

Summary
Intimate communication between neural and vascular cells is critical for normal brain development and function. Germinal matrix (GM), a key primordium for the brain reward circuitry, is unique among brain regions for its distinct pace of angiogenesis and selective vulnerability to hemorrhage during development. A major neonatal condition, GM hemorrhage can lead to cerebral palsy, hydrocephalus, and mental retardation. Here we identify a brain region-specific neural progenitor-based signaling pathway dedicated to regulating GM vessel development. This pathway consists of cell surface sphingosine-1-phosphate receptors, an intracellular cascade including Gα co-factor Ric8a and p38 MAPK, and target gene integrin β8, which in turn regulates vascular TGFβ signaling. These findings provide novel insights into region-specific specialization of neurovascular communication, with special implications for deciphering potent early-life endocrine as well as potential gut microbiota impacts on brain reward circuitry. They also identify tissue-specific molecular targets for GM hemorrhage intervention.
Keywords: neurovascular signaling, neural stem cell, angiogenesis, basal ganglia, germinal matrix, sphigosine-1-phosphate, G protein coupled receptor, integrin, TGFβ
eTOC Blurb
Germinal matrix vessels in the brain are particularly fragile and prone to hemorrhage. Ma et al uncover a neurovascular pathway specifically regulating germinal matrix vessel maturation, providing novel molecular-cellular insights and intervention venues.
Introduction
Communication between neural and vascular cells is crucial to normal brain development and function. It takes place in different forms throughout life and regions. As the most energy-intensive organ in the body, the adult brain is known to actively control blood flow to effectively deliver oxygen and nutrients. This is achieved through close coordination between neurons, glia, and vascular cells, which form the neurovascular unit and couple blood flow to levels of neural activity (Hall et al., 2014). In contrast, in the developing brain, neural activity appears to directly regulate angiogenesis (Andreone et al., 2015). To protect neural tissues, the brain also employs sophisticated endothelial junctions and specialized molecular import-export systems collectively known as the blood-brain barrier (BBB). BBB development, embarked on during embryogenesis, is regulated by a complex array of neurovascular signaling pathways (Andreone et al., 2015). It also strongly depends on pericytes, vascular support cells of a distinct origin (Armulik et al., 2011). Meanwhile, to facilitate information exchange, the brain also employs, at strategic locations along the neuraxis, unique endothelial openings for direct communication with the blood (Duvernoy and Risold, 2007). Thus, temporal and spatial specialization is a crucial feature of neurovascular communication. However, the underlying mechanisms have remained poorly understood.
The germinal matrix (GM) is a unique primordial brain tissue well known for its high susceptibility to perinatal hemorrhage. In the US alone, over 12,000 premature infants develop this condition each year (Ballabh, 2010). Among those who survive, at least half develop hydrocephalus, cerebral palsy, and cognitive deficits (Murphy et al., 2002; Pinto-Martin et al., 1999). Studies have uncovered a region-specific vascular fragility where endothelial cells (ECs) show paucity in pericyte coverage and immaturity in basal lamina differentiation, concomitant with a conspicuously low level of regional TGFβ (Ballabh, 2010; Braun et al., 2007). However, despite its persistent incidence and severe outcome, there is at present neither effective treatment nor prevention. The GM gives rise to the striatum, a critical part of the brain circuitry involved in, among others, habit formation, motivation, and reward-based learning. Defective striatal function is implicated in a large number of neuropsychiatric diseases including Huntington’s disease, Parkinson’s disease, schizophrenia, depression, and addiction. The striatum is also one of the few brain structures associated with adult neurogenesis, where the subventricular zone (SVZ) provides a continuous supply of neurons to the olfactory bulb throughout life in most mammals as well as a significant number of neurons to the striatum in humans (Ernst et al., 2014; Ihrie and Alvarez-Buylla, 2011). Interestingly, many striatum-based neurobiological processes are strongly influenced by blood-borne endocrine signals (Jang et al., 2013; Mak et al., 2007; Moaddab et al., 2015; Schoffelmeer et al., 2011), especially during early life (Lajud et al., 2013; Miller and Caldwell, 2015; Panagopoulos and Ralevski, 2014), and by vascular risk factors (Valkanova and Ebmeier, 2013), suggesting a unique role of regional vasculature. Gut microbiota-derived molecules have also been found to translocate into the brain and exert stage- and region-specific impacts on striatum (Arentsen et al., 2017; Diaz Heijtz et al., 2011). Thus, understanding GM angiogenesis may not only provide insights into mechanisms of region-specific neurovascular communication and GM hemorrhage, but may also have implications for understanding brain reward circuit development and function.
Sphingosine-1-phosphate (S1P) is a bioactive phospholipid metabolite with important functions in numerous developmental, physiological and pathological processes (Mendelson et al., 2014; Proia and Hla, 2015). A number of S1P receptors of the G protein coupled receptor (GPCR) family have been identified that mediate S1P function (Lee et al., 1998; Okamoto et al., 1998). Outside the nervous system, S1P signaling regulates, among others, vascular development and stability as well as immune cell trafficking. Inside the nervous system, it regulates neuronal survival, neurite outgrowth, and axon guidance (Herr et al., 2007; Strochlic et al., 2008). It also plays an evolutionarily conserved role in neurotransmitter release and synaptic plasticity (Shen et al., 2014). Importantly, in the striatum and related brain structures, several putative S1P receptors, including GPR3 and GPR6, are highly enriched and regulate development and function of the reward circuitry (Lobo et al., 2007; Tourino et al., 2012).
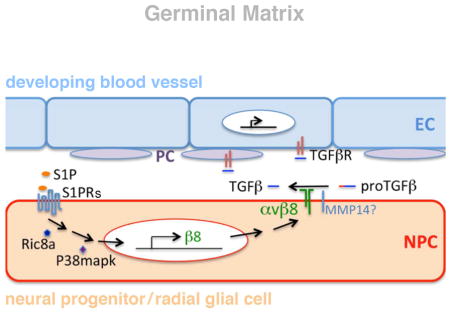
Here we report the identification of a region-specific, neural progenitor-based pathway of brain S1P signaling that selectively regulates angiogenesis in the GM. We first came across evidence for this while studying GPCR function using a ric8a mutation. Ric8a is an essential co-factor for several classes of Gα subunits (Gabay et al., 2011). In invertebrates, Ric8 regulates both asymmetric cell division and neurotransmission (Miller et al., 2000; Morin and Bellaiche, 2011). In mammals, it regulates mitotic spindle orientation (Woodard et al., 2010) as well as Bergmann glia-basement membrane adhesion (Ma et al., 2012). Here we show that in GM neural progenitors, S1PRs, Ric8a, and a downstream p38 MAP kinase form a novel pathway dedicated to regulating angiogenesis in this region. This pathway controls integrin β8 gene expression and consequently local TGFβ activity, which in turn signals to and regulates development of GM vasculature. Disruption of this pathway results in defective angiogenesis, hemorrhage, and enlarged ventricles, phenotypes similar to GM hemorrhage in humans. Restoring activity along this pathway rescues these deficits.
Results
ric8a mutation in neural progenitors results in vascular defects similar to human GM hemorrhage
To understand Ric8a function in brain development, we deleted a conditional ric8a allele (Ma et al., 2012) using nestin-cre, a cre line specifically expressed in neural cell types (Graus-Porta et al., 2001; Ma et al., 2013; Proctor et al., 2005; Stenman et al., 2008; Tronche et al., 1999). We found that ric8a flox/flox; nestin-cre mice (henceforth referred to as ric8a mutants) were born at the expected Mendelian ratio, but died within 12 hours after birth. Severe hemorrhage was observed, with 100% penetrance (10 out of 10 examined), specifically in the GM, but not other regions (Fig. 1A and B). To analyze vascular development, we employed isolectin B4 (IB4). In contrast to the wildtype where no similar structures were observed, ric8a mutant GM showed prominent glomeruloid vascular structures near the ventricle (Fig. 1D and E), indicating defective angiogenesis. Quantification confirmed consistent presence of glomeruloid areas in mutants (Fig. 1G). The vascular network was also severely compromised (Fig. 1D and E), with near 50% reductions in both vessel density (Fig. 1H) and branching frequency (wildtype, 152 ± 4/mm2; mutant, 79 ± 6/mm2; p < 0.001, n = 7). These abnormalities were brain region specific. The vascular network in the neocortex, for example, appeared completely normal, with neither vessel density (Fig. S1A, B, and G) nor branching frequency (wildtype, 76 ± 4/mm2; mutant, 83 ± 2/mm2; p = 0.21, n = 6) significantly affected. To further analyze hemorrhage, we employed Ter119 antibodies. While Ter119 positive red blood cells distributed exclusively within vessels in the wildtype GM (Fig. 1I), they frequently accumulated and formed large clusters outside vessels in mutants (Fig. 1J). Quantification confirmed large areas of hemorrhage (Fig. 1K). Thus, ric8a mutation results in defective angiogenesis specifically in the GM.
Figure 1. Ric8a is required in neural progenitors to regulate vessel development in the LGE.

See also Figure S1.
(A–C) Nissl staining of P0 striata from wildtype (A), ric8a; nestin-cre (B) and ric8a; dlx6a-cre (C) mutant animals. Note hemorrhages in B (arrows).
(D–F) Isolectin B4 (IB4) labeling of vessels in P0 striata of wildtype (D), ric8a; nestin-cre (E) and ric8a; dlx6a-cre (F) animals. Note glomeruloid structures in E (arrows).
