34+CACs and plasma of diabetic patients. A significant decrease in tube area was observed in HRECs from diabetic donors as compared to control HRECs. The tube formation deficiency was associated with increased expression of ASM in diabetic HRECs. Moreover, diabetic CD34+ CACs with high ASM showed defective incorporation into endothelial tubes. Diurnal release of CD34+ CACs was disrupted with the rhythmicity lost in diabetic patients.
Conclusion
Collectively, these findings support that diabetes-induced ASM upregulation has a marked detrimental effect on both retinal endothelial cells and CACs.
Keywords: Diabetic retinopathy, Acid sphingomyelinase, Circulating angiogenic cells, and revascularization
1. Introduction
Diabetic retinopathy (DR) is a microvascular disease that results from diabetes-induced retinal damage that is further exacerbated by bone marrow dysfunction. Bone marrow dysfunction leads to decreased release of cells into the circulation and changes in hematopoiesis resulting in increased circulating pro-inflammatory monocytes and diminished repair due to defective progenitor cells. Although DR influence all retinal cells, clinical manifestations of DR are mainly due to changes in retinal vessels, where early histological alterations include pericyte loss, thickening of basement membrane, capillary occlusion and endothelial cell degeneration (1,2). These are followed by break down of blood retinal barrier (BRB) and leaky vasculature leading to hemorrhages, hard exudates, and retinal edema; structural changes involving the vascular wall leading to microaneurysms; and finally neovascularization, vitreous hemorrhage and fibrous tissue formation (3). Impaired vision due to macular edema, or vision loss due to neovascularization-induced vitreous hemorrhage or tractional retinal detachment usually takes place in the later stages of the disease.
Circulating angiogenic cells (CACs), a population of vascular progenitors originated from HSC (4), are considered as key regulators for healthy maintenance of retinal vasculature. Diabetic metabolic abnormalities lead to defective vascular maintenance due, in part, to failed attempts by dysfunctional CACs to repair damaged endothelium.
HSCs isolated from bone marrow or CACs from peripheral blood of control (healthy) animals have been shown to repair ischemic damage and aid in reperfusion of ischemic tissues (4–6). Several studies have shown an association between DR risk and both reduced number (7–10) and function of CACs (11–16).
Several key hyperglycemia- and dyslipidemia-activated pathways leading to retinal endothelial cell and CAC dysfunction have been identified. Prominent among these are pathways that promote an increase of pro-inflammatory cytokines, pro-inflammatory lipids and pro-angiogenic factors (17–26). We have previously demonstrated activation of the central enzyme of sphingolipid metabolism, acid sphingomyelinase (ASM), as a key metabolic abnormality in diabetic retinal vasculature and CACs. ASM hydrolyzes sphingomyelin (SM) into pro-inflammatory and pro-apoptotic ceramide. Activation of ASM plays an important role in signal transduction in response to various stimuli including IL-1β (27,28) and TNF-α (29). Endothelial cells represent a major source of ASM (30–33). Inhibition of ASM exhibits protective effect in diabetes preventing diabetes-induced retinal inflammation and vascular degeneration (15,33,34).
Previously, we have identified key defects in circadian regulation of CACs. We showed that bone marrow denervation results in loss of circadian release of vascular reparative cells from the bone marrow and generation of increased numbers of proinflammatory cells. Using a rat model of T2D, we showed that the decrease in CACs release from diabetic bone marrow is caused by bone marrow neuropathy and that these changes precede the development of diabetic retinopathy. We observed a marked reduction in clock gene expression in the retina and in CACs. Denervation of the bone marrow resulted in progenitors being “trapped” within the bone marrow and in loss of the circadian release of these cells into the circulation. This reduction in the circadian peak of CAC release into the circulation led to diminished reparative capacity and resulted in development of acellular retinal capillaries (7). We also showed that Per2 mutant mice recapitulate key aspects of diabetes without the associated metabolic abnormalities. In Per2 mutant mice, we observed a threefold decrease in proliferation and 50% reduction in nitric oxide levels in CACs. Tyrosine hydroxylase-positive nerve processes and neurofilament-200 staining were reduced in Per2 mutant mice (suggestive of diabetic neuropathy) and increased acellular capillaries were identified (35). We also showed that as CD34+CACs acquired differentiation markers (towards the endothelial lineage), robust oscillations of clock genes are observed (36).
It is well accepted in diabetic complications field that cells isolated from diabetic tissue keep diabetic phenotype for several passages even when cultured in normal glucose. This is due to “metabolic memory”, or “legacy effect” for vascular disease in diabetes - the prolonged benefits of good glycemic control, as well as the prolonged harm poor control in diabetic patients(11,37–39). In this study we used HREC cells isolated from control and diabetic donor tissue as a model.
In the current study, we have focused exclusively on human CACs. We asked if the defect in circadian release observed in rodents with diabetes occurred in humans. We examined the effect of diabetes-induced ASM activity on the function of human CACs and retinal endothelial cells comparing the angiogenic ability of control (low ASM) and diabetic (high ASM) HRECs to form tube—like structures in vitro and determining the capacity of control (with low ASM) and diabetic (with high ASM) CACs to support endothelial tube formation.
2. Methods
Circadian study of human CD34+ CACs
The study was approved by the University of Florida IRB #411-2010. All study subjects provided informed consent. Individuals were brought into the Clinical Research Center at the University of Florida for 48 h During the first 24 h, individuals were evaluated and on the evening of the first day, a heparin lock was inserted into their forearm. During the second 24-h period, the individuals had 1 mL of blood removed every 2 h for a total of 24 h and analyzed for the number of CD34+ cells by flow cytometry. Clinical characteristics of the patients are presented in Table 1.
Table. 1.
