

Summary
Management of energy stores is critical during endurance exercise, with a shift in substrate utilization from glucose towards fat being a hallmark of trained muscle. Here we show that this key metabolic adaptation is both dependent on muscle PPARδ and stimulated by PPARδ ligand. Furthermore, we find that muscle PPARδ expression positively correlates with endurance performance in BXD mouse reference populations. In addition to stimulating fatty acid metabolism in sedentary mice, PPARδ activation potently suppresses glucose catabolism and does so without affecting either muscle fiber type or mitochondrial content. By preserving systemic glucose levels PPARδ acts to delay the onset of hypoglycemia and extends running time by ~100 minutes in treated mice. Collectively, these results identify a bifurcated PPARδ program that underlies glucose sparing and highlight the potential of PPARδ-targeted exercise mimetics in the treatment of metabolic disease, dystrophies and unavoidably, the enhancement of athletic performance.
eTOC
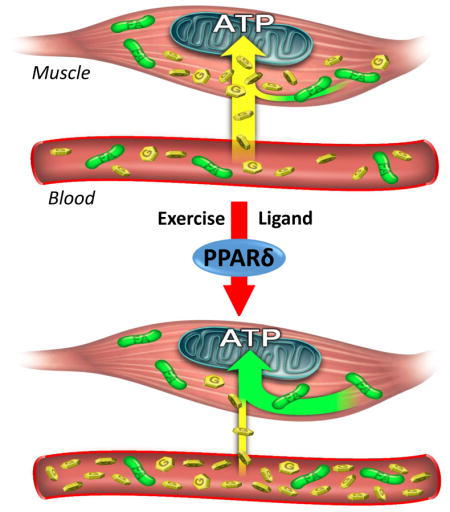
Carbohydrate depletion in endurance sports leads to “hitting the wall” phenomenon, which is mitigated through sports training. XXX et al show that muscle PPARδ actively suppresses glucose catabolism. Glucose sparing by PPARδ delays the onset of hypoglycemia and extends running time by ~100 minutes in agonist-treated mice.
In endurance sports such as marathon running and cycling, carbohydrate depletion, commonly known as “hitting the wall”, is a significant determinant of performance. Exercise training enhances endurance, in part, by delaying the depletion of carbohydrate stores (mainly glycogen in liver and muscle). The adaptive benefits of exercise training are commonly attributed to the glycolytic-to-oxidative fiber-type transformation and increased mitochondrial energetic capacity (Holloszy and Booth, 1976), programs in which the AMPK-PGC1α signaling pathway is now known to play a major role. At the same time, exercise also enhances muscle fatty acid (FA) oxidation (Mole et al., 1971), theoretically providing extra energy substrates for extended performance and reducing the dependence on glucose. This readily observed glucose sparing leads to the assumption that increased FA oxidation extends performance. However, the contribution of altered fat and glucose metabolism to endurance and the molecular mechanism underlying this metabolic shift are simply not known.
To address the above questions, we focused on the peroxisome proliferator-activated receptor delta (PPARδ), a nuclear receptor that serves as a key regulator of FA metabolism in muscle (Fan and Evans, 2014; Luquet et al., 2003; Wang et al., 2004). Muscle-specific over-expression of PPARδ not only induces an oxidative fiber-type transformation but also increases FA oxidation in skeletal muscle through the induction of two mitochondrial gatekeeper proteins, carnitine palmitoyl-transferase 1b (Cpt1b), the rate-limiting enzyme in the transport of FAs into the mitochondria, (Bruce et al., 2009; Kleiner et al., 2009) (Bruce et al., 2009; Kleiner et al., 2009) and pyruvate dehydrogenase kinase isozyme 4 (Pdk4), which negatively regulates the influx of glucose-derived pyruvate into the mitochondrial tricarboxylic acid (TCA) cycle (Luquet et al., 2003; Wang et al., 2004). Consequently, PPARδ transgenic mice run twice as long as the controls and represent a permanent strain of “marathon mice” (Wang et al., 2004). Conversely, muscle-specific PPARδ knockout mice show defects in fiber-type determination and mitochondrial biogenesis (Schuler et al., 2006).
In contrast to transgenesis, small molecule ligands that specifically activate PPARδ, including GW501516 (GW), have revealed multiple beneficial metabolic effects including 1) increased energy expenditure (Wang et al., 2003), 2) elevated FA oxidation (Dressel et al., 2003; Tanaka et al., 2003), 3) reduced obesity and insulin resistance (Tanaka et al., 2003; Wang et al., 2003), 4) exercise-induced muscle remodeling and collectively 5) enhanced running endurance by 80% or more (Narkar et al., 2008).
Despite the above 5 collectanea, the minimal components needed to enhanced running performance have eluded description. Here we show that activation of muscle PPARδ not only increases fat oxidation, but coordinately decreases glucose catabolism to forestall hypoglycemia and facilitate progressively longer running time. Unexpectedly, this substrate prioritization does not depend on either oxidative fiber-type transformation or mitochondrial biogenesis. Mechanistically, we identify a PPARδ-induced genomic signature in muscle as the transcriptional basis of glucose conservation and ultimately enhanced stamina. These findings identify muscle PPARδ as a key regulator of energetic resilience and offer a quantitative molecular approach in the development of new, safe and effective therapeutics for the treatment of metabolic disease and the promotion of metabolic health.
