

Abstract
Ageing is the main risk factor for human neurological disorders. Among the diverse molecular pathways that govern ageing, epigenetics can guide age-associated decline in part by regulating gene expression and also through the modulation of genomic instability and high-order chromatin architecture. Epigenetic mechanisms are involved in the regulation of neural differentiation as well as in functional processes related to memory consolidation, learning or cognition during healthy lifespan. On the other side of the coin, many neurodegenerative diseases are associated with epigenetic dysregulation. The reversible nature of epigenetic factors and, especially, their role as mediators between the genome and the environment make them exciting candidates as therapeutic targets. Rather than providing a broad description of the pathways epigenetically deregulated in human neurological disorders, in this review, we have focused on the potential use of epigenetic enzymes as druggable targets to ameliorate neural decline during normal ageing and especially in neurological disorders. We will firstly discuss recent progress that supports a key role of epigenetic regulation during healthy ageing with an emphasis on the role of epigenetic regulation in adult neurogenesis. Then, we will focus on epigenetic alterations associated with ageing-related human disorders of the central nervous system. We will discuss examples in the context of psychiatric disorders, including schizophrenia and posttraumatic stress disorders, and also dementia or Alzheimer’s disease as the most frequent neurodegenerative disease. Finally, methodological limitations and future perspectives are discussed.
Keywords: Epigenetics, DNA methylation, Histone modifications, Epidrug, Neurogenesis, Neurodegeneration, Psychiatric disorders
Background
Ageing, defined as the progressive functional decline of organisms at molecular, cellular and physiological level, is the main risk factor for major human diseases such as cancer, cardiovascular diseases or neurological disorders [1]. As a part of natural ageing, the human brain and nervous system go through natural changes that result in neuronal death and decline of memory, cognitive and coordination processes, among other functional impairments. The effects of ageing on the central nervous system are widespread, have multiple aetiologies and have different clinical manifestations depending on the person.
We must highlight that age-associated decline is part of the natural lifespan; however, this loss of neural function can also be associated with pathogenic conditions in a broad range of human disorders, including neurodevelopmental diseases (e.g. Rett syndrome), neurodegenerative disorders (dementia, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, etc.) or changes in behaviour leading to psychiatric diseases. Most of these complex disorders are the result of alterations in multiple molecular pathways together with the interaction of environmental factors.
It is clear that accumulating evidence of how these ageing-associated processes occur at molecular level will provide promising “druggable” targets for therapy in ageing-related disorders. In this way, much attention is paid to the molecular basis of ageing using many experimental cellular contexts, such as telomere shortening, DNA damage, loss of proteostasis and degeneration of cell or organ structures [1]. Nowadays, it is also widely accepted that changes in epigenetic modifications are a phenomenon associated with ageing throughout the control of gene expression and genomic instability [2, 3]. The dynamic and reversible nature of epigenetic alterations makes epigenetic mechanisms optimal targets for the development of novel treatment strategies in neurological disorders, a strategy that is currently used in the clinical management of other human complex disorders such as cancer [4].
In this review, we will summarize our current knowledge about the involvement of epigenetic factors in normal ageing (ageing-associated epigenome) and those environmental factors influencing the epigenetic landscape of an organism and that can be more easily modified with lifestyle (e.g. diet, stress or smoking). Since the use of agents and manipulations that boost neurogenesis is an important strategy to improve neurological function in human disorders with neural decline, we will also summarize the current uses of epigenetic-based treatments to improve adult neurogenesis. Additionally, we examine the preclinical studies about the use of pharmacological treatments to reverse the epigenetic signature and ameliorate neural dysfunction in human disorders, including common psychiatric disorders (schizophrenia and posttraumatic stress disorder), dementia and the most well-known neurological disorder, Alzheimer’s disease. In recent years, a lot of attention has been paid to the role of non-coding RNAs (ncRNAs) in the neural differentiation processes but also in the ethiopathology of neurological disorders [5]. However, due to current lack of ncRNA-based therapeutic strategies, we will focus on evidence accumulated with treatments targeting DNA methylation (and DNA demethylation) and histone modifications. In most cases, especially in psychiatric disorders, knowledge is still in its infancy and many questions about the epigenetic basis underlying the disease are yet to be addressed. Neurodegenerative diseases are a complex heterogeneous group of diseases, and the comprehensive understanding of the mechanisms involved in their initiation and progress should overpass some limitations in the research strategies. Some improvements are still needed, including increased sample size of the cohorts, more appropriate animal models for the diseases, multicentric validations or multivariable analysis. Elucidating the epigenetic signatures of brain diseases is imperative to developing and applying epigenetics-driven therapeutic approaches.
Main text
Age-related epigenetics
Studies of the epigenome have outlined a chromatin signature during human normal ageing. It is described that there is a general loss of histones [6] together with a massive alteration in the histone modification patterns. The global trends of the ageing- associated histone code are a loss of repressive marks and a gain of activating transcriptional marks, both actions resulting in gain and loss of heterochromatin regions. As examples, redistribution of the active histone mark H3K4me3 over tissue-specific genes [7] or gain of H4K16ac and H3K56ac [8] are hallmarks of ageing. As a consequence of the histone switch, widespread transcriptional deregulation occurs that includes global amplification of canonical transcripts [2]. Additionally, there are global and local changes of the methylome during mammalian ageing [9, 10]. Decreased CpG methylation was found in advanced aged blood samples, mainly affecting methylation spots into enhancers. In contrast, gain of methylation was also observed at specific loci at CpG islands [10], and interestingly in loci near tissue-specific genes, genes coding for transcription factors or genes associated with differentiation and development [2]. An important consideration for the role of CpG methylation in ageing is its tissue specificity, since methylation loci can vary from one cell type to another. In spite of potential divergences among tissues should be considered, a slow and gradual loss of genome-wide methylation (global hypomethylation) together with gain of methylation at specific loci (specific hypermethylation) during ageing was also reported in brain human tissues [11]. In summary, the altered pattern of CpG methylation during one’s lifespan is congruent with the changes in histone modifications marks at specific transcriptional networks. It remains to be elucidated what the roles of these specific genes are in the ageing process.
The relation between the effect of genetic variation and epigenetics should also be considered. The genetics underlying longevity have been widely explored [12, 13], but non-genetic contribution can be a confounding factor in these studies. Monozygotic twins are exceptional models for assessing the epigenetic effects of ageing on identical genomes [14, 15]. These studies showed that the epigenetic discordance between twins increased with ageing and support the idea that epigenetic drift is overcome by environmental factors during lifespan. Finally, it is important to consider that at present, it is unclear whether changes in epigenetic marks altered the expression of genes associated with ageing or whether the disturbance of molecular pathways during ageing results in epigenetic changes at higher scales in the genome. In other words, it is still unknown if epigenetic changes are drivers or just consequence of the ageing process. Recent technological advances provide useful tools for addressing these challenges, such as the use of CRISPR/dCas9 for enzyme targeting. In particular, epigenetic editing for rewriting the epigenome at specific loci will greatly contribute to the deciphering of the causative versus correlative changes in ageing [16].
Epigenetic as a bridge between environmental signals and genome response during early life and ageing
Internal and external environmental factors that are well-known contributors of ageing can be integrated into genome response by means of epigenetic responses (Fig. 1). Alterations in epigenetic modifications can be associated with caloric restriction, lower basal metabolic rate or stress, among others [17]. An increasing number of studies on the influence of the environment during in utero and in early stages of development have provided evidence of how external stimuli during stages of early life, such as exposure to toxins or nutritional deficiencies, govern the extent of disease vulnerability [18]. It has been proposed that environmental factors may interact with specific loci thereby modifying their expression through epigenetic mechanisms and increasing disease susceptibility in later life [19]. The effect of fetal nutrition, which depends on nourishment provided by the maternal system, has been widely described both in animal and human models [19, 20], and a positive relation between maternal diet and neurodegeneration has been supported in some human studies. The offspring of women exposed to the Dutch famine in 1944–1945 had significantly increased risk of several disorders, including schizophrenia [21, 22]. Although several components of the diet can mediate this effect, an association between vitamin B12 and folic acid supplementation in the mother’s diet during pregnancy and defects on the myelination in the nervous system of offspring has been described [23]. In a similar manner, the negative effect of fetal exposure to factors such as tobacco, alcohol consumption, stress or infections had also been investigated in the context of risk to neurological disorders in the offspring [24–26]. How epigenetics modulate changes in brain development and functions even across generations (the named “transgenerational inheritance”) will be the subject of future research in the field and surely will contribute to improve strategies supporting healthy development.
Fig. 1.
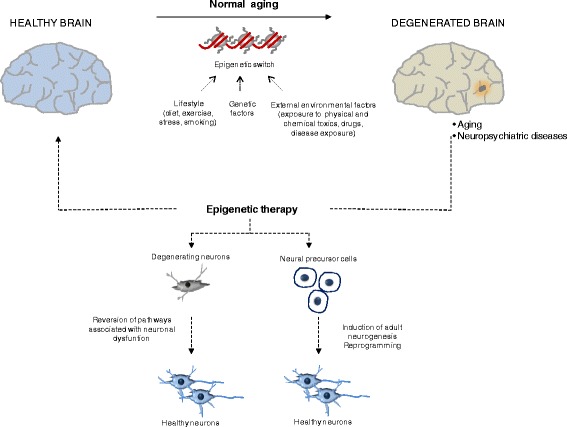
Epigenetic therapy in neuropsychiatric disorders. A combination of external and internal factors can induce epigenetic changes in the normal healthy brain during ageing but also under pathological conditions, including neurodegenerative disorders or psychiatric diseases. Epigenetic therapy, such as pharmacological treatment with DNA-demethylating drugs or HDAC inhibitors, can reverse the distorted epigenetic modifications and induce specific gene expression programs. Two main strategies can be achieved: first, the use of epigenetic drugs can induce adult neurogenesis by enhancing cellular reprogramming from neural precursor cells and, second, changes in epigenetic modifications can reverse gene expression of genes involved in neuronal dysfunction and relevant for the disease
It must be considered that embryonic development is the most susceptible period due to the high number of cell replication events and epigenetic drifts that take place during differentiation [18]. In postnatal development, several observations can also support the environment- epigenome connection. Diet is probably the best example of external stimuli affecting the epigenome and the ageing phenotype. Restrictions in nutrient intake (below the levels of malnutrition) extend lifespan and delay ageing in many organisms from yeast to humans [27]. One of the major factors activated under conditions of caloric restriction is Histone Deacetylase (HDAC) protein Sirtuin 1 (SIRT1) [3, 28]. The beneficial effects of activation of SIRT1 are exerted, at least in part, by improving mitochondrial function [29], but also because SIRT1 expression after caloric restriction results in decreased levels of inflammation-associated genes [28]. The activity of the sirtuin family of histone deacetylates is dependent on the cofactor NAD+ and NAD+ levels decline with age. Modulation of NAD+ availability, for example as a result of NAD+ donors in the diet, can result in altered SIRT1 function [30] and contrasts ageing effects. High NAD+ levels are also a consequence of an oxidative metabolic state induced by caloric restriction [3] and ameliorate ageing. Caloric restriction has been also associated with 5-methylcytosine contents and Dnmt3 activity in the hippocampus of mice models of ageing [31]. Similarly, it has also been shown that high nutrient intake mimics the CpG methylation profile of ageing cells in the liver [32].
Life stress has been also associated with health span and longevity and is a risk factor for late-life neurological and metabolic disorders [17]. Telomere shortening has been described in association with adult stress conditions [33]. In addition, life stressors induce alterations in CpG methylation in early stages of development in humans, affecting specific genes such as glucocorticoid stress response-mediators [34, 35]. Glucocorticoids can exert two complementary actions to decrease CpG methylation. On the one hand, they decrease the expression of the DNA methyltransferase DNMT1 in neurons [36], and on the other hand, exposure to glucocorticoids may upregulate the expression of DNA demethylases from the ten-eleven translocation (TET) family [37].