Clinical characteristics of control and diabetic subjects involved in the circadian study.
| Subject # | Gender | Age | Diabetes duration | HbA1C | CVD | Retinopathy | Medications |
|---|---|---|---|---|---|---|---|
| Diabetic 1 | Female | 67 | T2D 4 months | 7.5 | No | No | Glucophage |
| Diabetic 2 | Female | 65 | T2D 20 years | 8.8 | No | No | Lantus |
| Diabetic 3 | Male | 76 | T2D | 7.4 | No | No | Glucophage |
| Diabetic 4 | Female | 64 | T2D | 6.3 | No | No | Actos |
| Diabetic 5 | Female | 67 | T2D | 6.4 | No | No | Glucophage |
| Diabetic 6 | Female | 68 | T2D 9 years | 6.8 | No | No | Diabeta |
| Diabetic 7 | Female | 48 | T2D years | 6.0 | No | No | Metformin |
| Diabetic 8 | Female | 59 | T2D 10 years | 7.7 | No | No | Humulin |
| Control 1 | Female | 43 | No | No | |||
| Control 2 | Female | 57 | No | No | |||
| Control 3 | Female | 68 | No | No | |||
| Control 4 | Male | 43 | No | No |
Inclusion criteria: Individuals between the ages of 21 and 65 years were eligible to participate.
Exclusion criteria: Subjects were excluded for the following reasons: a) evidence of ongoing acute or chronic infection (HIV, Hepatitis B or C, or tuberculosis); b) ongoing malignancy; c) cerebral vascular accident or cerebral vascular procedure; d) current pregnancy; e) history of organ transplantation; f) presence of a graft; g) uremic symptoms, such as an estimated glomerular filtration rate of less than 20 cc/min (by Modification of Diet in Renal Disease equation), or an albumin of less than 3.6 (to avoid malnutrition as a confounding variable); h) a history of smoking; and i) anemia.
Postmortem imaging of human retina and cell culture
Primary cultures of HRECs were prepared from postmortem tissue obtained from National Disease Research Interchange, Philadelphia, PA and Midwest Eye-Banks, Ann Arbor, MI. The tissue was received within 36 h after death. The donor characteristics are provided in Table 2. Primary HRECs were isolated as previously described (40). As previously demonstrated the cells isolated from control and diabetic donors keep their phenotypes for 4–6 passages due to metabolic memory phenomenon (11, 37–39). On arriving in the laboratory, the eyes were placed on sterile gauze, and they were washed with povidone- iodine solution (Purdue Pharma L.P. Stamford, CT). After 10 minutes, the globe was punctured approximately 2 mm from the limbus, a circumferential incision was made and the anterior chamber was removed. A vitreous spatula was used to loosen the vitreous adherent to the anterior retina. When all the vitreous was removed, the retina was gently removed from the layer of retinal pigment epithelium and cut at the optic nerve. Before proceeding into the isolation of HRECs, retinas were rinsed, flatmounted and retinal imaging was taken using a Nikon SMZ-800 Stereo Microscope with Prior Proscan 3 Motorized XY System with Z Drive and MetaMorph Modules to perform image stitching, to properly determine the stage of retinopathy of the donors used for isolation of HRECs. Retinas included in this study have at least three signs of non-proliferative diabetic retinopathy (NPDR) such as microaneurysms, intraretinal haemorrhages and intraretinal microvascular abnormalities (IRMA). Retina was then placed on a 53-μm Nylon mesh filter (Sefar America, Buffalo, NY.), washed with a solution containing Glucose (5.5 mM), L-Glutamine (2 mM) and 1x MEM Non-Essential Amino Acids Solution (GIBCO, Life Technologies, Carlsbad, CA). The retinas were then placed into a 25-ml flask containing 100 U/ml of collagenase, Type 1 (Worthington Biochemical Co., Lakewood, NJ) in the above-mentioned solution containing 22% Bovine serum albumin (Sigma) and 0.01% Soybean Trypsin Inhibitor (Sigma). Retinas were then mechanically agitated using a shaker and allowed to digest at 37°C for approximately 60 minutes or until no tissue fragments could be seen. After digestion, cells were centrifuged at 1000 rpm for 5 minutes. The supernatant was removed and pellet was suspended in fresh media; 1:1 mix of low glucose Dulbecco’s modified Eagle’s medium (DMEM 1g/L)/F-12 nutrient mix (GIBCO, Life Technologies, Carlsbad, CA) supplemented with 10 % fetal calf serum (Hyclone, Logan, UT), 1% endothelial cell growth supplement (Millipore, MA), 1% insulin-transferrin- sodium selenite media supplement (Sigma) and 1% penicillin-streptomycin antimycotic (GIBCO, Invitrogen-Life Technologies, Carlsbad, CA). Glucose concentration in the final media was adjusted to 5.5 mM. The cells were maintained at 37°C in 95% air and 5% CO2 in a humidified cell culture incubator. Passages 3 to 5 were used in the experiments.
Table. 2.
