

Abstract
Systemic tumor necrosis factor-α (TNF-α) may contribute to the pathogenesis of cerebral malaria (CM) by promoting endothelial activation and parasite sequestration. However less is known about the role of central nervous system (CNS) TNF-α in CM. We assessed plasma (n=249) and cerebrospinal fluid (CSF) (n=167) TNF-α levels in Ugandan children with CM, plasma TNF-α in Ugandan community control children (n=198), and CSF TNF-α in North American control children who had recovered from leukemia (n=13). Plasma and CSF TNF-α were measured by magnetic bead assay. We compared plasma and CSF TNF-α levels in children with CM to mortality, acute and chronic neurologic deficits and long-term neurocognitive impairment. Plasma and CSF TNF-α levels were higher in CM than control children (P<0.0001 for both). CSF TNF-α levels were higher in children who had neurologic deficits at discharge or 6-months follow-up (P≤0.05 for both). Elevated CSF but not plasma TNF-α was associated with longer coma duration (Spearman’s rho 0.18, P=0.02) and deficits in overall cognition in children 5 years and older (β coefficient −0.74, 95%CI −1.35−−0.13, P=0.02). The study findings suggest that CNS TNF-α may be involved in the development of acute and chronic neurologic and cognitive sequelae in children with CM.
Keywords: pediatric cerebral malaria, tumor necrosis factor alpha, neurocognitive impairment
Introduction
Cerebral malaria (CM) is a leading cause of mortality from Plasmodium falciparum infection with a mortality rate of 13–15%(1–3). CM is also an important cause of short-(4) and long-term(5) neurocognitive impairment in African children. Tumor necrosis factor-alpha (TNF-α) is considered an important contributor to CM pathogenesis due to its role in promoting endothelial activation, which can further increase binding of infected erythrocytes (IE) to host endothelium(6, 7), an important hallmark of CM.
In mouse models of malaria, TNF-α has been shown to reduce parasitemia and protect against the early stages of infection(8–11) but has also been associated with disease severity when elevated at later stages(9). TNF-α has also been shown to be essential in the pathogenesis of experimental cerebral malaria(12, 13). This dual effect of TNF-α is thought to occur during human infections as well, as lower systemic TNF-α levels are seen at enrollment in patients with uncomplicated or mild malaria but higher levels are consistently detected in patients with severe malaria(14–18).
In humans two to ten-fold elevated systemic TNF-α levels have been associated with mortality in children with severe malaria(17, 19) and in children with CM specifically(16) in some studies, but not in others(14). High plasma levels of TNF-α have also been associated with hyperparasitemia and hypoglycemia(16, 19), deeper coma(20) and endothelial activation(17) in severe malaria. In addition, TNF-α polymorphisms associated with high TNF-α expression were more prevalent in patients with CM and fatal CM(21, 22). However, the use of antibodies against TNF-α did not reduce mortality and was associated with increased risk of neurologic deficits in children with CM(23), suggesting that more work is needed to understand the role of this cytokine not only systemically but also locally in the central nervous system (CNS).
In the CNS, TNF-α production by microglia and astrocytes has been associated with fatal murine cerebral malaria(24). The data from human studies of CNS TNF-α in CM patients is more limited, but also generally suggests a role for CNS TNF-α in CM: two studies have documented elevated CSF TNF-α levels in CM patients(20, 25) (another did not) (19), and autopsy studies of individuals who died of CM have shown TNF-α expression in the brain parenchyma (26–28). Our group has previously shown that high levels of TNF-α at enrollment in the CSF but not plasma were associated with neurologic deficits and impaired attention and working memory post-discharge in children five years and older (25) suggesting a role for local TNF-α in the neurologic outcomes of CM. Whether these findings would be more severe in the developing brains of children less than 5 years old is not known.
To better assess the role of systemic and CNS TNF-α in children with CM across the typical age spectrum in which CM is seen in Africa, we investigated how plasma and CSF TNF-α levels in Ugandan children 18 months to 12 years of age correlated with mortality, coma duration and acute and long-term neurologic and cognitive deficits, and assessed whether plasma and CSF TNF-α levels differed according to the presence of malaria retinopathy.
Materials and Methods
Study Design
This prospective cohort study of the effects of severe malaria on neurodevelopment was performed at Mulago Hospital, Kampala, Uganda between 2008 and 2015. Children with CM or community children (CC) were enrolled if they were between 18 months and 12 years of age. Cerebral malaria was defined as: 1) coma (Blantyre Coma Score ([BCS] ≤2); 2) Plasmodium falciparum on blood smear; and 3) no other known cause of coma (e.g., meningitis, a prolonged postictal state or hypoglycemia-associated coma reversed by glucose infusion). Children with CM were managed according to the Ugandan Ministry of Health treatment guidelines current at the time of the study. These included intravenous quinine treatment followed by oral quinine for severe malaria while admitted, and artemisinin combination therapy for outpatient follow-up therapy.
Eligible CC were 18 months to 12 years of age, currently healthy, and were recruited from the household or neighborhood of children with severe malaria. CC were recruited to be within the same age range as the children with severe malaria, using the age distribution from the first 45 children enrolled with severe malaria, but were not matched to specific children with severe malaria. Exclusion criteria for all children included: 1) known chronic illness requiring medical care; 2) known developmental delay; or 3) prior history of coma, head trauma, hospitalization for malnutrition, or cerebral palsy. Additional exclusion criteria for CC included: 1) illness requiring medical care within the previous 4 weeks; or 2) major medical or neurological abnormalities on screening physical exam.