Results
Exercise induces a PPARδ-dependent shift in muscle energy substrate utilization
While endurance exercise shifts muscle energy substrate usage from glucose to FAs (Hamilton and Booth, 2000) the dependence on PPARδ for this shift is not known. Accordingly, we compared the benefits of treadmill training in wild-type (WT) and muscle-specific PPARδ knockout (PDmKO) mice (Fig. S1A). After 4 weeks of daily running, WT mice show a clear shift in energy substrate usage from glucose to FA oxidation, as evidenced by the reduced respiratory exchange ratio (RER) (Fig. 1A–B, S1B–C) and increased palmitate-fueled mitochondrial oxygen consumption rate (OCR) (Fig. 1C). Notably, these metabolic changes are mostly abolished in PDmKO mice (Fig. 1A–C, S1B–C), demonstrating the dependence of exercise-induced metabolic adaptations on PPARδ. Mechanistically, we show that the exercise-induced up-regulation of mitochondrial gatekeeper genes (Fig. 1D) is heavily compromised (Pdk4 induction reduced by ~50 %, Fig. 1E) or completely absent (Cpt1b, Fig. 1F) in PDmKO mice. In terms of running endurance, the increase in performance of PDmKO mice after exercise training was only half that seen in WT controls (run-to-exhaustion treadmill test, Fig. 1G). Consistent with these results, PPARδ expression in skeletal muscle positively correlates with running distance, activity, and muscle mass in the BXD mouse genetic reference population (GRP) (Fig. 1H) (Andreux et al., 2012; Peirce et al., 2004). Additional negative correlations between Cpt1b and Pdk4 expression in muscle and plasma lactate (indicator for muscle glycolysis) and RER, respectively, further implicate PPARδ in the adaptive regulation of energy substrate utilization (Fig. S1D).
Figure 1. Exercise induces a PPARδ-dependent shift in muscle energy substrate utilization.

Experiments were performed in the same set of 4-month-old WT or PDmKO mice with or without 4 weeks of exercise training (n=5). (A) Oxygen consumption rate (OCR, VO2) and respiratory exchange ratio (RER, VCO2/VO2) measured over a 48-hour period. (B) Quantitative analysis of VO2 and RER using area under curve (AUC) of the data in (A). (C) OCR using palmitoyl-carnitine as the substrate in freshly isolated mitochondria from quadriceps muscle. (D) Diagram showing the two major types of fuel sources (glucose and FAs) and the gatekeeper enzymes that control their mitochondrial influx. (E–F) mRNA expression levels of Cpt1b and Pdk4 in soleus. (G) Total running time in a run-to-exhaustion endurance test. (H) Correlations between PPARδ expression in quadriceps and running distance, activity, and muscle mass. Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01).
Despite the requirement for muscle PPARδ in exercise-induced metabolic adaptations and endurance enhancement, the glycolytic-to-oxidative fiber-type switch and mitochondrial biogenesis are still achieved in PDmKO mice (Fig. S1A, S1E–J). Furthermore, sedentary PDmKO mice are indistinguishable from WT mice in terms of mitochondrial content and oxidative phosphorylation (OXPHOS) capacity (Fig. S1A, S1E–F), as well as muscle fiber-type composition (Fig. S1E, S1G–J). In addition, whole-body energy expenditure (measured as oxygen consumption rate (VO2), Fig. 1A–B, S1B–C), energy substrate utilization (RER, Fig. 1A–B, S1B–C), and mitochondrial fatty acid oxidation (Fig. 1C), are all independent of muscle PPARδ expression. This indicates a role for PPARδ in adaptive but not innate muscle activity and stands in contrast to previous reports (Schuler et al., 2006).
Ligand activation of muscle PPARδ prioritizes energy substrate usage to boost endurance
Previous studies showed that treatment of mice with the PPARδ agonist GW dramatically increased running endurance, but only when combined with daily exercise (Narkar et al., 2008). Based on the above, we re-examined the impact of GW on muscle energy substrate usage and endurance in fully sedentary mice. Unexpectedly, treatment of WT mice with GW (40 mg/kg in food) for a longer time (8 weeks compared to 4 weeks) reduced RER to a level similar to exercise training, indicative of increased FA metabolism (Fig. 2A–B, S2A–B). Consistent with this, palmitate-fueled respiration was ~50% higher in muscle mitochondria isolated from GW-treated mice, while succinate-fueled respiration was unchanged (Fig. 2C, S2C). GW treatment also more than doubled the palmitate-induced increase of OCR in cultured C2C12 myotubes (Fig. 2D–E). In addition, GW strongly induced the expression of the mitochondrial gatekeeper genes Pdk4 and Cpt1a/b both in vivo and in vitro (Fig. 2F–H) while serum lactate, an indicator of anaerobic glycolysis, was reduced ~40% in GW-treated mice (Fig. S2D). Notably, GW treatment of PDmKO mice failed to alter RER (Fig. 2A–B, S2A–B), FA oxidation in muscle mitochondria (Fig. 2C), expression of muscle Pdk4 and Cpt1b (Fig. 2F–G), or circulating lactate levels (Fig. S2D), establishing that the above effects are dependent on muscle PPARδ.
Figure 2. Ligand activation of muscle PPARδ induces substrate shift and boosts endurance by preserving glucose.