Clinical characteristics of control and diabetic HRECs donors and CD34+ CACs subjects involved in the study.
| HRECs donor # | Gender | Age | Diabetes duration | CVD | Retinopathy | Nephropathy | Neuropathy | Medications |
|---|---|---|---|---|---|---|---|---|
| Diabetic 1 | Male | 71 | T2D 18 years | Yes | Moderate NPDR | Yes | No | Insulin |
| Diabetic 2 | Male | 66 | T2D 16 years | Yes | Sever NPDR | Yes | No | Insulin Humalog and Humalin-N |
| Diabetic 3 | Female | 50 | T2D 15 years | No | Sever NPDR | No | No | Insulin |
| Diabetic 4 | Male | 73 | T2D 6 years | Yes | Mild NPDR | Yes | Yes | Insulin |
| Diabetic 5 | Male | 71 | T2D 20 years | Yes | Moderate NPDR | Yes | No | Insulin |
| Diabetic 6 | Male | 62 | T2D 22 years | Yes | Mild NPDR | Yes | No | Insulin |
| Diabetic 7 | Male | 70 | T2D 15 years | Yes | Moderate NPDR | Yes | No | Insulin |
| Control 1 | Female | 58 | No | No | No | No | No | |
| Control 2 | Male | 56 | No | Yes | No | No | No | |
| Control 3 | Male | 71 | No | No | No | No | No | |
| Control 4 | Female | 52 | No | No | No | No | No |
| CD34+ CACs subject # | Gender | Age | Diabetes duration | HbA1C | CVD | Retinopathy | Nephropathy | Neuropathy |
|---|---|---|---|---|---|---|---|---|
| Diabetic 1 | Male | 55 | T2D 10 years | 14 | No | Sever NPDR | No | No |
| Diabetic 2 | Female | 58 | T2D | 6.5 | No | Moderate NPDR | No | No |
| Diabetic 3 | Female | 59 | T2D | 7.1 | No | Moderate NPDR | No | No |
| Diabetic 4 | Male | 41 | T2D | 14 | No | PDR | No | No |
| Control 1 | Female | 57 | No | No | No | No | No | |
| Control 2 | Male | 49 | No | No | No | No | No | |
| Control 3 | Female | 39 | No | No | No | No | No | |
| Control 4 | Female | 58 | No | No | No | No | No |
Human CD34+ CACs isolation
Human peripheral blood samples (150 ml) were collected into Sodium Citrate-containing CPTTM glass vacuum tubes (BD, Franklin Lakes, NJ). Written informed consent was obtained from each patient, and all procedures were approved by the Institutional Review Board at the University of Florida (IRB # 408-2010). Peripheral blood mononuclear cells (MNCs) were isolated from the blood by density gradient centrifugation using Lympholyte (Cedarlane Laboratories Ltd., Ontario, Canada). The CD34+ cell fraction was then isolated from the MNCs using the EasySepTM CD34+ positive selection system according to the manufacturer’s instructions (Stem Cell Technologies, Vancouver, BC, Canada). Clinical characteristics of the patients are presented in Table 2.
Tube formation assay
Tube formation assay was performed using BD BioCoat Angiogenesis System-Endothelial Cells Tube Formation Matrigel Matrix 96-well plate (BD Biosciences Discovery Labware, Bedford, MA) according to the manufacturer’s instructions. Briefly, isolated CD34+ CACs and HRECs were labeled with Qtracker 655 and Qtracker 525 (Invitrogen); respectively. Control or diabetic HRECs were mixed in a 4:1 ratio with either control or diabetic CD34+ cells, seeded into Matrigel Matrix 96-well plate) and incubated for 16 to 18 h at 37°C (5% CO2). After incubation, wells were assessed for the presence of tube-like structures and images were taken in 10× magnifications using a Nikon TE2000 fluorescence microscope equipped with Photometrics CoolSNAP HQ2 camera. At least three different fields were randomly selected and captured to collect images for each well. Tube area and percentage of CD34+ incorporated into tubules were calculated using MetaMorph software system (Molecular Devices, Downingtown, PA). Statistics were performed on 3 independent wells per condition with minimum three images taken from each well.
Quantitative real-time PCR
Total RNA was extracted from HRECs, human CD34+ cells using QuickGene RNA (Fujifilm, Minato-Ku, Tokyo, Japan) or Qiagen RNeasy (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s instructions. NanoDrop 2000 (Thermo Scientific, IL, USA) was used to determine total RNA concentration. Total RNA was reverse transcribed into cDNA using superscript III first-strand synthesis system (Invitrogen, Carlsbad, CA). Human gene-specific primers for ASM were used. Expression levels were normalized to human cyclophilin. Sequence of specific primers used is given below:
Human ASM: caacctcgcgctgaagaa and tccaccatgtcatcctcaaa
Human Cyclophilin: aaggtcccaaagacagcaga and cttgccaccagtgccattat
ELISA assay
Blood samples were collected, centrifuged and plasma was stored at −80 °C. Samples were assayed for human ASM concentration using ELISA kit (Cloud–Clone Corp., Houston, TX, USA) according to the manufacturer’s protocol.
Statistical analyses
Data are presented as mean ± S.E.M. Results were analyzed for statistical significance by the Student’s t-test (GraphPad Prism 7, GraphPad Software, San Diego, CA). Significance was established at P < 0.05.
3. Results
ASM expression level is increased in HREC, CD34+ CACs and blood plasma of diabetic donors
To determine whether human diabetic tissues exhibited the same increase in ASM as we observed in animal models (15,33), we measured ASM expression level in human RECs, CD34+ CACs and plasma samples in both diabetic and control donors. ASM expression level was significantly increased in all three tissue types in diabetic compared to control donors (Fig. 1A, B and C).
FIG. 1. Diabetes induced increase in ASM expression.
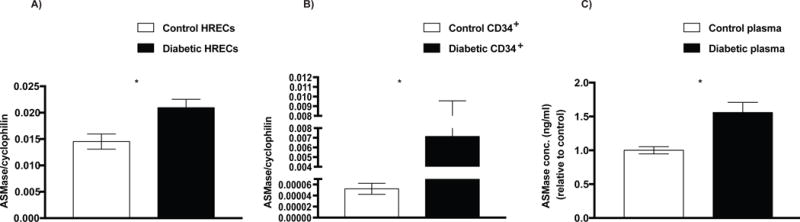
Total RNA was isolated and the transcript level of ASM was analyzed by qRT-PCR in (A) HRECs, (B) CD34+ cells and (C) plasma from diabetic donors (n=4–7) compared with control donors (n=3−4). Data are means ± SEM. *P < 0.05, significantly different as determined by Student’s t-test. Abbreviations: ASM, acid sphingomyelinase; HRECs, human retinal endothelial cells.