HIV-1 testing was performed if the parents or guardians of the child agreed to have this testing done. Three immunochromatographic tests (Determine, STAT-PAK and Uni-Gold) were used and a final diagnosis of HIV infection was made based on the Uganda National HIV testing algorithm.
The study was reviewed and approved by the institutional review boards for human studies at the Makerere University School of Medicine and the University of Minnesota. Written informed consent was obtained from parents or guardians of study participants.
Malarial retinopathy diagnosis
Children were assessed for malarial retinopathy by means of indirect and direct ophthalmoscopy as previously described(29). Ophthalmologic examination was done by medical officers trained by an experienced ophthalmologist. Ophthalmologic exams were performed in all children with CM on admission, and repeated every 24 hours for as long as the child remained comatose. Children were considered to have malarial retinopathy if one or more of the following findings were present: retinal hemorrhages, vessel changes, or macular or peripheral retinal whitening.
Neurological and cognitive assessment and follow-up
A neurologic deficit at discharge was defined as the presence of motor deficits, ataxia, movement disorder, behavior, or speech or visual disorders, in a child with no known prior deficits. Of the 79 children with CM that were discharged with neurologic deficits, 62 improved during the 6-months post-discharge, 11 had the neurologic deficits that persisted at the 6-months follow-up assessment and 6 of those children did not return for the follow-up.
Children had cognitive assessment a week after discharge (or at enrollment for CC) and then at 6 and 12 months after enrollment. For children younger than 5 years old, the Mullen Scales of Early Learning(30) were used to measure cognitive ability. Scores from fine motor, visual reception, receptive language, and expressive language scales were summed to give the early learning composite score, a measure of overall cognitive ability. Attention was assessed using the Early Childhood Vigilance Test (ECVT)(31), in which a child was required to focus his/her gaze on cartoons screened on a computer for about 7 minutes. The measure of attention is the percent time the child spent gazing at the screen. In children 5 years and older, the Kaufman Assessment Battery for Children (second edition) was used to measure overall cognitive ability(32). Lauria’s model was used to obtain a composite score including sequential processing, simultaneous processing, learning ability and planning ability. Attention in these children was assessed using the Test of Variables of Attention (TOVA)(33) to measure attention and impulse control in four main areas: response time variability, response time, impulse control (commission errors), and inattention (omission errors). Neuropsychology testers were blinded to the study groups.
TNF-α, endothelial activation marker and parasite biomass testing
In the prospective cohort study, cytokine testing was performed on admission plasma and CSF samples. CSF samples were obtained as part of standard work-up for children with CM in whom a lumbar puncture was not contraindicated. 0.5–2 ml of the CSF sample was stored at −80°C for later study testing. Since there was no non-infected population in Mulago Hospital from which CSF could ethically be obtained, CSF control samples were obtained from asymptomatic North American children successfully treated for prior leukemia who had CSF obtained after treatment to rule out return of malignancy (ruled out in all). Plasma samples from these North American children were not available.
Plasma and CSF levels of TNF-α were measured by magnetic cyometric bead assay (EMD-Millipore, Billerica, MA) according to manufacturer’s instructions with a BioPlex-200 system (Bio-Rad, Hercules, CA). Peripheral blood smears were assessed for Plasmodium species by microscopy with Giemsa staining using standard protocols. PfHRP-2 quantification was performed using the Malaria Ag CELISA (Cellabs, Brookvale, Australia). Total, circulating and sequestered parasite biomass were calculated as previously described (34). Plasma soluble intracellular adhesion molecule-1 (sICAM-1), vascular cellular adhesion molecule-1 (sVCAM-1), soluble E-selectin and soluble P-selectin were also measured by magnetic cytometric bead assay (R&D Systems, Minneapolis, MN) according to manufacturer’s instructions. Plasma angiopoietin-2 (Ang-2) levels and von Willebrand Factor (VWF) activity were quantified using the human angiopoietin 2 DuoSet ELISA kit (R&D Systems, Minneapolis, MN) and REAADS von Willebrand Factor activity ELISA kit (Corgenix, Broomfield, CO), respectively. Plasma and CSF albumin were quantified by the Advanced Research and Diagnostic Laboratory at the University of Minnesota.
Statistical Analysis
Demographic characteristics were compared using t-tests for continuous measures and Pearson’s χ2 test for categorical variables. Plasma and CSF TNF-α levels, endothelial activation markers, coma duration, and number of seizures had skewed distributions, so for these variables, Wilcoxon rank-sum testing was used for comparisons between groups (e.g., children with vs. without neurologic deficits), and Spearman’s rank correlation (rho) was used for assessment of correlations with continuous variables. Age-adjusted z-scores for cognitive outcomes were created using the scores of the community children (CC)(4, 5). For each outcome, the z-score was computed as (actual score minus average score for child’s age) / SD, where “average score for child’s age” and “SD” were computed by fitting a mixed linear model to data from all available visits for CC (allowing correlated errors for a child’s multiple visits). Z-scores have average of 0 and SD 1 in the reference population (CC) over all time points.
Results
Demographic characteristics of children with cerebral malaria and community children
268 children with cerebral malaria and 213 community controls (CC) were enrolled in this study. 249 children with CM and 198 CC had sufficient plasma for TNF-α testing. Of the 249 children with CM who had plasma available for testing, 167 had sufficient CSF for TNF-α testing.
Children with CM and the CC were of similar age, but a higher proportion of children with CM were male (Table 1). By definition, parasite densities were higher in children with CM (Table 1). HIV prevalence was low in our cohort of children with CM (2.2%), possibly reflecting the exclusion criterion of known chronic illness, and by definition, HIV infection was not present in the cohort of healthy community children.
