

Abstract
Recent studies describing the mutational landscape of head and neck squamous cell carcinoma (HNSCC) on a genomic scale by our group and others, including The Cancer Genome Atlas, have provided unprecedented perspective for understanding the molecular pathogenesis of HNSCC progression and response to treatment. These studies confirmed that mutations of the TP53 tumor suppressor gene were the most frequent of all somatic genomic alterations in HNSCC, alluding to the importance of the TP53 gene in suppressing the development and progression of this disease. Clinically, TP53 mutations are significantly associated with short survival time and tumor resistance to radiotherapy and chemotherapy in HNSCC patients, which makes the TP53 mutation status a potentially useful molecular factor for risk stratification and predictor of clinical response in these patients. In addition to loss of wild-type p53 function and the dominant-negative effect on the remaining wild-type p53, some p53 mutants often gain oncogenic functions to promote tumorigenesis and progression. Different p53 mutants may possess different gain-of-function properties. Therefore, mutant p53 is not just one protein but actually a variety of proteins that contribute to an exceptionally vast network of tumor-promoting processes. Herein we review the most up-to-date information about TP53 mutations available via The Cancer Genome Atlas-based analysis of HNSCC and discuss our current understanding of the potential tumor-suppressive role of p53, focusing on gain-of-function activities of p53 mutations. We also summarize our knowledge regarding use of the TP53 mutation status as a potential evaluation or stratification biomarker for prognosis and a predictor of clinical response to radiotherapy and chemotherapy in HNSCC patients. Finally, we discuss possible strategies for targeting HNSCCs bearing TP53 mutations.
Keywords: Head and neck squamous cell carcinomas, HNSCC, p53, P53 mutation, Gain-of-function
Head and neck squamous cell carcinoma (HNSCC) is the major form of head and neck cancer and the sixth most common cancer worldwide. The global number of new HNSCC cases annually is estimated to be more than 600,000, with about 355,000 deaths observed each year. In the United States, more than 60,000 new HNSCC cases and over 13,000 HNSCC deaths per year are recorded (Siegel et al., 2016). HNSCC occurs at various anatomical sites in the head and neck, including the oral cavity, larynx, pharynx, and nose/paranasal sinuses, and accounts for approximately 3% of all malignancies in the United States. The incidence and mortality rates for head and neck cancers are disproportionately distributed among different genders, race, and region of the world. For instance, head and neck cancers are more prevalent in male than in female individuals. Furthermore, owing to culturally or habitually related risk factors, the incidence of HNSCC at a particular anatomical site may be higher in a certain geographical area than in others. For example, mouth and tongue cancers are more common on the Indian subcontinent, where oral betel nut use is a common practice. On the other hand, in the United States, despite a decreasing incidence in black individuals over the past 2 decades, the mortality rate for HNSCC is still higher in that group than in white patients (DeSantis et al., 2013). Additionally, human papillomavirus (HPV)-associated oropharyngeal cancer has distinct epidemiological characteristics, as patients are likely nonsmokers, younger and less frequent alcohol users than those not infected with HPV, and certain sexual behaviors, exposure to HPV are correlated with development of HPV+ oropharyngeal cancer. Furthermore, HPV+ oropharyngeal cancer patients have much better prognoses than do matched staged HPV− oropharyngeal cancer patients.
The risk factors for HNSCC include smoking, alcohol consumption, viral infections (Epstein-Barr virus, HPV, and herpes simplex virus), betel nut chewing, occupational exposure to carcinogens, immunodeficiency, irradiation, diet, and genetic disposition. Some risk factors are closely linked with the molecular pathogenesis of HNSCC. For example, p53 protein, the translated product of the TP53 gene that is frequently mutated in HPV− HNSCC cells, can be bound and inactivated by the HPV E6 protein in HPV+ HNSCC patients, resulting in dysregulated cell-cycle progression and cellular proliferation.