Diabetes induces decrease in HREC tube formation
As shown above and previously demonstrated, HRECs isolated from diabetic donors have high ASM activity and expression level. To evaluate the effect of diabetes-induced increase in ASM on HRECs function, we performed tube formation assay to measure the ability of retinal endothelial cells to form blood-vessel-like tubular structure. Tube formation by HRECs isolated from healthy control retinas was compared to cells isolated from retinas with signs of NPDR as determined by post-mortem retinal imaging (Fig. 2A). A significant decrease in tube area was observed in HRECs from retinas with signs of NPDR as compared to control HRECs (Fig. 2B).
FIG. 2. Diabetes impairs tube formation capacity of HRECs.
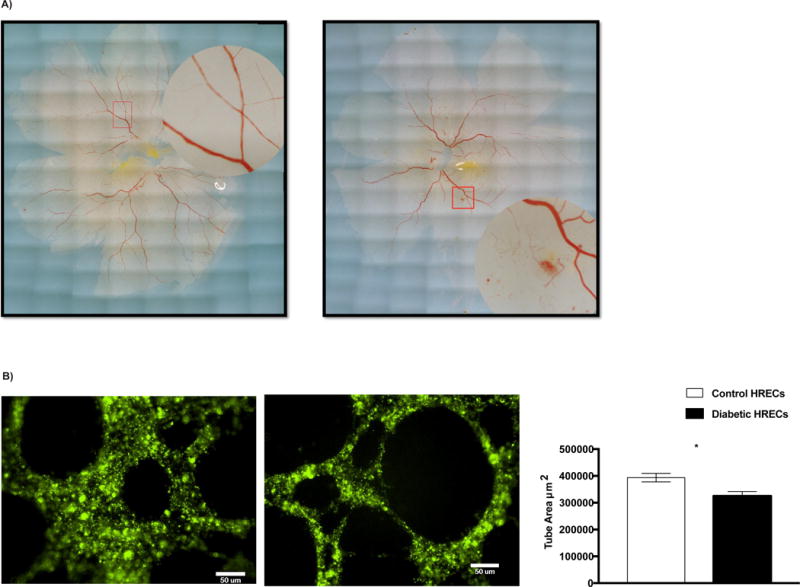
(A) Postmortem imaging of human retina. Control retina with well-organized blood vessels (left), Diabetic retina with signs of NPDR; intraretinal hemorrhages and microaneurysms (right). (B) An in vitro tube formation assay was performed in control (n=4) and diabetic (n=7) HRECs using Matrigel Matrix 96-well plate. Representative images of tube-like structures are shown. The cells were stained with Qtracker 525 (green), images were taken in 10× magnification and total tube areas were calculated using MetaMorph software system. Quantification of tube area is shown on far right. Data are means ± SEM. *P < 0.05, significantly different as determined by Student’s t -test. Scale bar = 50 μm.
Diabetes induced increase in ASM is associated with CD34+ CACs dysfunction
To determine the role of ASM expression in diabetes-induced defect in CD34+ CACs function, we seeded CD34+ CACs isolated from both control (low ASM) and diabetic (high ASM) subjects with HRECs and examined whether the level of ASM expression in CACs affects their ability to incorporate into the endothelial tubes formed by the HRECs. Interestingly, CD34+ CACs seeded alone did not form tube-like structures, but they did incorporate into tubes formed by HRECs when co-cultured with retinal endothelial cells (Fig. 3C). Increased incorporation into tubes formed by diabetic HRECs was observed for the control CD34+ CACs (low ASM) compared to diabetic CACs (high ASM) (Fig. 3B). As expected, control HRECs exhibited robust tube formation. Incorporation of CACs into control HREC tubes was not affected by the levels of ASM in CAC (Fig. 3A). These data demonstrate that high ASM expression levels in CD34+ CACs correlate with impaired incorporation ability, while CACs expressing lower levels of ASM display enhanced in vitro incorporation.
FIG. 3. Reduced incorporation of diabetic CD34+ CACs into diabetic HRECs tubes.
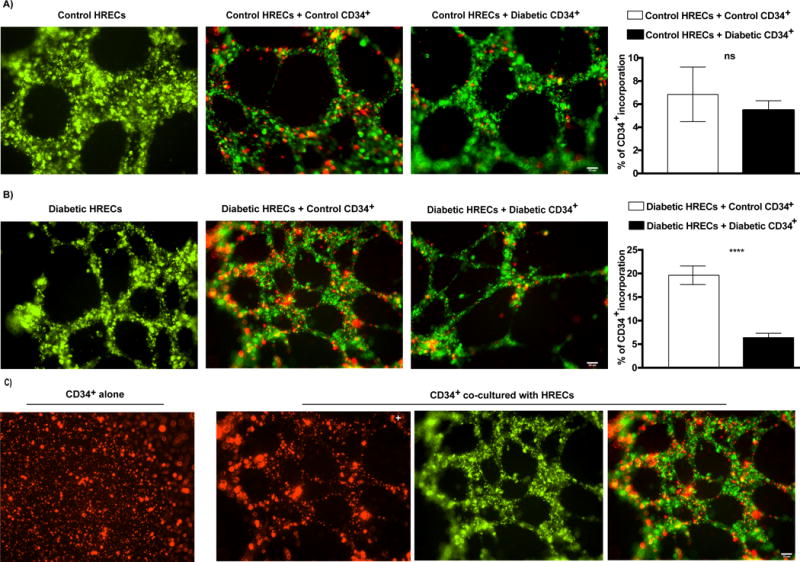
Tube formation by HRECs (Qtracker 525, green) isolated from control (A) or diabetic (B) donors either without CACs (left panel), or co-incubated with control (middle panel) or diabetic (right panel) CACs (Qtracker 655, red) is shown. Quantification of % of CD34+ CACs incorporation into HRECs tubes is shown on far right. Data are means ± SEM (n= 4−7). *** P < 0.0001, significantly different from control as determined by Student’s t -test; not significant at P > 0.05. Scale bar = 50 μm. (C) CD34+ CACs alone were not able to form tube-like structures (left panel), but incorporated into HREC tubes, forming tube-like structures, when co-cultured with HRECs (right three panels). Abbreviations: CACs, circulating angiogenic cells.