Healthy and non-healthy habits can also ameliorate or accelerate, respectively, ageing. Little is known about the underlying molecular mechanisms of the beneficial effect of exercise during ageing, but a few reports about the role of epigenetics exist. Decreased levels of the histone deacetylases HDAC4 and HDAC5 and increased levels of acetylated H3K36 were detected in human skeletal muscle in the brain after exercise [38, 39]. On the other hand, tobacco exposure can alter the CpG methylation status of genes associated with cellular homeostasis and development of ageing-associated disorders [40]. Furthermore, decreased H4K16Ac and increased H3K27me3 levels at specific locus have been also described in smokers [41]. Alcohol inhibition of the methionine synthase pathway, and consequently the S-adenosylmethionine levels, may be associated with hypomethylation of LINE sequences related to alcohol consumption [42].
Again, causal evidence that can directly link environmental factors and specific chromatin reordering through epigenetic mechanisms, and in consequence, affect ageing and health span, is still missing. Further research will open new avenues of chromatin-based strategies to delay (or even reverse) ageing and ageing-related diseases by manipulation of lifestyle conditions.
Epigenetic contributions to neurogenesis induction during ageing
One of the main end-point manifestations associated with ageing is loss of neuronal function that leads to impairment of memory and cognition. As aforementioned, epigenetic alterations contribute to the natural process of “healthy” ageing; however, the reversible nature of epigenetic marks adds extra value to them as potential targets for ameliorating neurological decline during ageing.
Although still far from clinical use, improving adult neurogenesis is a promising strategy to treat neurological disorders (Fig. 1). In the adult vertebrate brain, the formation of new neurons takes place in a specific population of cells referred as neural stem progenitor cells (NSPCs). Neurogenesis is generally not a frequent process under normal physiological conditions, but it is described to be induced after brain injury [43]. NSPCs can be found in local niches of the brain, such as the subventricular zone (SVZ) of the lateral ventricle or the subgranular zone (SGZ) of the dentate gyrus (DG) of the hippocampus [44]. Neurogenesis at SGZ has received a lot of attention due to its involvement in cognitive functions such as memory consolidation. Granule cells, the providers of excitatory input to the pyramidal cells of CA3 region, are the unique type of neuron that is generated from the NSPCs in the SGZ under physiological conditions [43]. Although it is still unclear how intrinsic and extrinsic mechanisms induce adult neurogenesis, different signals have been identified including presence of specific cytoplasmatic factors (growth factors, neurotrophins, cytokines and hormones, among others), transcriptional factor network and epigenetic regulators [44, 45].
In recent years, further evidence has demonstrated the role of epigenetic factors in the maintenance of neural stem cell renewal and also in the induction of new mature neurons. Although the contribution of ncRNAs to neural differentiation has been reported in different experimental systems, and especially for microRNAs, knowledge about their functional relevance is still in its infancy [46] and their therapeutic potential is largely unexplored. By contrast, the role of CpG methylation and histone modifications in neuronal cell fate and stem cell self-renewal has been widely explored [47–50]. For example, well-known pluripotency genes are epigenetically inactivated by CpG promoter methylation during adult neurogenesis from NSPCs [47]. Interestingly, changes in DNA methylation as a consequence of external stimuli and promotion of adult neurogenesis have been described. Physical exercise can induce neurogenesis, and during this process, an association with changes in the promoter methylation of the neurotrophic factor BDNF [48] were described. Loss of demethylation by active mechanisms should also be considered, such as the activation of the GADD45B demethylase in the DG cells during adult neurogenesis [49] or the transformation of 5-methylcytosine (5-mC) into 5-hydroxymethylcytosine (5-hmC) by enzymes of the TET family [50]. 5-hmC is enriched in adult neurons compared to NSPCs of the SVZ in the mouse developing brain and colocalize with MeCP2 and with the active chromatin histone modification H3K4me2 in mouse neurons [51]. Another mechanism by which CpG methylation results in transcriptional silence is by binding to methyl-CpG-binding proteins that recruit several chromatin remodelling proteins. As an example, it has been described that the Methyl-CpG-Binding Domain Protein 1 (MBD1) suppresses the expression of FGF-2 promoting differentiation during adult neurogenesis in the hippocampus [52]. MECP2 encodes an epigenetic factor that influences chromatin structure and considered to act mainly as a transcriptional repressor [53]. Furthermore, recent studies using induced pluripotent stem cells derived from Rett patients (a disorder causes generally by point mutations on the MECP2 gene) demonstrated the role of the MeCP2 protein in neuronal maturation [54]. In addition to DNA methylation, histone modifiers serve as important regulators in neuronal development. Mll1 (mixed-lineage leukaemia 1) is a histone methyltransferase (HMT) that is required for neuronal differentiation in the adult SVZ and its effect can be exerted by regulating the expression of DLX2 by increasing H3K27 methylation [55]. Histone acetylation-related enzymes, such as HDAC2, also impact the maturation and survival of adult neurons in the SVZ region [56].
The idea of effective stimulation of neuronal production by using epidrugs is highly attractive, and although in its infancy, it is supported by several lines of evidence (Table 1). Interestingly, pharmacological inhibition of HDAC activity alters neuronal differentiation. It has been reported that treatments with trichostatin A (TSA) or valproic acid (VPA) induced neuronal differentiation in adult progenitor cells [46, 57]. VPA treatment also improved the differentiation of sympathoadrenal progenitor cells into catecholaminergic neurons [58]. Epigenetic drugs targeting histone methylation are less extensively addressed. Pre-administration of Bix-01294, a G9a/GLP inhibitor, has a neuroprotective effect in a mouse model of neurodegeneration induced by ethanol and prevents deficits in long-term potentiation, memory and social recognition behaviour [59]. The underlying molecular mechanisms are still unclear although reactivation of specific genes involved in cell fate after epidrug treatments has been identified. For instance, TSA treatment of the PC12 cell line results in increased acetylation of Lys14 on histone H3 and upregulation of the expression of nur77 gene [60]. A neuroprotective effect of HDAC inhibitor (HDACi) treatment mediated by inflammation prevention has been also suggested [61]. It must be highlighted that multitargeting is also possible after treatment with epigenetic-based drugs due to the lack of isoform selectivity and also due to the off-target effects affecting non-histone proteins. As an example, treatment with the HDACi AR-42 restores the abnormalities in histone 4 acetylation observed in an in vitro model of Kabuki syndrome (with mutations in the KMT2D histone methyltransferase) and also alters methylation at H3K4 [62]. Pharmacological manipulation of chromatin complexes is also an alternative. The histone-interacting BET bromodomain proteins are downregulated during neurogenesis from NPCs, and the use of a bromodomain selective inhibitor (JQ-1) results in an increase in neuronal differentiation [63].
Table 1.
Epigenetic-based treatments associated with manipulation of neurogenesis in mammals
| Experimental system | Organism | Epigenetic drug | Functional effect | Ref. |
|---|---|---|---|---|
| Neural progenitor cells | Rat | VPA | Promotion of neuronal fate, inhibition of glial fate | [46] |
| Adrenomedullary sympathoadrenal progenitors | Bovine | VPA | Promotion of catecholaminergic neuronal differentiation | [58] |
| Brain neuroblastoma | Mouse | TSA | Induction of neurite extension | [201] |
| Cerebella granule neurons | Rat | TSA | Promotion of neuronal outgrowth | [202] |
| Adrenal medulla progenitors | Rat | TSA, VPA | Induction of neurite outgrowth | [60] |
| Neural progenitor cells | Mouse | JQ-1 | Promotion of neurogenesis, inhibition of gliogenesis | [63] |
| Neural progenitor cells | Mouse | SAHA, TSA | Reduction in neurogenesis in the ganglionic eminences, increase in neurogenesis in the cortex | [203] |
| Primary glial cultures and glioblastoma cells | Human | VPA | Alteration of glial cell morphology | [204] |
| Neural stem cells | Rat | 5-AZA | Reduction of migrated neurons and differentiation | [205] |
Representative examples of studies are included. 5-AZA 5-aza-cytidine, SAHA suberoylanilide hydroxamic acid, TSA trichostatin A, VPA valproic acid
Therapeutic epigenetic-driven approaches to treat psychiatric disorders
Epigenetic disequilibria could influence neurodevelopment and brain function at the level of neural circuits and behavioural outcome and be the trigger point of several psychiatric disorders [64–66] (Table 2). It is well known that genetic and environmental factors contribute to the underlying cause of schizophrenia (SZ) [66–73]. Recently, it was shown that the mammalian brain suffers a global epigenomic reconfiguration during fetal to young adult development which could influence SZ onset specifically before the age of 20 [11]. Epigenetic processes can be developmentally regulated, altered by drugs and environmental factors, and be tissue-specific [65, 66, 71, 74] and provide links between clinical manifestations of the psychiatric phenotype and lifestyle. For example, inhalation of nicotine by tobacco smoking (a confounding factor), regularly practised by SZ patients, could work as a self-medication. It is proposed to correct a cholinergic (nicotinic) neurotransmission deficit in those patients via epigenetic actions on GABAergic neurons [71, 75].
Table 2.
