

SUMMARY
Deamination of choline catalyzed by the glycyl radical enzyme choline trimethylamine-lyase (CutC) has emerged as an important route for the production of trimethylamine, a microbial metabolite associated with both human disease and biological methane production. Here, we have determined five high-resolution X-ray structures of wild-type CutC and mechanistically informative mutants in the presence of choline. Within an unexpectedly polar active site, CutC orients choline through hydrogen bonding with a putative general base, and through close interactions between phenolic and carboxylate oxygen atoms of the protein scaffold and the polarized methyl groups of the trimethylammonium moiety. These structural data, along with biochemical analysis of active site mutants, support a mechanism that involves direct elimination of trimethylamine. This work broadens our understanding of radical-based enzyme catalysis and will aid in the rational design of inhibitors of bacterial trimethylamine production.
Graphical abstract
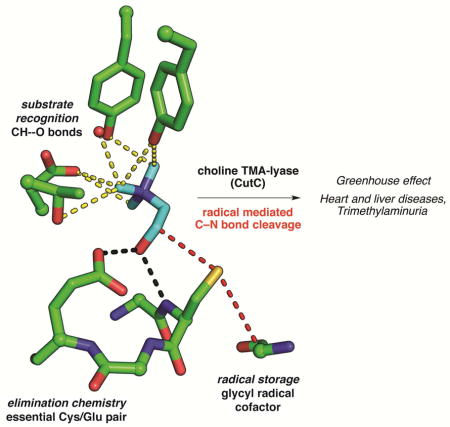
INTRODUCTION
Anaerobic cleavage of choline by microbes to form acetaldehyde and trimethylamine (TMA) impacts both the environment and human health (Figure 1A). Within the guts of ruminants and in marine sediments, choline-derived TMA is converted to the powerful greenhouse gas methane by archaea (Brugere et al., 2014, Hippe et al., 1979). TMA has been estimated to account for between 35% and 60% of the methane produced in marine sediments (Hippe et al., 1979), thus playing a major role in the greenhouse effect, as well as in global nitrogen and carbon cycles. Although choline is an important nutrient for humans, conversion of choline to TMA by human gut microbiota and its further oxidation to trimethylamine N-oxide by liver enzymes (Krueger and Williams, 2005) is implicated in diseases, including nonalcoholic fatty liver disease (Dumas et al., 2006), atherosclerosis (Tang et al., 2013, Wang et al., 2011, Wang et al., 2014), and the metabolic disorder trimethylaminuria (fish malodor syndrome) (Christodoulou, 2012). A choline utilization (cut) gene cluster was recently discovered in Desulfovibrio species and characterized (Craciun and Balskus, 2012). The key enzyme in this pathway is the C–N-bond-cleaving glycyl radical enzyme (GRE) choline trimethylamine-lyase (CutC), which converts choline into TMA and acetaldehyde (Craciun et al., 2014). Bioinformatic analyses have revealed that choline utilization is prevalent in human stool metagenomes and is widely distributed among both environmental and human-associated bacteria (Martinez-del Campo et al., 2015), highlighting the importance of CutC for TMA production in diverse anoxic microbial habitats.
Figure 1. Anaerobic metabolism of choline into trimethylamine (TMA) is a disease-associated microbial activity that utilizes the glycyl radical enzyme (GRE) CutC.

(A) Anaerobic microbes generate TMA from choline in the human gut and in the environment. TMA is further metabolized to the greenhouse gas methane by archaea, or to the disease-associated metabolite trimethylamine N-oxide by a human liver monooxygenase. (B) General mechanistic hypothesis for GRE function: the 5′-deoxyadenosyl radical (Ado•) is used by an AdoMet radical enzyme to produce a stable glycyl radical within GREs. A thiyl radical is proposed to serve as the active oxidant in all GREs and would be generated transiently within the active site. S = substrate, P = product. Following initial hydrogen atom abstraction to form an α-hydroxy radical, (C) CutC may perform base-catalyzed direct elimination of TMA. (D) Alternately, choline cleavage could involve a 1,2-migration of the trimethylammonium moiety followed by decomposition of the resulting carbinolamine.
The glycyl radical enzyme (GRE) family has emerged as a remarkable class of biocatalysts that play central roles in anaerobic microbial metabolic pathways (Selmer et al., 2005). GREs are post-translationally modified by a radical S-adenosylmethionine (AdoMet) activating protein (CutD within the cut gene cluster), which installs a stable, α-carbon glycyl radical. This glycyl radical species is proposed to generate a transient thiyl radical that initiates radical catalysis and is regenerated upon product formation (Figure 1B). We previously envisioned two general mechanisms for choline cleavage by CutC (Craciun et al., 2014) based on either the adenosylcobalamin (AdoCbl)-dependent ethanolamine ammonia lyase (EAL) (Toraya, 2003) or the AdoMet-dependent radical C–N lyase DesII (Ruszczycky and Liu, 2015) and a related GRE, glycerol dehydratase (GDH), that performs an analogous C–O bond scission (O’Brien et al., 2004). Both potential mechanisms begin with the cleavage of the C1 C–H bond of choline by the transient thiyl radical, generating a substrate-based α-hydroxyalkyl radical. The following steps diverge; either a base-catalyzed 1,2-elimination of TMA (Figure 1C) or a 1,2-migration followed by elimination (Figure 1D) could yield the final products. The active site environment is expected to control which of these mechanisms is possible through the interactions of the protein with the substrate and radical intermediates. Understanding the structure of CutC is therefore important not only for enhancing our knowledge of radical enzymes, but also for designing inhibitors of TMA generation. Here we report a high-resolution structure of CutC from Desulfovibrio alaskensis G20 bound to choline, which, along with mutagenesis experiments, structures of CutC mutants, and in vitro biochemical analyses, provides new insight into the mechanism of this C–N cleaving radical enzyme.