Mouse experiments were performed in the same set of 4-month-old WT or PDmKO mice with or without 8 weeks of oral GW treatment (n=8). (A) RER measured over a 48-hour period. (B) Quantitative RER analysis using AUC of the data in (A). (C) OCR using palmitoyl-carnitine as the substrate in freshly isolated mitochondria from quadriceps. (D) Change of OCR upon palmitate injection measured in C2C12 myotubes treated with vehicle or GW. (E–F) mRNA expression levels of Pdk4 and Cpt1b in white quadriceps. (G) mRNA expression levels of Pdk4 and Cpt1a/b in C2C12 myotubes. Data normalized to the level of U36 (U36b4). (H) Total running time in the endurance test. (J) Blood glucose (solid lines) and lactate (dotted lines) monitored during the run-to-exhaustion endurance test in mice treated with or without GW. (K) Diagram showing the PPARδ-controlled energy substrate shift induced by its ligands or exercise. Yellow marks glucose and its usage while green marks FAs and their usage. *p < 0.05, **p < 0.01, ***p < 0.001.
Consistent with an energy substrate shift, the longer 8 week GW treatment of sedentary mice was sufficient to confer ~1.5 hrs longer running time than untreated controls (Fig. 2I). This endurance benefit is lost in PDmKO mice and thus dependent on muscle PPARδ activation (Fig. 2I). Unexpectedly, muscular glycogen content (Fig. S2E–F), mitochondrial quantity and OXPHOS activity (Fig. S2C, S2G–I), and muscle fiber-type composition (Fig. S2J–K) – changes commonly associated with endurance enhancement (Holloszy and Booth, 1976) – were not affected by GW treatment. Thus, while establishing that ligand activation of PPARδ can enhance endurance in sedentary mice, these findings implicate a novel mechanism of action.
PPARδ increases running endurance by preserving glucose
The above data suggested a PPARδ-controlled metabolic shift that preserves systemic glucose contributing to a GW-induced endurance enhancement. To determine the magnitude and timing of these changes, we monitored blood glucose in mice treated with or without GW during a run-to-exhaustion test. As expected, both control and GW-treated mice showed time-dependent reductions in blood glucose to <70mg/dL, at which point the mice stopped running and often lost consciousness (Fig. 2J and S2L). By increasing blood glucose to >120mg/dL via IP injection (250mg/kg), exhausted mice were able to resume running for another ~20 minutes, confirming that blood glucose is the major determinant of endurance. Interestingly, while the blood glucose in the control mice started dropping after 90–120 minutes of running, GW-treated mice were able to maintain normal glycemic levels for extended periods and delay the onset of blood glucose reduction even after 180 minutes of running (Fig. 2J). It is important to note that the glucose-sparing effects of GW treatment parallel those seen with exercise training, suggesting a common underlying mechanism (Fig. S2M). Blood lactate was also monitored during our run-to-exhaustion tests, which showed minimal fluctuation in both control and GW-treated mice (Fig. 2J dashed lines, and S2N), indicating that the endurance regimen did not exceed the aerobic threshold of the tested mice. In combination, our data describe a PPARδ-controlled muscle reprogramming that boosts exercise endurance by inversely regulating fat and glucose metabolism, thereby preserving circulating glucose to support other tissues such as the brain (Fig. 2K).
PPARδ orchestrates mitochondrial gene networks
Global transcriptional analyses in the glycolytic white quadriceps muscle (WQ) identified 975 genes with altered expression upon GW treatment, with 492 up- and 483 down-regulated, respectively. In addition to the key mitochondrial genes Pdk4 and Cpt1b described above (Fig. 2F–G), gene ontology (GO) analysis of up-regulated genes revealed significant enrichment in the PPAR signaling pathway as well as lipid and FA catabolism (including Lpl, Lipe, Acadl, Acads, and Acaa2) (Fig. 3A–B and S3A). Interestingly, lipogenic genes including PPARγ (master adipogenic regulator), and fatty acid synthase (Fasn) were also induced (Fig. 3A–B and S3A), which would theoretically lead to a futile cycle of lipid catabolism and anabolism. Additionally, genes involved in antioxidant defense and glutathione synthesis (including Cat, Sod3, and Gpx1) were highly up-regulated (Fig. 3A–B and S3A). Counter-intuitively, pathways in carbohydrate metabolism, including the hexose metabolic process, pentosephosphate shunt, and insulin signaling were also significantly enriched (Fig. 3A–B and S3A). However, the induction of Fbp2, Pck1, and Pcx (Fig. 3B) is consistent with the induction of an anabolic program, suggesting a possible role in muscle repair.
Figure 3. PPARδ gene network orchestrates opposing changes on fat and sugar metabolism.

RNA-seq experiments were performed in white quadriceps from 4-month-old WT mice with or without 8 weeks of GW treatment. (A) Heat maps showing individual gene expression changes in gene ontology (GO) terms identified in RNA-seq results. Data are presented as log2(fold change). (B) GW-induced fold induction of the expression of genes involved in FA catabolism (Lpl, Lipe, Acadl, Acads, and Acaa2), lipogenesis (PPARg and Fasn), antioxidant (Cat, Sod3, and Gpx1), and gluconeogenesis (Fbp2, Pck1, and Pcx). (C) GW-induced fold repression of the expression of genes involved in insulin signaling (Irs2), glucose uptake (Slc2a3), glycolysis (Hk2, Gck, Pgk1, and Pfkm), and mitochondrial pyruvate entry (Mpc1). (D) Diagram showing the metabolism of glucose and FAs, the two major cellular energy substrates, as well as GW-induced gene expression changes that regulate their metabolism. Red and blue labels represent up- and down-regulation induced by GW treatment, respectively. p < 0.01 or 0.001 in all genes listed in (B) & (C).
Conversely, pathways of insulin signaling, glycolysis, and carbohydrate catabolism were significantly enriched in the down-regulated gene set (including Irs2, Slc2a3, and Hk2) (Fig. 3A, 3C, and S3B). Notably, these transcriptional changes, combined with the suppression of the recently identified mitochondrial pyruvate carrier Mpc1, coordinately reduce muscle glucose catabolism (Fig. 3A, 3C, and S3B). These studies reveal that PPARδ reprograms muscle metabolism for endurance by reciprocal regulation of gene programs promoting FA oxidation and suppressing glucose metabolism (Fig. 3D).