Epigenetic dysregulation in schizophrenia, PTSD (post-traumatic stress disorders) and Alzheimer’s disease
| Disorder | Gene | Molecular effect | Specie | Method | References |
|---|---|---|---|---|---|
| Schizophrenia | |||||
| EPHA4, PKNOX1, ESR1, among others | DNA methylation, hsa-miR-219a-5p | Human | Gene set enrichment analysis | [206] | |
| NUBP1, PRKCE, HLA-DQA1, HLA-B, FRK, IL12RB1, among others | DNA methylation | Human | DNA methylation array | [74] | |
| FAM63B, among others | DNA methylation | Human | Methylome-wide association study, targeted pyrosequencing of bisulfite-converted DNA | [207] | |
| GAD67 | DNA methylation, H3 acetylation | Mouse | MeDIP, ChIP, qPCR, Western Blotting | [75, 77, 85] | |
| REELIN | DNA methylation, H3 acetylation | Mouse, Human | MeDIP, ChIP, qPCR, Western Blotting, Gel Shift Binding Assays, Methylome-wide association study, targeted pyrosequencing of bisulfite-converted DNA | [77, 85, 207, 208] | |
| BDNF | DNA methylation, H3 acetylation | Mouse, Human | MeDIP, ChIP, qPCR, Microarray-based DNA methylation profiling | [76, 209] | |
| mGlu2 | DNA methylation, H3 acetylation | Mouse | Bisulfite sequencing, ChIP, qPCR | [210] | |
| HLA genes, among others | Histone acetylation | Mouse | Microarray studies, qRT-PCR, Western Blotting | [114] | |
| COMT | DNA methylation | Human | Bisulfite, DNA sequencing, Methylated SpecificPCR and bisulfite sequencing, qRT-PCR | [211, 212] | |
| S-COMT | DNA methylation | Human | Pyrosequencing | [213] | |
| FOSP2 | DNA methylation | Human | Bisulfite DNA sequencing | [214] | |
| HTR2A | DNA methylation | Human | Bisulfite DNA sequencing | [215] | |
| SOX10 | DNA methylation | Human | Bisulfite DNA sequencing | [216] | |
| 5HTR1A | DNA methylation | Human | High-resolution melt assay | [217] | |
| MAOA | DNA methylation | Human | Methylated Specific PCR | [218] | |
| MEK1 | DNA methylation | Human | Microarray-based DNA methylation profiling | [219] | |
| CAMKIIγ | miR-129 | Mouse | miRNA array profiling, miRNA specific RT-PCR | [220] | |
| PTSD | |||||
| BDNF | H4 acetylation | Mouse, Rat | Western Blotting, ChIP, qPCR | [221, 222] | |
| FKBP5 | DNA methylation/hydroxymethylation | Mouse | Bisulfite sequencing, Pyrosequencing | [34, 223] | |
| HDAC1, cFos | H3 acetylation and methylation | Mouse | ChIP, qPCR | [224] | |
| NR2B | H3 and H4 acetylation | Rat | Western Blotting, ChIP, RT-PCR | [225] | |
| CBP, p300, PCAF | H2B and H4 acetylation | Rat | Western Blotting, ChIP, PCR | [226] | |
| Calcineurin (CaN) | DNA methylation | Rat | Bisulfite sequencing | [227] | |
| PP1 | DNA methylation | Rat | DNA methylation assay | [228] | |
| REELIN | DNA demethylation | Rat | DNA methylation assay | [228] | |
| IGF2, cFOS, ARC | H3 acetylation | Mouse | ChIP, qPCR | [229] | |
| TLR1, IL8, CNTN2, among others | DNA methylation | Human | DNA methylation array | [230, 231] | |
| MAN2C1 | DNA methylation | Human | DNA methylation array | [232] | |
| IGF2, H19, IL8, IL16, IL18 | DNA methylation | Human | Pyrosequencing | [233] | |
| COMT | DNA methylation | Human | DNA methylation array | [234] | |
| ADCYAP1R1 | DNA methylation | Human | DNA methylation array | [235] | |
| NR3C1 | DNA methylation | Human | Pyrosequencing and clonal sequencing | [236–238] | |
| SLC6A3 | DNA methylation | Human | DNA methylation array | [239] | |
| SLC6A4 | DNA methylation | Human | DNA methylation array | [240] | |
| APC5, TPR, CLEC9A, ANXA2, TLR8 | DNA methylation | Human | DNA methylation array | [241] | |
| FKBP5 | miR-511 | Mouse | qPCR | [242] | |
| Alzheimer’s disease | |||||
| APP | DNA methylation | Human | MSRE-SB, pyrosequencing | [125–127] | |
| BACE1 | DNA methylation | Cell lines | HPLC | [129] | |
| PSEN1 | DNA methylation | Cell lines | HPLC | [129] | |
| Neprylisin | DNA methylation | Cell lines | MS-PCR | [131] | |
| DUSP22 | DNA methylation | Human | BS-array, pyrosequencing | [117] | |
| DUSP22 | DNA hydroxymethylation | Human | WG 5-hmC-enriched seq | [117] | |
| SORBS3 | DNA methylation | Human | BS-array, pyrosequencing, MS-PCR | [116, 132] | |
| NF-kB | DNA methylation | Human and Cell lines | Specific Methylation Assay | [133, 134] | |
| COX2 | DNA methylation | Human and Cell lines | Specific Methylation Assay | [134] | |
| BDNF | DNA methylation | Human | MSRE-PCR | [133] | |
| CREB | DNA methylation | Human | MSRE-PCR | [133] | |
| TBXA2R | DNA methylation | Human | BS-array, pyrosequencing, MS-PCR | [116] | |
| ANK1 | DNA methylation | Human | WGBS, BS-array, pyrosequencing | [121, 122] | |
| BACE1 | H3 acetylation | Human | FAIRE/ChIP | [130] | |
| Neprylisin | H4 acetylation | Cell lines | ChIP | [144] | |
| miR-9 | downregulation | Human | PCR | [179] | |
| miR-26a | upregulation | Human | PCR | [161] | |
| miR-29a/b-1 | downregulation | Human | miRNA microarray | [164] | |
| miR-29c | downregulation | Mouse | RT-PCR | [164, 168] | |
| miR-34c | downregulation | Human | RNA sequencing | [178] | |
| miR-101 | downregulation | Cell lines | PCR | [158] | |
| miR-106b | downregulation | Human | miRNA microarray, Northern blot | [157] | |
| miR-107 | downregulation | Human | miRNA microarray | [163] | |
| miR-124 | downregulation | Human | RT-PCR | [160] | |
| miR-125 | upregulation | Human | Northern blot | [161, 174, 183] | |
| miR-132 | downregulation | Human | Northern blot | [161, 174, 183] | |
| miR-137 | downregulation | Human | PCR | [162] | |
| miR-153 | downregulation | Human | PCR | [158] | |
| miR-181c | downregulation | Human | PCR | [161, 162, 179] | |
| miR-132 | downregulation | Human | Northern blot | [174] | |
| miiR-219 | downregulation | Human | RNA sequencing | [170] | |
| miR-339-5p | downregulation | Human and Cell lines | RT-PCR | [167] | |
| BACE1-AS | upregulation | Human and Cell lines | RT-PCR | [169] | |
| lnc-17A | upregulation | Human | RT-PCR | [184] | |
DNA methylation, histone alterations and microRNA expression is summarized. BS-array bisulfite-modified DNA based arrays, ChIP chromatin immunoprecipitation, FAIRE formaldehyde-assisted isolation of regulatory elements, HPLC high-performance liquid chromatography, MeDIP methylated DNA immunoprecipitation, MS-PCR methylation specific PCR, MSRE-PCR methylation-sensitive restriction enzyme-PCR, qPCR quantitative real-time PCR, WG 5-hmC-enriched seq whole-genome sequencing analysis of 5-hydroxymethylcytosine-enriched sequences, WGBS whole-genome bisulphite sequencing
It is also remarkable that pathways relevant for actual therapeutic management of SZ are regulated by epigenetic mechanisms. Clinically, the main antipsychotic treatments target the dopaminergic, serotoninergic and monoaminergic receptor systems that exert therapeutic effects in SZ patients [67]. The aetiology of SZ and lifetime antipsychotic use has been associated with DNA methylation changes in MEK1 gene promoter in the frontal cortex of the post-mortem brain. Around 30% of people with SZ have treatment-resistant SZ, and in this case, clozapine is the only effective treatment [73]. In mice, GADD45b mRNA is increased by clozapine [76], but not haloperidol, via stimulation of H3K9 acetylation [77]. Clozapine may exert its therapeutic actions by acting on GABAergic and glutamatergic gene promoters [76, 78, 79], in part targeting DNA methylation via GADD45b, as well as histone methylation and chromatin relaxation [6].
Research during the last two decades suggested that abnormal RELN, DNMT1 and glutamic acid decarboxylase 67 (GAD67) neuronal expression are a feature observed in animal and human brains of SZ patients [71, 80]. Human post-mortem studies show that RELN is downregulated in GABAergic neurons of psychotic patients due to promoter hypermethylation of RELN gene that is associated with an increase in DNMT1 and DNMT3a which is consistent with the “epigenetic GABAergic theory of major psychosis” [71, 80]. Interestingly, early life stress can impact the methylation levels of selected promoters; accordingly, behavioural programming is possible and potentially reversible, at least, in animal models [81]. In a mouse model of prenatal restraint stress that induces epigenetic remodelling in offspring, clozapine but not haloperidol reduces the increased DNMT1 and TET1 levels in frontal cortex of adult prenatal restraint stress mice and also reduces DNMT1 binding to RELN, GAD67 and BDNF promoters [82].
In SZ, HDACis also have a lot of potential as pharmacological treatments. In this context, histone H3 phosphorylation is increased in peripheral blood mononuclear cells when compared to healthy controls [83]. It is known that some HDACis facilitate DNA demethylation [84]. Moreover, combinations of various antipsychotics (e.g. clozapine, olanzapine, quetiapine) and valproic acid (VPA), a HDACi that reduces MeCP2 binding to RELN and GAD67 promoters [85], might prove beneficial in the treatment of SZ based on results from animal and clinical studies [73].
Lastly, disequilibria in microglia and mitochondrial function must also be taken into consideration when discussing SZ. Microglia is important for immune defence in the central nervous system, and the HDACi butyrate influences microglial function and has potential therapeutic functions in SZ [86]. It is known that butyrate among other functions in the CNS reinforces memory function [87] and synaptic plasticity [64, 88]. Mitochondrial dysfunction and cellular energy dysfunction are also associated with SZ. In this context, the butyrate and several carnitinoids could have potential as therapeutic agents to treat SZ and other brain disorders [88].
Posttraumatic stress disorder (PTSD) can develop as a result of a terrifying and traumatic event that can have short-term or long-lasting effects on neuronal function, brain plasticity and behavioural adaptations to psychosocial stressors [89, 90]. Excessive fear and anxiety are some of the main hallmarks of PTSD where extinction training leads to a gradual reduction of fear called “fear extinction” in animals and “exposure-based therapy” in humans. This extinction mechanism and its molecular mechanisms are well conserved across species [90–92]. Considerable progress has recently been made in the preclinical development of cognitive enhancers (e.g. D-cycloserine, yohimbine and glucocorticoids) that potentiate fear extinction. As a result, several targets had been identified, including diverse neurotransmitter systems but also proteins from the IGF2, BDNF and FGF2 pathway or epigenetic modifications and their downstream signalling pathways [90, 91]. The PTSD phenotype is complex and, as many other brain diseases, emerges from interactions between multiple genetic and epigenetic factors [89, 90]. We will focus on the most-well studied epigenetic modifications in fear extinction, DNA methylation and acetylation of histone proteins. However, the importance of ncRNAs in post-transcriptional regulation of gene expression in PTSD is well established [89, 93, 94]. It can be mentioned as an example the role of miR-138b that inhibits the original fear memory and downregulates plasticity-related genes (e.g. creb1 and sp1) in the infralimbic prefrontal cortex of mice [95]. Based on current literature, successful fear extinction is mediated by epigenetic mechanisms, which include enhancement of HAT activity, reduction of HDAC activity (e.g. HDAC2), DNA methylation and DNA demethylation by TET proteins [89, 90]. There are several animal and human studies supporting the role of DNA methylation in PTSD [89]. In mice, for example, DNA methylation is increased in BDNF exon IV in females resistant to fear extinction, which leads to a decrease in BDNF expression in the prefrontal cortex [96]. Interestingly, it has been demonstrated in mice that IGF2/IGFBP7 signalling regulates fear extinction via an upregulation of IGF2 and downregulation of IGFBP7, which promotes survival of 17–19-day-old newborn hippocampal neurons [97]. Both IGF2 and IGFBP7 genes are regulated via DNA methylation and other epigenetic mechanisms [91, 98]. This signalling pathway might have potential as therapeutic target for PTSD, although this possibility will need to be studied further.
In humans, several genes associated with stress response (e.g. NR3C1, FKBP5), neurotransmitter activity (e.g. SLC6A4), immune regulation (e.g. IGF2, H19, IL8, IL16, IL18) and repetitive genomic elements (e.g. LINE-1, Alu) were studied in peripheral blood using either a candidate genetic locus or a genome-wide approach. It was found that their methylation levels are altered in PTSD patients [89]. The TET family of methylcytosine dioxygenases enzymes (TET1, TET2 and TET3) undergoes DNA demethylation (i.e. reverses DNA methylation), which seems to also play an important role during fear extinction [99]. One recent study demonstrates that gene knockdown of TET1 impairs extinction [100]. Furthermore, another study shows that 5-hmC and TET3 occupancy undergo genome-wide redistribution on extinction-related genes and that Gephyrin mRNA expression is increased in the infralimbic prefrontal cortex [101]. These preclinical findings have made TET enzymes and DNA demethylation promising therapeutic targets to potentiate fear extinction; however, compounds that target the TET enzymes and subsequently DNA demethylation are not yet available.