RESULTS
A conserved GRE architecture is maintained in CutC
We determined the structure of CutC from D. alaskensis G20 by molecular replacement with AdoCbl-independent GDH (PDB ID 1R8W) (O’Brien et al., 2004) as the search model. CutC crystallizes as a dimer of dimers (Figure 2A) whose tetrameric assembly resembles that of other GRE enzymes (Figure S1). As expected from the recently determined structure of a proteolytic fragment of a homologous CutC from the opportunistic pathogen Klebsiella pneumoniae (Kalnins et al., 2015), each CutC monomer adopts the canonical GRE fold: a ten-stranded β/α barrel (β1-β10) with two loops containing catalytic residues inserted into the center of the barrel (Figure 2B). The so-called Gly loop is located at the tip of the C-terminal glycyl radical domain, a structural unit believed to undergo conformational changes that can close or expose the active site during installation of the glycyl radical by the activating enzyme (Vey et al., 2008). A universally conserved glycine (Gly821 in CutC) is found at the tip of the Gly loop, fully in the interior of the barrel. Next to the Gly loop in the enzyme interior is the Cys loop, which contains a universally conserved cysteine residue (Cys489 in CutC) and other residues that contribute to catalysis and are unique to specific GREs (Lehtio and Goldman, 2004). The active site cavity is found directly above the Cys loop, consistent with reaction initiation by a transient thiyl radical generated on Cys489. In agreement with these proposed roles in CutC, C489A and G821A mutants were found to be completely inactive in the production of TMA in an end point assay (Craciun et al., 2014).
Figure 2. Overall structure of CutC from D. alaskensis G20.

(A) CutC crystallizes as a dimer of dimers; the dimeric unit is shown below rotated 90°. (B) Within a single monomer, the active site cavity is found at the center of the (β/α)10 barrel (solid ribbon), above the Cys loop. Residues Cys489 (location of putative, transient thiyl radical) and Gly821 (location of the stable glycyl radical) are shown in spheres. The glycyl radical domain (yellow) harbors the active site glycyl radical on the Gly loop in post-translationally modified proteins. (C) Cross-section of the CutC active site displaying a ring of aromatic residues (sticks) at the top of the active site, including two residues on a helix that caps the top of the barrel (gray). Choline is shown in the active site (cyan sticks). See Figure S1 for structural comparisons.
A notable feature of the CutC active site also observed in other GREs (Funk et al., 2014, Funk et al., 2015) is a hydrophobic cap formed by a helix extending from β3 (Figure 2C). This cap packs against two tyrosine residues that participate in choline binding (described below) and appears to block access to the active site from the exterior of the protein; simple rotation of the side chains of Phe389 and/or Trp379 into adjacent, solvent-filled regions may allow access to the active site. Surprisingly, we find choline bound in the active site and hypothesize that it bound during expression and was retained through purification. We were unable to acquire a structure of the ligand-free wild-type enzyme, despite dialysis of the protein after purification.
Selective binding of choline involves unexpected interactions
The CutC active site is formed by residues from the Cys loop and the top face of the barrel (Figure 3). The Cys loops of CutC and GDH are quite similar (Figure 3D), both sharing the GCVEP sequence motif that is common in GRE enzymes (Figure S2). In CutC, the Cys loop employs the backbone amide nitrogen of Gly488/Cys489 and the carboxylate of Glu491 to form hydrogen bonds with the choline hydroxyl group, positioning C1 of choline 3.6 Å from the Cys489 thiol, a distance suitable for pro-S hydrogen atom abstraction by the putative thiyl radical (Figure 3A). C2 of choline is also in proximity, 4.3 Å, to Cys489 potentially facilitating reformation of the thiyl radical following choline cleavage. The choline trimethylammonium group extends away from the Cys loop and is oriented gauche to the hydroxyl, with an average dihedral angle of 61° in the four molecules in the asymmetric unit. The aromatic ring of β3 residue Phe395 appears to stabilize this conformation of choline, establishing a face-on cation-π-like interaction with C2 (C2 to ring center: 3.8 Å), which would be expected to have a partial positive charge due to the adjacent quaternary ammonium (Figure 3A, C).
Figure 3. CutC binds substrate in a pocket above the Cys loop.

(A) Stereoimage of CutC bound to its substrate, choline (cyan). The proposed hydrogen atom transfer pathway from Gly821 to Cys489 to C1 of choline is marked (red dashes). Hydrogen bonds (2.5–3.2 Å) are shown for residues within the active site (black dashes). The average HO–C1–C2–N(Me3) dihedral angle is 61°. (B) Stereoimage of the CH---O hydrogen bonds (yellow dashes) present in the CutC-choline complex. (C) Diagram of protein and water interactions with the trimethylammonium moiety of choline with CH–O and hydrogen bond distances (Å). CH---O hydrogen bonds are indicated for C to O distances of 3.8 Å or less (yellow). Hydrogen bonds (black) and presumed cation-π interactions (gray) are shown between protein and substrate atoms. Distances (Å) are given in the diagram. The maximum-likelihood-estimated coordinate error is 0.20 Å. (D) Comparison of CutC (green) and GDH (magenta) active site residues involved in binding C1 and C2 (sticks). The Cys loop GCVEP motif is conserved between the two enzymes. An aromatic residue contributed from β3 coordinates C2 of choline (gray dashes). The proposed hydrogen transfer pathways (red dashes) between the active site Gly, Cys, and C1 are virtually identical in the two structures. See Figure S2 for sequence alignments and Figure S3 for additional CH---O hydrogen bonding analysis.
The primary choline-selective interactions are made to the trimethylammonium substituent of choline. Unexpectedly, these interactions involve the side chain oxygen atoms of Tyr208, Tyr506, and Asp216 (Figure 3B). These oxygen atoms approach the trimethylammonium methyl groups closely, displaying C to O distances of 3.3 and 3.5 Å, shorter than a typical van der Waals distance. Such contacts, previously described as CH---O hydrogen bonds, are ubiquitous features in protein and nucleic acid structures and are proposed to play an important, but underappreciated, role in stabilizing macromolecule structure (Adhikari and Scheiner, 2013, Horowitz and Trievel, 2012, Horowitz et al., 2013). In cases where a partial positive charge is localized on the carbon atom, CH---O hydrogen bonds contribute an estimated ~1.2 kcal/mol to the binding energy, comparable to weak hydrogen bonds (Musah et al., 1997). Within CutC, there are at least four close CH---O hydrogen bonds (3.2–3.5 Å) and another three longer interactions (3.5–3.7 Å) (Figure 3C). A single water molecule held in the active site by Tyr506 also participates in a close CH---O hydrogen bond. A survey of proteins that interact with trimethylammonium-containing metabolites reveals that CH---O hydrogen bonds are, in fact, quite common (Figure S3). In this analysis, these contacts appear more abundant than the better-known cation-π interactions from aromatic residues that have been proposed to be a major contributor to binding energy for trimethylammonium ligands (Dougherty and Stauffer, 1990). We also expect that a deprotonated Asp216 will contribute to substrate binding through electrostatic interactions with the positively charged trimethylammonium group (Figure 3C).