Table 1.
Demographic characteristics of children with cerebral malaria and healthy community control children
| CM (n=249) | CC (n=198) | Pa | |
|---|---|---|---|
| Age (months), median (IQR) | 41.5 (30.3–56.4) | 43.6 (32.6–56.5) | 0.35 |
| Sex (male), n (%) | 147 (59.0) | 91 (46.0) | 0.006 |
| Parasite density (/μl), median (IQR) | 48080 (11360–234360), n=242 | 0 (0–0), n=193 | <0.0001 |
| HIV positive, n (%) | 5 (2.2), n=229 | 0 (0), n=198 | 0.04 |
Age and parasite density were compared by Wilcoxon rank-sum (Mann-Whitney) test and sex by χ2test
The 19 children who did not have plasma available for testing did not differ from the 249 children with plasma available for testing in terms of age, mortality, neurologic deficits at discharge, or coma duration (all P values >0.05).
The 82 children with CM who had plasma but no CSF available for testing differed from the 167 children who had CSF available for testing in having higher mortality (21/82, 25.6%, vs. 9/167, 5.4%, P<0.001), but lower neurologic deficits at discharge (16/61 survivors, 26.2%, 63/156 survivors, 40.4%, P=0.05) and shorter coma duration during admission (median 44 vs. 59 hours, P=0.03). Age did not differ between children who did not vs. did have CSF available for testing (median 44.7 vs. 39.7 months, P=0.09). The difference in mortality reflects that many of the children for whom no CSF was available were too unstable to have a lumbar puncture performed during their acute illness.
Plasma and CSF TNF-α levels in children with CM do not correlate with age, sex, nutrition or HIV status
Plasma and CSF TNF-α levels were not associated with age (P>0.11 for both), sex (P>0.20 for both), nutritional status (weight for age z-score, P>0.3 for both), or HIV status (P>0.10 for both) in children with CM, and as a result, these factors were not adjusted for in the following analysis of TNF-α with acute and long-term outcomes in CM.
Plasma TNF-α levels in children with CM correlate with parasite biomass and endothelial activation
Plasma TNF-α levels correlated strongly and positively with all markers of endothelial activation (P<0.0005 for all, Table 2) except VWF. Plasma TNF-α levels were also associated with increased PfHRP-2 levels, as well as total, circulating and sequestered parasite biomass (P<0.0001 for all, Table 2).
Table 2.
Association of plasma TNF-α levels with endothelial activation markers and parasite biomass in children with CM
| Plasma TNF-α | |||
|---|---|---|---|
|
| |||
| N | Spearman’s rho | P | |
| sP-selectin | 195 | 0.29 | <0.0001 |
| sE-selectin | 205 | 0.36 | <0.0001 |
| sICAM-1 | 205 | 0.24 | 0.0004 |
| sVCAM-1 | 205 | 0.33 | <0.0001 |
| Ang2 | 152 | 0.51 | <0.0001 |
| VWF | 199 | 0.04 | 0.56 |
| PfHRP-2 | 249 | 0.57 | <0.0001 |
| Total parasite biomass | 249 | 0.55 | <0.0001 |
| Sequestered parasite biomass | 242 | 0.49 | <0.0001 |
| Circulating parasite biomass | 242 | 0.30 | <0.0001 |
Plasma TNF-α levels are higher in children with CM than community children, and correlate weakly with CSF TNF-α levels in children with CM
Plasma TNF-α levels in children with CM were significantly higher than CC (Figure 1a) recruited from the same households or neighborhoods of children with CM. Median levels of plasma and CSF TNF-α were similar between the two age-groups in CM (plasma, n=194, 107pg/ml for <5 years, n=55, 83.2pg/ml for ≥5 years, P= 0.07; CSF, n=136, 1.33pg/ml for < 5 year, and n=31, 1.34pg/ml for ≥5 years, P=0.84).
Figure 1. Plasma and CSF TNF-α levels are higher in children with cerebral malaria at enrollment compared to controls.
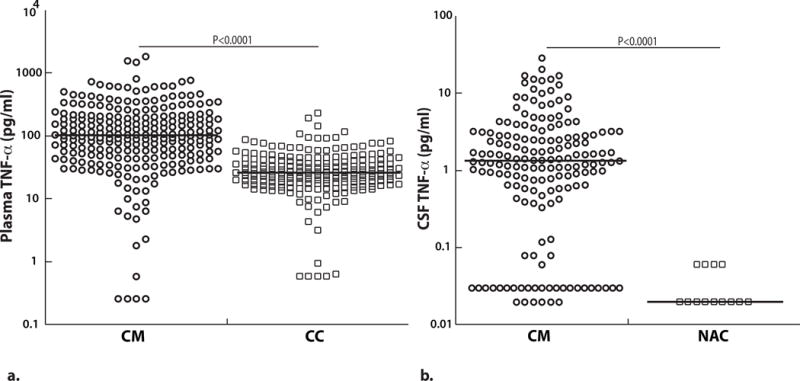
(A) Plasma (CM, n=249 and CC, n=198) and (B) CSF TNF-α (CM, n=167 and NAC, n=13) at enrollment (values on a logarithmic scale). The horizontal line represents median values. Two-sample Wilcoxon rank-sum (Mann-Whitney) test was used to compare median levels between groups. Cerebral malaria (CM), Ugandan community children (CC), North American control children (NAC). NAC samples were obtained from asymptomatic children successfully treated for prior leukemia who had CSF obtained after treatment to rule out return of malignancy (ruled out in all).