(G) Quantification of areas with glomeruloid structures in P0 ventricular zones (VZs) shows significant increases in nestin-cre and foxg1-cre, but not dlx6a-cre mutants. **, p < 0.01; n = 5.
(H) Quantification of vessel density in P0 striata shows severe reductions in nestin-cre and foxg1-cre, but not dlx6a-cre mutants. ***, p < 0.001; n = 7.
(I–K) Hemorrhage in ric8a; nestin-cre mutant striata. Anti-Ter119 labeling of red blood cells (I–J) shows large accumulations outside vessels in mutants (arrows in L). (K) Quantification of hemorrhage areas. **, p < 0.01; n = 6.
(L–N) Cell death in ric8a; nestin-cre mutant striata. Anti-cleaved caspase3 labeling (L–M) shows increased numbers of apoptotic cells in mutants (arrows in M). (N) Quantification of apoptotic cell numbers per field. ***, p < 0.001; n = 5.
Scale bars: 200 μm for A–F, I, J, L, and M.
To corroborate the observed phenotype, we employed foxg1-cre, another cre line specifically targeting forebrain neural cell types (Hebert and McConnell, 2000). Similar to nestin-cre, foxg1-cre mutant GM also consistently showed glomeruloid vascular structures near the ventricle (Fig. 1G, S1I and J). It also showed severe reductions in vessel density (Fig. 1H, S1I and J) and branching frequency (wildtype, 152 ± 4/mm2; mutant, 92 ± 4/mm2; p < 0.001, n = 7). In contrast, no such defects were observed in the neocortex (Fig. S1G). Thus, these results further indicate Ric8a is specifically required in forebrain neural cell types for regulating GM angiogenesis.
Since nestin-cre is expressed in neural progenitors, it also deletes ric8a from post-mitotic neurons. To determine cell type-specific requirement, we next deleted ric8a from GM neurons using dlx6a-cre, a cre line expressed in post-mitotic neurons but not neural progenitors in the ventral forebrain (Monory et al., 2006). In contrast to nestin-cre, dlx6a-cre mutants showed neither hemorrhage nor vascular abnormalities (Fig. 1C and F). Quantification confirmed a lack of glomeruloid structures as well as normal GM vessel density (Fig. 1G and H) and branching frequency (wildtype, 152 ± 4/mm2; mutant, 156 ± 4/mm2; p = 0.58, n = 7). Thus, loss of Ric8a from post-mitotic neurons does not affect GM angiogenesis. Ric8a is likely required in GM neural progenitors for regulating angiogenesis.
The ric8a; nestin-cre mutant phenotype is reminiscent of the neonatal condition of GM hemorrhage in humans, where bleeding in this region results in cell death, gliosis, and severe neurological outcomes (Ballabh, 2010). To evaluate effects of ric8a mutation, we next used an antibody against active Caspase-3 for assessing apoptosis. We found that ric8a mutation resulted in a dramatic increase, by >10 folds, in the number of Caspase3-positive cells (Fig. 1L–N), indicating increased cell death. Consistently, we also observed dramatically increased GFAP staining (Fig. S1C–D), indicating gliosis. Since GM hemorrhage frequently leads to hydrocephalus, we further evaluated ventricular size. We found that ric8a mutation resulted in a nearly 4-fold increase in the size of lateral ventricles (Fig. S1E, F, and H). Thus, ric8a mutation results in perturbed vessel development, hemorrhage, cell death, and ventricular dilation, similar to symptoms of human GM hemorrhage.
ric8a mutation results in primary vascular defects in the embryonic GM
To determine how ric8a mutant defects arise, we next examined vessel development in the embryonic GM (also known as LGE, lateral ganglionic eminence) (Fig. 2A–H). At embryonic day 13.5 (E13.5), the overall vascular network pattern, vessel density, and branching all appeared normal in the mutant GM (Fig. 2M, S2A, B, and D). Beginning at E14.5, however, significant reductions in vessel density (by ~30%) and branching frequency (by >50%) were observed (Fig. 2A, B, M, and S2D). This suggests that Ric8a dependent neurovascular signaling is required as early as E14.5. These defects become even more severe at E15.5, when vessel density and branching were reduced by ~50% and 60%, respectively (Fig. 2M and S2D). Similar to the neonatal brain, we also observed prominent glomeruloid structures and enlarged vessels at E16.5 (Fig. 2C and D). In addition, anti-Ter119 staining revealed clear local hemorrhages at E17.5 (Fig. S2C), indicating compromised vessel integrity during embryogenesis. Thus, ric8a mutation results in defective angiogenesis from early stages of GM development.
Figure 2. Ric8a mutation results in primary vascular defects in the embryonic LGE.

See also Figure S2.
(A–B) IB4 labeling shows LGE vascular defects in ric8a; nestin-cre mutants (B) in comparison to wildtype (A) at E14.5.
(C–D) Anti-laminin staining shows glomeruloid structures (arrows) in mutant LGE (D) at E16.5.
(E–F) Anti-PDGFRβ staining (red) shows similar pericyte (arrows) coverage along vessel (IB4, green) in wildtype (E) and mutant LGE (F).
(G–H) Anti-collagen IV staining shows defective basement membrane maturation in mutants (H) in comparison to wildtype (G).
(I–J) Anti-Nestin staining shows comparable radial glial scaffold between wildtype (I) and mutant (J) LGE at E14.5.
(K–L) Anti-Tubulin βIII (Tuj) staining shows comparable neuronal development between wildtype (K) and mutant (L) LGE at E14.5.
(M) Quantification of vessel density in wildtype and mutant LGE during embryogenesis. **, p < 0.01; n = 3.
(N) Quantification of perciyte density along vessels in wildtype and mutants (p = 0.59, n = 5).
(O) Quantification of collagen IV staining intensity shows significant reductions in mutants. *, p < 0.05; n = 4. AU, arbitrary units.
Scale bar: 200 μm for A, B, K, L, N and O, 100 μm for D, E, J and I, and 50μm in F and G.
Pericytes are major support cells with a key role in vessel integrity (Armulik et al., 2011). To determine potential pericyte defects, we employed PDGFRβ as a marker. We found that pericytes were recruited normally to mutant vessels (Fig. 2E and F) and their density, even though lower, as expected, than that in the neocortex, were quantitatively unaffected (Fig. 2N). This suggests that pericyte recruitment may not play a primary role in the vascular deficits. In contrast, unlike the uniform pattern observed in the wildtype, staining of Collagen IV, a basement membrane component regulated by TGFβ signaling, appeared patchy and discontinuous along mutant vessels (Fig. 2G and H). Quantification further showed a >80% reduction in intensity (Fig. 2O), indicating defective vessel maturation. Interestingly, we also frequently observed enhanced expression of laminin, another basement membrane component, along mutant vessels, suggesting potential compensation (see also Fig. 2C and D). However, the BBB marker Glut1 appeared normal (Fig. S2E and F), suggesting a specific effect of ric8a mutation. Thus, these results indicate that ric8a mutation disrupts GM angiogenesis from early development with a major effect on vessel basement membrane maturation.
To determine potential effects of ric8a mutation on GM neurogenesis, we evaluated expression of a series of neural cell type markers. First, we examined neural progenitor proliferation. We found that at E15.5, mutant GM neural progenitors in the VZ showed a normal pattern of BrdU labeling (Fig. S2I and J). Anti-Nestin staining also revealed an intact radial glial scaffold (Fig. 2I and J), suggesting normal progenitor fate. Furthermore, Tubulin β3, a post-mitotic neuronal marker, was expressed in a pattern indistinguishable from the wildtype (Fig. 2K and L), suggesting unaffected neuronal differentiation. Consistently, Ctip2, a transcriptional factor expressed by GM neurons, was also expressed in a normal pattern (Fig. S2G, and H). Thus, these results indicate that GM neurogenesis is largely normal at the onset of vascular defects. ric8a mutation likely results in primary defects in GM angiogenesis, which in turn leads to secondary defects such as cell death and gliosis.
ric8a mutation results in compromised integrin β8 expression and TGFβ signaling
The vascular phenotypes are reminiscent of those in mice mutant for integrin αv and β8 (McCarty et al., 2002; Zhu et al., 2002). Compelling evidence indicates that a major function of brain integrin αvβ8 is to promote activation of latent TGFβ ligands, which in turn regulate vessel development (Arnold et al., 2012; Hirota et al., 2011; Mu et al., 2002; Yang et al., 2007). To determine whether this process is affected, we first evaluated effects of ric8a mutation on EC TGFβ signaling. To this end, we employed an anti-phospho-Smad3 (p-Smad3) antibody to evaluate the activity of Smad3, a key mediator of TGFβ signaling. We found that p-Smad3 intensity in mutant ECs was notably lower that that in the wildtype (Fig. 3A, A′, B, and B′). Quantification showed a >50% reduction in p-Smad3 positive ECs along GM vessels (Fig. 3G). This indicates that endothelial TGFβ signaling is compromised. Importantly, consistent with the regional specificity of vascular defects (Figs. 1 and S1), endothelial p-Smad3 staining was normal in the mutant cortex (Fig. S3A, B, and G). To directly test levels of active TGFβ, we next employed a cell-based assay where a luciferase reporter is placed under control of a TGFβ-responsive promoter (Abe et al., 1994). Consistent with diminished p-Smad3, mutant GM extracts induced substantially lower levels of luciferase activity (Fig. 3H). In contrast, the total level of (active and latent) TGFβ in GM extracts was unaffected (Fig. S3H). Thus, these results indicate that ric8a mutation results in a severely diminished production of active but not total TGFβ, and consequently compromised TGFβ signaling and defective GM angiogenesis.