Diabetes induced increase in ASM is associated with loss of circadian release of CD34+ CACs
We have previously demonstrated that normal diurnal pattern of CACs release from the bone marrow is critical for efficient repair of retinal vasculature in rodents (7). Increase in ASM activity in CACs in diabetic animal models was associated with decreased membrane fluidity and impaired migration leading to increased CAC retention and loss of circadian release from the bone marrow (34). We next determined the effect of diabetes on circadian release of CD34+ CACs in diabetic patients. Peripheral blood of type 2 diabetic individuals was collected every 2 h for 24 h and analyzed for the number of CD34+ CACs by flow cytometry and compared with control subjects. The dash line is the model fitted curve for individual subjects and bold curve is the fitted curve for population (Fig. 4 A and B). In agreement with previous studies, healthy individuals had a peak of circulating CD34+ cells in the middle of the night, representing the rest phase for humans (Fig. 4A), however this peak of CD34+ release was lost in T2D subjects (Fig. 4B).
FIG. 4. Loss of circadian release of diabetic CD34+ CACs.
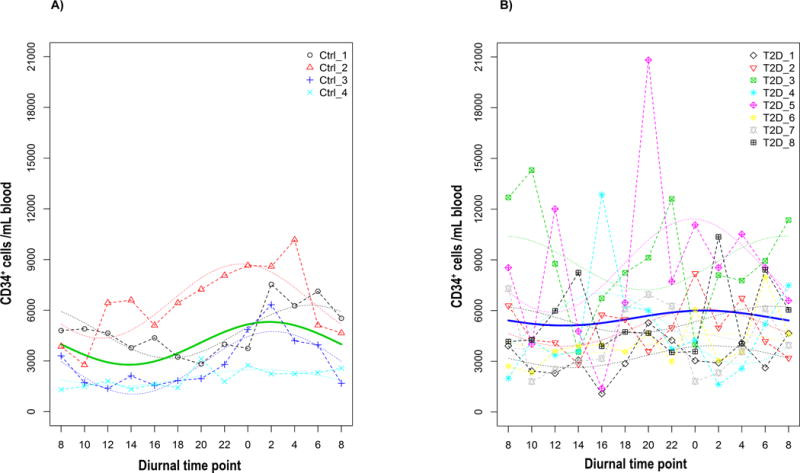
Peripheral blood was collected every 2 h for a total of 24 h from (A) control (n=4) and (B) diabetic (n=8) subjects and was analyzed for the number of CD34+ CACs by flow cytomtery. (A) In control subjects, there is a clear peak of circulating CD34+ CACs that occurred in the middle of the night. (B) Rhythmic CD34+ CACs release pattern was blunted in Type 2 diabetic patients. The dash line is the model fitted curve for individual patients and bold curve is the fitted curve for population.
4. Discussion
Diabetic retinopathy is a sight threatening complication of diabetes with limited treatment strategies. Understanding the key molecular factors involved in the disease is important for developing therapeutic targets to prevent progression into ocular neovascularization and blindness. ASM is shown to be a key element in inflammatory signaling through ceramide-mediated signal transduction (41,42). Diabetes-induced increase in ASM activity has been shown to modulate inflammatory response in mature retinal endothelial cells (43), however, there is no direct experimental evidence showing ASM effect on endothelial function. Here we demonstrated that HRECs isolated from type 2 diabetic subjects with signs of NPDR had altered retinal endothelial cell function with impaired capacity to form tube-like structures when compared with control HRECs. The deficiency in tube formation was associated with increased expression of ASM in diabetic HRECs. This is consistent with previous studies showing ASM–mediated endothelial cell apoptosis in various tissues including retina, lungs and gastrointestinal tract (30,31,33,44), and ASM antiangiogenic effect in tumor treatment (45). Endothelium is the major source of ASM production in the body with endothelial cells synthesizing 20 times as much ASM as any other cell type (32); thus, it is not surprising that ASM plays a major role in endothelial cells function. In agreement with previous studies (32), endothelial cells had very high ASM expression level, which was further increased in diabetes. Moreover, increased ASM level in plasma further reflects the increased production and secretion of ASM by activated endothelial cells.
Retinal vascular repair and revascularization is aided by CACs (4,13,16,46–49). We examined the effect of diabetes-induced increase in ASM on the ability of CD34+ CACs to incorporate and thus help in repair of defective vascular-like tube structures formed by diabetic HRECs. We demonstrated that diabetic CD34+ CACs, with high ASM, showed minimal incorporation into the defective tubes formed by diabetic HRECs. Non-diabetic CD34+ CACs with low ASM showed robust incorporation. These results are in line with other studies showing that diabetic CACs are defective in proliferation, migration, adhesion, differentiation, and participation in vascular regeneration process (13,14,16,46,50,51). Interestingly, no significant difference was observed between non-diabetic and diabetic CD34+ CACs incorporation into control HRECs. This is consistent with our previous studies demonstrating lack of incorporation of control CACs into healthy vasculature that does not require repair in non-diabetic (13). We have previously demonstrated that high level of ASM inversely correlates with migration and reparative capacity of CACs, with the inhibition of diabetes-induced ASM resulting in improved migration and retinal vascular regeneration in diabetic mouse model (15,34).