Disrupting enzyme-substrate CH---O hydrogen bonds impairs binding and catalysis
Based on the crystal structure of CutC, Tyr208 and Tyr506 mutants were constructed to determine if the proposed CH---O hydrogen bonds are important for binding choline or C–N bond cleavage (Table 1). The activation of these mutants by CutD occurred to a lesser extent than for wild-type CutC, perhaps due to disruption of the conformational changes required for glycyl radical installation. The fraction of the enzyme in radical form, however, appears stable over time (Figure S4A). Kinetic analyses revealed little change in KM for both CutC-Y208F and CutC-Y506F, but the catalytic activity of CutC-Y208F was more impaired than CutC-Y506F. Interestingly, the double mutant CutC-Y208F/Y506F is similar to CutC-Y208F in kcat, but it has a pronounced defect (~7 fold with respect to CutC-Y208F and ~11 fold with respect to CutC-WT) in KM, and thus diminished catalytic proficiency. To further investigate the contribution of CH---O contacts to choline binding, we crystallized CutC-Y208F after incubation with 10 mM choline. A mostly unperturbed active site is observed in the structure, with choline positioned similarly to wild-type CutC (Figure 4A), suggesting that the loss of activity for CutC-Y208F is not due to changes in how choline is bound.
Table 1. Activation and kinetic parameters of CutC mutants.
Activation was measured by double integration of the EPR signal from each mutant after activation by CutD (see Experimental Procedures). The values reported with standard deviation are the average of three replicates. Activity for the mutants was initially detected by analysis of TMA production in an overnight end point assay. With the exception of CutC-D216N, mutants with detectable activity were kinetically characterized using a coupled assay as described in the methods. The un-normalized kcat is given based on the total concentration of dimeric CutC. A normalized kcat was estimated by dividing kcat by the proportion of activated enzyme.
| protein | radicals per polypeptide (%) | detectable activity (TMA) | KM (mM) | kcat (s−1) | glycyl-radical-normalized kcat (s−1) |
|---|---|---|---|---|---|
| wild type | 20.8 | yes | 0.13 ± 0.01 | 157 ± 2 | 755 ± 11 |
| E491Q | 3.9 | no | ND | ND | ND |
| E491A | 10.5 | no | ND | ND | ND |
| T502A | <1 | yes | ND | 0.036 ± 0.003 | ND |
| D216N | <1 | yes | ND | ND | ND |
| Y208F | 5.5 | yes | 0.22 ± 0.03 | 0.73 ± 0.01 | 13.3 ± 0.3 |
| Y506F | 1.9 | yes | 0.12 ± 0.03 | 3.27 ± 0.11 | 172 ± 6 |
| Y208F/Y506F | 1.7 | yes | 1.53 ± 0.12 | 0.15 ± 0.01 | 9.1 ± 0.2 |
Figure 4. Structures of mutant CutC enzymes.

(A,B,C,D) Composite omit density (light blue) is contoured at 1.5σ around active site residues with changes to each mutant structure highlighted (yellow). Hydrogen bonds (black dashes), steric clashes (pink dashes), and hydrogen-atom transfer pathways (red dashes) are indicated. Crystals were grown in the presence of 10 mM choline. (A) CutC-Y208F (1.90-Å resolution) closely resembles the wild-type enzyme, with the only difference being the generation of a new partially-occupied water binding site (HOH*). (B) E491A at 1.90-Å resolution contains active site density consistent with disordered water and/or partial occupancy of choline. (C) The E491Q mutant (1.60-Å resolution) has no density for choline. Instead, two new water molecules are bound in the active site (HOH*). The orientation of the side chain amide is deduced based on the contacts to hydrogen bond donors within the Cys loop. (D) T502A at 1.85-Å resolution is virtually identical to the wild-type CutC structure. See Table 1 for activity data on mutant proteins.
Structure and mutagenesis support catalytic role for Glu491
The CutC mechanistic hypotheses in Figure 1C and 1D require an acceptor for the choline hydroxyl proton, and Glu491 of the conserved GCVEP motif appears positioned to perform this function. The equivalent Glu has been well studied in the E. coli class Ia ribonucleotide reductase (RNR) and was found to play a modest role in substrate binding and a critical role as an acid/base catalyst (Persson et al., 1997, Lawrence et al., 1999). By analogy, mutation of Glu491 would be expected to have an effect on both substrate binding and catalysis. Indeed, we find that both CutC-E491A and CutC-E491Q are completely inactive in an overnight end point assay (Figure S4C, Table 1). To further investigate the source of the inactivity, we determined crystal structures of CutC-E491A and CutC-E491Q. The wild-type and mutant enzymes are virtually identical in structure [root mean square deviation (RMSD) of 0.20 or 0.18 Å for CutC-E491A and CutC-E491Q, respectively] including the arrangement of residues in the active site (RMSD of 0.11 or 0.09 for Cys loop residues), confirming that inactivity of mutant enzymes is not due to a disruption in protein structure. However, substrate binding does appear to be impaired. Despite inclusion of 10 mM choline in the protein solution prior to crystallization, no clear density is observed for choline within either active site (Figure 4B, C). For E491A, density in the active site is consistent with disordered water molecules and/or choline bound at partial occupancy. For E491Q, there is no indication that choline is bound even at low occupancy. Instead, two new water molecules are bound: one replaces the hydroxyl group of choline and the other makes contacts to Asp216 and Thr502.
Analysis of predicted hydrogen orientations in wild-type CutC suggests that Glu491 acts as a hydrogen bond acceptor and that the Gly488/Cys489 peptide bond acts as a hydrogen bond donor to the choline C1 hydroxyl (Figure 5A). Whereas E491A cannot participate in hydrogen bonding, E491Q could serve as a donor or acceptor depending on the side chain orientation. Inspection of the E491Q mutant structure indicates that E491Q is positioned such that the side chain amide nitrogen, which is a hydrogen bond donor, is pointing toward the choline-binding site (Figure 5B). In particular, the close interaction between E491Q side chain and a backbone amide nitrogen of the Cys loop is indicative of a hydrogen bond between a backbone amide and a side chain carbonyl. Thus, the E491Q mutant replaces the hydrogen bond acceptor for the choline hydroxyl with a hydrogen bond donor, which seems to impair choline binding possibly more than the simple absence of a side chain given the observed electron density (Figure 4B, C). Although, we cannot determine the severity of the defect in catalysis for E491Q or E491A and cannot sort out the contribution of this residue to substrate binding versus catalysis, the complete absence of activity even at high choline concentrations (100 mM) prompt us to propose that Glu491, like the structurally similar Glu441 in class Ia RNR, is a catalytic base that also contributes to substrate binding affinity.