Figure 3. Compromised vascular TGFβ signaling and neural progenitor β8 expression in ric8a mutant LGE.

See also Figure S3.
(A–B′) Phospho-Smad3 (red) staining shows reduced TGFβ signaling along LGE vessels (IB4, green) in mutants (B & B′) compared to wildtype (A & A″). Phospho-Smad3 staining alone is shown in A′-B′.
(C–D) Phospho-p38 staining shows reduced p38 activity in LGE VZ in mutants (D) compared to wildtype (C).
(E–F) In situ hybridization shows reduced β8 mRNA levels in LGE VZ in mutants (F) compared to wildtype (E). Note similar levels in cortical VZ (*).
(G) Quantification of phospho-Smad3 EC density shows reductions in mutants. *, p < 0.05; n = 6.
(H) TGFβ activity, assayed using a reporter cell line, is reduced in mutant striata. **, p < 0.01; n = 6.
(I–J) RT-qPCR analysis shows significant reductions of β8 but not αv mRNA in mutant LGE neurospheres. **, p < 0.01; n = 5.
(K–L) RT-qPCR analysis shows no significant changes in β8 mRNA in either mutant cortical (K) or mutant MGE (L) neurospheres (p > 0.58; n = 4-10).
(M–N) RT-qPCR analysis shows no significant changes of β3 (M) or β5 (N) mRNA in mutant cortical LGE neurospheres (p > 0.25; n = 4).
Scale bar: 150μm for A–B and 100 μm for I–L, O, and P.
Integrin αvβ8 directly participates in latent TGFβ activation (Mu et al., 2002). To determine whether changes in αvβ8 expression may be responsible, we next isolated GM neural progenitors for in vitro analysis. We found that size and morphology of mutant GM neurospheres were indistinguishable from the wildtype, suggesting a lack of ric8a requirement for proliferation. This is consistent with normal BrdU incorporation in vivo (Fig. S2). RT-qPCR also showed normal levels of gsx2, a transcription factor specific to GM progenitors (Fig. S3I). In contrast, β8 mRNA levels were severely reduced, by >70%, in ric8a mutant neurospheres (Fig. 3I). Consistently, we also observed loss of β8 from the GM by in situ hybridization (Fig. 3E–F), in contrast to the cortex where it was unaffected (Fig. S3E–F). In contrast to β8, however, mRNA levels of αv were not significantly altered (Fig. 3J). Nor was mmp14, a metalloproteinase involved in αvβ8-dependent TGFβ activation (Mu et al., 2002) (Fig. S3O). Thus, ric8a mutation results in severe loss of integrin β8 expression in GM neural progenitors, which may in turn lead to compromised TGFβ signaling and defective vessel development.
To further evaluate effects of ric8a mutation, we examined β8 mRNA levels in neurospheres derived from the embryonic neocortex and medial ganglion eminence (MGE). RT-qPCR showed that, in contrast to GM, β8 transcript was not significantly affected in either cortical or MGE cells (Fig. 3K and L). This indicates a brain region specific effect, consistent with our observation of vascular and TGFβ signaling defects specific to the GM. Furthermore, mRNA levels of several other known αv partners, including integrin β3, β5 and β6, were not also significantly affected (Fig. 3M and N, S3J). Nor were the levels of several other common factors involved in brain angiogenesis (Fig. S3K–N). Thus, these results indicate that ric8a mutation results in a specific loss of integrin β8 expression from GM but not other regional neural progenitors.
Studies have shown that p38 MAPK pathway is essential for β8 expression in cultured cells (Markovics et al., 2010). To evaluate its potential role in GM progenitors, we examined levels of active p38 using an anti-phospho antibody. We found that in the wildtype brain, GM neural progenitors were strongly positive for phospho-p38. In contrast, in ric8a mutants, the level was severely reduced (Fig. 3C and D). Importantly, phospho-p38 level was not notably affected in cortical neural progenitors (Fig. S3C–D). Thus, Ric8a is specifically required for p38 activation in GM neural progenitors and may regulate integrin β8 through p38 MAPK.
S1P regulates integrin β8 via Ric8a and p38 in GM neural progenitors
Ric8a is an essential co-factor for several classes of Gα proteins (Gabay et al., 2011). In the brain, S1P-GPCR signaling through striatal neurons has been suggested to regulate development and function of the reward circuitry (Lobo et al., 2007; Tourino et al., 2012). Importantly, blood-borne endocrine signals also exert powerful impacts on striatal circuit function (Lajud et al., 2013; Miller and Caldwell, 2015; Panagopoulos and Ralevski, 2014). This raises the question of whether brain S1P signaling may coordinate direct central regulation of the reward circuitry with control of peripheral endocrine access, via regional vasculature. In support, among S1P receptors, S1PR1 is highly expressed in GM neural progenitors (McGiffert et al., 2002). To test the role of S1P, we first examined its effects on β8 in GM neurospheres. RT-qPCR showed that S1P dramatically up-regulated β8 mRNA, by >9 folds, in wildtype neurospheres (Fig. 4A). S1P also up-regulated levels of αv moderately, by ~60% (Fig. S4A). On the other hand, W146, an inhibitor of S1PR1, dramatically decreased β8, by >70%, while having no effects on αv (Fig. 4C). This parallels the distinct effects observed of ric8a mutation on αv and β8 (Fig. 3I and J). Importantly, W146 also abolished S1P induction of β8 (Fig. 4B). Thus, these results indicate that S1P-S1PR1 signaling plays a critical role in regulating β8 mRNA in GM neural progenitors.
Figure 4. S1P regulates integrinβ8 mRNA in LGE neural progenitors in a region-specific manner dependent on Ric8a/p38MAPK.

See also Figure S4.
(A) S1P dramatically up-regulates β8 mRNA in LGE neurospheres, an effect blocked by p38 inhibitor and ric8a mutation. *, p < 0.05; n = 5-7.
(B) S1PR1 antagonist W146 suppresses basal and S1P-induced β8 mRNA in LGE neurospheres. **, p < 0.01; n = 4.
(C) W146 has no effects on αv mRNA in LGE neurospheres (p > 0.05; n = 4).
(D–E) S1P does not induce β8 mRNA in MGE (D) or cortical (E) neurospheres (p > 0.45; n = 4).
(F) s1pr1/ric8a compound mutation suppresses basal and S1P-induced β8 mRNA in LGE neurospheres. *, p < 0.05; **, p < 0.01; n = 3.
(G) S1P treatment (10 minutes) specifically induces p38 phosphorylation in LGE, but not cortical or MGE neurospheres.
To evaluate the role of Ric8a, we next treated ric8a mutant GM neurospheres with S1P. In contrast to wildtype neurospheres, we found S1P fail to up-regulate β8 in mutant cells (Fig. 4A). This indicates that S1P induces β8 through a Ric8a-dependent cascade. As mentioned, p38 MAPK has been found to regulate β8 (Markovics et al., 2010). We also found its activity dependent on Ric8a (Fig. 3C and D). To test p38 requirement, we next employed p38 inhibitor SB202190, which we found abolished β8 induction by S1P (Fig. 4A). Thus, these results indicate that Ric8a and p38 form an intracellular cascade that mediates S1P regulation of β8.
To further evaluate S1P regulation of β8, we determined tissue and target specificity of this pathway. Similar to the region-specific effects of ric8a mutation (Fig. 3I, K, and L), we found that S1P failed to up-regulate β8 in either MGE or cortical progenitors (Fig. 4D and E). Similarly, while S1P induced strong phospho-p38 in GM neurospheres, it failed to do so in cortical and MGE neurospheres (Fig. 4G). Thus, both S1P and Ric8a regulate p38 and β8 in a region-specific manner. Furthermore, S1P had no significant effects on several regional factors, including gsx2 and mash1 (Fig. S4G and H). This demonstrates target gene specificity. Interestingly, although β3 and β6 were not significantly affected, we found S1P also elevate β5 mRNA (Fig. S4B–D). This suggests potentially coordinated gene regulation. Thus, these results indicate that the S1PR-Ric8a-p38 pathway is specifically involved in regulating integrin β8 in GM neural progenitors.
To account for the regional specificity, we examined S1PR1-5 and Ric8a expression across brain regions. We found that, besides S1PR1, several other S1PRs are also expressed and functionally involved in β8 regulation in GM neurospheres (Fig. S4I–J). Consistently, S1P can substantially up-regulate β8, by >4 folds, in the absence of S1PR1 (p < 0.001, n = 3) (Fig. 4F). While expression of these receptors is relatively low in the MGE, it is significant in cortical progenitors. Furthermore, Ric8a is expressed at similar levels across these regions (Fig. S4K). Thus, S1PR1-3 likely all contribute to β8 regulation in GM NPCs, while distinct wiring of other intracellular signaling and/or transcriptional regulators may play a significant role in regional specificity.