CSF TNF-α was weakly correlated with plasma TNF-α levels in children with CM (Spearman’s rho 0.15, P=0.06), suggesting that CSF levels of TNF-α may reflect local CNS production of TNF-α. To investigate this further, we assessed the association of CSF-to-plasma TNF-α ratio (CSF TNF-α x1000/Plasma TNF-α (pg/ml)) with CSF-to-plasma albumin ratio (CSF albumin x1000/Plasma albumin (mg/L)). The TNF-α ratio correlated positively with the albumin index (Spearman’s rho=0.29, P=0.0004), suggesting that blood-brain barrier (BBB) leakage affects the levels of TNF-α seen in the CSF. However, CSF-to-plasma TNF-α ratios (n=167, median [25th percentile, 75th percentile], 13.0 [3.1, 35.9]) were higher than the values for albumin index (n=149, 5.3 [3.1, 10.2]), suggesting some local production of TNF-α in the CNS.
Plasma and CSF TNF-α levels in children with CM do not differ according to malaria retinopathy
The presence of malarial retinopathy at admission has been associated with brain sequestration post-mortem in cerebral malaria (35), but it is unclear if children with clinical CM and no retinopathy have a milder form of CM or an alternative cause of coma. We compared TNF-α levels in children with CM with vs. without retinopathy. Plasma TNF-α levels did not differ significantly between retinopathy positive and retinopathy negative children with CM (P=0.67, Figure 2a). CSF TNF-α levels also did not differ significantly between RP and RN children with CM (P=0.30, Figure 2b).
Figure 2. Plasma and CSF TNF-α levels are not significantly different between retinopathy positive and negative cerebral malaria children.
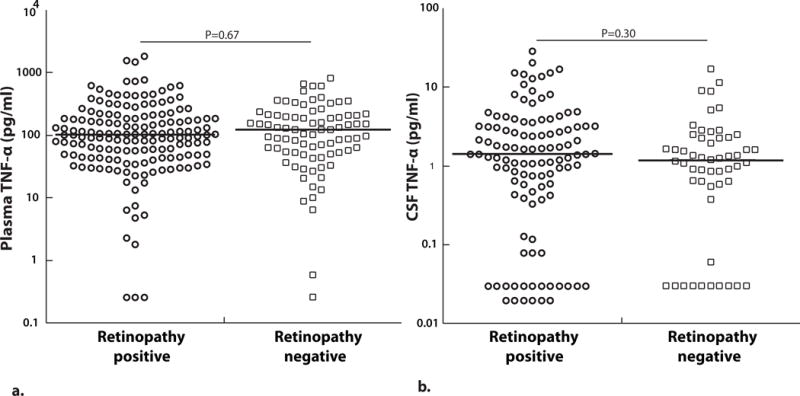
(A) Plasma (RP, n=158 and RN, n=81) and (B) CSF TNF-α (RP, n=110 and RN, n=53) at enrollment based on retinopathy characteristics (values on a logarithmic scale). The horizontal line represents median values. Two-sample Wilcoxon rank-sum (Mann-Whitney) test was used to compare median levels between groups.
Plasma and CSF TNF-α levels in children with CM do not differ according to acute mortality
Plasma TNF-α levels did not differ significantly between children with CM who died vs. those that survived (P=0.28, Figure 3a). CSF TNF-α levels also did not differ significantly between children with CM who died and those who survived (P=0.59, Figure 3b).
Figure 3. Plasma and CSF TNF-α levels are not significantly different between cerebral malaria children that died vs. those that survived.
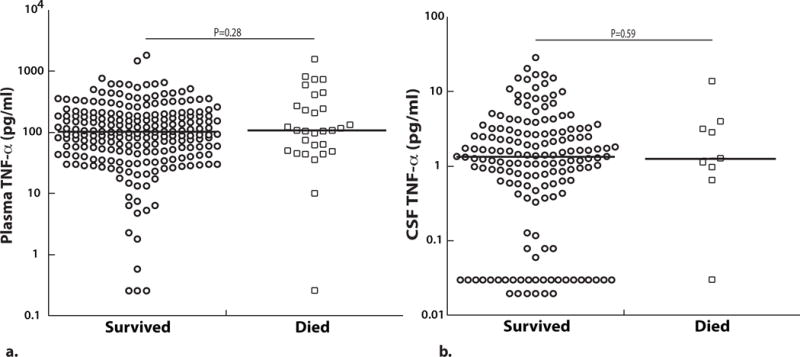
(A) Plasma (survived, n=219 and died, n=30) and (B) CSF TNF-α (survived, n=158 and died, n=9) at enrollment based on survival outcome (values on a logarithmic scale). The horizontal line represents median values. Two-sample Wilcoxon rank-sum (Mann-Whitney) test used to compare median levels between groups.
Elevated CSF TNF-α levels are associated with prolonged coma during admission, neurologic deficits at discharge and 6-months follow-up and deficits in overall cognition at 12-months follow-up
Plasma TNF-α levels did not differ significantly between children with vs. without neurologic deficits at discharge (P=0.41, Figure 4a) and between children with CM with or without neurologic deficits at 6 months follow-up (P=0.63, Figure 4a). However, levels of TNF-α in the CSF were significantly higher in children with CM who were discharged with neurologic deficits (P=0.04, Figure 4b). CSF TNF-α levels at enrollment were also higher in children that had neurologic deficits at 6-months follow-up compared to those who did not (P=0.05, Figure 4b). Similarly, CSF TNF-α, but not plasma TNF-α was positively associated with coma duration in children with CM (Table 3). Neither plasma nor CSF TNF-α levels were associated with number of seizures during admission (Table 3).