Ligand Remodeling of the PPARδ cistrome
Chromatin immuno-precipitation coupled with deep sequencing (ChIP-Seq) was used to establish the PPARδ cistrome in both the presence and absence of ligand. In C2C12 cells, 8474 and 10482 PPARδ binding sites were identified in vehicle and GW treated cells, respectively (Fig. 4A). Motif analysis identified a consensus PPAR response element significantly enriched in both conditions (Fig. 4A), confirming the quality of the ChIP. 5586 binding sites were common to both vehicle and GW treated cells, with GW recruiting an additional 4896 unique sites and dismissing 2888 pre-ligand sites. PPARδ binding sites were predominantly located in the intergenic and intronic regions, with only 4% in gene promoter regions (Fig. 4B), as seen with many transcription factors.
Figure 4. Ligand Remodeling of the PPARδ cistrome.

Experiments were performed in 5-day-differentiated C2C12 myotubes with 6-hour treatment of DMSO or GW. (A) Venn diagram showing distinct and overlapping binding sites of PPARδ in C2C12 myotubes with DMSO or GW treatment. PPRE motif (top) was significantly enriched. (B) Pie chart illustrating genomic locations of PPARδ binding sites in the DMSO-treated condition. GW treatment gave similar results. (C) Pie chart showing the number of PPARδ-bound or non-bound genes from all 975 genes that are changed by GW treatment. GO and pathway analysis of up- and down-regulated genes is listed. (D) ChIP-qPCR showing PPARδ binding sites that are enhanced by GW treatment or not. IgG: IgG control; PD-DM: PPARδ-ChIP in DMSO treatment; PD-GW: PPARδ-ChIP in GW treatment. (E–F) ChIP-Seq and RNA-seq reads aligned to Pdk4 (left) and Slc25a20 (right). (G–H) ChIP-qPCR showing the changes of NcoR (E) and Pgc1α (F) binding sites by GW treatment. IgG: IgG control; Pgc1α/Ncor-DM: Pgc1α/Ncor-ChIP in DMSO treatment; Pgc1α/Ncor-GW: Pgc1α/Ncor-ChIP in GW treatment. *p < 0.05, **p < 0.01.
Correlating the PPARδ cistrome with the ligand-induced transcriptional changes (Fig. 3 and S3) revealed that ~50% of GW-regulated genes (457 of 975 genes) were direct PPARδ target genes (based on proximity of binding sites to the closest transcription start site), with roughly equivalent numbers of these genes up and down-regulated (223 and 234, respectively, Fig. 4C). Furthermore, GO analysis of the 457 directly regulated genes showed enrichment of the same categories as seen in the entire GW transcriptome, including FA metabolism in up-regulated and glucose catabolism in down-regulated gene sets (Fig. 3A, S3, and 4C). In contrast, the 518 indirect PPARδ target genes failed to show any significant GO enrichment. The marked overlap between the transcriptomic and cistromic findings support a direct role for PPARδ-regulated transcriptional changes in both exercise-and ligand-induced adaptations in energy substrate utilization.
Interestingly, PPARδ employs distinct mechanisms to regulate target genes. Only a subset of genes showed increased PPARδ chromatin binding upon ligand treatment (e.g. Pdk4 and Mlycd, in contrast to Cpt1a, Slc25a20, Angptl4, and Ucp3, where PPARδ binding was independent of ligand) (Fig. 4D–F). Further investigation of the latter gene set revealed both ligand-induced loss of co-repressor binding (loss of NCoR binding on Angptl4 and Ucp3) as well as ligand-induced recruitment of co-activator (increased Pgc1α binding on Cpt1a and Slc25a20 (Fig. 4G–H).
AMPK activation is insufficient to induce a metabolic shift
Previously, a functional interaction between PPARδ and AMPK was suggested based on a synergistic induction of FA metabolism genes by co-treatment with GW and the AMPK activator AICAR; an effect that was dependent on PPARδ (Narkar et al., 2008). To explore the regulatory hierarchy controlling metabolic substrate utilization, we determined the effects of AICAR treatment on PPARδ target genes (Fig. 3A–B). AICAR alone did not significantly activate PPARδ target genes involved in FA metabolism, except for the induction of Pparγ, in agreement with an earlier study (Fig. S4A–B). Furthermore, AICAR failed to repress glucose catabolism genes (Fig. S4A). Consistent with this lack of transcriptional changes, 8 weeks of AICAR treatment did not alter substrate utilization in mice (Fig. S4C). AICAR treatment did increase energy expenditure in mice, presumably through mitochondrial biogenesis (Fig. S4C–D). Notably, the ability of AICAR to induce mitochondrial biogenesis is independent of PPARδ (Fig. S4D), in line with the data that PPARδ activation or depletion did not affect mitochondrial biogenesis (Fig. S2G).
Discussion
In endurance sport competitions such cycling, marathon runs, race walking, and cross-country skiing, ‘hitting the wall’ is a dramatic demonstration of sudden and complete exhaustion. It is thought to be due to the depletion of liver and muscle glycogen and can be averted by training that promotes mitochondrial biogenesis, increased type I fibers and enhanced FA burning. In this study, we show that PPARδ expression correlates with endurance and its activation by exercise mimetics, such as GW, is sufficient to increase running time by ~100 minutes without changes in either muscle fiber type or mitochondrial biogenesis. Thus, the foundational core of endurance enhancement appears to be purely metabolic. Furthermore, even though the GW impact appears to be achieved via increased FA metabolism, the strongest correlation to endurance is maintenance of blood glucose above 70mg/dl. Indeed, expression of key glycolytic genes such as Hk2, Gck and the recently described mitochondrial pyruvate carrier (Mcp1) is dramatically reduced.