SPV106, a HAT p300/CBP-associated factor (PCAF) activator, facilitates fear extinction and protects against fear renewal when injected in rodent infralimbic prefrontal cortex [102]. On the other hand, if we inhibit HAT p300 in the infralimbic cortex, strengthen fear extinction is enhanced [103]. Thus, HAT modulators affect fear extinction in different ways, and additional work is required to unravel their mechanisms of action. Alternatively, gene transcription of extinction-relevant genes that can be enhanced by HDAC inhibitors such as TSA, sodium butyrate, entinostat (MS-275), vorinostat (SAHA), VPA and Cl-944 can strengthen fear extinction displaying better selectivity towards class-I HDACs [89, 90]. In this context, targeting specific HDAC isoforms could be a useful therapeutic approach to modulating fear extinction [104]. Moreover, MS-275, SAHA and Cl-944 rescue fear extinction deficits in various rodent models [89, 90], and HDAC2 seems to play a crucial function in this rescue [105]. Bahari-Javan et al. observed in rodents that HDAC1 is required for extinction learning that comprises H3K9 deacetylation followed by H3K9 trimethylation of target genes [92, 106]. These facilitating effects on fear extinction are likely due to initiation of various extinction-related gene transcription programs. For example, SAHA and VPA increase acetylation in the promoter of GRIND2B (NMDA receptor subunit 2B) and histone H4 acetylation in the promoter IV of BDNF; Cl-994 increases histone H3 acetylation in the promoter region of plasticity associated genes (e.g. IGF2, ARC, C-FOX), and some neurotransmitter systems increase H3 acetylation in the promoter of certain genes (e.g. BDNF, CAMK2A, CREB) [90, 105].
Epigenetic link between psychiatric disorders and dementia
Gene expression in the human brain changes with age [106], and it is known that some psychiatric disorders (e.g. SZ, PTSD and depression) may trigger or accelerate the progression of dementia, a neurodegenerative disease [92, 107–109]. Although the specific molecular link has not been properly established, epigenetic effects on disease phenotypes may explain how early life stressors (e.g. a psychiatric disorder) can account for the susceptibility of dementia later in life [11]. For example, in this context, there is evidence showing that childhood abuse induces differential DNA methylation and gene expression patterns in PTSD patients compared to PTSD patients without childhood abuse [110]. IGFBP7, one of the seven IGFBPs identified in the mammalian genome that is used to transport and regulate the bioavailability of IGF1 and IGF2, is deregulated in PTSD and dementia via Igfbp7 promoter DNA methylation in mouse and human brains [91, 98]. In this context, epigenetic pharmacology emerges as an appealing alternative to treat multifactorial diseases with deregulation in multiple signalling pathways in the framework of personalized medicine [111].
Epigenetic mechanisms are essential for normal brain function (i.e. learning and memory processes), and on the basis of the literature presented in this review, disruptions of these mechanisms are closely related to the molecular alterations associated with disorders such as depression or Alzheimer’s disease. Alzheimer’s disease (AD) is the most prevalent form of dementia currently affecting more than 48 million people worldwide with devastating consequences for the affected patients, their relatives and health systems. Its incidence is expected to double every 20 years (from 74.7 million in 2030 to 131.5 million in 2050, according to the World Alzheimer Report 2015). Increasing life expectancy calls for the urgent development of strategies to delay, attenuate or prevent Alzheimer’s disease, since therapeutic approaches directed only at the moderate-to-late stages have been disappointing in clinical trials so far. AD is divided in two subtypes: familial and sporadic cases. Familial AD (FAD) accounts for only approximately 5% of all AD cases and is associated with inherited mutations in the amyloid precursor protein (APP) gene and the presenilin 1 and 2 (PSEN1/2) genes [112]. This subtype is characterized by presentation of the major hallmarks of the disease before the age of 55 years (early onset AD (EOAD)). Sporadic cases of AD usually present a later age of onset (≥65 years; late onset AD (LOAD)), and the scientific community has still not associated this subtype to any genetic mutation. Thus, identification of non-genetic factors that trigger sporadic forms of the disease is crucial in terms of prevention, and knowledge of the underlying etiopathogenic mechanisms will aid the development of timely interventions.
Epigenetic deregulation in neurodegenerative disorders: Alzheimer’s disease as a model
Since recent studies have described that gene–environment interactions may underlie neuropsychiatric disorders [113–115], many research efforts have been oriented into the study of the alterations of the “physiological epigenome” associated with AD [116, 117]. Epigenetic mechanisms, such as modifications of DNA-structure or of associated histones, regulate gene transcription and may contribute substantially in the interplay of genetic and environmental factors in the determination of the human phenotype [118–120]. Alterations of the levels of 5-mC and 5-hmC and other epigenetic marks during the lifespan have been associated with the progression of AD. To date, several groups [121–123] have identified, by epigenome-wide analysis, several genes regulated by DNA methylation in human brain AD samples. These studies reflect that AD, as well as dementias in general, has specific epigenetic signatures [124]. In particular, several research teams have described age-dependent methylation changes of a number of AD-related genes. APP gene expression is partially regulated through methylation of the multiple CpG sites of its promoter, and hypomethylation events have been described in association with AD in patients over 70 years of age [125–127]. However, these data could not be confirmed by another study with bigger cohorts [128]. Other APP related genes, such as BACE1 (that codes for an enzyme responsible for the misprocessing of APP towards toxic Aβ generation), can be regulated via epigenetic mechanisms and hypomethylation states of that promoter have been associated with AD [129, 130]. Another enzyme associated with Aβ, PSEN1, has also been shown to have an aberrant methylation status in AD [129]. Importantly, Aβ itself has been described as an epigenetic modulator by inducing global DNA hypomethylation and specific hypermethylation of Neprilysin, an enzyme associated with its degradation [131], thus reducing its expression. Therefore, despite still not being well defined, it seems that the Aβ generation mechanisms are associated with DNA methylation patterns in a bidirectional manner. Tau protein, the other major pathomolecular change in AD, has also been shown to be regulated epigenetically. DUSP22 is a phosphatase with the ability to dephosphorylate abnormal tau and is downregulated in AD brain samples by hypermethylation of its promoter [117].
Besides the principal molecules associated with AD, several studies have indicated the importance of epigenetic processes in gene expression regulation that occur in AD. SORBS3, or Vinexin, encodes for a cell adhesion protein involved in synaptic function, and several groups have found a hypermethylation of its promoter [116, 132]. This process normally appears in an age-dependent fashion but is accelerated in AD. Other genes such as the gene NF-kB or some regions of the promoter of the gene Cyclooxygenase-2 (COX-2), both proinflammatory and associated with inflammatory events in AD, have been reported to be hypomethylated [133, 134]. Hypermethylation of the promoters of BDNF and cAMP response element-binding protein (CREB) were found in the frontal cortex of AD patients [133]. Both proteins are critical for neuronal survival [135, 136] and have been associated with AD [137]. The importance of the CREB signalling in AD is evident since other authors have also described alterations in genes related to this molecular pathway, such as hypermethylation of Thromboxane A2 Receptor (TBXA2R) [116], a G-protein receptor regulating CREB [138]. Recent epigenome-wide association studies (EWAS) identify another gene called Ankyrin1 (an adaptor protein; ANK1) in hypermethylated state in cortex samples of AD patients [121, 122]. Importantly, the epigenetic state of ANK1 showed a strong correlation both with early and late stages of the disease, suggesting its possible validity as a biomarker.
Besides DNA methylation, the role of histone modifications has been also dissected in AD [5]. However, few studies have focused on human brain samples. Of all the histone modifications described so far, lysine acetylation and lysine methylation are the most common [139]. For example, histone acetylation has been described to be reduced both in the human brain tissue and in AD mouse models [140, 141]. Importantly, the transcriptional activity of AD associated genes has been associated with specific histone marks, such as increased acetylation of H3 at the BACE1 promoter [130]. This histone mark activates gene transcription by relaxing the chromatin structure. One of the proteolytic fragments of APP is the APP C-terminal fragment (AICD). Several studies have demonstrated the ability of AICD to recruit, directly or indirectly, the histone acetyltransferase TIP60 [142, 143]. The consequences of this interaction are the repression of Neprilysin expression in NB7 cells by acetylation of lysines on histone H4 [144]. Also in human samples, other histone marks were found to be altered. For example, an increase of phosphorylation of histone H2AX, a histone variant, in the hippocampus was found in AD samples [145], as well as increased global H3 phosphorylation in the frontal cortex [133] and hippocampus [146].
However, most current knowledge on the involvement of the histone code derives from work done using transgenic mouse models. Decreased levels of H4 acetylation were found in APP/PS1 of the mouse model hippocampus after a learning task [147]. However, deeper analysis showed an increased H4 acetylation in the CA1 region of hippocampus together with increased H3 acetylation and phosphorylation in the prefrontal cortex of another APP model, the Tg2575 [148]. These data emphasize the necessity of restricting epigenetic analysis to small brain areas or even single-cell analysis to completely understand the role of the epigenetic processes in AD. H3 and H4 acetylation was increased in neuronal cell cultures from an AD mouse model expressing mutations in APP and in Tau (3xTg) [149]. Regarding other hallmarks of AD, hyperacetylation of H3 on the promoter of BACE1 in 3xTg brains [130] leading to increased transcriptional activity of the gene, as well as on the promoter of BACE1 and PS1 in N2a cells expressing a mutated form of APP, has been described [150]. Other modifications have not been sufficiently studied although experiments in animal models have explored histone methylation [151, 152] and ubiquitination [153] involvement in learning and memory processes, also indicating possible implications in cognitive impairments.
Although the AD “histone code” remains to be deciphered, it is evident that histone alterations play important roles both by altering histone marks and by changing the levels of histone modifying enzymes [141, 154] in dementias and are suitable targets for pharmacological approaches.
Non-coding RNAs [155] have also been associated with AD [156]. Several studies have analysed alterations of miRNA expression in several areas of post-mortem AD brains, showing a broad spectrum of changes in a multitude of miRNAs. Some of the most relevant changes occur in miRNAs targeting mechanisms implicated in APP and/or its misprocessing towards the amyloidogenic pathway. miR-106b [157] and -153 [158] are downregulated in AD (temporal cortex and frontal cortex, respectively), and one of its multiple targets is the mRNA of APP [159]. Other miRNAs with the ability to regulate APP are miR-101 [158] and miR-124 [160], and both are described as downregulated in AD brains. miR-137 and -181c are also downregulated in AD [161, 162], and their downregulation promotes APP processing into neurotoxic forms of Ab. Other key molecules of the amyloidogenic cascade like BACE1 are also targeted by several miRNAs. Numerous miRNAs with the capability of reducing BACE1 levels were found to be reduced in several areas of AD brains, for example the miR-29a/b-1 cluster, -29c, -107, -339-5p and -485-5-p [163–168]. Other ncRNAs also target BACE1, including the long ncRNA BACE1-AS, by regulating BACE1 RNA stability. BACE1-AS was described as being in an upregulated state in AD brains samples suggesting its role in incrementing BACE1 levels [169]. Tau is also regulated via miRNAs. miR-219 was found downregulated in the AD human frontal cortex [170] and is thought to regulate tau mRNA directly. Another enzyme involved in the aberrant phosphorylation of tau is Glycogen Synthase Kinase 3β (GSK3β) that is considered the major modulator of tau phosphorylation in the brain [132]. Additional studies have demonstrated that GSK3β is negatively regulated by miR-26a, a microRNA deregulated in AD [161, 171]. Several miRNAs impact on several hallmarks of AD simultaneously. miR-107 is another miRNA targeting BACE1 [166], but it is also capable of deregulating cdk5 [172], a kinase related to tau phosphorylation. In the case of miR-124 and -137, both target APP metabolism as well as the differential splicing of tau [161, 173]. miR-9 and miR-132 can also regulate tau splicing mechanisms [174–176]. Interestingly, both also have the ability to reduce SIRT1, a sirtuin that can deacetylate tau [154, 177]. Together with those, miR-34c and miR-181c can reduce the levels of SIRT1 and all of them are reduced in AD [162, 178, 179].