Figure 4. CSF TNF-α levels are higher in children with cerebral malaria who had neurologic deficits at discharge or 6-months follow-up.
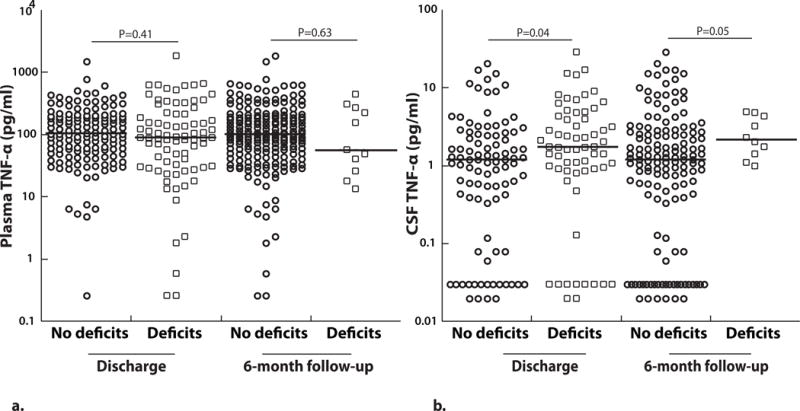
(A) Plasma (no deficits at discharge, n=138 and deficits at discharge, n=79; no deficits at 6-months follow-up, n=198 and deficits at 6-months follow-up, n=11) and (B) CSF TNF-α (no deficits at discharge, n=93 and deficits at discharge, n=63; no deficits at 6-months follow-up, n=141 and deficits at 6-months follow-up, n=10) at enrollment based on neurologic outcomes at discharge or 6-months follow-up (values on a logarithmic scale). The horizontal line represents median values. Two-sample Wilcoxon rank-sum (Mann-Whitney) test was used to compare median levels between groups.
Table 3.
Association of plasma and CSF TNF-α levels with number of seizures and duration of coma during hospitalization
| Plasma TNF-α | CSF TNF-α | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| N | Spearman’s rho | P | n | Spearman’s rho | P | |
| Coma duration (hours) | 219 | −0.04 | 0.59 | 158 | 0.18 | 0.02 |
| Number of seizures | 249 | 0.01 | 0.85 | 167 | 0.02 | 0.80 |
Plasma TNF-α levels were not associated with deficits in the areas of overall cognition and attention at 12 month follow-up in children <5 years or children ≥5 years of age (P >0.59 for all, Table 4). In children ≥5 years of age, elevated CSF TNF-α levels were associated with lower z-score values for overall cognition but not attention at 12 months follow-up (Table 4). However, there was no association between CSF TNF-α levels and cognition or attention in children <5 years of age. Overall cognition and attention scores at 12 months follow-up were not significantly different between children with CM who had HIV and neurocognitive testing (n=4) compared to those who did not have HIV (n=170 for overall cognition and n=173 for attention) (all P >0.07).
Table 4.
Association of plasma and CSF TNF-α levels with cognitive outcomes at 12 months follow-up
| Younger than 5 years old | 5 years old or older | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Overall cognition | Attention | Overall cognition | Attention | |||||
|
|
||||||||
| β* coefficient (95% CI), n |
P | β* coefficient (95% CI), n |
P | β* coefficient (95% CI), n |
P | β* coefficient (95% CI), n |
P | |
| Plasma TNF-α (pg/ml) | −0.008 (−0.51–0.50), 130 | 0.98 | 0.08 (−0.24–0.39), 134 | 0.64 | −0.18 (−0.87–0.51), 47 | 0.60 | −0.07 (−0.61–0.48), 46 | 0.81 |
| CSF TNF-α (pg/ml) | 0.05 (−0.27–0.37), 98 | 0.78 | −0.09 (−0.30–0.12), 102 | 0.39 | −0.74 (−1.35– −0.13), 29 | 0.02 | −0.16 (−0.67–0.35), 28 | 0.52 |
TNF-α levels were log-transformed (log base 10)
CSF TNF-α levels are higher in Ugandan children with CM than in currently healthy North American control children with prior neoplastic disease
In order to estimate whether CSF TNF-α levels were in the normal range in children with CM, we compared CSF TNF-α levels in CM with CSF TNF-α levels in North American children with prior neoplastic disease (see Methods) as there was no healthy population from which we could ethically obtain CSF samples at Mulago hospital. Children with CM had significantly higher CSF levels of TNF-α as compared to control North American children with prior neoplastic disease (P<0.0001 Figure 1b). Unfortunately, plasma samples from the North American children were not available, so we could not assess the association of plasma with CSF TNF-α in this group. However, considering that most CSF TNF-α levels were at the limit of detection (Figure 1b) and that the North American children were healthy at the time of sample collection, we expect plasma levels of TNF-α in the North American children would be <50 pg/ml, as in studies of other healthy children from high-income countries (36, 37), and similar to our study’s Ugandan CC controls (18–42pg/ml).
Discussion
In the present study, we show that CSF but not plasma TNF-α correlate with key clinical outcomes in children with CM, including coma duration, acute and long-term neurologic deficits and long-term cognitive outcomes. The study findings demonstrate the importance of CNS TNF-α in the neurologic outcomes in children with CM, and suggest that CNS TNF-α and factors, such as sequestered parasite biomass, that appear to affect CNS TNF-α production, may be good targets for adjunctive therapy to reduce neurologic morbidity in CM.