Figure 5. Putative hydrogen bonding networks in wild-type and mutant CutC enzymes.

The orientation is shifted ~90° around the y-axis from Figure 4. Tyr506 and the Gly loop are not shown for clarity. Polar hydrogen atoms are shown within the networks based on inferred position given the available donor and acceptor groups, and considering that the pH of the crystallization was 8.0. Riding hydrogen atoms were added to the model during the final rounds of refinement. (A) In wild-type CutC, two networks of hydrogen bonds are highlighted (black dashes). Interactions between Glu491 and Thr502 and a backbone amide suggest that it is deprotonated and able to act as a general base to accept a proton from choline (cyan). Asp216 is also likely to be deprotonated, sharing a single proton in a hydrogen bond (O–O distance of 2.7 Å) with Tyr208. A proposed connection (white dashes) between Thr502-Asp216 would be possible if either of these residues were to change conformation. CH---O hydrogen bonds to Tyr208 and Aps216 are shown as yellow dashes. (B) In CutC-E491Q, the side chain carbonyl of Gln491 is likely to be pointing down toward the backbone amide, leaving the side chain nitrogen to point toward Thr502. (C) In CutC-T502A, no connection is available between Glu491 and Asp216. See Figure S4 and Table 1 for activity data on mutant CutC proteins.
A putative proton transfer network in the active site of CutC
In order to complete the catalytic cycle, the active site base that removed the choline C1 proton must be deprotonated. In the absence of an obvious proton pathway leading away from the active site to the exterior of the protein, we suggest that TMA may be the ultimate acceptor for the C1 hydroxyl proton. Direct deprotonation of the putative base, Glu491, by TMA is unlikely as the nitrogen is 4.6 Å away from the side chain of Glu491 and the movement of Glu491 is sterically blocked by both the C1 of choline and Thr502 (Figure 5A). Instead, Asp216 is positioned close to the trimethylammonium moiety such that it could serve as the general acid. However, Asp216 is also too far from Glu491 for direct proton transfer (5.8 Å) and its movement is additionally blocked by Thr502 (Figure 5A). Our structures suggest Thr502 may bridge the gap between Glu491 and Asp216 by simultaneously accepting and donating a proton. Thr502 is already in position to hydrogen bond with Glu491 (2.5 Å), and a simple rotation of Thr502 around χ1 by 10–15° should allow the formation of a new hydrogen bond between Thr502 and Asp216. In fact, in the structure of CutC-E491Q, which may serve as a mimic for the protonated state of Glu491, the hydroxyl group of Thr502 has moved closer to Asp216 (3.6 Å) (Figure 5A, B).
To further explore the roles of Asp216 and Thr502, which are both absolutely conserved in CutC enzymes (Figure S2), we generated CutC-D216N and CutC-T502A mutant proteins. The activation of both mutant proteins was below the limit of detection of our EPR instrument, but both enzymes were active in an overnight endpoint assay for TMA detection (Figure S4D). Unfortunately, the low activity of CutC-D216N in the overnight assay precluded further kinetic analysis of this mutant. For CutC-T502A, a kinetic assay was performed and low activity was observed; however, this activity started to diminish after 30 min (Table 1, Figure S4B). We were able to determine the structure of CutC-T502A, which shows choline binding at full occupancy with no discernable differences from wild-type (Figure 4D), except for the fact that the hydrogen bonding network is interrupted (Figure 5C).
DISCUSSION
The C–N lyase chemistry performed by CutC is unique among GREs, but resembles that of other radical enzymes that promote 1,2-elimination reactions: the C–O cleaving GRE GDH, the AdoCbl dependent enzymes EAL and diol dehydratase, and the AdoMet radical enzyme DesII. Based on previous studies of these enzymes, we have considered two mechanisms for CutC (Figure 1C, D): a direct elimination of TMA from the substrate-based radical, similar to the one favored for GDH (Feliks and Ullmann, 2012) and DesII (Ruszczycky and Liu, 2015) or a 1,2-migration, similar to the proposed mechanism for EAL (Toraya, 2003) (Figure S5). The biochemical and structural data presented here appear to complement the elegant mechanistic studies performed with DesII using deuterated and fluorinated substrate analogs (Lin et al., 2015), and the structural and computational studies for GDH (Feliks and Ullmann, 2012, O’Brien et al., 2004). As explained below, on the basis of these experiments, we favor a direct elimination mechanism for choline cleavage.
Catalysis is initiated by the formation of a transient thiyl radical (Figure 6). The position of choline within the active site of CutC indicates that abstraction of a hydrogen atom from C1 (likely the pro-S) by a Cys489 thiyl radical is the probable first step in choline cleavage (Figure 6, I). The second step (Figure 6, II) would either involve base-catalyzed deprotonation of C1-OH followed by heterolytic C–N bond cleavage or formation of a carbinolamine through migration of the trimethylammonium group from C2 to C1, followed by a decomposition of that species. Here, the structure and mutagenesis data, along with literature precedent, support the former mechanism. First, E491 is ideally positioned to catalyze the deprotonation of C1-OH. The pKa of E491 should be sufficient for it to act as a base given that the pKa of the choline hydroxyl [~13.9 (Dawson, 1969)] is already lower than that of a typical alcohol, and the pKa values of α-hydroxy radicals are often further depressed by an additional 4–8 pH units (Laroff and Fessenden, 1973). Importantly, E491 occupies a position in the GRE fold that is often associated with acid/base chemistry (Lehtio and Goldman, 2004, Wei et al., 2014), and here we show that the conservative mutation of this residue to Gln in CutC leads to a complete loss of activity. Additionally, a computational study of the GDH mechanism found that proton transfer between the C1 hydroxyl group of glycerol and the analogous active site Glu triggers dehydration by direct elimination (Feliks and Ullmann, 2012). For class Ia RNR, proton transfer between the 3′ hydroxyl of the ribonucleotide substrate and the active site Glu occurs prior to elimination of water from the 2′ nucleotide position (Persson et al., 1997, Lawrence et al., 1999), and for DesII, EPR and kinetic isotope effect studies implicate a general base in the direct elimination of ammonia from TDP-4-amino-4,6-dideoxy-D-glucose (Ruszczycky and Liu, 2015), but the lack of a crystal structure for DesII precludes a specific assignment of this residue. Thus, we propose that in CutC, Glu491 acts as a general base to form a transient ketyl radical, a species similar to those postulated in the 1,2-elimination of water by analogous radical dehydratases (Buckel and Golding, 1998, Hans et al., 2002, Lenz and Giese, 1997) and in the elimination of ammonia by DesII (Ruszczycky and Liu, 2015), and also similar to the ketyl radical species that are known to form in RNR (Persson et al., 1997, Lawrence et al., 1999).