S1PR1 and Ric8a act in the same pathway to regulate GM angiogenesis in vivo
Our in vitro results strongly suggest that the S1PR-Ric8a pathway regulates integrin β8 and TGFβ signaling during GM angiogenesis. To determine whether S1PR1 function is required in vivo, we employed mouse genetics. Earlier work has shown that S1PR1 is required in ECs for vessel development in vivo (Allende et al., 2003; Gaengel et al., 2012; Jung et al., 2012). However, its role in neural progenitors has not been specifically determined. To this end, we used nestin-cre to delete s1pr1. We first evaluated effects of s1pr1 mutation on β8 in GM neurospheres. We found that it resulted in a significant, over 60%, reduction (wildtype, 100 ± 7%; mutant, 31 ± 4%; p < 0.001, n = 4), consistent with results using S1PR1 inhibitors (Fig. 4B). The s1pr1 flox/flox; nestin-cre mice (henceforth referred to as s1pr1 mutants) were born at the expected Mendelian ratio and survived into adulthood. At P0, we observed distinct, though subtle, alterations in vessel morphology (Fig. 5A and B). There was also strong trend in increased glomeruloid areas (p = 0.08, n = 5) (Fig. 5D). Furthermore, quantification showed a significant, though modest (~10%), reduction in vessel density (Fig. 5E). However, vessel branching frequency (wildtype, 155 ± 4/mm2; mutant, 159 ± 8/mm2; p = 0.90, n = 6) was not significantly affected. Thus, these results indicate that s1pr1 deletion from neural progenitors significantly affects GM angiogenesis, supporting the interpretation of S1PR1 function in vivo.
Figure 5. S1PR1 and Ric8a act in the same pathway in regulating LGE angiogenesis.

See also Figure S5.
(A–C′) s1pr1 single mutation (s1pr1 flox/flox; nestin-cre) results in subtle (B & B′), while s1pr1/rci8a compound mutation (s1pr1 flox/flox; ric8a flox/+; nestin-cre) results in obvious vessel (IB4, green) defects in P0 striata (C & C′), in comparison to wildtype (A & A′). Note altered vessel morphology in single (arrows in B) and glomeruloid structures in compound mutants (arrows in C).
(D) Quantification of glomeruloid areas shows significant increases in compound mutants. **, p < 0.01; n = 6.
(E) Quantification of vessel density shows severe reductions in s1pr1/ric8a compound mutants to similar those in ric8a single mutants (see Fig. 1H). **, p < 0.01; ***, p < 0.001; n = 17.
(F–H) Hemorrhage in compound mutant striata. Anti-Ter119 labeling of red blood cells (F–G) shows large accumulations outside vessels (arrows in G). (H) Quantification of hemorrhage areas. *, p < 0.05; n = 6.
(I–K) Cell death in compound mutant striata. Anti-caspase3 labeling (I–J) shows large numbers of apoptotic cells in compound mutants (arrows in J). (K) Quantification of apoptotic cell numbers. ***, p < 0.001; n = 5.
Scale bar: 200μm for A–C, F–G, and I–J.
The subtle defects in s1pr1 mutants suggest potential compensation by other receptors, although we found expression of none of the other S1PRs significantly altered despite severe loss of s1pr1 mRNA (Fig. S5A–E). To further determine S1PR1 function, we next attenuated signal transduction along the pathway, by deleting one copy of ric8a gene in s1pr1 conditional mutant background and generated s1pr1 flox/flox; ric8a flox/+; nestin-cre mice (henceforth referred to as s1pr1/ric8a compound mutants). Previously, we had observed ric8a heterozygosity result in a reduction (~66%) in ric8a mRNA (Fig. S5F). Thus, we anticipated that ric8a heterozygosity in s1pr1 mutant background might further compromise this pathway. Indeed, compared to ~60% reduction in s1pr1 single mutants, compound mutation resulted in a more severe, ~80%, reduction in β8 mRNA (Fig. 4F). Importantly, this also resulted in more severe defects. Prominent glomeruloid structures as well as a spare vascular network were consistently observed (Fig. 5C), a phenotype significantly more severe than that in s1pr1 single mutants while similar to that in ric8a single mutants (Fig. 5A and B). Quantification confirmed a significant increase in glomeruloid areas (Fig. 5D) as well as severe reductions in vessel density (Fig. 5E) and branching (wildtype, 155 ± 4/mm2; compound mutant, 79 ± 4/mm2; p < 0.001, n = 6). Similar to ric8a mutants, we also observed GM-specific hemorrhage (Fig. 5F–H). In contrast, no such phenotypes were observed in ric8a flox/+; nestin-cre animals (referred to as ric8a +/− heterozygotes) (Fig. 5D–E. S5J–K). This indicates that the effects arise from genetic interactions between S1PR1 and Ric8a. Furthermore, as in ric8a mutants, we observed elevated apoptosis (Fig. 5I–K) as well as GFAP expression (Fig. S5G–I). Unlike ric8a mutants, however, the compound mutants survived postnatally, likely because of largely normal GPCR signaling in much of the rest of the nervous system. Nevertheless, they displayed a dramatic, close to 4 fold, enlargement of the lateral ventricle (Fig. S5L–N). Thus, these results strongly demonstrate that S1PR1 and Ric8a act in the same pathway in vivo to regulate GM angiogenesis.
p38 MAPK activation rescues ric8a and s1pr1/ric8a compound mutant phenotypes
So far we have shown that disruption of S1PR-Ric8a pathway in neural progenitors results in defective β8 expression, TGFβ signaling, and vascular development in the GM. We have also shown that this pathway regulates β8 through p38 MAPK (Fig. 3C and D, 4A). If the p38-β8 axis is indeed a key mediator, one would predict that activating p38 should induce β8 and rescue vascular defects. To test this, we employed a pharmacological approach. Anisomycin, a cellular stress inducer, is known to activate p38 (Hazzalin et al., 1996). To test its effects, we delivered a low dose of anisomycin at E14.5 and analyzed one day later. We found anisomycin greatly elevate p38 activity in mutant neurospheres, to a level similar to wildtype (Fig. 6A and B, compare to Fig. 3C and D). This demonstrates in vivo efficacy of anisomycin. In vitro, we found anisomycin also dramatically elevate β8 mRNA in wildtype neurospheres (control, 100 ± 10%; treated, 349 ± 44%; p < 0.01, n = 4). Importantly, it similarly elevated β8 in mutant neurospheres, by close to 5 folds (Fig. 6C). This demonstrates p38 efficacy and sufficiency in inducing β8, even in the absence of upstream pathway activity. Furthermore, anisomycin fully restored EC Smad3 activity in mutant GM (Fig. 6D–F). Thus, these results demonstrate that p38 activation is sufficient to induce β8 and restore TGFβ signaling in the absence of upstream signaling.
Figure 6. p38 activation rescues vascular defects in ric8a single and s1pr1/ric8a compound mutants.

See also Figure S6.
(A–B) Treatment with a low dose of anisomycin at E14.5 dramatically increases phospho-P38 (red) levels in mutant LGE (B) at E15.5 in comparison to the untreated (A).
(C) Anisomycin elevates integrin β8 mRNA in mutant LGE neurospheres despite ric8a deficiency. *, p < 0.05; n = 4.
(D–F) Anisomycin restores levels of phospho-Smad3 (red) along LGE vessels (IB4, green) in ric8a mutants (see Fig. 3B for untreated mutants) (D–E). Quantification shows a density of p-Smad3+ ECs indistinguishable from wildtype (F) (p > 0.68; n = 6).
(G, J, M, P) Anisomycin treatment at E14.5 restores vessel morphology (IB4, green) in P0 ric8a single (J) and s1pr1/ric8a compound mutant (M) striata, to levels similar to wildtype (G). Quantification confirms rescue of vessel density (P) (***, p < 0.001; n = 17).
(H, K, N, Q) Anisomycin treatment at E14.5 prevents hemorrhage (Ter119 for red blood cell, red) in P0 ric8a single (K) and s1pr1/ric8a compound mutant (L) striata. Quantification confirms suppression of hemorrhage (Q) (*, p < 0.05; n = 6).
(I, L, O, R) Anisomycin treatment at E14.5 suppresses cell death (caspase 3, red) in P0 ric8a single (L) and s1pr1/ric8a compound mutant (O) striata, to levels similar to wildtype (I). Quantification confirms suppression effects (R) (***, p < 0.001; n = 5).
Scale bars: 200μm in A, B, G, J, M, H, K, and N. 100μm in D, E, I, L, and O.
To determine effects of p38 activation on GM angiogenesis, we next analyzed neonatal brains of both ric8a single and s1pr1/ric8a compound mutants, following treatment at E14.5. We found that anisomycin restored GM vessel morphology in ric8a mutants (Fig. 6J, compare to G). Quantification showed a complete recovery of vessel density (Fig. 6P and S6A) as well as a suppression of glomeruloid areas (Fig. S6B). A similar effect was observed in compound mutants on both vessel density (Fig. 6M and P) and glomeruloid area (untreated mutant, 781 ± 99 μm2; treated mutant, 0 ± 0 μm2; p < 0.001, n = 6). Furthermore, anisomycin eliminated hemorrhage from both classes of mutants (Fig. 6H, K, N and Q), and drastically suppressed apoptosis (Fig. 6I, L, O and R). Lastly, anisomycin suppressed gliosis-associated GFAP expression (Fig. S6C–E). Thus, these results indicate that p38 activation is sufficient to rescue GM angiogenesis and prevent hemorrhage in S1PR1-Ric8a pathway mutants. p38 MAPK and integrin β8 are likely major mediators of brain S1P regulation of GM angiogenesis.