In this study, we compared effect of ASM on functional capacity of healthy and diabetic CACs in human on control and diabetic HRECs. We revealed that the defective incorporation of diabetic CD34+ CACs was associated with high ASM. As previously shown, high ASM activity leads to accumulation of membrane ceramides. Accumulation of ceramide results in decreased membrane fluidity, cell rigidity and defective migration which can explain defective incorporation of diabetic CACs (15,34,52) and likely supports their defective release from the bone marrow into the systemic circulation.
Although several lines of evidence demonstrate that ASM expression is an important factor for progenitor cell release from the marrow, migration, proliferation and homing to the injured vasculature; other factors beyond ASM activation are also known to be involved in diabetes-induced CAC dysfunction. These include bone marrow neuropathy and low level of neurotransmitters production; increase in TGF beta leading to decreased NO bioavailability and diminished expression of CXCR4 (7,53). These factors affect chemoattraction to SDF1 and lead to increase in stem cell quiescence in the bone marrow niche. In health ASM works in concert with other factors to maintain optimal bone marrow stem cell production and release, however activation of ASM along with other factors disrupts this balance in diabetes (7,34,54,55).
The physiological release of bone marrow progenitor cells including CD34+ CACs into the circulation is not constant, but follows circadian oscillations. These oscillations are regulated by sympathetic signaling to bone marrow stromal cells, which results in CXCL12 (SDF-1) down regulation and progenitor egress from the bone marrow; all of these processes occur in circadian pattern under control of clock genes (56). Diabetic bone marrow neuropathy with disruption of circadian rhythm may contribute to endothelial progenitor cell dysfunction. Wang et al. have demonstrated that mutation in circadian gene Per2 leads to reduced endothelial cell progenitor mobilization and revascularization (57). We investigated whether circadian release of CD34+ CACs is altered in diabetic subjects. Peripheral blood was collected every 2 h for 24 h from both control and diabetic subjects. In agreement with previous studies (58–60), normal rhythmic oscillations were observed in control subjects with a clear peak of circulating CD34+ CACs cells in the middle of the night (resting phase), optimal time of regeneration in humans. Similar to our observations in diabetic rat model (7). This study revealed that circadian fluctuation of CD34+ CACs was disrupted with rhythmicity lost in diabetic individuals. Importantly, we have previously demonstrated that, similar to endothelial cells, the increase in ASM activity in CACs in diabetic animal models was associated with increased ceramide levels, decreased membrane fluidity and impaired migration. This impairment in migration, combined with diminished sympathetic signaling to BM may lead to increased CAC retention in the bone marrow and loss of circadian release of the progenitor cells as demonstrated in this study.
5. Conclusion
This study underscores the deleterious effect of high ASM levels on the vascular repair potential of both mature retinal endothelial cells and circulating angiogenic cells in diabetes. Correcting this defect could treat vasodegeneration, enhancing vessel repair and thus preventing progression into PDR stage.
Highlights.
Increase of acid sphingomyelinase (ASM) in diabetes damages retinal vasculature.
CD34+CACs from diabetic patients with high ASM have dysfunctional vascular repair.
Diabetic individuals showed disrupted diurnal release pattern of CD34+CACs.
Acknowledgments
Research was supported by MEAS Grant MICL02163 to JVB, NIH grants: RO1EY016077 to JVB, R01EY025383 to JVB and MBG, EY012601-15, EY007739-25 to MBG, and Juvenile Diabetes Research Foundation (JDRF) Fellowship JDRF 3-PDF-2014-108-A-N to Q.W. We acknowledge National Research Disease Interchange and Michigan Eye-Bank for providing human retina tissue.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Nermin Kady: designed the experiments, performed the co-culture study and wrote the manuscript. Yuanqing Yan: performed the diurnal CD34+ CAC release study, wrote and edited the manuscript, Tatiana Salazar: performed human CD34+ CAC part of the study, edited the manuscript. Qi Wang, Harshini Chakravarthy, Chao Huang, Eleni Beli and Svetlana Navitskaya: performed HREC isolation and culture, and CD34+ CAC isolation and co-culture experiments, edited the manuscript. Maria B. Grant: contributed to conception, design, financial and administrative support, maintained IRB approval for research involving human subjects, edited the manuscript. Julia V. Busik: designed the study, provided financial and administrative support, performed pilot experiments, and reviewed and edited the manuscript. All authors have approved the final article.
Disclosure of Interest:
The authors indicate that ‘Conflicts of interest: none’.