Another important alteration in AD is synaptic dismantling and alteration of synaptic transmission [180, 181]. While several miRNAs have been associated with those mechanisms [182], miR-132 and miR-125b in particular have been associated with AD. It has been described that miR-132 is reduced in the hippocampus, cerebellum and medial frontal gyrus, whereas miR-125 is increased in these areas [161, 174, 183]. Another ncRNA, the long non-coding RNA 17A, is elevated in AD brains and regulates GABA transmission [184]. Due to the role of microRNAs in synaptic plasticity [185] and increasing evidence that dysregulation of miRNAs biogenesis is implicated in AD, these epigenetic effectors seem critical not only in the normal gene expression pattern of neurons but also in the pathophysiology of AD. The complexity of ncRNA function and their intricate patterns of expression in nervous system demand further investigation, which may eventually lead to the discovery of new druggable targets to delay or prevent AD.
AD’s pharmacoepigenomics
Overall, it is evident that epigenomic alterations in AD make suitable targets for therapeutic interventions (Table 2). However, so far, only histone alterations have been properly studied in pharmacoepigenomics.
The most common histone modifications (acetylation and methylation) are performed by the balanced activity of HATs and HDACs on the one hand and histone methyltransferases and demethylases (HDMTs) on the other hand [153, 186]. Due to the availability of drugs targeting those enzymes, most research performed to describe its role has been performed in mouse models of AD. The induction of histone acetylation through inhibition of HDACs has been proposed as a candidate approach to treat AD based on of several lines of evidence using such models [187]. One of the first demonstrations of the role of the potential use of HDACi to treat AD showed that administration of the unspecific HDACi sodium butyrate in an AD mice model (CK-p25 mice) was able to restore cognitive capabilities initially decreased in the transgenic mice [188]. An increased expression of HDAC2 in human AD samples (hippocampal area CA1 and entorhinal cortex) and also in the hippocampal area CA1 and in prefrontal cortex of the CK-p25 mice and the 5XFAD model has been described [141], reinforcing the idea of using HDACi to treat AD. Administration of another HDAC inhibitor TSA also improved memory formation in APP/PS1 mice by increasing H4 acetylation in brain [147]. VPA, another inhibitor of HDAC1, was useful in order to reduce Aβ levels and plaques in the hippocampus of an APP model (PDAPP) [189] and also was able to improve learning capabilities of an AD mice model. Similar data was obtained in a Tg2576 model treated with the HDAC inhibitor sodium phenylbutyrate, where chronic treatment reduced tau hyperphosphorylation but could not revert Aβ accumulation [190]. In subsequent studies, the same team treated younger animals with the same compound and found decreased Aβ accumulation and reduced immunoinflammatory events [190] indicating the importance of the disease stage chosen for treatment. Other drugs, such as SAHA, also improved cognitive capabilities of APP/PS1delta9 mice [191], and MS-275, a specific HDAC1 antagonist [192], showed the same improvement in cognition of APP/PS1 mice together with a reduction of amyloid plaques in the hippocampus of treated animals [193]. Although we still do not completely understand the role of HDAC inhibition in the brain [194], it seems evident that selective pharmacological inhibition of some of the multiple HDAC members is a promising area of research for treating early stages of AD.
Conclusions
The involvement of epigenetic factors as key players in the ageing process in the brain and in age-related neurodegenerative and psychiatric disorders is widely accepted and provides important insights as to how they can potentially mediate interactions between genetic and environmental risk factors. In spite of epigenetic-based therapy emerging as an appealing alternative approach to the treatment of neuropsychiatric diseases with deregulation in multiple signalling pathways, many unresolved questions still hinder the progression of candidate therapies to clinical trials.
Future translational research approaches to the development of epigenetic therapeutics in neuropsychiatric diseases must overcome a number of limitations. One of the first bottlenecks is the heterogeneity in the design of studies that frequently use different experimental models, as well as in the particular regions of the brain analysed and in the variable sensitivity and resolution of the epigenetic methods employed. Most studies have been performed with small sample sizes and thus have low statistical power and have only addressed a few epigenetic marks in a few specific tissues. With this level of heterogeneity, it is very difficult to infer broad conclusions about the implications of epigenetics in neuronal development and its alterations in neuropsychiatric diseases. It is also important to note that studies in living humans, as opposed to those in in vitro and animal models, are very scarce. Other limitations regarding the design of the studies are the phenomenological and dichotomous definitions of the disorders, the multiple clinical manifestations, the inability to control lifestyle factors and the inability to distinguish chronological correlations between environmental exposure, epigenetic modifications and disease progression.
In order to address many of the questions mentioned above and increase the reproducibility of existing epigenetic findings, there are several challenges that must be confronted. Some important ones include the following: (1) to carry out larger, longitudinal, multicenter and prospective studies in order to investigate brain diseases and their interactions; (2) to consider tissue and cell-type specificity by using dissection of brain tissues; and (3) to include multiple epigenetic marks, genome-wide studies and integrate the results into specific chromatin contexts.
We must also consider the new challenges in epigenetic research. There is no doubt about the importance of non-coding RNAs in post-transcriptional regulation of gene expression in neural differentiation and their deregulation in several human disorders, including neuropsychiatric diseases [5, 195]. Furthermore, we must add new layers of complexity such as the epigenetic regulation of RNA (i.e. RNA methylation) [196], three-dimensional chromatin structure as a key regulator of transcription [197] and the epigenetic control of the mitochondrial genome that can explain the mitochondrial dysfunction observed in neuropsychiatry diseases [198].
Emerging technologies for epigenetic research can also improve our knowledge. As one prominent example, the use of CRIPSR/Cas9 technology and its adaptations to different models (for example, epigenetic editing) can demonstrate the causal role of epigenetics in instructing gene expression [16]. In addition, computational modelling can accelerate the search for new epigenetic therapeutic approaches to treat neurological disorders, map them to clinical predictions and further our understanding of complex brain diseases at the individual and population levels [199]. In light of the latest advances in induced pluripotent stem cell (iPSC) technology, future epigenomic brain approaches will involve the study of specific neuronal populations derived from patient-cells, allowing a better understanding of the disorder by disease modelling and a faster drug screening/repurposing in a personalized manner [200].
It is clear that current knowledge of the epigenetic changes that occur during healthy ageing and pathological conditions in the brain is increasing, but much research is still required before translating the findings to clinical practice. This is of particular relevance due to the numbers of elderly people in third world societies and the social effects of cognitive impairment. In summary, we need to overcome important challenges to identify new epigenetic therapeutic targets and to develop appropriate, randomised and controlled trials with human subjects.
Acknowledgements
We thank COST Action CM1406 “Epigenetic Chemical Biology” for their financial support regarding the publication fee.
Funding
Research in the group of MB is supported by EU Joint Programme–Neurodegenerative Disease Research (JPND; EPI-AD Consortium), RecerCaixa Foundation, Federación Española de Enfermedades Raras (FEDER), Federación Española de Enfermedades Neuromusculares (ASEM), Fundación Isabel Gemio, Spanish Association Against Cancer (AECC) and the Spanish Ministry of Health (FIS). RCA-B is supported by a Ramón & Cajal grant (RYC-2014-15246) and by a Galicia Innovation Agency (GAIN) grant (IN607D-2016/003).
Availability of data and materials
Not applicable.
Authors’ contributions
RD-M, RCA-B, ME and MB contribute to specific content of the review. MB prepares the main text, table and figure. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 5-hmC
5-Hydroxymethylcytosine
- 5-mC
5-Methylcytosine
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- DG
Dentate gyrus
- DNMT
DNA methyltransferase
- EOAD
Early-onset Alzheimer’s disease
- FAD
Familiar Alzheimer’s disease
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylase
- HDACi
Histone deacetylase inhibitor
- HDMT
Histone demethylase
- HMT
Histone methyltransferase
- LOAD
Late-onset Alzheimer’s disease
- ncRNAs
Non-coding RNA
- NSPCs
Neural stem progenitor cells
- PTSD
Posttraumatic stress disorder
- SAHA
Suberoylanilide hydroxamic acid
- SGZ
Subgranular zone
- SVZ
Subventricular zone
- SZ
Schizophrenia
- TET
Ten-eleven translocation
- TSA
Trichostatin A
- VPA
Valproic acid
Contributor Information
Raúl Delgado-Morales, Email: rdelgado@idibell.cat.
Roberto Carlos Agís-Balboa, Email: roberto.carlos.agis.balboa@sergas.es.
Manel Esteller, Email: mesteller@idibell.cat.
María Berdasco, Phone: 34-93-2607247, Email: mberdasco@idibell.cat.