FTY720 rescues s1pr1/ric8a compound mutant phenotypes
FTY720 is a sphingosine analog approved by FDA for treatment of multiple sclerosis (Chun and Brinkmann, 2011). Following phosphorylation in vivo, it acts as an agonist and induces S1P receptor internalization at high dosages, which in turn alters lymphocyte trafficking and attenuates autoimmunity. As mentioned, we found that despite the mutant background, S1P can substantially up-regulate β8, by >4 folds, in s1pr1/ric8a compound mutant neurospheres (Fig. 4F). This indicates that, though attenuated, the S1PR-Ric8a pathway in compound mutants can be activated by receptor engagement. Furthermore, several genes in S1P production and secretion are expressed in GM neural progenitors (Fig. S6F–G). As such, one would predict that at a low dosage, FTY720 may be processed and in turn boost S1PR/Ric8a signaling and rescue GM angiogenesis. To test this, we first evaluated FTY720 efficacy in inducing β8. We found that at 1 μM, FTY720 substantially up-regulated β8, by close to 3 folds in GM neurospheres (Fig. S4E). This indicates FTY720 as an effective S1PR agonist. To test effects in vivo, we next delivered FYT720 at E14.5 and analyzed s1pr1/ric8a compound mutants at P0. We found that FTY720 completely restored GM vessel morphology (Fig. S6H, K, and N). It also eliminated hemorrhage (Fig. S6I, L and O), and drastically suppressed apoptosis (Fig. S6J, M and P). Importantly, no similar rescues were observed in ric8a mutants (Fig. S6Q–R), where the S1P pathway is more severely disrupted (Figs. 3–4). This suggests that FTY720 acts though GM neural progenitors. Thus, these results further implicate the neural progenitor S1P pathway in regulating GM angiogenesis, and identify a drug for potential tissue-specific GM hemorrhage intervention.
Exogenous integrin β8 rescues ric8a mutant vascular defects
Our results above also strongly indicate that integrin β8 is a key mediator of brain S1P regulation of GM angiogenesis. If so, one would also predict that exogenous expression of β8 should rescue GM vessel defects. To test this, we packaged retroviruses expressing either EGFP alone or both EGFP and integrin β8, delivered them into E14.5 lateral ventricles (Fig. S7A–D), and examined effects at P0. We found that expression of EGFP alone had no obvious effects in ether wild type or ric8a mutant brains (Fig. 7A and B). In contrast, β8 expression strongly suppressed glomeruloid areas and restored vessel density in ric8a mutants (Fig. 7D), while having minimal effects on wild type brains (Fig. 7C). Quantification confirmed a near complete suppression of glomeruloid areas (Fig. 7I), as well as restoration of vessel density (Fig. 7J) in ric8a mutants. We also observed a trend of decreased hemorrhage areas (Fig. S7E–H and S7M), as well as a significant reduction in the number of hemorrhage foci (Fig. S7N). These partial effects may result from viral infection of only a subset of neural progenitors, in contrast to drug treatment. Nonetheless, the number of apoptotic figures in ric8a mutants was also significantly suppressed by β8 (Fig. 7E–H and 7K). Furthermore, the severity of gliosis was similarly substantially reduced (Fig. S7I–L and S7O). Thus, these results indicate that integrin β8 is a major mediator of brain S1P regulation of GM angiogenesis.
Figure 7. Exogenous integrin β8 rescues vascular defects in ric8a mutants.

See also Figure S7.
(A–D) Exogenous GFP has no obvious effects on vessel morphology in wildtype striata (A) and does not rescue defects in ric8a mutants (B). Exogenous integrin β8 also has no effects on wildtype striata (C), but rescues mutant defects (D). Note absence of glomeruloid structures in (D) in comparison to (B) (arrows).
(E–H) Exogenous GFP has no obvious effects on apoptosis (arrows) in wildtype striata (E) and does not rescue apoptosis in ric8a mutants (F). Exogenous integrin β8 also has no effects on wildtype striata (G), but suppresses apoptosis in mutants (H).
(I) Quantification of glomeruloid areas shows near complete suppression by β8 expression in ric8a mutants. **, p < 0.01; n = 12.
(J) Quantification of vessel density shows restoration to a level indistinguishable from wildtype by β8 expression in ric8a mutants. **, p < 0.01; n = 10.
(K) Quantification of apoptotic cell number shows significant suppression by β8 expression in ric8a mutants. *, p < 0.05; n = 12.
Scale bars: 200μm in A–D and 100μm in E–H.
Discussion
In contrast to organ-wide processes such as BBB formation, essentially nothing is known about region-specific mechanisms of brain angiogenesis. Among brain regions, the GM is unique for its distinct program of angiogenesis and selective vulnerability to hemorrhage. Here we have elucidated a GM-specific neurovascular pathway through which neural progenitors regulate angiogenesis, a pathway that likely underlies key angiogenic features unique to this region. We have delineated, from cell surface S1PRs to intracellular Ric8a and p38 MAPK, the signaling cascade through which brain S1P regulates integrin β8 expression. We also showed that this cascade interfaces with vessel development through αvβ8-dependent TGFβ activation. Furthermore, we showed disruption of this pathway result in region-specific vascular defects and hemorrhage similar to human GM hemorrhage, and demonstrated p38 and β8 as key mediators of this pathway. Together, these results elucidate a GM-specific neural progenitor pathway dedicated to the regulation of vessel development. This provides mechanistic insights into regional specialization of neurovascular communication during brain angiogenesis. In light of the potential effects on striatal function of blood-borne endocrine factors and gut microbiota-derived molecules, it has also significant implications for better understanding the brain reward circuitry. Clinically, it sheds light on the molecular basis of the unique susceptibility of GM to hemorrhage, and identifies tissue-specific targets for intervention.
Neural progenitor-based regulation of angiogenesis by S1P
Outside the nervous system, S1P signaling is well known for its critical role in angiogenesis. Acting directly on ECs (Allende et al., 2003; Kono et al., 2004), S1P promotes EC survival and migration and enhances cell-cell junctions (Lee et al., 1999; Lee et al., 2001). Blood-borne S1P also suppresses vessel sprouting though inhibiting VEGFR signaling (Ben Shoham et al., 2012; Gaengel et al., 2012; Jung et al., 2012). In this study, we showed that, distinct from these effects, brain S1P regulates GM angiogenesis via a completely different route, through regulating neural progenitor gene expression. This is consistent with the polarized nature of vascular ECs, the receptors on which brain S1P may lack access to. Although their specific roles remain to be further elucidated, we implicated several S1PRs. We also substantially elucidated the molecular cascade through which S1P regulates GM angiogenesis. We determined that a key target gene is integrin β8, which in turn engages TGFβ, a secreted factor critical to angiogenesis throughout the body. Previous studies have shown that a major role of integrin αvβ8 is regulating TGFβ ligand activation (Arnold et al., 2012; Hirota et al., 2011; Mu et al., 2002; Yang et al., 2007). However, how αvβ8 is itself regulated had remained unclear. We showed that in GM neural progenitors β8 activity is regulated by S1P signaling at the mRNA level. This identifies a direct molecular link through which the S1P and TGFβ pathways intersect.
Region-specific angiogenesis in the brain
Brain vessels are well known for organ-wide features including shared origin from outside the neural tube and development of the BBB. These processes are also frequently linked at the molecular level. For example, vessel ingression and BBB formation are both regulated by canonical Wnt/β-catenin signaling, and mutations in this pathway result in widespread deficits in both processes along the neuraxis (Daneman et al., 2009; Stenman et al., 2008; Zhou and Nathans, 2014). This strongly suggests common signaling regulating angiogenesis throughout the brain. Interestingly, recent studies indicate that this situation is more complex. For example, despite convergence on β-catenin, canonical Wnt signaling depends on distinct components, such as Norrin/Tetraspanin12 vs. Wnt7a/b/GPR124, in different regions (Junge et al., 2009; Zhou and Nathans, 2014). Although this may reflect a case of convergent evolution, it also raises the possibility of regional specialization of neurovascular communication, perhaps concordant with regionalization of brain function. Our finding that integrin β8 is differentially regulated along the forebrain dorsoventral axis provides further direct evidence. We found that β8 is regulated by S1P only in the GM, but not in cortical or other regional neural progenitors, even though its function is required in all regions. Disruption of S1P regulation also results in vascular deficits only in the GM. Thus, these findings elucidate a mechanism for region-specific control of brain angiogenesis, through coupling a common, brain-wide regulator to local cell-cell signaling. Our results further suggest that region-specific wiring of intracellular cascades likely plays a major role. It will be interesting to determine how this is achieved and whether similar adaptations may have evolved in other brain regions.