Plasma TNF-α levels were 4-fold higher in children with CM as compared to healthy community controls, and correlated with elevated levels of endothelial activation markers, parasite biomass and sequestered parasite biomass. It is difficult to know whether greater parasite burden led to greater TNF-α production from activated immune cells, or whether increased TNF-α led to greater endothelial activation and parasite binding, leading to increased sequestration and parasite biomass. Likely both phenomena are involved in the association. However, plasma TNF-α levels did not correlate with any clinical outcomes.
In contrast, CSF TNF-α levels correlated with acute (coma duration, neurologic deficits at discharge) and chronic (neurologic deficits at 6 months, worse neurocognitive scores at 12 months) CNS complications of CM. Elevated levels of TNF-α in the CNS can lead to tissue damage in cerebral ischemic stroke (38), spontaneous demyelination (39), death of neurons and oligodendrocytes(40), and are involved in a number of other CNS disorders such as bacterial meningitis(41), multiple sclerosis(42), Parkinson’s disease(43), and murine(24) and human cerebral malaria(26–28). Most human studies of CM that have looked at local TNF-α have done so in brain tissue. These studies allow for careful assessment of areas of the brain affected by TNF-α, but are limited in sample size and do not permit assessment of the role of CNS TNF-α in children who survive CM. This study and a prior study by our group(25) are, to our knowledge, the only studies to correlate CSF TNF-α levels with long-term sequelae. In both studies, plasma TNF-α levels were not correlated with any outcome. Together, the studies provide evidence that CNS TNF-α is associated with long-term neurologic and neurocognitive impairment in CM.
Parasite sequestration and systemic inflammation are thought to lead to BBB damage and leakage in CM, which could expose the brain parenchyma to plasma proteins and promote astrocyte and microglial activation(44). In the present study, the albumin index and CSF to plasma TNF-α ratio were strongly correlated, but the absolute values for TNF-α ratios were higher than the albumin index, suggesting that TNF-α found in the CSF may be partially produced in the CNS. Though we could not determine the cellular source of TNF-α in this study, the most likely sources are activated astrocytes and microglia (40, 45, 46).
It is unclear why CSF TNF-α levels were associated with impaired overall cognition only in children ≥5 years of age. It is possible that the effects of CNS TNF-α are better detected by the types of neurocognitive testing done in children ≥5 years of age as compared to those in done in children <5 years of age. Although the tests used are designed to test similar overall domains, the testing components and specific functions tested are different. Neural plasticity could allow better recovery from TNF-α-related CNS injury in younger children, but differences in cognitive scores between community children and children with CM overall were as large or larger in children <5 years and children ≥5 years of age (data not shown), so this explanation seems less likely as a reason for the differences. A recent study in a Malawian pediatric cohort saw a high prevalence of HIV (15%) in children with CM(47). CM children who were HIV positive were significantly older and had higher intravascular platelets and monocytes compared to children with CM but no HIV(47), suggesting that HIV could be an additional complication in these older children with CM. Perhaps due to the exclusion of children with known chronic illness in the present study, the prevalence of HIV infection in children with CM in this cohort was much lower (2.2%). Levels of CSF TNF-α were not significantly different between children with CM who had HIV infection vs. those that did not, and neurocognitive outcomes also did not differ between children with CM who had HIV infection vs. those who did not. Therefore, in the present study, HIV was not a major factor affecting neurocognitive outcomes. TNF-α in the CNS can have double functionality depending on the receptor it acts on and the stage of development. As an example, TNF-α by acting on TNFR2 promotes oligodendrocyte progenitor cell proliferation and remyelination but as oligodendroglia mature, TNF-α by acting on TNFR1 can reduce oligodendroglia maturation (48, 49). How the local events during a CM episode affect TNFR1 and TNFR2 expression and the expression of other cytokine and chemokines in the CNS needs further investigation.
Children in whom we did not have sufficient CSF to test for TNF-α were sicker than those for whom we were able to test CSF. This was not surprising, because the primary reason for not obtaining CSF was that the patient was too unstable to have a lumbar puncture or had a contraindication to lumbar puncture. As a result, the study findings likely underestimate the true associations of CNS TNF-α with mortality and acute and long-term complications of CM.
We did not see a significant difference in plasma TNF-α levels in CM children with vs. without malarial retinopathy. These findings differ from a recent study in which higher neutrophil activation and higher plasma TNF-α levels were seen in retinopathy positive vs. negative CM patients (50). The reasons for this difference could be differences in the diagnosis of malarial retinopathy in the two sites or differences in the prevalence of TNF-α promoter polymorphisms, which were not identified in either study. Importantly, we also found that CSF TNF-α levels did not significantly differ between retinopathy positive and negative CM, suggesting that TNF-α is important in CM pathogenesis regardless of presence of retinopathy.
In conclusion, our study data show that plasma and CSF TNF-α levels are elevated in children with cerebral malaria, and that elevated CSF TNF-α levels in children with CM are associated with prolonged coma, acute and long-term neurologic deficits, and long-term cognitive impairment. Our results emphasize the importance of studying both the peripheral and CNS immune responses since they do not always tell the same story. The data suggest that CNS TNF-α, as opposed to systemic TNF-α, is particularly important in neurologic outcomes in children with CM, and demonstrate the need for assessment of how age may alter the effects of risk factors for neurologic impairment. Further studies that aim to understand the role and regulation of CNS TNF-α in children with CM, with the goal of specifically targeting the CNS production and effects of TNF-α, could lead to adjunctive treatments that decrease neurologic morbidity in CM.
Acknowledgments
We thank the study teams at Makerere University, Mulago Hospital and University of Minnesota for their essential work on this study. We are grateful to the children and parents who took part in the study.