Activation of muscle PPARδ either genetically or pharmacologically is sufficient to dramatically improve endurance capacity. However, fiber type changes and mitochondrial biogenesis found in the PPARδ transgenic models (Luquet et al., 2003; Wang et al., 2004) were not seen in ‘ligand only’ activation. Instead, we find that PPARδ ligand prioritizes energy substrate usage to increase FA catabolism while lowering glycolysis with the net effect of preserving systemic glucose (Fig. 2K).
This PPARδ-directed metabolic shift is driven by the induction of a metabolic gene signature change, which includes not only the induction of the mitochondrial gatekeeper genes Cpt1b and Pdk4, but also the up-regulation of genes in FA transport, FA oxidation, lipogenesis, and gluconeogenesis, and the down-regulation of genes in glucose uptake, glycolysis, and mitochondrial pyruvate entry. Overall, these gene expression changes promoted the metabolic shift in energy substrate from glucose to FA in the muscle. Antioxidant genes, as well as genes in glutathione synthesis, were also significantly induced by GW treatment, which could protect against increased reactive oxidative species (ROS) due to elevated mitochondrial energy metabolism.
Our study reveals the molecular mechanism underlying muscle PPARδ regulation of its target genes. We show that ligand activation recruits PPARδ to new cis regulatory elements on its target genes. In addition, at sites where PPARδ is naturally bound, ligand binding modifies its interaction with coactivators or co-repressors for target gene regulation (Yamamoto et al., 2011). Our data also suggested PPARδ as the direct executor in GW-induced gene expression changes, since ~50% of the GW-induced genes contain PPARδ binding sites. In those PPARδ bound genes, about half were up-regulated and half down-regulated upon GW activation, suggesting PPARδ could both directly activate and suppress downstream genes in a ligand-dependent manner, a circumstance observed for other nuclear receptor members such as PPARγ (Duan et al., 2008).
The importance of muscle PPARδ in regulating energy metabolism has been further confirmed by the correlation study in 38 BXD mouse lines. We observed strong correlations between PPARδ target gene expression and metabolic fitness. As a nuclear receptor, the transcription activity of PPARδ is regulated by its endogenous ligands. Its activity, rather than its own expression level, controls its target gene expression. In support of this, exercise training highly induced the expression of PPARδ target genes such as Cpt1b and Pdk4 but had no effect on PPARδ itself in skeletal muscle.
This work identifies PPARδ as both the master regulator and key executor of adaptive changes in energy substrate use in skeletal muscle. Notably, pharmacologic activation of PPARδ replicates the exercise-induced changes in substrate utilization to preserve systemic glucose and thereby delay the onset of hypoglycemia, or “hitting the wall”. While exercise-induced muscle remodeling is well documented, the health benefits have been largely attributed to mitochondrial biogenesis and fiber-type transformation (Fan and Evans, 2016). In contrast, pharmacophores that activate PPARδ promote endurance through preserving glucose, essentially “pushing back the wall”, without affecting mitochondrial biogenesis or fiber-type transformation. This ability to chemically activate energetic circuits regulated by PPARδ has the potential to confer health benefits in a variety of human disease.
STAR METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ronald Evans (evans@salk.edu).
Experimental model and subject details
Mouse models
PDmKO mice were generated by crossing Pparδfl/fl mice (Barak et al., 2002) with Acta1-Cre mice (JAX 006149). Male mice of 2–4 months of age were used in all experiments. Age-matched Cre-mice were used as controls.
Cell lines
C2C12 myoblasts were cultured in DMEM supplemented with 20% FBS. All C2C12 in vitro experiments were performed in differentiated myotubes, after 5 days of differentiation in DMEM supplemented with 2% horse serum. Primary myoblasts were isolated from Pparδfl/fl mice and the knockout of Pparδ was achieved by the transfection of adenovirus expressing Cre recombinase (Vector Biolabs Cat 1700). Primary myotube formation was induced by 2 days of culture in DMEM supplemented with 2% horse serum.
Method details
Animal studies
Prior to training or exhaustion running, mice were pre-adapted to the treadmill for 20 minutes per day for 3 days at a gradually increased speed (5 to 10 meter/min). Daily exercise training was achieved with one hour of forced running on a rodent treadmill (Harvard Apparatus) at a speed of 10 meter/min. The same treadmill was used for the run-to-exhaustion test, which included 10 minutes of adaptive period with a gradually increasing speed from 10 to 15 meter/min followed by the exhaustion run at 15 meter/min till mice failed. Blood glucose and lactate levels were measured using a glucometer (Nova max) and a Lactate Scout analyzer (EKF Diagnostic) with tail-nip bleeding. GW501516 was administered through customized diets (Envigo) at the concentration of 40mg/kg. Energy expenditure (VO2), RER (VCO2/VO2), and activity were monitored with the CLAMS/Oxymax system (Columbus Instruments). Body temperature was measured with a rectal thermometer (Thermoworks) and an infrared camera (FLIR). All animal protocols were reviewed and approved by the Institute of Animal Care and Use Committee (IACUC) of their respective institutes, and studies were conducted in compliance with institutional and national guidelines.