Figure 6.

Mechanistic proposal for choline cleavage by CutC.
Following deprotonation of the α-hydroxyalkyl radical, we propose that rapid heterolytic C–N bond cleavage allows the direct elimination of TMA (Figure 6, III, IV). This proposal draws support from radiolysis experiments in which choline-derived radicals undergo fragmentation very rapidly under neutral conditions (Foster and West, 1974). EPR studies of the substrate-based radical of TDP-D-quinovose generated by DesII have revealed the importance of establishing overlap between the partially filled p-orbital at the radical center and the σ* orbital of the adjacent functional group to be eliminated (Ruszczycky et al., 2011). Within the active site of CutC, choline is oriented such that the pro-S C1 hydrogen atom is expected to be antiperiplanar to the trimethylammonium group, potentially allowing hyperconjugation between the p-orbital of a C1 choline radical and the C–N σ* orbital, and thereby facilitating TMA elimination.
The observed CH---O hydrogen bonds between the trimethylammonium group and the polar residues lining the back of the active site (Asp216, Tyr208, Tyr506) would also be expected to weaken the C–N bond of choline. Our structure suggests these CH---O hydrogen bonds likely play a role in both choline binding selectivity and in catalysis, consistent with the ~83-fold reduced catalytic efficiency of the CutC double mutant Y208F/Y506F. Although perturbation of these CH---O contacts diminishes activity, this double mutant is nonetheless active. Were migration of the trimethylammonium group key to catalysis (the EAL model), one would predict that residues that could facilitate this migration through direct contacts with the trimethylammonium moiety would be catalytically essential, as has been observed for ammonia migration in ethanolamine ammonia-lyase (Mori et al., 2014). Whereas a 1,2-migration reaction requires that exquisite control be exerted over the migrating group by active site residues, no such requirement is necessary for a 1,2-elimination; indeed, this reaction proceeds very rapidly in solution (Foster and West, 1974). Thus, the retention of activity for the Y208F/Y506F double mutant supports a 1,2-elimination mechanism. Finally, migration of the bulky trimethylammonium moiety appears to be restricted by Thr502, which packs against this part of the substrate (Figure S6).
Additional mechanistic steps to consider include deprotonation of the general base Glu491 to reset the CutC active site for the next round of catalysis (Figure 6, IV) and leaving group protonation (Figure 6, V). In GDH, the proposed general base for deprotonation of the C1 hydroxyl group is Glu435 (equivalent to Glu491), and the proposed general acid for protonation of the departing C2 hydroxyl group is His164 (analogous to Asp216) (Figure 3D). Proton transfer between these two residues is thought to occur directly (Feliks and Ullmann, 2012), with no equivalent of Thr502 blocking the requisite conformational changes. In CutC, Glu491 and Asp216 are too distant to interact directly and movement is blocked by Thr502. Thus, we propose that the bridging/blocking residue Thr502 mediates proton transfer (Figure 6, IV), akin to what has been proposed previously for Ser and Thr residues in other systems (Erez et al., 2011, Patton et al., 2011). We anticipate computational studies may provide additional insights into this putative proton transfer network, which has been challenging to study experimentally.
Once the C–N bond is broken, the product-based radical species re-abstracts the hydrogen atom from Cys489 to reform the thiyl radical and generate acetaldehyde (Figure 6, V). For AdoCbl enzymes, this re-abstraction step to reform the 5′-deoxyadenosyl radical (Ado•) is energetically challenging. It has been proposed for EAL that 1,2-migration facilitates re-abstraction of a hydrogen atom from Ado, as this step was calculated to be exothermic by 1.2 kcal/mol (Wetmore et al., 2002). If base-catalyzed direct elimination were to occur in AdoCbl enzymes, the vinoxy radical formed would be stabilized by resonance (C–H bond enthalpy, ~95.5 kcal/mol (da Silva et al., 2006)) and could not effectively regenerate Ado• (Ado–H bond enthalpy, 99.9 kcal/mol (Wetmore et al., 2001)). In contrast to Ado•, the putative thiyl radical in GREs can be much more easily regenerated by the product radical (cysteine S–H bond enthalpy, ~87 kcal/mol (Rauk et al., 1998)), removing the necessity for the migration mechanism.
SIGNIFICANCE
Although it has long been known that the microbial communities in the human gut affect human health, the recent wealth of sequencing data on the human gut microbiome has allowed for the discovery of novel enzymes and pathways that link the microbiota to human disease. The recently identified microbial glycyl radical enzyme CutC cleaves choline to generate TMA, a metabolite associated with cardiovascular disease. By dissecting the mechanistic details of CutC catalyzed C–N cleavage and elucidating the importance of active site residues for binding and catalysis, our work facilitates the rational design of specific inhibitors of CutC, and thus of TMA production.
EXPERIMENTAL PROCEDURES
Source of materials, general methods, cloning, expression, and purification of wild-type and mutant CutC enzymes are described in detail in the Supplemental Experimental Procedures.
Crystallization of wild-type CutC
Initial screening of non-post-translationally modified wild-type CutC protein with the intact N-terminal hexahistidine tag and 18-residue truncation was performed exposed to air with the aid of an Art Robbins Phenix micro-pipetting robot and a Formulatrix Rock Imager. Numerous initial conditions were found, with optimization yielding a well solution containing 17% (w/v) polyethylene glycol (PEG) 8000, 0.3 M lithium chloride, and 0.1 M Tris pH 8.0. Diffraction-quality crystals were obtained in hanging drop vapor diffusion trays at 21°C. Protein at 15 mg/mL in buffer containing 50 mM potassium phosphate pH 8.0, 50 mM potassium chloride, and 10% (v/v) glycerol was mixed with well solution in a 1:1 ratio. Plate-like crystals formed after 2–3 days of equilibration and grew to maximum size over one week. Crystals were cryoprotected by soaking for 15–30 min in solution containing 20% (v/v) glycerol, 25% (w/v) PEG 8000, 0.5 M lithium chloride, and 0.1 M Tris pH 8.0. Choline was not added to the protein prior to crystallization but was included in the cryoprotection solution at 10 mM.