References
- 1.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benayoun BA, Pollina E, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16(10):593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166(4):822–39. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19(5):698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, Mastroeni D, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64. doi: 10.1016/j.pneurobio.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’ Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17(10):1218–25. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27(16):1787–99. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15(12):1495–506. doi: 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell. 2015;14(6):924–32. doi: 10.1111/acel.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109(26):10522–7. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cournil A, Kirkwood TB. If you would live long, choose your parents well. Trends Genet. 2001;17(5):233–5. doi: 10.1016/S0168-9525(01)02306-X. [DOI] [PubMed] [Google Scholar]
- 13.Schoenmaker M, de Craen AJ, de Meijer PH, Beekman M, Blauw GJ, Slagboom PE, et al. Evidence of genetic enrichment for exceptional survival using a family approach: the Leiden Longevity Study. Eur J Hum Genet. 2006;14(1):79–84. doi: 10.1038/sj.ejhg.5201508. [DOI] [PubMed] [Google Scholar]
- 14.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cano-Rodriguez D, Rots MG. Epigenetic editing: on the verge of reprogramming gene expression at will. Curr Genet Med Rep. 2016;4(4):170–9. doi: 10.1007/s40142-016-0104-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gassen NC, Chrousos GP, Binder EB, Zannas AS. Life stress, glucocorticoid signaling, and the aging epigenome: implications for aging-related diseases. Neurosci Biobehav Rev. 2016. doi: 10.1016/j.neubiorev.2016.06.003. [DOI] [PubMed]
- 18.Toraño EG, García MG, Fernández-Morera JL, Niño-García P, Fernández AF. The impact of external factors on the epigenome: in utero and over lifetime. Biomed Res Int. 2016;2016:2568635. doi: 10.1155/2016/2568635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Modgil S, Lahiri DK, Sharma VL, Anand A. Role of early life exposure and environment on neurodegeneration: implications on brain disorders. Transl Neurodegener. 2014;3:9. doi: 10.1186/2047-9158-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104(32):13056–61. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown AS, Susser ES. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr Bull. 2008;34(6):1054–63. doi: 10.1093/schbul/sbn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, de Rooij SR. Hungry in the womb: what are the consequences? Lessons from the Dutch famine. Maturitas. 2011;70(2):141–5. doi: 10.1016/j.maturitas.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Lövblad K, Ramelli G, Remonda L, Nirkko AC, Ozdoba C, Schroth G. Retardation of myelination due to dietary vitamin B12 deficiency: cranial MRI findings. Pediatr Radiol. 1997;27(2):155–8. doi: 10.1007/s002470050090. [DOI] [PubMed] [Google Scholar]
- 24.Rachdaoui N, Sarkar DK. Transgenerational epigenetics and brain disorders. Int Rev Neurobiol. 2014;115:51–73. doi: 10.1016/B978-0-12-801311-3.00002-0. [DOI] [PubMed] [Google Scholar]
- 25.Babenko O, Kovalchuk I, Metz GA. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci Biobehav Rev. 2015;48:70–91. doi: 10.1016/j.neubiorev.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Weber-Stadlbauer U. Epigenetic and transgenerational mechanisms in infection-mediated neurodevelopmental disorders. Transl Psychiatry. 2017;7(5):e1113. doi: 10.1038/tp.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fontana L, Partridge L, Longo VD. Extending healthy life span—from yeast to humans. Science. 2010;328(5976):321–6. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meydani SN, Das SK, Pieper CF, Lewis MR, Klein S, Dixit VD, et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: a randomized controlled trial in non-obese humans. Aging (Albany NY). 2016;8(7):1416-31. [DOI] [PMC free article] [PubMed]
- 29.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–38. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cantó C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22(1):31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chouliaras L, van den Hove DL, Kenis G, Keitel S, Hof PR, van Os J, Steinbusch HW, Schmitz C, Rutten BP. Prevention of age-related changes in hippocampal levels of 5-methylcytidine by caloric restriction. Neurobiol Aging. 2012;33(8):1672–81. doi: 10.1016/j.neurobiolaging.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A. 2014;111(43):15538–43. doi: 10.1073/pnas.1412759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boks MP, van Mierlo HC, Rutten BP, Radstake TR, De Witte L, Geuze E, et al. Longitudinal changes of telomere length and epigenetic age related to traumatic stress and post-traumatic stress disorder. Psychoneuroendocrinology. 2015;51:506–12. doi: 10.1016/j.psyneuen.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16(1):33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zannas AS, Arloth J, Carrillo-Roa T, Iurato S, Röh S, Ressler KJ, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015;16:266. doi: 10.1186/s13059-015-0828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang X, Ewald ER, Huo Y, Tamashiro KL, Salvatori R, Sawa A, et al. Glucocorticoid-induced loss of DNA methylation in non-neuronal cells and potential involvement of DNMT1 in epigenetic regulation of Fkbp5. Biochem Biophys Res Commun. 2012;420(3):570–5. doi: 10.1016/j.bbrc.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bose R, Spulber S, Kilian P, Heldring N, Lönnerberg P, Johnsson A, et al. Tet3 mediates stable glucocorticoid-induced alterations in DNA methylation and Dnmt3a/Dkk1 expression in neural progenitors. Cell Death Dis. 2015;6:e1793. doi: 10.1038/cddis.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol. 2009;587(Pt 24):5951–8. doi: 10.1113/jphysiol.2009.181065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abel JL, Rissman EF. Running-induced epigenetic and gene expression changes in the adolescent brain. Int J Dev Neurosci. 2013;31(6):382–90. doi: 10.1016/j.ijdevneu.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeilinger S, Kühnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, et al. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009;69(8):3570–8. doi: 10.1158/0008-5472.CAN-08-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schernhammer ES, Giovannucci E, Kawasaki T, Rosner B, Fuchs CS, Ogino S. Dietary folate, alcohol and B vitamins in relation to LINE-1 hypomethylation in colon cancer. Gut. 2010;59(6):794–9. doi: 10.1136/gut.2009.183707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gould E. How widespread is adult neurogenesis in mammals? Nat Rev Neurosci. 2007;8:481–8. doi: 10.1038/nrn2147. [DOI] [PubMed] [Google Scholar]
- 44.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun J, Sun J, Ming GL, Song H. Epigenetic regulation of neurogenesis in the adult mammalian brain. Eur J Neurosci. 2011;33:1087–93. doi: 10.1111/j.1460-9568.2011.07607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci. 2004;101:16659–64. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30:755–66. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–51. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–7. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, et al. 5-hmCmediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–16. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diotel N, Mérot Y, Coumailleau P, Gueguen MM, Sérandour AA, Salbert G, et al. 5-hydroxymethylcytosine marks postmitotic neural cells in the adult and developing vertebrate central nervous system. J Comp Neurol. 2017;525(3):478–97. doi: 10.1002/cne.24077. [DOI] [PubMed] [Google Scholar]
- 52.Liu C, Teng ZQ, Santistevan NJ, Szulwach KE, Guo W, Jin P, et al. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell. 2010;6:433–44. doi: 10.1016/j.stem.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Della Ragione F, Vacca M, Fioriniello S, Pepe G, D’Esposito M. MECP2, a multi-talented modulator of chromatin architecture. Brief Funct Genomics. 2016;15(6):420–31. doi: 10.1093/bfgp/elw023. [DOI] [PubMed] [Google Scholar]
- 54.Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J. Rett syndrome induced pluripotent stem cell derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 2012;17:1261–71. doi: 10.1038/mp.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim DA, Huang YC, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J, et al. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 2009;458:529–33. doi: 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jawerka M, Colak D, Dimou L, Spiller C, Lagger S, Montgomery RL, et al. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010;6:93–107. doi: 10.1017/S1740925X10000049. [DOI] [PubMed] [Google Scholar]
- 57.Chu T, Zhou H, Lu L, Kong X, Wang T, Pan B, et al. Valproic acid-mediated neuroprotection and neurogenesis after spinal cord injury: from mechanism to clinical potential. Regen Med. 2015;10(2):193–209. doi: 10.2217/rme.14.86. [DOI] [PubMed] [Google Scholar]
- 58.Vukićević V, Qin N, Balyura M, Eisenhofer G, Wong ML, Licinio J, et al. Valproic acid enhances neuronal differentiation of sympathoadrenal progenitor cells. Mol Psychiatry. 2015;20(8):941–50. doi: 10.1038/mp.2015.3. [DOI] [PubMed] [Google Scholar]
- 59.Subbanna S, Basavarajappa BS. Pre-administration of G9a/GLP inhibitor during synaptogenesis prevents postnatal ethanol-induced LTP deficits and neurobehavioral abnormalities in adult mice. Exp Neurol. 2014;261:34–43. doi: 10.1016/j.expneurol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tomioka T, Maruoka H, Kawa H, Yamazoe R, Fujiki D, Shimoke K, et al. The histone deacetylase inhibitor trichostatin A induces neurite outgrowth in PC12 cells via the epigenetically regulated expression of the nur77 gene. Neurosci Res. 2014;88:39–48. doi: 10.1016/j.neures.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 61.Ziemka-Nalecz M, Jaworska J, Sypecka J, Polowy R, Filipkowski RK, Zalewska T. Sodium butyrate, a histone deacetylase inhibitor, exhibits neuroprotective/neurogenic effects in a rat model of neonatal hypoxia-ischemia. Mol Neurobiol. 2016. doi:10.1007/s12035-016-0049-2. [DOI] [PMC free article] [PubMed]
- 62.Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC, et al. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med. 2014;6(256):256ra135. doi: 10.1126/scitranslmed.3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Ma J, Meng G, Lin H, Wu S, Wang J, et al. BET bromodomain inhibition promotes neurogenesis while inhibiting gliogenesis in neural progenitor cells. Stem Cell Res. 2016;17(2):212–21. doi: 10.1016/j.scr.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 64.Fischer A. Epigenetic memory: the Lamarckian brain. EMBO J. 2014;33(9):945–67. doi: 10.1002/embj.201387637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 66.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8(5):355–67. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 67.Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet. 2016;388(10039):86–97. doi: 10.1016/S0140-6736(15)01121-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Modai S, Shomron N. Molecular risk factors for schizophrenia. Trends Mol Med. 2016;22(3):242–53. doi: 10.1016/j.molmed.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 69.Akbarian S. Epigenetic mechanisms in schizophrenia. Dialogues Clin Neurosci. 2014;16(3):405–17. doi: 10.31887/DCNS.2014.16.3/sakbarian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19(11):1442–53. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38(1):138–66. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petronis A. The origin of schizophrenia: genetic thesis, epigenetic antithesis, and resolving synthesis. Biol Psychiatry. 2004;55(10):965–70. doi: 10.1016/j.biopsych.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 73.Lally J, Gaughran F, Timms P, Curran SR. Treatment-resistant schizophrenia: current insights on the pharmacogenomics of antipsychotics. Pharmgenomics Pers Med. 2016;9:117–29. doi: 10.2147/PGPM.S115741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alelú-Paz R, Carmona FJ, Sanchez-Mut JV, Cariaga-Martínez A, González-Corpas A, Ashour N, et al. Epigenetics in schizophrenia: a pilot study of global DNA methylation in different brain regions associated with higher cognitive functions. Front Psychol. 2016;7:1496. doi: 10.3389/fpsyg.2016.01496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci U S A. 2008;105(42):16356–61. doi: 10.1073/pnas.0808699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matrisciano F, Dong E, Gavin DP, Nicoletti F, Guidotti A. Activation of group II metabotropic glutamate receptors promotes DNA demethylation in the mouse brain. Mol Pharmacol. 2011;80(1):174–82. doi: 10.1124/mol.110.070896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci U S A. 2008;105(36):13614–9. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guidotti A, Auta J, Chen Y, Davis JM, Dong E, Gavin DP, et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology. 2011;60(7-8):1007–16. doi: 10.1016/j.neuropharm.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27(42):11254–62. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guidotti A, Grayson DR, Caruncho HJ. Epigenetic RELN dysfunction in schizophrenia and related neuropsychiatric disorders. Frontiers Cell Neurosci. 2016;10:89. doi: 10.3389/fncel.2016.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 82.Dong E, Tueting P, Matrisciano F, Grayson DR, Guidotti A. Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl Psychiatry. 2016;6:e711. doi: 10.1038/tp.2015.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sharma RP, Feiner B, Chase KA. Histone H3 phosphorylation is upregulated in PBMCs of schizophrenia patients in comparison to healthy controls. Schizophr Res. 2015;169(1-3):498–9. doi: 10.1016/j.schres.2015.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kundakovic M, Chen Y, Guidotti A, Grayson DR. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol. 2009;75(2):342–54. doi: 10.1124/mol.108.051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong E, Agis-Balboa RC, Simonini MV, Grayson DR, Costa E, Guidotti A. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc Natl Acad Sci U S A. 2005;102(35):12578–83. doi: 10.1073/pnas.0505394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moos WH, Maneta E, Pinkert CA, Irwin MH, Hoffman ME, Faller DV, et al. Epigenetic treatment of neuropsychiatric disorders: autism and schizophrenia. Drug Dev Res. 2016;77(2):53–72. doi: 10.1002/ddr.21295. [DOI] [PubMed] [Google Scholar]