Western blotting
Freshly frozen quadriceps muscle was pulverized in liquid nitrogen. About 50mg of muscle powder was weighed and proteins extracted with the ProPrep protein extraction solution (Bulldog Bio, 17081). Protein concentration was measured with the Pierce BCA protein assay kit (Thermo Fisher, 23225). 20μg of total proteins was loaded onto 4–12% gradient NuPAGE Bis-Tris gels (Invitrogen) and run for 1.5 hours at 120V. Proteins were transferred to nitrocellulose membranes (Biorad). After one-hour blocking in 5% milk PBST (0.1% Tween 20) solution, membranes were incubated overnight at 4°C with in-house developed guinea pig anti-PPARδ and commercial PGC1α (Abcam ab54481), Tom20 (Cell Signaling 42406), and HSP90 (Cell Signaling 4877) antibodies, followed by 3x 5-minute washes in PBST, 1-hour incubation in secondary HRP antibodies, and 3x 5-minute washes in PBST. SuperSignal West Pico (Thermo 34080) was used to develop the signals.
Seahorse Bioanalyzer
For Seahorse analysis (XF96, Agilent Technologies), C2C12 myoblasts were seeded at a density of 104/well in XF96 plates. After 4 days of differentiation, cells were treated overnight with vehicle (DMSO) or GW501516 (0.2μM). OCR and ECAR were next measured following manufacturer’s instructions with the injection of Seahorse XF Palmitate-BSA FAO Substrate (Agilent Technologies) and Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies).
Immunohistochemistry and histochemistry
Gastrocnemius, soleus, and plantaris muscles were dissected and freshly frozen in liquid N2-cooled isopentane. 10μm cryosections were prepared using a cryostat (Zeiss). Sections were dried at RT (room temperature) for 30 minutes and stored in −80°C or directly used for stainings. For immunohistochemistry, sections were blocked in 5% GS/PBST (5% goat serum in PBST) for 1 hour at RT and incubated overnight at 4°C with a mixture of three primary mouse monoclonal antibodies in 5% GS/PBST against MYH7 (BA-F8, IgG2b), MYH2 (SC-71, IgG1), and MYH4 (BF-F3, IgM), obtained from DSHB at the University of Iowa. After 3x 5-minute washes in PBST, sections were incubated for one hour with a mixture of three goat anti-mouse secondary antibodies against IgG2b (Alexa 350), IgG1 (Alexa 488), and IgM (Alexa 555) in 5% GS/PBST, followed by 3x 5-minute washes in PBST. Sections were then mounted with ProLong Gold Anti-fade mountant (Thermo Fisher) and sealed with nail polish. For histochemical mitochondrial complex I and IV activity staining, sections were incubated at RT for 15 minutes in freshly prepared complex I assay solution (100mM Tris (pH 7.1), 1mg/ml nitro blue tertrazolium, and 1mg/ml NADH) or complex IV assay solution (200mM NaH2PO4 (pH 7.4), 7.5mg/ml sucrose, 2ug/ml catalase, 1.5mg/ml cytochrome c, and 1mg/ml DAB). Sections were washed in distilled water and mounted in Permount (Thermo Fisher). For glycogen staining, a Periodic Acid-Schiff (PAS) staining system (MilliporeSigma, 395B) was used following the manufacturer’s manual and mounted with Permount. All sections were imaged with an Olympus VS-120 Virtual Slide Scanning Microscope.
Mitochondrial isolation and analysis
Mitochondria were isolated from freshly dissected quadriceps muscle using a mitochondrial isolation kit following the manufacturer’s manual (MilliporeSigma, MITOISO1). Mitochondrial protein concentration was measured using a BCA assay. 50ug of freshly isolated mitochondria was added to each well in a Seahorse XF96 plate and pelleted by 30 minutes of centrifugation at 4°C at the speed of 3000g. OCR was measured using the Seahorse XF96 bioanalyzer and the XF96 assay kit (Agilent Technologies) following manufacturer’s protocol with either succinate or palmitoyl-carnitine as the energy substrate. The activities of citrate synthase and mitochondrial complex I, II+III, and IV, were measured using spectrophotometric enzyme assays (Fan et al., 2012). Specifically, freshly isolated mitochondria were disrupted with 3 cycles of freeze-and-thaw and immediately used to detect complex I, II+III, IV, and citrate synthase activities by spectrophotometric assays. Complex I activity was measured as the rate of NADH oxidation at 340 nm. A reaction containing 400 μl water, 500 μl 2x Buffer (500 mM sucrose, 2 mM EDTA, 100 mM Tris-HCl, pH 7.4), 10 μl 10 mM decylubiquinone (DB, Sigma), 1 μl 2M KCN, and 20ug mitochondria, was incubated at 30°C for 5 minutes. 50 μl 1mM NADH was then added to trigger the reaction, and absorption at 340 nm was measured for 5 minutes for total complex I activity. A parallel experiment was performed in the presence of 5 μl 1 mg/ml rotenone to measure rotenone-insensitive complex I activity. Complex II+III activity was measured as the rate of cytochrome c (cyt c) reduction at 550 nm. A reaction was set up containing 550 μl water, 400 μl 100 mM potassium phosphate buffer (pH 7.4), 20 μl 1 M succinate, 1 μl 0.5 M EDTA, 1 μl 2M KCN, and 30 μl 1 mM cyt c. 10μg mitochondria was then added and the absorption at 550 nm was monitored for 2 minutes for complex II+III activity. Complex IV activity was measured as the rate of cytochrome c (cyt c) oxidation at 550 nm. A reaction was set up containing 850 μl water, 100 μl 100 mM potassium phosphate buffer (pH 7.4), and 50 μl mM reduced cyt c. 2 μg mitochondria was then added and the absorption at 550 nm was monitored for 2 minutes for complex IV activity. To reduce cyt c, 2 μl 1 M dithiothreitol (DTT) was added to 1 ml 1 mM cyt c and was ready to use after a 15-minute incubation. Citrate synthase activity was measured as the reduction of DTNB at 412 nm. A reaction was set up containing 800 μl water, 100 μl 1 M Tris-HCl (pH 8.0), 50 μl 6 mM Acetyl-CoA, and 10 μl 10 mM DTNB. 10 μg mitochondria was then added and the absorption at 412 nm was monitored for 2 minutes for citrate synthase activity.