Crystallization of CutC mutants
Initial attempts to crystallize CutC mutants in the same condition as wild-type CutC were unsuccessful. Therefore, new crystals of CutC mutants (E491Q, E491A, T502A, or Y208F) with an N-terminal hexahistine tag and an N-terminal 52-residue truncation were identified in screening trays using the same setup as for wild-type. Optimized crystals were grown using hanging drop vapor diffusion at 21°C (room temperature). CutC mutant protein at 8 mg/mL in buffer containing 50 mM potassium phosphate pH 8.0, 50 mM potassium chloride, and 10 mM choline was mixed in a 1:1 ratio with well solution containing 1.0–1.2 M sodium malonate pH 7.0–8.0. Crystals were rod-like and grew within 7 days. Crystals were cryoprotected by brief transfer into a solution containing 3.4 M sodium malonate pH 7.0 and 10 mM choline and cryocooled by plunging in liquid nitrogen.
Structure determination of wild-type CutC
Data and refinement statistics can be found in the Supplemental Experimental Procedures. Crystals were indexed in space group P21 with cell edges a = 79.6 Å, b = 234.9 Å, c = 105.0 Å, β = 109.6°, and diffraction images were collected at the Advanced Photon Source beamline 24ID-C at a wavelength of 0.9795 Å on a Pilatus 6M detector (Dectris). Data were indexed, integrated, and scaled in HKL2000 (Otwinowski and Minor, 1997). The structure of CutC was solved by molecular replacement in the Phenix implementation of Phaser (McCoy et al., 2007). The structure of AdoCbl-independent glycerol dehydratase (PDB code 1R8W, 37% identity) (O’Brien et al., 2004) was used to construct a search model after trimming of nonidentical side chains in Phenix Ensembler (Adams et al., 2010). A solution with four molecules per asymmetric unit was found with an initial Rfree of 0.48 at 2.8-Å resolution. Several rounds of initial refinement in phenix.refine (Adams et al., 2010) with tight NCS restraints were sufficient to reduce Rfree values below 0.4, and manual building of side chains further reduced Rfree to ~0.3. NCS restraints were removed after initial refinement and addition of water molecules. Inclusion of explicit riding hydrogen atoms once the model was close to convergence improved both Rfree and the model geometry. Positional and B factor refinement continued at the full resolution until the model was complete. Choline was fit into difference density and verified with simulated annealing composite omit maps. Parameter files for choline were generated in Phenix eLBOW. Water molecules were placed automatically after ligands were refined and were verified manually. The final model contains all native residues at the C terminus in all four molecules. No density is observed for the N-terminal hexahistidine tag or residues 19–52 in the 18-residue truncated construct, but all other residues (53–846) are present and well-ordered in the model. Despite different crystal contacts for each molecule in the asymmetric unit, the structures of each monomer were essentially indistinguishable, with root-mean-square deviations of 0.11–0.14 Å. Structural figures were made in PyMOL v1.4.1 (Schrodinger).
Structure determination of CutC mutants
Data processing and model refinement statistics can be found in the Supplemental Experimental Procedures. Diffraction images were indexed in space group P42212 with cell edges approximately a = b = 230 Å, c = 79 Å. All data were collected and processed as for wild-type CutC. The structure of each mutant was solved by molecular replacement in the Phenix implementation of Phaser (McCoy et al., 2007) using the wild-type monomer as a search model. Two molecules are present per asymmetric unit, but the dimer interface is crystallographic, resulting in an overall tetrameric structure similar to the wild type. Positional and B factor refinement was conducted with phenix.refine (Adams et al., 2010) with Rfree flags propagated to each dataset. NCS and reference model restraints were used initially to reduce overfitting of the model but were removed after convergence with no change in the Rfree values. Riding hydrogen atoms were included at the end of refinement, resulting in an improvement of Rfree and geometry. Each final model contains native protein residues 53–846 in each chain; residues 1–52 were omitted in the construct. The N-terminal thrombin cleavage tag and linker are clearly visible in all chains; the hexahistidine tag is not ordered, but a single histidine residue is visible in one chain. Choline and active site waters were modeled into omit maps after refinement of the remainder of the protein. Three sodium ions and eight malonate ions are present in the final model, likely as a result of the high concentration of sodium malonate in the crystallization solution. No sodium ions were found in the wild-type structure. All ligands and mutated residues were verified rigorously with composite omit maps.
Generation of glycyl radical in CutC
Activation assays were set up in an MBraun glove box in 1.5 mL polypropylene Eppendorf tubes and contained 50 mM potassium phosphate pH 8, 50 mM potassium chloride, 150 μM sodium dithionite (NaDT), 200 μM AdoMet, 10 μM CutC dimer, and 30 μM CutD in a total volume of 200 μL. A mixture of buffer, CutD and NaDT was incubated for 20 min prior to addition of AdoMet and CutC, then glycyl radical formation was carried out for 1 h at room temperature. This incubation time was determined as optimal based on the stability of the glycyl radical signal for wild type CutC and CutC-Y208F (Figure S4A). The conditions used for CutC activation were optimized as described below. Despite additional attempts at optimization, no glycyl radical signal was observed for CutC-T502A when the activation assay was incubated for 30 min or 60 min. X-band EPR spectroscopy and spin concentration measurements were performed as previously described (Craciun et al., 2014), without taking into consideration the difference in receiver gain for standard and enzymatic assays as this difference is already accounted for by the spectrometer. The limit of detection for this technique was estimated at ~0.5 μM.
The activation assay for glycyl radical formation was optimized with wild-type CutC using a coupled, spectrophotometric assay for acetaldehyde, as described for steady state kinetics. The following parameters were surveyed for the activation reaction: incubation time (30 to 90 min), buffer (25 mM Tris pH 8 vs. 50 mM potassium phosphate pH 8), ratio of CutC to CutD (from 1:0.4 to 1:4), using size exclusion chromatography-purified CutC, using size exclusion chromatography-purified CutD, reductant (NaDT, E. coli flavodoxin (Hall et al., 2000) reduced with NaDT, acriflavin, methyl viologen, titanium (III) citrate), concentration of NaDT (2–20 equivalents), and concentration of AdoMet (10–80 equivalents). For CutC-T502A, no glycyl radical signal was observed after 30 or 60 min incubation using the optimized activation conditions. In addition, no signal was observed after this mutant was purified by size exclusion chromatography, when Tris buffer was used instead of potassium phosphate, or when acriflavin (6 equivalents) was used instead of NaDT as reductant. For CutC-D216N, no EPR signal was observed when this mutant was assayed with Tris buffer after purification by size exclusion chromatography.