- 87.Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J Alzheimers Dis. 2011;26(1):187–97. doi: 10.3233/JAD-2011-110080. [DOI] [PubMed] [Google Scholar]
- 88.Graff J, Tsai LH. The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol. 2013;53:311–30. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- 89.Zannas AS, Provencal N, Binder EB. Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry. 2015;78(5):327–35. doi: 10.1016/j.biopsych.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 90.Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther. 2015;149:150–90. doi: 10.1016/j.pharmthera.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agis-Balboa RC, Fischer A. Generating new neurons to circumvent your fears: the role of IGF signaling. Cell Mol Life Sci. 2014;71(1):21–42. doi: 10.1007/s00018-013-1316-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bahari-Javan S, Sananbenesi F, Fischer A. Histone-acetylation: a link between Alzheimer’s disease and post-traumatic stress disorder? Front Neurosci. 2014;8:160. doi: 10.3389/fnins.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Giridharan VV, Thandavarayan RA, Fries GR, Walss-Bass C, Barichello T, Justice NJ, et al. Newer insights into the role of miRNA a tiny genetic tool in psychiatric disorders: focus on post-traumatic stress disorder. Transl Psychiatry. 2016;6(11):e954. [DOI] [PMC free article] [PubMed]
- 94.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 95.Lin Q, Wei W, Coelho CM, Li X, Baker-Andresen D, Dudley K, et al. The brain-specific microRNA miR-128b regulates the formation of fear-extinction memory. Nat Neurosci. 2011;14(9):1115–7. doi: 10.1038/nn.2891. [DOI] [PubMed] [Google Scholar]
- 96.Baker-Andresen D, Flavell CR, Li X, Bredy TW. Activation of BDNF signaling prevents the return of fear in female mice. Learn Mem. 2013;20(5):237–40. doi: 10.1101/lm.029520.112. [DOI] [PubMed] [Google Scholar]
- 97.Agis-Balboa RC, Arcos-Diaz D, Wittnam J, Govindarajan N, Blom K, Burkhardt S. A hippocampal insulin-growth factor 2 pathway regulates the extinction of fear memories. EMBO J. 2011;30(19):4071–83. doi: 10.1038/emboj.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agbemenyah HY, Agis-Balboa RC, Burkhardt S, Delalle I, Fischer A. Insulin growth factor binding protein 7 is a novel target to treat dementia. Neurobiol Dis. 2014;62:135–43. doi: 10.1016/j.nbd.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 99.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14(6):341–56. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79(6):1109–22. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, et al. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A. 2014;111(19):7120–5. doi: 10.1073/pnas.1318906111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, et al. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. J Neurosci. 2012;32(35):11930–41. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marek R, Coelho CM, Sullivan RK, Baker-Andresen D, Li X, Ratnu V, et al. Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J Neurosci. 2011;31(20):7486–91. doi: 10.1523/JNEUROSCI.0133-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Whittle N, Singewald N. HDAC inhibitors as cognitive enhancers in fear, anxiety and trauma therapy: where do we stand? Biochem Soc Trans. 2014;42(2):569–81. doi: 10.1042/BST20130233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsai LH, Gräff J. On the resilience of remote traumatic memories against exposure therapy-mediated attenuation. EMBO Rep. 2014;15(8):853-61. [DOI] [PMC free article] [PubMed]
- 106.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429(6994):883–91. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 107.Yaffe K, Vittinghoff E, Lindquist K, Barnes D, Covinsky KE, Neylan T, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608–13. doi: 10.1001/archgenpsychiatry.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF., 3rd Late-life depression and risk of vascular dementia and Alzheimer’s disease: systematic review and meta-analysis of community-based cohort studies. Br J Psychiatry. 2013;202(5):329–35. doi: 10.1192/bjp.bp.112.118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ribe AR, Laursen TM, Charles M, Katon W, Fenger-Grøn M, Davydow D, et al. Long-term risk of dementia in persons with schizophrenia: a Danish population-based cohort study. JAMA Psychiat. 2015;72(11):1095–101. doi: 10.1001/jamapsychiatry.2015.1546. [DOI] [PubMed] [Google Scholar]
- 110.Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2013;110(20):8302–7. doi: 10.1073/pnas.1217750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.de Lera AR, Ganesan A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin Epigen. 2016;8:105. doi: 10.1186/s13148-016-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006296. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–72. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 114.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194–204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pena CJ, Bagot RC, Labonte B, Nestler EJ. Epigenetic signaling in psychiatric disorders. J Mol Biol. 2014;426:3389–412. doi: 10.1016/j.jmb.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanchez-Mut JV, Aso E, Panayotis N, Lott I, Dierssen M, Rabano A, et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain. 2013;136:3018–27. doi: 10.1093/brain/awt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sanchez-Mut JV, Aso E, Heyn H, Matsuda T, Bock C, Ferrer I, et al. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus. 2014;24:363–8. doi: 10.1002/hipo.22245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Reichenberg A, Mill J, MacCabe JH. Epigenetics, genomic mutations and cognitive function. Cogn Neuropsychiatry. 2009;14:377–90. doi: 10.1080/13546800902978417. [DOI] [PubMed] [Google Scholar]
- 119.Amin V, Harris RA, Onuchic V, Jackson AR, Charnecki T, Paithankar S, et al. Epigenomic footprints across 111 reference epigenomes reveal tissue-specific epigenetic regulation of lincRNAs. Nat Commun. 2015;6:6370. doi: 10.1038/ncomms7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317-30 [DOI] [PMC free article] [PubMed]
- 121.De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–63. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17:1164–70. doi: 10.1038/nn.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry. 2016;6:e718. doi: 10.1038/tp.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Delgado-Morales R, Esteller M. Opening up the DNA methylome of dementia. Mol Psychiatry. 2017; doi: 10.1038/mp.2016.242. [DOI] [PMC free article] [PubMed]
- 125.West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci. 1995;6:141–6. doi: 10.1007/BF02736773. [DOI] [PubMed] [Google Scholar]
- 126.Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, Ukitsu M. Reduction with age in methylcytosine in the promoter region −224 approximately −101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70:288–92. doi: 10.1016/S0169-328X(99)00163-1. [DOI] [PubMed] [Google Scholar]
- 127.Iwata A, Nagata K, Hatsuta H, Takuma H, Bundo M, Iwamoto K, et al. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum Mol Genet. 2014;23:648–56. doi: 10.1093/hmg/ddt451. [DOI] [PubMed] [Google Scholar]
- 128.Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J Neuropathol Exp Neurol. 2009;68:880–91. doi: 10.1097/NEN.0b013e3181af2e46. [DOI] [PubMed] [Google Scholar]
- 129.Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 130.Marques SC, Lemos R, Ferreiro E, Martins M, de Mendonca A, Santana I, et al. Epigenetic regulation of BACE1 in Alzheimer’s disease patients and in transgenic mice. Neuroscience. 2012;220:256–66. doi: 10.1016/j.neuroscience.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 131.Chen KL, Wang SS, Yang YY, Yuan RY, Chen RM, Hu CJ. The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Commun. 2009;378:57–61. doi: 10.1016/j.bbrc.2008.10.173. [DOI] [PubMed] [Google Scholar]
- 132.Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry. 2012;2:e132. doi: 10.1038/tp.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gu X, Sun J, Li S, Wu X, Li L. Oxidative stress induces DNA demethylation and histone acetylation in SH-SY5Y cells: potential epigenetic mechanisms in gene transcription in Abeta production. Neurobiol Aging. 2013;34:1069–79. doi: 10.1016/j.neurobiolaging.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 135.Arthur JS, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, et al. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24:4324–32. doi: 10.1523/JNEUROSCI.5227-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lipsky RH, Marini AM. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci. 2007;1122:130–43. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 137.Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–22. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- 138.Saura CA, Valero J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev Neurosci. 2011;22:153–69. doi: 10.1515/rns.2011.018. [DOI] [PubMed] [Google Scholar]
- 139.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics. 2012;12:1261–8. doi: 10.1002/pmic.201200010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–6. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/S0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 143.Kim HS, Kim EM, Kim NJ, Chang KA, Choi Y, Ahn KW, et al. Inhibition of histone deacetylation enhances the neurotoxicity induced by the C-terminal fragments of amyloid precursor protein. J Neurosci Res. 2004;75:117–24. doi: 10.1002/jnr.10845. [DOI] [PubMed] [Google Scholar]
- 144.Belyaev ND, Nalivaeva NN, Makova NZ, Turner AJ. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 2009;10:94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Myung NH, Zhu X, Kruman II, Castellani RJ, Petersen RB, Siedlak SL, et al. Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age (Dordr) 2008;30:209–15. doi: 10.1007/s11357-008-9050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ogawa O, Zhu X, Lee HG, Raina A, Obrenovich ME, Bowser R, et al. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathol. 2003;105:524–8. doi: 10.1007/s00401-003-0684-3. [DOI] [PubMed] [Google Scholar]
- 147.Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J Alzheimers Dis. 2009;18:131–9. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lithner CU, Hernandez CM, Nordberg A, Sweatt JD. Epigenetic changes related to beta-amyloid-implications for Alzheimer’s disease. Alzheimers Dement. 2009;5:P304. doi: 10.1016/j.jalz.2009.04.457. [DOI] [Google Scholar]
- 149.Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet. 2015;386:1683–97. doi: 10.1016/S0140-6736(15)00462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu X, Deng Y, Yu D, Cao H, Wang L, Liu L, et al. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer’s disease. PLoS One. 2014;9:e103067. doi: 10.1371/journal.pone.0103067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, et al. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–99. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Koshibu K, Graff J, Beullens M, Heitz FD, Berchtold D, Russig H, et al. Protein phosphatase 1 regulates the histone code for long-term memory. J Neurosci. 2009;29:13079–89. doi: 10.1523/JNEUROSCI.3610-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 154.Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr, Bennett DA, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guil S, Esteller M. RNA-RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem Sci. 2015;40:248–56. doi: 10.1016/j.tibs.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 156.Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, De Jager PL. Epigenomics of Alzheimer’s disease. Transl Res. 2015;165:200–20. doi: 10.1016/j.trsl.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hebert SS, Horre K, Nicolai L, Bergmans B, Papadopoulou AS, Delacourte A, et al. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis. 2009;33:422–8. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 158.Long JM, Ray B, Lahiri DK. MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J Biol Chem. 2012;287:31298–310. doi: 10.1074/jbc.M112.366336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Patel N, Hoang D, Miller N, Ansaloni S, Huang Q, Rogers JT, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener. 2008;3:10. doi: 10.1186/1750-1326-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Smith PY, Delay C, Girard J, Papon MA, Planel E, Sergeant N, et al. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum Mol Genet. 2011;20:4016–24. doi: 10.1093/hmg/ddr330. [DOI] [PubMed] [Google Scholar]
- 161.Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14:27–41. doi: 10.3233/JAD-2008-14103. [DOI] [PubMed] [Google Scholar]
- 162.Geekiyanage H, Chan C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J Neurosci. 2011;31:14820–30. doi: 10.1523/JNEUROSCI.3883-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, et al. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–23. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–20. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Faghihi MA, Zhang M, Huang J, Modarresi F, Van der Brug MP, Nalls MA, et al. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11:R56. doi: 10.1186/gb-2010-11-5-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nelson PT, Wang WX. MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J Alzheimers Dis. 2010;21:75–9. doi: 10.3233/JAD-2010-091603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Long JM, Ray B, Lahiri DK. MicroRNA-339-5p down-regulates protein expression of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J Biol Chem. 2014;289:5184–98. doi: 10.1074/jbc.M113.518241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lei X, Lei L, Zhang Z, Zhang Z, Cheng Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int J Clin Exp Pathol. 2015;8:1565–74. [PMC free article] [PubMed] [Google Scholar]