GM-specific neurovascular interaction and striatal reward circuit function
The brain has a complex relationship with the blood. On one hand, it employs BBB to protect neural cells; on the other, it depends on and receives direct input from the blood. Blood-borne endocrine factors have powerful effects on the brain. A well-known example is the hypothalamus, where the vasculature forms specialized openings allowing direct blood-brain communication. As a result, local neurons can directly sense blood-borne signals. For example, pancreatic islet-derived insulin, adipose tissue-derived leptin, and stomach-derived ghrelin all feed directly into the hypothalamic energy-regulating circuit (Sohn et al., 2013). Interestingly, recent studies show that many of these signals also directly regulate neuronal function in the striatum, the GM-derived brain structure involved in habit formation, motivation, and reward-related behaviors. Insulin increases dopamine clearance and modulates impulsive behavior (Schoffelmeer et al., 2011), while ghrelin enhances locomotor activity induced by cocaine (Jang et al., 2013). Several other endocrine factors have also been implicated. Oxytocin, best known for promoting social bonding, robustly activates striatal neurons (Moaddab et al., 2015), while prolactin affects social behavior (Mak et al., 2007). In light of the unique role played by vascular specialization in the hypothalamus, these findings suggest that the unique program of GM angiogenesis may have also evolved to serve the unique neurobiology of striatum. It may, for example, play an as yet unrecognized role in regulating endocrine access to striatal circuitry. Consistent with this, abnormal neonatal oxytocin and prolactin signaling has been found to result in altered social, reproductive, and depressive-like behaviors in adult animals (Lajud et al., 2013; Miller and Caldwell, 2015), reminiscent of the long-lasting effects of neonatal insulin, leptin, and ghrelin on the hypothalamic circuit and adult feeding behavior (Ralevski and Horvath, 2015). Accumulating evidence also implicates ghrelin in mediating effects of drug abuse that lead to addiction (Panagopoulos and Ralevski, 2014). Furthermore, epidemiological studies have discovered a strong relationship between vascular risks and depression (Valkanova and Ebmeier, 2013). In parallel, recent findings indicate that gut microbiota-derived molecules can also translocate into the brain and have life stage- as well as brain region-specific effects on the striatum (Arentsen et al., 2017; Diaz Heijtz et al., 2011). Thus, region-specific neurovascular interaction may have a unique role in the development and function of brain reward circuitry and, as a result, implications for a large number of neuropsychiatric diseases.
GM angiogenesis and GM hemorrhage
The GM is unique in its susceptibility to hemorrhage during the perinatal period, affecting about 1 in 300 births especially among premature infants (Ballabh, 2010). Among those who survive, at least half develop hydrocephalus, cerebral palsy, and cognitive deficits later in life (Murphy et al., 2002; Pinto-Martin et al., 1999). Yet there is neither effective treatment nor effective prevention. Studies of postmortem specimens have uncovered a GM-specific vascular fragility, concomitant with a conspicuously low level of regional TGFβ activity (Braun et al., 2007), suggesting a link to hemorrhage vulnerability. However, the underlying mechanisms had remained obscure. In this study, we have uncovered a GM-specific pathway dedicated to regulating TGFβ ligand activation and regional angiogenesis. We also substantially elucidated the molecular cascade. Most importantly, we found that boosting the activity of this pathway, through manipulating several components along this cascade, is sufficient to rescue GM hemorrhage. We also found that fingolimod, an FDA-approved S1PR agonist (Chun and Brinkmann, 2011), can effectively rescue the phenotypes. These findings thus open up venues for tissue-specific intervention in GM hemorrhage.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhen Huang (zhuang3@wisc.edu). Requests will be handled according to UW-Madison policy regarding MTA and related matters.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
S1pr1 and/or Ric8a conditional alleles were bred with nestin-cre, foxg1-cre, or dlx6a-cre lines, and maintained in a mixed 129/B6 background. Stage-matched animals (from embryos to neonates) were analyzed in blind regarding sex and littermates were used for comparative analysis whenever possible. Animal use was in accordance with institutional and national guidelines and regulations, and approved by IACUC at UW-Madison.
Primary cell cultures
Neural progenitor cells were isolated, in blind with regard to sex, from the cerebral cortices, lateral or medial ganglion eminences of E14.5 mouse embryos. Cells were cultured in serum-free neural stem cell medium containing B27, 10 μg/ml EGF, and 10 μg/ml FGF at 37°C.
METHOD DETAILS
Generation of mouse with ric8a conditional allele
Standard molecular biology techniques were employed for generating the conditional ric8a allele. Briefly, genomic fragments, of 4.5 and 2.5 kilobases and flanking exons 2–4 of the ric-8a locus at the 5′ and 3′ side, respectively, were isolated by PCR using high fidelity polymerases. Targeting plasmid was constructed by flanking the genomic fragment containing exons 2–4 with two loxP sites together with a neomycin positive selection cassette, followed by 5′ and 3′ genomic fragments as homologous recombination arms and a pgk-DTA gene as a negative selection cassette. ES cell clones were screened by Southern blot analysis using external probes at 5′ and 3′ sides. For derivation of conditional allele, the neomycin cassette was removed by crossing to an actin-flpe transgenic line after blastocyst injection and germ line transmission. The primer set for genotyping ric8a conditional allele, which produces a wildtype band of ~110bp and a mutant band of ~200bp, is: 5′-cctagttgtgaatcagaagcacttg-3′ and 5′-gccatacctgagttacctaggc-3′. Animals homozygous for the conditional ric8a allele are viable and fertile, without obvious phenotypes.
Animal Breeding
Ric8a conditional allele was generated as describe above. S1pr1 conditional allele as well as nestin-cre, foxg1-cre, or dlx6a-cre lines were purchased from the Jackson lab (Jax). S1pr1 and/or Ric8a conditional alleles were bred with nestin-cre, foxg1-cre, or dlx6a-cre lines, and maintained in a mixed 129/B6 background. Stage-matched animals were analyzed in blind regarding sex and littermates were used whenever possible.
Immunohistochemistry
Vibratome sections (40–50um) from mouse brains were fixed in 4% paraformaldehyde overnight at 4°C. The following primary antibodies were used at respective dilutions/concentrations: mouse anti-BrdU supernatant (clone G3G4, Developmental Studies Hybridoma Bank (DSHB), University of Iowa, IA; 1:40), mouse anti-Nestin supernatant (DSHB; 1:20), rabbit anti-laminin (Sigma; 1:2000), and rabbit anti-phospho-Smad2/3 (Ser463/465) (41D10; Cell Signaling, 1:200). Biotinylated Isolectin B4 (Vector Laboratories, 1:200), Rat anti-Terr119 (BD Pharmingen, 1:200), Rabbit ant-cleaved caspase 3 (Cell Signaling, 1:300), Rabbit anti-PDGFRβ (Cell Signaling, 1:400), Rabbit anti-Phospho-p38 (Cell Signaling, 1:400), Rabbit anti-GFP (Life Technologies, 1:200), Rabbit anti-GFAP (1:500), Rabbit anti-Glut1(Thermo Scientific, 1:300), FITC and Cy3 conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Peroxidase conjugated secondary antibodies were purchased from Santa Cruz Biotech. Staining procedures were performed as described previously (Ma et al., 2013). Briefly, primary antibodies were incubated at 4 degree for overnight followed by three washes with normal serum, and secondary antibodies were incubated at room temperature for 4 hours followed by DAPI 1: 2000 for 20 mins. For phospho-Smad2/3 staining, we used a tyramide signal amplification (TSA) plus Cy3 kit (PerkinElmer, Waltham, MA, 1:1000) based on manufacturer’s instruction. After secondary antibody and DAPI staining, sections were mounted with Fluoromount G medium (Southern Biotech, Birmingham, AB) and analyzed under a Nikon eclipse Ti microscope or an Olympus confocal microscope.
Neurosphere culture
Cerebral cortices and lateral and medial ganglion eminences were isolated from E14.5 mouse embryos. Tissues were mechanically dissociated with a P1000 micropipette. Dissociated cells were incubated in serum-free neural stem cell medium containing B27, 10 μg/ml EGF, and 10 μg/ml FGF at 37°C. All experiments were performed after ~7 day culture when neurospheres with reasonable size were formed. For S1P- and FTY720 experiments, the neurospheres were starved for 2 to 6 hours with serum-free medium minus EGF and FGF. S1P (1 μM) and FTY720 (1 μM) was then added for 75 minutes and 2hourr, respectively, before RNA isolation. For SB202190 (5 μM), W146 (10 μM), JTE-013 (2 μM), TY52156 (10 μM), and anisomycin (10 nM) experiments, no starvation was required. Cultures were treated overnight (SB202190, W146, JTE-013, TY52156) or for 6 hours (anisomycin). Total RNA was prepared using TRIzol reagents (Invitrogen).