Kinetic analysis of choline cleavage
A spectrophotometric coupled assay monitoring oxidation of nicotinamide adenine dinucleotide (NADH) by yeast alcohol dehydrogenase (YADH) was used to determine the kinetics of acetaldehyde production and thereby choline cleavage. Activation of CutC was carried out as described above. All assays contained 200 μM NADH, 50 mM potassium phosphate pH 8, 50 mM potassium chloride, diluted activation mixture (4000-fold for wild type, 33-fold for Y208F, 150-fold for Y506F, 10-fold for Y208F/Y506F, 2.5-fold for T502A), YADH (0.4–4 μM) and choline (0–10 mM, except for T502A, for which 100 mM choline was used) in a total volume of 200 μL. The assay was carried out in triplicate in a 96-well plate and the NADH absorbance at 340 nm was monitored for 7 min. The absorbance decreased linearly, indicating a constant concentration of activated enzyme during the course of our measurements.
LC-MS/MS analysis of trimethylamine production
For E491A and E491Q mutant enzymes, glycyl radical formation on CutC was carried out on a 40 μL scale as described above, then 100 mM choline was added to the assays, followed by overnight (16 h) incubation in an Mbraun glove box at room temperature. For CutC-wild-type, CutC-T502A and CutC-D216N purified by size exclusion chromatography, activation assays were carried out in a similar manner, but contained 25 mM Tris buffer pH 8, 50 mM sodium chloride, 200 μM NaDT, 200 μM AdoMet, 10 μM CutC dimer, and 40 μM CutD in a total volume of 50 μL. Choline was added to a final concentration of 20 mM, and the assays were incubated overnight (16 h) in an Mbraun glove box at room temperature. For LC-MS/MS analysis, 4 μL of each assay were diluted into buffer R (20% 5 mM ammonium formate pH 4 and 80 % acetonitrile). This mixture was centrifuged at 13,000 rpm for 10 min to remove particulates, diluted 20 fold with buffer R, then 3 μL were analyzed by LC-MS/MS using a previously-described method for underivatized TMA detection (precursor-product ion pair: m/z 60.1 → m/z 45, collision energy: 21 V, limit of detection after dilution: ~ 1 μM) (Martinez-del Campo et al., 2015).
Acknowledgments
S.B. is supported by an HHMI International Student Research Fellowship. M.A.F. is supported in part by the National Science Foundation Graduate Research Fellowship under Grant No. 0645960. E.P.B. is supported by the Richard and Susan Smith Family Foundation Award for Excellence in Biomedical Research and the Packard Fellowship for Science and Engineering. C.L.D. is a Howard Hughes Medical Institute Investigator. This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by award GM103403 from the National Center for Research Resources at the National Institute of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors declare no competing financial interests.
Footnotes
ACCESSION CODES
Protein coordinates and structure factors have been submitted to the Protein Data Bank under accession codes 5FAU (wild type), 5KDP (E491A), 5FAV (E491Q mutant), 5FAW (T502A mutant), and 5FAY (Y208F mutant).
AUTHOR CONTRIBUTIONS
S.B. purified proteins and carried out biochemical assays. M.A.F. performed crystallization, X-ray data collection, and structure determination. S.B. and M.A.F. analyzed structures and biochemical data with input from E.P.B. and C.L.D. S.B., M.A.F., E.P.B. and C.L.D. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari U, Scheiner S. Magnitude and mechanism of charge enhancement of CH··O hydrogen bonds. J Phys Chem A. 2013;117:10551–62. doi: 10.1021/jp4081788. [DOI] [PubMed] [Google Scholar]
- Brugere JF, Borrel G, Gaci N, Tottey W, O’Toole PW, Malpuech-Brugere C. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes. 2014;5:5–10. doi: 10.4161/gmic.26749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckel W, Golding BT. Radical species in the catalytic pathways of enzymes from anaerobes. FEMS Microbiol Rev. 1998;22:523–541. [Google Scholar]
- Christodoulou J. Trimethylaminuria: an under-recognised and socially debilitating metabolic disorder. J Paediatr Child Health. 2012;48:E153–5. doi: 10.1111/j.1440-1754.2010.01978.x. [DOI] [PubMed] [Google Scholar]
- Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci USA. 2012;109:21307–12. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9:1408–13. doi: 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- da Silva G, Kim CH, Bozzelli JW. Thermodynamic properties (enthalpy, bond energy, entropy, and heat capacity) and internal rotor potentials of vinyl alcohol, methyl vinyl ether, and their corresponding radicals. J Phys Chem A. 2006;110:7925–34. doi: 10.1021/jp0602878. [DOI] [PubMed] [Google Scholar]
- Dawson RMC. Data for biochemical research. London/New York: Oxford Univ. Press (Clarendon); 1969. [Google Scholar]
- Dougherty DA, Stauffer DA. Acetylcholine binding by a synthetic receptor: implications for biological recognition. Science. 1990;250:1558–60. doi: 10.1126/science.2274786. [DOI] [PubMed] [Google Scholar]
- Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA. 2006;103:12511–6. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez Y, Gepshtein R, Presiado I, Trujillo K, Kallio K, Remington SJ, Huppert D. Structure and excited-state proton transfer in the GFP S205A mutant. J Phys Chem B. 2011;115:11776–85. doi: 10.1021/jp2052689. [DOI] [PubMed] [Google Scholar]
- Feliks M, Ullmann GM. Glycerol dehydratation by the B12-independent enzyme may not involve the migration of a hydroxyl group: a computational study. J Phys Chem B. 2012;116:7076–87. doi: 10.1021/jp301165b. [DOI] [PubMed] [Google Scholar]
- Foster T, West PR. Photolysis of aqueous solutions of hydrogen peroxide containing β-ammonio alcohols. Can J Chem. 1974;52:3589–3598. [Google Scholar]
- Funk MA, Judd ET, Marsh EN, Elliott SJ, Drennan CL. Structures of benzylsuccinate synthase elucidate roles of accessory subunits in glycyl radical enzyme activation and activity. Proc Natl Acad Sci USA. 2014;111:10161–6. doi: 10.1073/pnas.1405983111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk MA, Marsh EN, Drennan CL. Substrate-bound structures of benzylsuccinate synthase reveal how toluene is activated in anaerobic hydrocarbon degradation. J Biol Chem. 2015;290:22398–408. doi: 10.1074/jbc.M115.670737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Jordan-Starck TC, Loo RO, Ludwig ML, Matthews RG. Interaction of flavodoxin with cobalamin-dependent methionine synthase. Biochemistry. 2000;39:10711–9. doi: 10.1021/bi001096c. [DOI] [PubMed] [Google Scholar]