This work was supported by grants from the National Institute of Neurologic Disorders and Stroke and the Fogarty International Center (R01 NS05534, D43 NS078280). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Footnotes
Disclosures: None
E.S. and G.S.P. performed testing of study biomarkers. E.S. analyzed the data, and wrote the first draft of the manuscript; R.O.O. and C.C.J. designed the study and supervised its conduct; B.J.O, R.I., G.S.P. and A.C. helped to interpret study data; C.C.J. designed the research, supervised biomarker testing and edited the manuscript. All authors contributed to critical revision of the manuscript.
References
- 1.Bangirana P, Opoka RO, Boivin MJ, et al. Severe malarial anemia is associated with long-term neurocognitive impairment. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2014;59(3):336–44. doi: 10.1093/cid/ciu293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seydel KB, Kampondeni SD, Valim C, et al. Brain swelling and death in children with cerebral malaria. The New England journal of medicine. 2015;372(12):1126–37. doi: 10.1056/NEJMoa1400116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy SC, Breman JG. Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. The American journal of tropical medicine and hygiene. 2001;64(1–2 Suppl):57–67. doi: 10.4269/ajtmh.2001.64.57. [DOI] [PubMed] [Google Scholar]
- 4.Boivin MJ, Bangirana P, Byarugaba J, et al. Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics. 2007;119(2):e360–6. doi: 10.1542/peds.2006-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.John CC, Bangirana P, Byarugaba J, et al. Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics. 2008;122(1):e92–9. doi: 10.1542/peds.2007-3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341(6237):57–9. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- 7.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. The New England journal of medicine. 1996;334(26):1717–25. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 8.Clark IA, Hunt NH, Butcher GA, Cowden WB. Inhibition of murine malaria (Plasmodium chabaudi) in vivo by recombinant interferon-gamma or tumor necrosis factor, and its enhancement by butylated hydroxyanisole. Journal of immunology. 1987;139(10):3493–6. [PubMed] [Google Scholar]
- 9.Jacobs P, Radzioch D, Stevenson MM. A Th1-associated increase in tumor necrosis factor alpha expression in the spleen correlates with resistance to blood-stage malaria in mice. Infection and immunity. 1996;64(2):535–41. doi: 10.1128/iai.64.2.535-541.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevenson MM, Ghadirian E. Human recombinant tumor necrosis factor alpha protects susceptible A/J mice against lethal Plasmodium chabaudi AS infection. Infection and immunity. 1989;57(12):3936–9. doi: 10.1128/iai.57.12.3936-3939.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taverne J, Tavernier J, Fiers W, Playfair JH. Recombinant tumour necrosis factor inhibits malaria parasites in vivo but not in vitro. Clinical and experimental immunology. 1987;67(1):1–4. [PMC free article] [PubMed] [Google Scholar]
- 12.Grau GE, Fajardo LF, Piguet PF, et al. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science. 1987;237(4819):1210–2. doi: 10.1126/science.3306918. [DOI] [PubMed] [Google Scholar]
- 13.Rudin W, Eugster HP, Bordmann G, et al. Resistance to cerebral malaria in tumor necrosis factor-alpha/beta-deficient mice is associated with a reduction of intercellular adhesion molecule-1 up-regulation and T helper type 1 response. The American journal of pathology. 1997;150(1):257–66. [PMC free article] [PubMed] [Google Scholar]
- 14.Shaffer N, Grau GE, Hedberg K, et al. Tumor necrosis factor and severe malaria. The Journal of infectious diseases. 1991;163(1):96–101. doi: 10.1093/infdis/163.1.96. [DOI] [PubMed] [Google Scholar]
- 15.Kern P, Hemmer CJ, Van Damme J, Gruss HJ, Dietrich M. Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. The American journal of medicine. 1989;87(2):139–43. doi: 10.1016/s0002-9343(89)80688-6. [DOI] [PubMed] [Google Scholar]
- 16.Kwiatkowski D, Hill AV, Sambou I, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336(8725):1201–4. doi: 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 17.Tchinda VH, Tadem AD, Tako EA, et al. Severe malaria in Cameroonian children: correlation between plasma levels of three soluble inducible adhesion molecules and TNF-alpha. Acta tropica. 2007;102(1):20–8. doi: 10.1016/j.actatropica.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Walther M, Jeffries D, Finney OC, et al. Distinct roles for FOXP3 and FOXP3 CD4 T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS pathogens. 2009;5(4):e1000364. doi: 10.1371/journal.ppat.1000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grau GE, Taylor TE, Molyneux ME, et al. Tumor necrosis factor and disease severity in children with falciparum malaria. The New England journal of medicine. 1989;320(24):1586–91. doi: 10.1056/NEJM198906153202404. [DOI] [PubMed] [Google Scholar]
- 20.Esamai F, Ernerudh J, Janols H, et al. Cerebral malaria in children: serum and cerebrospinal fluid TNF-alpha and TGF-beta levels and their relationship to clinical outcome. Journal of tropical pediatrics. 2003;49(4):216–23. doi: 10.1093/tropej/49.4.216. [DOI] [PubMed] [Google Scholar]
- 21.McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371(6497):508–10. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 22.Knight JC, Udalova I, Hill AV, et al. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nature genetics. 1999;22(2):145–50. doi: 10.1038/9649. [DOI] [PubMed] [Google Scholar]