RT-qPCR
Total RNAs were isolated using Trizol (Invitrogen) and cDNAs prepared using a reverse transcription kit and random hexamer primers (Applied Biosystems). qPCR reactions were performed using the GoTaq qPCR master mix (Promega). mRNA levels were normalized to those of GAPDH and the delta-delta CT method used to determine expression level. See Key Resources Table for primer information.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-BrdU supernatant, clone G3G4 | Developmental Studies Hybridoma Bank (DSHB), University of Iowa, IA | Cat # AB_2314035 |
| Mouse anti-Nestin supernatant | Developmental Studies Hybridoma Bank (DSHB), University of Iowa, IA | Cat # AB_2235915 |
| Rabbit anti-laminin | Sigma-Aldrich | Cat # L9393 |
| Rabbit anti-phospho-Smad2/3 (Ser463/465) | Cell Signaling | Cat # #8828 |
| Biotinylated Isolectin B4 | Vector Laboratories | Cat # B-1205 |
| Rat anti-Terr119 | BD Pharmingen | Cat # 553986 |
| Rabbit ant-cleaved caspase 3 | Cell Signaling | Cat # 9664 |
| Rabbit anti-PDGFRβ | Cell Signaling | Cat # 3169 |
| Mouse anti- class III β-Tubilin (Tuj) | Covance | MMS-435P |
| Rabbit anti-Phospho-p38 | Cell Signaling | Cat # 4511 |
| Rabbit anti-GFP | Life Technologies | Cat # A11122 |
| Rabbit anti-Collagen IV | AbD Serotec | Cat # 21501470 |
| Rabbit anti-GFAP | Dako | Cat # Z 0334 |
| Rabbit anti-Glut1 | Thermo Scientific | Cat# RB-9052-P0 |
| Rat anti-Ctip2 | Abcam | Cat # Ab18465 |
| FITC conjugated secondary antibodies | Jackson ImmunoResearch Laboratories | Cat # 111-095-003 |
| Cy3 conjugated secondary antibodies | Jackson ImmunoResearch Laboratories | Cat # 711-165-152 Cat # 111-165-144 |
| FITC conjugated streptavidin | Jackson ImmunoResearch Laboratories | Cat # 016-010-084 |
| Peroxidase conjugated secondary antibodies | Santa Cruz Biotech | Cat # SC-2004 Cat # SC-2005 |
| Alkaline phosphatase-conjugated antibody against digoxygenin | Roche | Cat # 11093274910 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Anisomycin | Cayman Chemical | Cat # 11308 |
| FTY720 | Cayman chemical | Cat # 162359-56-0 |
| S1P | Cayman Chemical | Cat # 62570 |
| W-146 | Cayman Chemical | Cat # 10009109 |
| JTE-013 | Cayman Chemical | Cat # 10009458 |
| TY 52156 | Tocris | Cat # 5328 |
| SB202190 | AdipoGen | Cat # AG-CR1-0028 |
| Integrin β8 probe | Reichardt lab, UCSF | Zhu et al., 2002 |
| Critical Commercial Assays | ||
| Tyramide signal amplification (TSA) plus Cy3 kit | PerkinElmer, Waltham, MA | Cat # NEL744E001KT |
| GoTaq qPCR master mix | Promega | Cat # A6002 #TM318 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| MLEC TGFbeta luciferase reporter cell lines | Dr. Rifkin lab at NYU, New York, USA | Abe et al., 1994 |
| Experimental Models: Organisms/Strains | ||
| Ric8a conditional allele | Our own lab | Ma et al., 2012 |
| S1pr1 conditional allele | JAX | JAX: 019141 Allende et al., 2003 |
| Nestin-cre | JAX | JAX: 003771 Tronche et al., 1999 |
| Foxg1-cre | JAX | JAX: 004337 Hebert and McConnell, 2000 |
| Dlx6a-cre | JAX | JAX: 008199 Monory et al., 2006 |
| Oligonucleotides (see also Table S1) | ||
| GAPDH: F-tgcccccatgtttgtgatg, R-tgtggtcatgagcccttcc | Our own lab | |
| integrin β8: F-ccagtactgtgagaaggatgac, R-agttgacacagtgctgtgctga | Our own lab, this paper | |
| integrin β3: F- tggagacacctgtgtgagaag, R-ttcacctgctcgatgtcatc | Our own lab, this paper | |
| integrin β5: F-tcctgcttcgagagtgagtt, R-ttctccacagtgacactctcc | Our own lab, this paper | |
| integrin β6: F-cgtgtacatctgaggatggag, R-acaggtagcactcgatgcaac | Our own lab, this paper | |
| integrin αv: F- gagtacttctcggtggtcct, R-ggggggctccaataaacaca | Our own lab, this paper | |
| Gsx2: F-gccgccactacctacaacat, R-atgtcgcgatctcgattctc | Our own lab, this paper | |
| MMP14: F-ccctaggcctggaacattct, R-tttgggcttatctgggacag | Our own lab, this paper | |
| Mash1: F-tctcctgggaatggactttg, R-ggttggctgtctggtttgtt | Our own lab, this paper | |
| Ric8a: F-gcactgcgtgatggttgaagc, R-ctggtgcagacgtttctctagg | Our own lab, this paper | |
| Recombinant DNA | ||
| Integrin β8 IMAGE cDNA clone | Transomic | |
| Software and Algorithms | ||
| N/A | ||
| Other | ||
| N/A | ||
In situ hybridization
E14.5 brains were dissected and fixed in 4% paraformaldehyde in DEPC-PBS at 4°C overnight. The brains were then treated with 10μg/ml proteinase K for 40–60 minutes at room temperature, followed by pre-hybridization at 65°C to permeabilize tissues and to minimize background staining. Probes were then added and hybridized at 65°C overnight. After extensive washes (6 washes, 1–1.5hr per wash), signals were visualized using and antibody against digoxygenin (Roche). The integrin β8 probe plasmids were obtained from the lab of Dr. L. F. Reichardt, UCSF.
Pharmacology
For drug injection, pregnant females (14 days after fertilization) were intraperitoneally injected with chemical compounds: anisomycin (0.1mg/kg body weight (gbw)), or FTY720 (0.3mg/kg body weight, gbw). Embryos or pups were collected for analyses as described in the Results section. BrdU was injected at 100 μg/gbw for 4 hours before embryos are collected for cell proliferation analysis.
In utero lateral ventricle viral injection
Timed pregnant mice were anesthetized using isoflurane (1–3% for induction, 0.5–3% for maintenance). Preemptive antibiotic and analgesic were then administered subcutaneously (s.c.), with the dose depending on the weight of the animal: Meloxicam (1–2 mg/kg s.c. of 5mg/ml) and Buprenorphine (0.05–.1 mg/kg s.c. 0.3 mg/ml). Once the animal was anesthetized, it was kept warm on a heating pad. On each eye, a drop of eye ointment is applied. The abdomen near the site of incision was then sprayed with 70% ethanol and then wiped with betadine to sterilize. A toe pinch was used to ensure that the mouse was the correct anesthetic plane and is ready for surgery. A small sagittal incision was made across the abdomen skin exposing the body wall. A similar incision was made in the body wall taking care to avoid blood vessels by lifting the skin and body wall. The uterine horns was gently pulled from the abdomen using blunt forceps and placed on the gauze pad. Hydration of the tissues, organs, and embryos was maintained by regular application of sterile saline (0.9% NaCl). Embryos were injected with ~1 μl of virus plus fast green dye (sterilized by passing through a 0.22 μm filter) into the lateral ventricle of the brain on one side for each fetus. After the injection, the uterine horns were gently pushed back into the abdomen using two blunt forceps. The body wall was then closed with absorbable sutures. The skin was closed with non-absorbable or absorbable sutures and antibiotic ointment was applied to the incision. The mouse was kept in the surgery room on a heating pad to recover, and monitored once every 24 hours for 3 days for signs of pain, discomfort or infection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Vessel morphology and immunofluorescence were quantified using Nikon NIS-Elements BR 3.0 and Image J software as previously published (Ma et al. 2013). Student’s t test and ANOVA followed by Tukey’s post hoc test were employed for data involving two and more conditions, respectively. p < 0.05 is considered statistically significant in all cases. Sample sizes and p-values are included in the figure legends. Error bars represent SEM.
Supplementary Material
Highlights.
GPCR signaling disruption in neural progenitors leads to germinal matrix hemorrhage
GM neural progenitors regulate regional angiogenesis through integrin β8 and TGFβ
Sphingosin-1-phosphate signaling via GPCRs regulates neural β8 and GM angiogenesis
Pharmacological and genetic activation of S1P pathway prevents GM hemorrhage
Acknowledgments
We thank Dr. L. F. Reichardt (Simons foundation) for critical reading of the manuscript. This work was supported in part by a Basil O’Connor award and a National Institute of Health (NIH) grant NS076729 to Z. H. S.M. was supported in part by an AHA pre-doctoral fellowship 14PRE19080006. We thank Dr. D. Rifkin (NYU) for the TGFβ reporter cell line, Dr. L. F. Reichardt for β8 in situ probes, Dr. E. Bresnick (UW-Madison) for access to a qPCR machine, and Drs. K. Taylor and E. Dent (UW-Madison) for assistance with mouse surgery.