- Hans M, Bill E, Cirpus I, Pierik AJ, Hetzel M, Alber D, Buckel W. Adenosine triphosphate-induced electron transfer in 2-hydroxyglutaryl-CoA dehydratase from Acidaminococcus fermentans. Biochemistry. 2002;41:5873–82. doi: 10.1021/bi020033m. [DOI] [PubMed] [Google Scholar]
- Hippe H, Caspari D, Fiebig K, Gottschalk G. Utilization of trimethylamine and other N-methyl compounds for growth and methane formation by Methanosarcina barkeri. Proc Natl Acad Sci USA. 1979;76:494–8. doi: 10.1073/pnas.76.1.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S, Dirk LM, Yesselman JD, Nimtz JS, Adhikari U, Mehl RA, Scheiner S, Houtz RL, Al-Hashimi HM, Trievel RC. Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J Am Chem Soc. 2013;135:15536–48. doi: 10.1021/ja407140k. [DOI] [PubMed] [Google Scholar]
- Horowitz S, Trievel RC. Carbon-oxygen hydrogen bonding in biological structure and function. J Biol Chem. 2012;287:41576–82. doi: 10.1074/jbc.R112.418574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalnins G, Kuka J, Grinberga S, Makrecka-Kuka M, Liepinsh E, Dambrova M, Tars K. Structure and function of CutC choline lyase from human microbiota bacterium Klebsiella pneumoniae. J Biol Chem. 2015;290:21732–40. doi: 10.1074/jbc.M115.670471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106:357–87. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroff GP, Fessenden RW. Equilibrium and kinetics of the acid dissociation of several hydroxyalkyl radicals. J Phys Chem. 1973;77:1283–1288. [Google Scholar]
- Lawrence CC, Bennati M, Obias HV, Bar G, Griffin RG, Stubbe J. High-field EPR detection of a disulfide radical anion in the reduction of cytidine 5′-diphosphate by the E441Q R1 mutant of Escherichia coli ribonucleotide reductase. Proc Natl Acad Sci USA. 1999;96:8979–84. doi: 10.1073/pnas.96.16.8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtio L, Goldman A. The pyruvate formate lyase family: sequences, structures and activation. Protein Eng Des Sel. 2004;17:545–52. doi: 10.1093/protein/gzh059. [DOI] [PubMed] [Google Scholar]
- Lenz R, Giese B. Studies on the mechanism of ribonucleotide reductases. J Am Chem Soc. 1997;119:2784–2794. [Google Scholar]
- Lin GM, Choi SH, Ruszczycky MW, Liu HW. Mechanistic investigation of the radical S-adenosyl-L-methionine enzyme DesII using fluorinated analogues. J Am Chem Soc. 2015;137:4964–7. doi: 10.1021/jacs.5b02545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio. 2015:6. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Oiwa T, Kawaguchi S, Kondo K, Takahashi Y, Toraya T. Catalytic roles of substrate-binding residues in coenzyme B12-dependent ethanolamine ammonia-lyase. Biochemistry. 2014;53:2661–71. doi: 10.1021/bi500223k. [DOI] [PubMed] [Google Scholar]
- Musah RA, Jensen GM, Rosenfeld RJ, McRee DE, Goodin DB, Bunte SW. Variation in strength of an unconventional CH to O hydrogen bond in an engineered protein cavity. J Am Chem Soc. 1997;119:9083–9084. [Google Scholar]
- O’Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, Lanzilotta WN. Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemistry. 2004;43:4635–45. doi: 10.1021/bi035930k. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Patton GC, Stenmark P, Gollapalli DR, Sevastik R, Kursula P, Flodin S, Schuler H, Swales CT, Eklund H, Himo F, Nordlund P, Hedstrom L. Cofactor mobility determines reaction outcome in the IMPDH and GMPR (beta-alpha)8 barrel enzymes. Nat Chem Biol. 2011;7:950–8. doi: 10.1038/nchembio.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AL, Eriksson M, Katterle B, Potsch S, Sahlin M, Sjoberg BM. A new mechanism-based radical intermediate in a mutant R1 protein affecting the catalytically essential Glu441 in Escherichia coli ribonucleotide reductase. J Biol Chem. 1997;272:31533–41. doi: 10.1074/jbc.272.50.31533. [DOI] [PubMed] [Google Scholar]
- Rauk A, Yu D, Armstrong D. Oxidative damage to and by cysteine in proteins: an ab initio study of the radical structures, CH, SH, and CC bond dissociation energies, and transition structures for H abstraction by thiyl radicals. J Am Chem Soc. 1998;120:8848–8855. [Google Scholar]
- Ruszczycky MW, Choi SH, Mansoorabadi SO, Liu HW. Mechanistic studies of the radical S-adenosyl-L-methionine enzyme DesII: EPR characterization of a radical intermediate generated during its catalyzed dehydrogenation of TDP-D-quinovose. J Am Chem Soc. 2011;133:7292–5. doi: 10.1021/ja201212f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszczycky MW, Liu HW. Mechanistic enzymology of the radical SAM enzyme DesII. Isr J Chem. 2015;55:315–324. doi: 10.1002/ijch.201400130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer T, Pierik AJ, Heider J. New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol Chem. 2005;386:981–8. doi: 10.1515/BC.2005.114. [DOI] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New Engl J Med. 2013;368:1575–84. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem Rev. 2003;103:2095–127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc Natl Acad Sci U S A. 2008;105:16137–41. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 2014;35:904–10. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Funk MA, Rosado LA, Baek J, Drennan CL, Stubbe J. The class III ribonucleotide reductase from Neisseria bacilliformis can utilize thioredoxin as a reductant. Proc Natl Acad Sci USA. 2014;111:E3756–65. doi: 10.1073/pnas.1414396111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore SD, Smith DM, Bennett JT, Radom L. Understanding the mechanism of action of B12-dependent ethanolamine ammonia-lyase: synergistic interactions at play. J Am Chem Soc. 2002;124:14054–65. doi: 10.1021/ja027579g. [DOI] [PubMed] [Google Scholar]
- Wetmore SD, Smith DM, Golding BT, Radom L. Interconversion of (S)-glutamate and (2S,3S)-3-methylaspartate: a distinctive B12-dependent carbon-skeleton rearrangement. J Am Chem Soc. 2001;123:7963–72. doi: 10.1021/ja004246f. [DOI] [PubMed] [Google Scholar]