- 23.van Hensbroek MB, Palmer A, Onyiorah E, et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. The Journal of infectious diseases. 1996;174(5):1091–7. doi: 10.1093/infdis/174.5.1091. [DOI] [PubMed] [Google Scholar]
- 24.Medana IM, Hunt NH, Chaudhri G. Tumor necrosis factor-alpha expression in the brain during fatal murine cerebral malaria: evidence for production by microglia and astrocytes. The American journal of pathology. 1997;150(4):1473–86. [PMC free article] [PubMed] [Google Scholar]
- 25.John CC, Panoskaltsis-Mortari A, Opoka RO, et al. Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. The American journal of tropical medicine and hygiene. 2008;78(2):198–205. [PMC free article] [PubMed] [Google Scholar]
- 26.Udomsangpetch R, Chivapat S, Viriyavejakul P, et al. Involvement of cytokines in the histopathology of cerebral malaria. The American journal of tropical medicine and hygiene. 1997;57(5):501–6. doi: 10.4269/ajtmh.1997.57.501. [DOI] [PubMed] [Google Scholar]
- 27.Brown H, Turner G, Rogerson S, et al. Cytokine expression in the brain in human cerebral malaria. The Journal of infectious diseases. 1999;180(5):1742–6. doi: 10.1086/315078. [DOI] [PubMed] [Google Scholar]
- 28.Armah H, Dodoo AK, Wiredu EK, et al. High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Annals of tropical medicine and parasitology. 2005;99(7):629–47. doi: 10.1179/136485905X51508. [DOI] [PubMed] [Google Scholar]
- 29.Villaverde C, Namazzi R, Shabani E, Opoka RO, John CC. Clinical Comparison of Retinopathy-Positive and Retinopathy-Negative Cerebral Malaria. The American journal of tropical medicine and hygiene. 2017 doi: 10.4269/ajtmh.16-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullen EM. In: Mullen Scales of Early Learning manual. AGS, editor. Circle Pines, MN: American Guidance Service; 1995. p. ix.p. 85. [Google Scholar]
- 31.Goldman DZ, Shapiro EG, Nelson CA. Measurement of vigilance in 2-year-old children. Developmental Neuropsychology. 2004;25(3):227–50. doi: 10.1207/s15326942dn2503_1. [DOI] [PubMed] [Google Scholar]
- 32.Kaufman AS, Kaufman NL. KABC-II : Kaufman Assessment Battery for Children. 2nd. Circle Pines, MN: AGS Pub; 2004. [Google Scholar]
- 33.Greenberg LM, Waldman ID. Developmental normative data on the test of variables of attention (T.O.V.A.) Journal of child psychology and psychiatry, and allied disciplines. 1993;34(6):1019–30. doi: 10.1111/j.1469-7610.1993.tb01105.x. [DOI] [PubMed] [Google Scholar]
- 34.Dondorp AM, Desakorn V, Pongtavornpinyo W, et al. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS medicine. 2005;2(8):e204. doi: 10.1371/journal.pmed.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor TE, Fu WJ, Carr RA, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nature medicine. 2004;10(2):143–5. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 36.Kleiner G, Marcuzzi A, Zanin V, Monasta L, Zauli G. Cytokine levels in the serum of healthy subjects. Mediators of inflammation. 2013;2013:434010. doi: 10.1155/2013/434010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sack U, Burkhardt U, Borte M, et al. Age-dependent levels of select immunological mediators in sera of healthy children. Clinical and diagnostic laboratory immunology. 1998;5(1):28–32. doi: 10.1128/cdli.5.1.28-32.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu T, Clark RK, McDonnell PC, et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke; a journal of cerebral circulation. 1994;25(7):1481–8. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 39.Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(24):11294–8. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prasad R, Kapoor R, Srivastava R, Mishra OP, Singh TB. Cerebrospinal fluid TNF-alpha, IL-6, and IL-8 in children with bacterial meningitis. Pediatric neurology. 2014;50(1):60–5. doi: 10.1016/j.pediatrneurol.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Hofman FM, Hinton DR, Johnson K, Merrill JE. Tumor necrosis factor identified in multiple sclerosis brain. The Journal of experimental medicine. 1989;170(2):607–12. doi: 10.1084/jem.170.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boka G, Anglade P, Wallach D, et al. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neuroscience letters. 1994;172(1–2):151–4. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- 44.Brown H, Hien TT, Day N, et al. Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathology and applied neurobiology. 1999;25(4):331–40. doi: 10.1046/j.1365-2990.1999.00188.x. [DOI] [PubMed] [Google Scholar]
- 45.Frei K, Siepl C, Groscurth P, et al. Antigen presentation and tumor cytotoxicity by interferon-gamma-treated microglial cells. European journal of immunology. 1987;17(9):1271–8. doi: 10.1002/eji.1830170909. [DOI] [PubMed] [Google Scholar]
- 46.Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. Journal of immunology. 1990;144(8):2999–3007. [PubMed] [Google Scholar]
- 47.Hochman SE, Madaline TF, Wassmer SC, et al. Fatal Pediatric Cerebral Malaria Is Associated with Intravascular Monocytes and Platelets That Are Increased with HIV Coinfection. mBio. 2015;6(5):e01390–15. doi: 10.1128/mBio.01390-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arnett HA, Mason J, Marino M, et al. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nature neuroscience. 2001;4(11):1116–22. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 49.Probert L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience. 2015;302:2–22. doi: 10.1016/j.neuroscience.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 50.Feintuch CM, Saidi A, Seydel K, et al. Activated Neutrophils Are Associated with Pediatric Cerebral Malaria Vasculopathy in Malawian Children. mBio. 2015;7(1) doi: 10.1128/mBio.01300-15. [DOI] [PMC free article] [PubMed] [Google Scholar]