Footnotes
Author Contributions
Z. H. and S. M. conceived the project and designed the experiments. Z. H. generated the ric8a allele, S. M. performed most experiments, D. S. and P. K. T. performed β8 rescue and other experiments, all participated in data analysis, and Z. H. and S. M. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Analytical biochemistry. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- Andreone BJ, Lacoste B, Gu C. Neuronal and vascular interactions. Annual review of neuroscience. 2015;38:25–46. doi: 10.1146/annurev-neuro-071714-033835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arentsen T, Qian Y, Gkotzis S, Femenia T, Wang T, Udekwu K, Forssberg H, Diaz Heijtz R. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Molecular psychiatry. 2017;22:257–266. doi: 10.1038/mp.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Developmental cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Arnold TD, Ferrero GM, Qiu H, Phan IT, Akhurst RJ, Huang EJ, Reichardt LF. Defective retinal vascular endothelial cell development as a consequence of impaired integrin alphaVbeta8-mediated activation of transforming growth factor-beta. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:1197–1206. doi: 10.1523/JNEUROSCI.5648-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatric research. 2010;67:1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Shoham A, Malkinson G, Krief S, Shwartz Y, Ely Y, Ferrara N, Yaniv K, Zelzer E. S1P1 inhibits sprouting angiogenesis during vascular development. Development. 2012;139:3859–3869. doi: 10.1242/dev.078550. [DOI] [PubMed] [Google Scholar]
- Braun A, Xu H, Hu F, Kocherlakota P, Siegel D, Chander P, Ungvari Z, Csiszar A, Nedergaard M, Ballabh P. Paucity of pericytes in germinal matrix vasculature of premature infants. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:12012–12024. doi: 10.1523/JNEUROSCI.3281-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Brinkmann V. A mechanistically novel, first oral therapy for multiple sclerosis: the development of fingolimod (FTY720, Gilenya) Discovery medicine. 2011;12:213–228. [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernoy HM, Risold PY. The circumventricular organs: an atlas of comparative anatomy and vascularization. Brain research reviews. 2007;56:119–147. doi: 10.1016/j.brainresrev.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, Possnert G, Druid H, Frisen J. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156:1072–1083. doi: 10.1016/j.cell.2014.01.044. [DOI] [PubMed] [Google Scholar]
- Gabay M, Pinter ME, Wright FA, Chan P, Murphy AJ, Valenzuela DM, Yancopoulos GD, Tall GG. Ric-8 proteins are molecular chaperones that direct nascent G protein alpha subunit membrane association. Science signaling. 2011;4:ra79. doi: 10.1126/scisignal.2002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, Niaudet C, Hagikura K, Lavina B, Muhl L, Hofmann JJ, Ebarasi L, Nystrom S, Rymo S, Chen LL, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Developmental cell. 2012;23:587–599. doi: 10.1016/j.devcel.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazzalin CA, Cano E, Cuenda A, Barratt MJ, Cohen P, Mahadevan LC. p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Current biology: CB. 1996;6:1028–1031. doi: 10.1016/s0960-9822(02)00649-8. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Developmental biology. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Herr DR, Grillet N, Schwander M, Rivera R, Muller U, Chun J. Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:1474–1478. doi: 10.1523/JNEUROSCI.4245-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S, Liu Q, Lee HS, Hossain MG, Lacy-Hulbert A, McCarty JH. The astrocyte-expressed integrin alphavbeta8 governs blood vessel sprouting in the developing retina. Development. 2011;138:5157–5166. doi: 10.1242/dev.069153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrie RA, Alvarez-Buylla A. Lake-front property: a unique germinal niche by the lateral ventricles of the adult brain. Neuron. 2011;70:674–686. doi: 10.1016/j.neuron.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang JK, Kim WY, Cho BR, Lee JW, Kim JH. Microinjection of ghrelin in the nucleus accumbens core enhances locomotor activity induced by cocaine. Behavioural brain research. 2013;248:7–11. doi: 10.1016/j.bbr.2013.03.049. [DOI] [PubMed] [Google Scholar]
- Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Developmental cell. 2012;23:600–610. doi: 10.1016/j.devcel.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. The Journal of biological chemistry. 2004;279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- Lajud N, Gonzalez-Zapien R, Roque A, Tinajero E, Valdez JJ, Clapp C, Torner L. Prolactin administration during early postnatal life decreases hippocampal and olfactory bulb neurogenesis and results in depressive-like behavior in adulthood. Hormones and behavior. 2013;64:781–789. doi: 10.1016/j.yhbeh.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Paik JH, Sapkota GP, Ancellin N, Chae SS, Wu M, Morales-Ruiz M, Sessa WC, Alessi DR, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Molecular cell. 2001;8:693–704. doi: 10.1016/s1097-2765(01)00324-0. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S, Hla T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Cui Y, Ostlund SB, Balleine BW, Yang XW. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nature neuroscience. 2007;10:1395–1397. doi: 10.1038/nn1987. [DOI] [PubMed] [Google Scholar]
- Ma S, Kwon HJ, Huang Z. Ric-8a, a guanine nucleotide exchange factor for heterotrimeric G proteins, regulates bergmann glia-basement membrane adhesion during cerebellar foliation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:14979–14993. doi: 10.1523/JNEUROSCI.1282-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Kwon HJ, Johng H, Zang K, Huang Z. Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS biology. 2013;11:e1001469. doi: 10.1371/journal.pbio.1001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak GK, Enwere EK, Gregg C, Pakarainen T, Poutanen M, Huhtaniemi I, Weiss S. Male pheromone-stimulated neurogenesis in the adult female brain: possible role in mating behavior. Nature neuroscience. 2007;10:1003–1011. doi: 10.1038/nn1928. [DOI] [PubMed] [Google Scholar]
- Markovics JA, Araya J, Cambier S, Jablons D, Hill A, Wolters PJ, Nishimura SL. Transcription of the transforming growth factor beta activating integrin beta8 subunit is regulated by SP3, AP-1, and the p38 pathway. The Journal of biological chemistry. 2010;285:24695–24706. doi: 10.1074/jbc.M110.113977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, Rubin K, Shani M, Dvorak HF, Wolburg H, Bader BL, et al. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Molecular and cellular biology. 2002;22:7667–7677. doi: 10.1128/MCB.22.21.7667-7677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGiffert C, Contos JJ, Friedman B, Chun J. Embryonic brain expression analysis of lysophospholipid receptor genes suggests roles for s1p(1) in neurogenesis and s1p(1–3) in angiogenesis. FEBS letters. 2002;531:103–108. doi: 10.1016/s0014-5793(02)03404-x. [DOI] [PubMed] [Google Scholar]
- Mendelson K, Evans T, Hla T. Sphingosine 1-phosphate signalling. Development. 2014;141:5–9. doi: 10.1242/dev.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, McManus JR, Rand JB. RIC-8 (Synembryn): a novel conserved protein that is required for G(q)alpha signaling in the C. elegans nervous system. Neuron. 2000;27:289–299. doi: 10.1016/s0896-6273(00)00037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TV, Caldwell HK. Oxytocin during Development: Possible Organizational Effects on Behavior. Frontiers in endocrinology. 2015;6:76. doi: 10.3389/fendo.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moaddab M, Hyland BI, Brown CH. Oxytocin excites nucleus accumbens shell neurons in vivo. Molecular and cellular neurosciences. 2015;68:323–330. doi: 10.1016/j.mcn.2015.08.013. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Bellaiche Y. Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Developmental cell. 2011;21:102–119. doi: 10.1016/j.devcel.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. The Journal of cell biology. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BP, Inder TE, Rooks V, Taylor GA, Anderson NJ, Mogridge N, Horwood LJ, Volpe JJ. Posthaemorrhagic ventricular dilatation in the premature infant: natural history and predictors of outcome. Archives of disease in childhood Fetal and neonatal edition. 2002;87:F37–41. doi: 10.1136/fn.87.1.F37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Takuwa N, Gonda K, Okazaki H, Chang K, Yatomi Y, Shigematsu H, Takuwa Y. EDG1 is a functional sphingosine-1-phosphate receptor that is linked via a Gi/o to multiple signaling pathways, including phospholipase C activation, Ca2+ mobilization, Ras-mitogen-activated protein kinase activation, and adenylate cyclase inhibition. The Journal of biological chemistry. 1998;273:27104–27110. doi: 10.1074/jbc.273.42.27104. [DOI] [PubMed] [Google Scholar]
- Panagopoulos VN, Ralevski E. The role of ghrelin in addiction: a review. Psychopharmacology. 2014;231:2725–2740. doi: 10.1007/s00213-014-3640-0. [DOI] [PubMed] [Google Scholar]
- Pinto-Martin JA, Whitaker AH, Feldman JF, Van Rossem R, Paneth N. Relation of cranial ultrasound abnormalities in low-birthweight infants to motor or cognitive performance at ages 2, 6, and 9 years. Developmental medicine and child neurology. 1999;41:826–833. doi: 10.1017/s0012162299001644. [DOI] [PubMed] [Google Scholar]
- Proctor JM, Zang K, Wang D, Wang R, Reichardt LF. Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. The Journal of clinical investigation. 2015;125:1379–1387. doi: 10.1172/JCI76369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralevski A, Horvath TL. Developmental programming of hypothalamic neuroendocrine systems. Frontiers in neuroendocrinology. 2015;39:52–58. doi: 10.1016/j.yfrne.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Schoffelmeer AN, Drukarch B, De Vries TJ, Hogenboom F, Schetters D, Pattij T. Insulin modulates cocaine-sensitive monoamine transporter function and impulsive behavior. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:1284–1291. doi: 10.1523/JNEUROSCI.3779-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Giordano F, Wu Y, Chan J, Zhu C, Milosevic I, Wu X, Yao K, Chen B, Baumgart T, et al. Coupling between endocytosis and sphingosine kinase 1 recruitment. Nature cell biology. 2014;16:652–662. doi: 10.1038/ncb2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Elmquist JK, Williams KW. Neuronal circuits that regulate feeding behavior and metabolism. Trends in neurosciences. 2013;36:504–512. doi: 10.1016/j.tins.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- Strochlic L, Dwivedy A, van Horck FP, Falk J, Holt CE. A role for S1P signalling in axon guidance in the Xenopus visual system. Development. 2008;135:333–342. doi: 10.1242/dev.009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourino C, Valjent E, Ruiz-Medina J, Herve D, Ledent C, Valverde O. The orphan receptor GPR3 modulates the early phases of cocaine reinforcement. British journal of pharmacology. 2012;167:892–904. doi: 10.1111/j.1476-5381.2012.02043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature genetics. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Valkanova V, Ebmeier KP. Vascular risk factors and depression in later life: a systematic review and meta-analysis. Biological psychiatry. 2013;73:406–413. doi: 10.1016/j.biopsych.2012.10.028. [DOI] [PubMed] [Google Scholar]
- Woodard GE, Huang NN, Cho H, Miki T, Tall GG, Kehrl JH. Ric-8A and Gi alpha recruit LGN, NuMA, and dynein to the cell cortex to help orient the mitotic spindle. Molecular and cellular biology. 2010;30:3519–3530. doi: 10.1128/MCB.00394-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. The Journal of cell biology. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nathans J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Developmental cell. 2014;31:248–256. doi: 10.1016/j.devcel.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.