- 169.Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14:723–30. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Santa-Maria I, Alaniz ME, Renwick N, Cela C, Fulga TA, Van Vactor D, et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J Clin Invest. 2015;125:681–6. doi: 10.1172/JCI78421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mohamed JS, Lopez MA, Boriek AM. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3b. J Biol Chem. 2010;285:29336–47. doi: 10.1074/jbc.M110.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Moncini S, Salvi A, Zuccotti P, Viero G, Quattrone A, Barlati S, et al. The role of miR-103 and miR-107 in regulation of CDK5R1 expression and in cellular migration. PLoS One. 2011;6:e20038. doi: 10.1371/journal.pone.0020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang WX, Huang Q, Hu Y, Stromberg AJ, Nelson PT. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: white matter versus gray matter. Acta Neuropathol. 2011;121:193–205. doi: 10.1007/s00401-010-0756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–4. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 175.Smith P, Al Hashimi A, Girard J, Delay C, Hebert SS. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J Neurochem. 2011;116:240–7. doi: 10.1111/j.1471-4159.2010.07097.x. [DOI] [PubMed] [Google Scholar]
- 176.Smith PY, Hernandez-Rapp J, Jolivette F, Lecours C, Bisht K, Goupil C, et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. 2015;24:6721–35. doi: 10.1093/hmg/ddv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Strum JC, Johnson JH, Ward J, Xie H, Feild J, Hester A, et al. MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol Endocrinol. 2009;23:1876–84. doi: 10.1210/me.2009-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, Rao P, et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011;30:4299–308. doi: 10.1038/emboj.2011.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schonrock N, Humphreys DT, Preiss T, Gotz J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-beta. J Mol Neurosci. 2012;46:324–35. doi: 10.1007/s12031-011-9587-2. [DOI] [PubMed] [Google Scholar]
- 180.Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–93. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 181.Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schratt G. microRNAs at the synapse. Nat Rev Neurosci. 2009;10:842–9. doi: 10.1038/nrn2763. [DOI] [PubMed] [Google Scholar]
- 183.Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- 184.Massone S, Vassallo I, Fiorino G, Castelnuovo M, Barbieri F, Borghi R, Tabaton M, Robello M, Gatta E, Russo C, et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol Dis. 2011;41:308–17. doi: 10.1016/j.nbd.2010.09.019. [DOI] [PubMed] [Google Scholar]
- 185.Aksoy-Aksel A, Zampa F, Schratt G. MicroRNAs and synaptic plasticity—a mutual relationship. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130515. [DOI] [PMC free article] [PubMed]
- 186.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–17. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 188.Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–38. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 189.Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry. 2004;43:6899–908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 190.Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology. 2009;34:1721–32. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 191.Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2010;35:870–80. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 193.Zhang ZY, Schluesener HJ. Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J Neuropathol Exp Neurol. 2013;72:178–85. doi: 10.1097/NEN.0b013e318283114a. [DOI] [PubMed] [Google Scholar]
- 194.Lopez-Atalaya JP, Ito S, Valor LM, Benito E, Barco A. Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 2013;41:8072–84. doi: 10.1093/nar/gkt590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Riva P, Ratti A, Venturin M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr Alzheimer Res. 2016;13(11):1219-231. [DOI] [PubMed]
- 196.Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013;11(1):8–17. doi: 10.1016/j.gpb.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ntziachristos P, Abdel-Wahab O, Aifantis I. Emerging concepts of epigenetic dysregulation in hematological malignancies. Nat Immunol. 2016;17(9):1016–24. doi: 10.1038/ni.3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Devall M, Roubroeks J, Mill J, Weedon M, Lunnon K. Epigenetic regulation of mitochondrial function in neurodegenerative disease: new insights from advances in genomic technologies. Neurosci Lett. 2016;625:47–55. doi: 10.1016/j.neulet.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Stephan KE, Schlagenhauf F, Huys QJ, Raman S, Aponte EA, Brodersen KH, et al. Computational neuroimaging strategies for single patient predictions. Neuroimage. 2016;145(Pt B):180–99. doi: 10.1016/j.neuroimage.2016.06.038. [DOI] [PubMed] [Google Scholar]
- 200.Horvath P, Aulner N, Bickle M, Davies AM, Nery ED, Ebner D, et al. Screening out irrelevant cell-based models of disease. Nat Rev Drug Discov. 2016;15(11):751–69. doi: 10.1038/nrd.2016.175. [DOI] [PubMed] [Google Scholar]
- 201.Inokoshi J, Katagiri M, Arima S, Tanaka H, Hayashi M, Kim YB, et al. Neuronal differentiation of neuro 2a cells by inhibitors of cell cycle progression, trichostatin A and butyrolactone I. Biochem Biophys Res Commun. 1999;256:372–6. doi: 10.1006/bbrc.1999.0316. [DOI] [PubMed] [Google Scholar]
- 202.Gaub P, Tedeschi A, Puttagunta R, Nguyen T, Schmandke A, Di Giovanni S. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 2010;17:1392–408. doi: 10.1038/cdd.2009.216. [DOI] [PubMed] [Google Scholar]
- 203.Shakèd M, Weissmüller K, Svoboda H, Hortschansky P, Nishino N, Wölfl S, et al. Histone deacetylases control neurogenesis in embryonic brain by inhibition of BMP2/4 signaling. PLoS One. 2008;3(7):e2668. doi: 10.1371/journal.pone.0002668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Leng Y, Wang J, Wang Z, Liao HM, Wei M, Leeds P, et al. Valproic acid and other HDAC inhibitors upregulate FGF21 gene expression and promote process elongation in glia by inhibiting HDAC2 and 3. Int J Neuropsychopharmacol. 2016;12:19(8). doi: 10.1093/ijnp/pyw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Singh RP, Shiue K, Schomberg D, Zhou FC. Cellular epigenetic modifications of neural stem cell differentiation. Cell Transplant. 2009;18(10):1197–211. doi: 10.3727/096368909X12483162197204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hass J, Walton E, Wright C, Beyer A, Scholz M, Turner J, et al. Associations between DNA methylation and schizophrenia-related intermediate phenotypes—a gene set enrichment analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2015;59:31–9. doi: 10.1016/j.pnpbp.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Aberg KA, McClay JL, Nerella S, Clark S, Kumar G, Chen W, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiat. 2014;71(3):255–64. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Grayson DR. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102(26):9341–6. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57(5):500–9. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 210.Kurita M, Holloway T, García-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–54. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Abdolmaleky HM, Cheng KH, Faraone SV, Wilcox M, Glatt SJ, Gao F, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolardisorder. Hum Mol Genet. 2006;15(21):3132–45. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lott SA, Burghardt PR, Burghardt KJ, Bly MJ, Grove TB, Ellingrod VL. The influence of metabolic syndrome, physical activity and genotype on catechol-O-methyl transferase promoter-region methylation in schizophrenia. Pharmacogenomics J. 2013;13(3):264–71. doi: 10.1038/tpj.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Melas PA, Rogdaki M, Ösby U, Schalling M, Lavebratt C, Ekström TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012;26(6):2712–8. doi: 10.1096/fj.11-202069. [DOI] [PubMed] [Google Scholar]
- 214.Tolosa A, Sanjuán J, Dagnall AM, Moltó MD, Herrero N, de Frutos R. FOXP2 gene and language impairment in schizophrenia: association and epigenetic studies. BMC Med Genet. 2010;11:114. doi: 10.1186/1471-2350-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Abdolmaleky HM, Yaqubi S, Papageorgis P, Lambert AW, Ozturk S, Sivaraman V, Thiagalingam S. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr Res. 2011;129(2-3):183–90. doi: 10.1016/j.schres.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 216.Iwamoto K, Bundo M, Yamada K, Takao H, Iwayama-Shigeno Y, Yoshikawa T, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J Neurosci. 2005;25(22):5376–81. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Carrard A, Salzmann A, Malafosse A, Karege F. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J Affect Disord. 2011;132(3):450–3. doi: 10.1016/j.jad.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 218.Chen Y, Zhang J, Zhang L, Shen Y, Xu Q. Effects of MAOA promoter methylation on susceptibility to paranoid schizophrenia. Hum Genet. 2012;131(7):1081–7. doi: 10.1007/s00439-011-1131-5. [DOI] [PubMed] [Google Scholar]
- 219.Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82(3):696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kocerha J, Faghihi MA, Lopez-Toledano MA, Huang J, Ramsey AJ, Caron MG, Sales N, Willoughby D, Elmen J, Hansen HF, Orum H, Kauppinen S, Kenny PJ, Wahlestedt C. MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc Natl Acad Sci U S A. 2009;106(9):3507–12. doi: 10.1073/pnas.0805854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14(4):268–76. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Solanki N, Alkadhi I, Atrooz F, Patki G, Salim S. Grape powder prevents cognitive, behavioral, and biochemical impairments in a rat model of posttraumatic stress disorder. Nutr Res. 2015;35(1):65–75. doi: 10.1016/j.nutres.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sawamura T, Klengel T, Armario A, Jovanovic T, Norrholm SD, Ressler KJ, et al. Dexamethasone treatment leads to enhanced fear extinction and dynamic Fkbp5 regulation in amygdala. Neuropsychopharmacology. 2016;41(3):832–46. doi: 10.1038/npp.2015.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bahari-Javan S, Maddalena A, Kerimoglu C, Wittnam J, Held T, Bähr M, et al. HDAC1 regulates fear extinction in mice. J Neurosci. 2012;32(15):5062–73. doi: 10.1523/JNEUROSCI.0079-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fujita Y, Morinobu S, Takei S, Fuchikami M, Matsumoto T, Yamamoto S, et al. Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J Psychiatr Res. 2012;46(5):635–43. doi: 10.1016/j.jpsychires.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 226.Bousiges O, Vasconcelos AP, Neidl R, Cosquer B, Herbeaux K, Panteleeva I, et al. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology. 2010;35(13):2521–37. doi: 10.1038/npp.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, et al. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13(6):664–6. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–69. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 229.Gräff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell. 2014;156(1-2):261–76. doi: 10.1016/j.cell.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los SR, Goldmann E, Galea S. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2010;107(20):9470–5. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Uddin M, Galea S, Chang SC, Koenen KC, Goldmann E, Wildman DE, Aiello AE. Epigenetic signatures may explain the relationship between socioeconomic position and risk of mental illness: preliminary findings from an urban community-based sample. Biodemography Soc Biol. 2013;59(1):68–84. doi: 10.1080/19485565.2013.774627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Uddin M, Galea S, Chang SC, Aiello AE, Wildman DE, de los Santos R, et al. Gene expression and methylation signatures of MAN2C1 are associated with PTSD. Dis Markers. 2011;30(2-3):111–21. doi: 10.1155/2011/513659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rusiecki JA, Byrne C, Galdzicki Z, Srikantan V, Chen L, Poulin M, et al. PTSD and DNA methylation in select immune function gene promoter regions: a repeated measures case-control study of U.S. military service members. Front Psychiatry. 2013;4:56. doi: 10.3389/fpsyt.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Norrholm SD, Jovanovic T, Smith AK, Binder E, Klengel T, Conneely K, et al. Differential genetic and epigenetic regulation of catechol-O-methyltransferase is associated with impaired fear inhibition in posttraumatic stress disorder. Front Behav Neurosci. 2013;7:30. doi: 10.3389/fnbeh.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470(7335):492–7. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Vukojevic V, Kolassa IT, Fastenrath M, Gschwind L, Spalek K, Milnik A, et al. Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J Neurosci. 2014;34(31):10274–84. doi: 10.1523/JNEUROSCI.1526-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Labonté B, Azoulay N, Yerko V, Turecki G, Brunet A. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. Transl Psychiatry. 2014;4:e368. doi: 10.1038/tp.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, et al. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry. 2015;77(4):356–64. doi: 10.1016/j.biopsych.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 239.Chang SC, Koenen KC, Galea S, Aiello AE, Soliven R, Wildman DE, Uddin M. Molecular variation at the SLC6A3 locus predicts lifetime risk of PTSD in the Detroit Neighborhood Health Study. PLoS One. 2012;7(6):e39184. doi: 10.1371/journal.pone.0039184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Koenen KC, Uddin M, Chang SC, Aiello AE, Wildman DE, Goldmann E, et al. SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depress Anxiety. 2011;28(8):639–47. doi: 10.1002/da.20825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, et al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(6):700–8. doi: 10.1002/ajmg.b.31212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zheng D, Sabbagh JJ, Blair LJ, Darling AL, Wen X, Dickey CA. MicroRNA-511 binds to FKBP5 mRNA, which encodes a chaperone protein, and regulates neuronal differentiation. J Biol Chem. 2016;291(34):17897–906. doi: 10.1074/jbc.M116.727941. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.