References
- 1.Hammes HP, Feng Y, Pfister F, Brownlee M. Diabetic retinopathy: Targeting vasoregression. Diabetes. 2011;60(1):9–16. doi: 10.2337/db10-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44(7):791–804. doi: 10.1007/s001250100544. [DOI] [PubMed] [Google Scholar]
- 3.Wong K. Defining diabetic retinopathy severity. Diabet Retin Evidence-Based Manag. 2010;10:105–20. [Google Scholar]
- 4.Grant MB, May WS, Caballero S, Brown GaJ, Guthrie SM, Mames RN, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8(6):607–12. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 5.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, et al. Neovascular ization of ischemic myocardium by human bone - marrow — derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. NAt Med. 7(4):430–6. doi: 10.1038/86498. 200. [DOI] [PubMed] [Google Scholar]
- 6.Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A. 2000;97(7):3422–7. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Busik JV, Tikhonenko M, Bhatwadekar A, Opreanu M, Yakubova N, Caballero S, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206(13):2897–906. doi: 10.1084/jem.20090889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusuyama T, Omura T, Nishiya D, Enomoto S, Matsumoto R, Takeuchi K, et al. Effects of treatment for diabetes mellitus on circulating vascular progenitor cells. J Pharmacol Sci. 2006;102:96–102. doi: 10.1254/jphs.fp0060256. [DOI] [PubMed] [Google Scholar]
- 9.Fadini GP, Pucci L, Vanacore R, Baesso I, Penno G, Balbarini A, et al. Glucose tolerance is negatively associated with circulating progenitor cell levels. Diabetologia. 2007;50(10):2156–63. doi: 10.1007/s00125-007-0732-y. [DOI] [PubMed] [Google Scholar]
- 10.Hazra S, Jarajapu YPR, Stepps V, Caballero S, Thinschmidt JS, Sautina L, et al. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia. 2013;56(3):644–53. doi: 10.1007/s00125-012-2781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53(1):195–9. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 12.Loomans C, van Haperen R, Duijs JM, Verseyden C, de Crom R, et al. Diffrentaion of bone marrow-derived endothelial progenitor cells is shifted into a proinflammatory phenotype by hyperglycemia. Mol Med. 2009;15(5–6):152–9. doi: 10.2119/molmed.2009.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caballero S, Sengupta N, Afzal A, Chang KH, Li Calzi S, Guberski DL, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56(4):960–7. doi: 10.2337/db06-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segal MS, Shah R, Afzal A, Perrault CM, Chang K, et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelail progenitor cell migratory defect associated with diabtes. Diabetes. 2006;55(1):102–9. [PubMed] [Google Scholar]
- 15.Tikhonenko M, Lydic TA, Opreanu M, Li Calzi S, Bozack S, McSorley KM, et al. N-3 Polyunsaturated Fatty Acids Prevent Diabetic Retinopathy by Inhibition of Retinal Vascular Damage and Enhanced Endothelial Progenitor Cell Reparative Function. PLoS One. 2013;8(1):1–10. doi: 10.1371/journal.pone.0055177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamarat R, Silvestre J-S, Le Ricousse-Roussanne S, Barateau V, Lecomte-Raclet L, Clergue M, et al. Impairment in ischemia-induced neovascularization in diabetes: bone marrow mononuclear cell dysfunction and therapeutic potential of placenta growth factor treatment. Am J Pathol. 2004;164(2):457–66. doi: 10.1016/S0002-9440(10)63136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowluru RA. Role of interleukin-1β in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88(10):1343–7. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kowluru RA, Odenbach S. Role of interleukin-1β in the development of retinopathy in rats: Effect of antioxidants. Investig Ophthalmol Vis Sci. 2004;45(11):4161–6. doi: 10.1167/iovs.04-0633. [DOI] [PubMed] [Google Scholar]
- 20.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alph suppression. FASEB J. 2002;16(13):438–40. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 21.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009 Aug;15(2007):1418–28. [PMC free article] [PubMed] [Google Scholar]
- 22.Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye (Lond) 2006;20(12):1366–9. doi: 10.1038/sj.eye.6702138. [DOI] [PubMed] [Google Scholar]
- 23.Joussen AM, Poulaki V, Qin W, Kirchhof B, Mitsiades N, Wiegand SJ, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160(2):501–9. doi: 10.1016/S0002-9440(10)64869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adamiec-Mroczek J, Oficjalska-Młyńczak J. Assessment of selected adhesion molecule and proinflammatory cytokine levels in the vitreous body of patients with type 2 diabetes—role of the inflammatory-immune process in the pathogenesis of proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2008;246(12):1665–70. doi: 10.1007/s00417-008-0868-6. [DOI] [PubMed] [Google Scholar]
- 25.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96(19):10836–41. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semeraro F, Cancarini A, Dell’Omo R, Rezzola S, Romano MR, Costagliola C. Diabetic retinopathy: Vascular and inflammatory disease. J Diabetes Res. 2015;2015:1–16. doi: 10.1155/2015/582060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu P, Anderson RG. Compartmentalized production of ceramide at the cell surface. J Biol Chem. 1995;270(45):27179–85. doi: 10.1074/jbc.270.45.27179. [DOI] [PubMed] [Google Scholar]
- 28.Mathias S, Younes A, Kan C, Orlow I, Joseph C, Kolesnick RN, et al. Activation of the Sphingomyelin Signaling Pathway in Intact EL4 Cells and in a Cell-Free System by IL-1β. Science. 1993;259(5094):519–22. doi: 10.1126/science.8424175. [DOI] [PubMed] [Google Scholar]
- 29.Schutze S, Potthoff K, Machleidt T. TNF Activates Phospholipase Sphingomyelin NF-KB by Phosphatidylcholine-Specific C-Induced “Acidic” Breakdown. Cell. 1992;71(5):765–76. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- 30.Santana P, Peña LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, et al. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell. 1996;86(2):189–99. doi: 10.1016/s0092-8674(00)80091-4. [DOI] [PubMed] [Google Scholar]
- 31.Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, Garzotto M, et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186(11):1831–41. doi: 10.1084/jem.186.11.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, et al. Human Vascular Endothelial Cells Are a Rich and Regulatable Source of Secretory Sphingomyelinase. J Biol Chem. 1998;273(7):4081–8. doi: 10.1074/jbc.273.7.4081. [DOI] [PubMed] [Google Scholar]