RNA analysis
Total RNAs were extracted from the glycolytic white quadriceps muscle or C2C12 myotubes with Trizol (Invitrogen) and the RNeasy Mini kit with on-column DNase treatment (Qiagen). RNA quality was confirmed using the Agilent 2100 Bioanalyzer and RNA-Seq libraries prepared using the TruSeq RNA Sample Preparation Kit v2 according to Illumina protocol. Multiplexed libraries were validated using the Agilent BioAnalyzer, normalized and pooled for sequencing. High-throughput sequencing was performed on the Hiseq 2000 system (Illumina) with a 100-bp read length. For quantitative PCR (qPCR) analysis, 1μg of RNA was reverse transcribed using the iScript supermix (Biorad) and qPCR performed with specific primer pairs and the SYBR Green Master Mix on the CFX384 system (Biorad).
Chromatin immunoprecipitation (ChIP) analysis
Each ChIP was performed in 4x107 cells using a previously described protocol (Barish et al., 2010). Briefly, C2C12 myotubes were crosslinked with 1% freshly prepared formaldehyde in PBS for 10 minutes at room temperature. After glycine neutralization, cells were harvested and nuclei were isolated, lysed and sheared with the Diagenode Bioruptor to yield DNA fragment sizes of 200–500 base pairs followed by immunoprecipitation using the following antibodies: normal guinea pig IgG, in-house developed guinea pig anti-PPARδ and NcoR antibodies, normal rabbit IgG, and rabbit anti-PGC1α antibody (Abcam ab54481). DNA was next extracted and subjected to either qPCR or ChIP-seq library preparation using the NEXTflex Illumina ChIP-Seq Library Prep Kit (Bioo Scientific). High-throughput sequencing was performed on the Hiseq 2000 system (Illumina) with a 50-bp read length.
Glycogen assay
Glycogen was measured using a commercial kit (BioVision K646-100) following manufacturer’s instructions. Snap-frozen muscles were ground using a mortar and pestle in liquid N2, and weighed tissue powder lysed in the provided buffer. Tissue lysate was then assayed for glycogen content.
Quantification and statistical analysis
ANOVA and post-hoc analysis was used to evaluate statistical significance in studies using four groups of samples. For studies with only two samples, unpaired t-test was used instead. For RNA-Seq, image analysis and base calling were done with Illumina CASAVA-1.8.2. Short read sequences were mapped to a UCSC mm9 reference sequence using the RNA-Seq aligner STAR (Dobin et al., 2013). Known splice junctions from mm9 were supplied to the aligner and de novo junction discovery was also permitted. Differential gene expression analysis, statistical testing and annotation were performed using Cuffdiff 2 (Trapnell et al., 2013). Transcript expression was calculated as gene-level relative abundance in fragments per kilobase of exon model per million mapped fragments and employed correction for transcript abundance bias (Roberts et al., 2011). Results for genes of interest were also explored visually using the UCSC Genome Browser. For ChIP-Seq, short DNA reads were demultiplexed using Illumina CASAVA v1.8.2. Reads were aligned against the mouse mm9 using the Bowtie aligner allowing up to 2 mismatches in the read. Only tags that map uniquely to the genome were considered for further analysis. Subsequent peak calling and motif analysis were conducted using HOMER, a software suite for ChIP-Seq analysis. The methods for HOMER, which are described below, have been implemented and are freely available at http://biowhat.ucsd.edu/homer/. One tag from each unique position was considered to eliminate peaks resulting from clonal amplification of fragments during the ChIP-Seq protocol. Peaks were identified by searching for clusters of tags within a sliding 200 bp window, requiring adjacent clusters to be at least 1 kb away from each other. The threshold for the number of tags that determine a valid peak was selected for a false discovery rate of <0.01, as empirically determined by repeating the peak finding procedure using randomized tag positions. Peaks are required to have at least 4-fold more tags (normalized to total count) than input or IgG control samples and 4-fold more tags relative to the local background region (10 kb) to avoid identifying regions with genomic duplications or non-localized binding. Peaks are annotated to gene products by identifying the nearest RefSeq transcriptional start site. Visualization of ChIP-Seq results was achieved by uploading custom tracks onto the UCSC genome browser. Gene ontology and pathway analysis was performed with the DAVID functional annotation tool (https://david.ncifcrf.gov). The BXD correlation analysis was based on the Spearman’s rho and performed in R as previously described (Andreux et al., 2012).