- 33.Opreanu M, Tikhonenko M, Bozack S, Lydic TA, Reid GE, McSorley KM, et al. The unconventional role of acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes. 2011;60(9):2370–8. doi: 10.2337/db10-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakravarthy H, Navitskaya S, O’Reilly S, Gallimore J, Mize H, et al. Role of Acid Sphingomyelinase in Shifting the Balance Between Proinflammatory and Reparative Bone Marrow Cells in Diabetic Retinopathy. Stem cells. 2016;34(4):972–83. doi: 10.1002/stem.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhatwadekar AD, Yan Y, Qi X, Thinschmidt JS, Neu MB, Li Calzi S, et al. Per2 mutation recapitulates the vascular phenotype of diabetes in the retina and bone marrow. Diabetes. 2013;62(1):273–82. doi: 10.2337/db12-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhatwadekar AD, Yan Y, Stepps V, Hazra S, Korah M, Bartelmez S, et al. MiR-92a corrects CD34+ cell dysfunction in diabetes by modulating core circadian genes involved in progenitor differentiation. Diabetes. 2015;64(12):4226–37. doi: 10.2337/db15-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy S, Sala R, Cagliero E, Lorenzi M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci U S A. 1990;87(1):404–8. doi: 10.1073/pnas.87.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grant MB, Caballero S, Tarnuzzer RW, Bass KE, Ljubimov AV, Spoerri PE, et al. Matrix metalloproteinase expression in human retinal microvascular cells. Diabetes. 1998;47(8):1311–7. doi: 10.2337/diab.47.8.1311. [DOI] [PubMed] [Google Scholar]
- 39.Kowluru RA, Santos JM, Mishra M. Epigenetic modifications and diabetic retinopathy. Biomed Res Int. 2013;2013:635284. doi: 10.1155/2013/635284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busik JV, Mohr S, Grant MB. Hyperglycemia-Induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57(7):1952–65. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gulbins E, Grassmé H. Ceramide and cell death receptor clustering. Biochim Biophys Acta. 2002;1585(2–3):139–45. doi: 10.1016/s1388-1981(02)00334-7. [DOI] [PubMed] [Google Scholar]
- 42.Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. 2004;82(1):27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- 43.Opreanu M, Lydic TA, Reid GE, McSorley KM, Esselman WJ, Busik JV. Inhibition of cytokine signaling in human retinal endothelial cells through downregulation of sphingomyelinases by docosahexaenoic acid. Invest Ophthalmol Vis Sci. 2010;51(6):3253–63. doi: 10.1167/iovs.09-4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Haimovitz-friedman A, et al. Endothelial apoptosis as the the Primary Lesion Initiating Intestinal Radiation Damage in Mice. Science. 2001;293(5528):293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-barros AM, Paris F, Cordon-cardo C, Lyden D. Tumor Response to Radiotherapy Regulated by Endothelial Cell. Apoptosis Science. 2003;300(5622):1155–9. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 46.Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest. 2000;106(4):571–8. doi: 10.1172/JCI9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85(3):221–8. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 48.Asahara T, Murohara T, Sullivan A, Silver M, Van R, Asahara T, et al. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, et al. Ischemia - and cytokine - induced mobilization of bone marrow- derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5(4):434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 50.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106(22):2781–6. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Lin S, Lin F, Wu T, Tsao C. High Glucose Impairs Early and Late Endothelial Progenitor Cells by Modifying Nitric Oxide — Related but Not Oxidative Stress — Mediated Mechanisms. Diabetes. 2007 Jun;56:1559–68. doi: 10.2337/db06-1103. [DOI] [PubMed] [Google Scholar]
- 52.Catapano ER, Arriaga LR, Espinosa G, Monroy F, Langevin D, López-Montero I. Solid character of membrane ceramides: A surface rheology study of their mixtures with sphingomyelin. Biophys J. 2011;101(11):2721–30. doi: 10.1016/j.bpj.2011.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhatwadekar AD, Guerin EP, Jarajapu YPR, Caballero S, Sheridan C, Kent D, et al. Transient inhibition of transforming growth factor-β1 in human diabetic CD34+ cells enhances vascular reparative functions. Diabetes. 2010;59(8):2010–9. doi: 10.2337/db10-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakravarthy H, Beli E, Navitskaya S, O’Reilly S, Wang Q, Kady N, et al. Imbalances in mobilization and activation of pro-inflammatory and vascular reparative bone marrow-derived cells in diabetic retinopathy. PLoS One. 2016;11(1):1–21. doi: 10.1371/journal.pone.0146829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Q, Navitskaya S, Chakravarthy H, Huang C, Kady N, Lydic TA, et al. Dual Anti-Inflammatory and Anti-Angiogenic Action of miR-15a in Diabetic Retinopathy. EBioMedicine. 2016;11:138–50. doi: 10.1016/j.ebiom.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452(7186):442–7. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 57.Wang CY, Wen MS, Wang HW, Hsieh IC, Li Y, et al. Increased vascular senescence and impaired endothelial progenitor cell function mediated by mutation of circadian gene Per2. Circulation. 2008;118(21):2166–73. doi: 10.1161/CIRCULATIONAHA.108.790469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsinkalovsky O, Smaaland R, Rosenlund B, Sothern RB, Hirt A, Steine S, et al. Circadian variations in clock gene expression of human bone marrow CD34+ cells. J Biol Rhythms. 2007;22(2):140–50. doi: 10.1177/0748730406299078. [DOI] [PubMed] [Google Scholar]
- 59.Mendez-Ferrer S, Andrew C, Merad M, Frenett PS. Circadian rhytms influence hematopoietic stem cells. Curr Opin Hematol. 2009;16(4):235–42. doi: 10.1097/MOH.0b013e32832bd0f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abrahamsen JF, Smaaland R, Sothern RB, Laerum OD. Variation in cell yield and proliferative activity of positive selected human CD34+ bone marrow cells along the circadian time scale. Eur J Haematol. 1998;60(1):7–15. doi: 10.1111/j.1600-0609.1998.tb00990.x. [DOI] [PubMed] [Google Scholar]