Data and software availability
RNA-Seq data reported in this paper have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database, Accession # SRP070441. ChIP-Seq data can be accessed at SRP081099.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-PPARδ | This paper | N/A |
| Guinea pig anti-NCOR | Yamamoto et al., 2011 | N/A |
| Rabbit anti-PGC1α | Abcam | ab54481 |
| Rabbit anti-Tom20 | Cell Signaling | 42406 |
| Rabbit anti-HSP90 | Cell Signaling | 4877 |
| Mouse IgG2b anti-MYH7 | DSHB | BA-F8 |
| Mouse IgG1 anti-MYH2 | DSHB | SC-71 |
| Mouse IgM anti-MYH4 | DSHB | BF-F3 |
| Goat anti-Mouse IgG2b, Alexa 350 | Thermo Fisher | A-21140 |
| Goat anti-Mouse IgG1, Alexa 488 | Thermo Fisher | A-21121 |
| Goat anti-Mouse IgM, Alexa 555 | Thermo Fisher | A-21426 |
| Bacterial and Virus Strains | ||
| Cre-GFP adenovirus | Vector Biolabs | Cat 1700 |
| Chemicals | ||
| GW501516 | Cayman Chemical | 317318-70-0 |
| Critical Commercial Assays | ||
| Palmitate-BSA FAO Substrate | Agilent | 102720-100 |
| Cell Mito Stress Test Kit | Agilent | 103015-100 |
| Glycogen assay kit | BioVision | K646-100 |
| Mitochondria isolation kit | MilliporeSigma | MITOISO1-1KT |
| PERIODIC ACID-SCHIFF (PAS) STAINING SYSTEM | MilliporeSigma | 395B |
| Deposited data | ||
| RNA-seq | NCBI SRA database | SRP070441 |
| ChIP-seq | NCBI SRA database | SRP081099 |
| Experimental Models: Cell Lines | ||
| C2C12 | ATCC | CRL-1772 |
| Experimental Models: Organisms/Strains | ||
| Pparδfl/fl | Barak et al., 2002 | N/A |
| ACTA-Cre | JAX | 006149 |
| Oligonucleotides | ||
| Full sequences in Table S2 | IDT | N/A |
Supplementary Material
Table S1. Microarray analysis of AICAR treated muscle (related to Figure 4).
HIGHLIGHTS.
Exhaustion of systemic glucose limits endurance exercise
PPARδ regulates substrate utilization without mitochondrial biogenesis
PPARδ represses glycolytic genes in muscle to slow glucose consumption
Glucose sparing by PPARδ dramatically extends running time
Acknowledgments
We thank C. Brondos and E. Ong for administrative assistance, J. Nery for assistance with DNA sequencing, C. Benner for assistance with HOMER software, and H. Juguilon and J. Alvarez for technical assistance. This work was funded by grants from NIH (DK057978, HL105278, DK090962, HL088093, ES010337 and CA014195) and National Health and Medical Research Council of Australia Project Grants 512354 and 632886 (C.L. and M.D.), as well as the Leona M. and Harry B. Helmsley Charitable Trust (#2012-PG-MED002), the Samuel Waxman Cancer Research Foundation and Ipsen/Biomeasure. R.M.E. and M.D. are supported in part by a Stand Up to Cancer Dream Team Translational Cancer Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0509). R.M.E is an investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute.
Footnotes
Author Contributions
W.F., M.D., and R.M.E. designed the study. W.F., W.W., C.S.L., M.H., and C.E.W. conducted all experiments. V.S., H.L., and J.A. performed the BXD correlation analysis. C.L. and R.T.Y. analyzed genomic data. W.F., A.R.A., M.D., and R.M.E. drafted and revised the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreux PA, Williams EG, Koutnikova H, Houtkooper RH, Champy MF, Henry H, Schoonjans K, Williams RW, Auwerx J. Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell. 2012;150:1287–1299. doi: 10.1016/j.cell.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, Boland R, Evans RM. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:303–308. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, Cooney GJ, Febbraio MA, Kraegen EW. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes. 2009;58:550–558. doi: 10.2337/db08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GE. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Molecular endocrinology. 2003;17:2477–2493. doi: 10.1210/me.2003-0151. [DOI] [PubMed] [Google Scholar]
- Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circulation research. 2008;102:283–294. doi: 10.1161/CIRCRESAHA.107.164384. [DOI] [PubMed] [Google Scholar]
- Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Current opinion in cell biology. 2014;33C:49–54. doi: 10.1016/j.ceb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Evans R. Exercise mimetics in muscle: influence on health and performance. Cell metabolism. 2016 doi: 10.1016/j.cmet.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MT, Booth FW. Skeletal muscle adaptation to exercise: a century of progress. J Appl Physiol. 2000;88:327–331. doi: 10.1152/jappl.2000.88.1.327. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annual review of physiology. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- Kleiner S, Nguyen-Tran V, Bare O, Huang X, Spiegelman B, Wu Z. PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function. The Journal of biological chemistry. 2009;284:18624–18633. doi: 10.1074/jbc.M109.008797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003;17:2299–2301. doi: 10.1096/fj.03-0269fje. [DOI] [PubMed] [Google Scholar]
- Mole PA, Oscai LB, Holloszy JO. Adaptation of muscle to exercise. Increase in levels of palmityl Coa synthetase, carnitine palmityltransferase, and palmityl Coa dehydrogenase, and in the capacity to oxidize fatty acids. The Journal of clinical investigation. 1971;50:2323–2330. doi: 10.1172/JCI106730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC genetics. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, Desvergne B, Wahli W, Chambon P, Metzger D. PGC1 alpha expression is controlled in skeletal muscles by PPAR beta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metabolism. 2006;4:407–414. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan WW, Downes M, Heligon C, Barish GD, Desvergne B, Evans RM, et al. NCoR1 Is a Conserved Physiological Modulator of Muscle Mass and Oxidative Function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Microarray analysis of AICAR treated muscle (related to Figure 4).