Given the significant diversity of the anatomical sites and molecular pathogenesis of HNSCC, treatment of it often integrates multimodal approaches, including chemotherapy, radiotherapy (particularly intensity-modulated radiotherapy), and surgery. Despite several decades of advances that have improved diagnosis and management of HNSCC, approximately half of all patients die of it. Currently, treatment plans for HNSCC are still selected using the well-established prognostic parameters of tumor site, primary TNM stage, and histological tumor characteristics. However, owing to marked heterogeneity in the biology and molecular pathogenesis of HNSCCs, even tumors at similar stages respond very differently to the same treatment. As a result, identifying reliable biomarkers is imperative to assist with prognosis and stratify patients for customized treatment plans that do not have unnecessary toxic effects or costs and are more effective than standard chemotherapy and radiotherapy. A large body of evidence, backed by the latest results of genomic sequencing analyses of and The Cancer Genome Atlas (TCGA) data on HNSCC, suggests that genetic alterations in specific signaling pathways, including those of the TP53 gene, play critical roles in HNSCC tumorigenesis and progression.
In this review, we describe the latest advances in understanding the molecular pathogenic mechanisms that drive the progression of HNSCC and highlight the impact of TP53 mutations on the development and progression of HNSCC and the importance of these mutations as prognostic and predictive biomarkers for HNSCC in patients undergoing treatment. Understanding TP53 mutations and their impact on p53 signaling and interaction with other cellular growth and survival signaling pathways can help with the design of new, more effective therapeutic strategies that target TP53 mutation-bearing HNSCC, providing a precision medicine approach to the treatment of this disease.
THE MOLECULAR PATHOGENESIS OF HNSCC
Previous cytogenetic and single-gene studies established a model of the tumorigenesis of HNSCC, in which accumulation of chromosomal rearrangements such as deletions and duplications drives the carcinogenic transformation of epithelial squamous cells (Califano et al., 1996). In this model, progressive loss of heterozygosity in chromosomal regions 9p21, 3p, and 17p13 along with amplification of 11q13 may drive the transition from hyperplasia in normal mucosa to dysplasia and subsequent carcinoma in situ, finally resulting in malignant invasion and metastasis of HNSCC (Figure 1). Consistent with this model, some critical tumor suppressor genes and oncogenes are found in genetically altered chromosomal regions, including p16 at 9p21, TP53 at 17p13, and cyclin D1 at 11q13.
Figure 1.
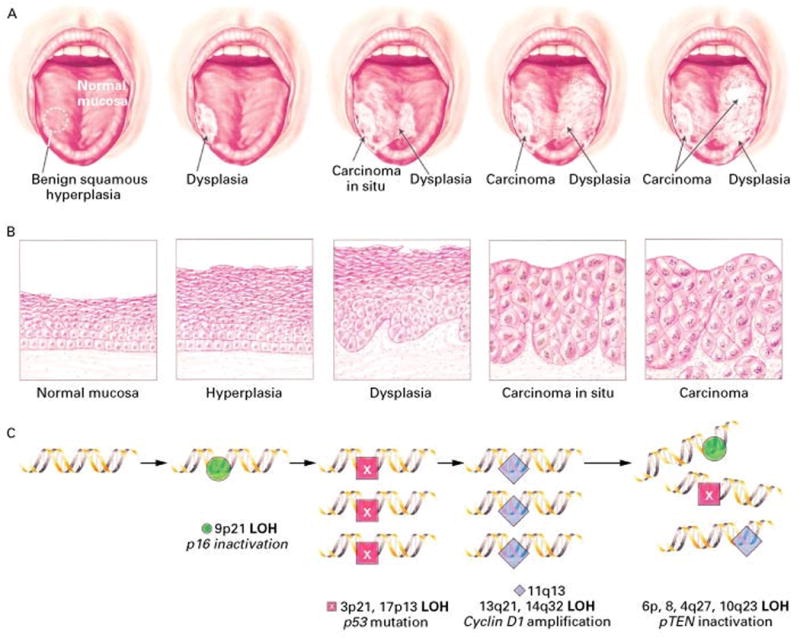
The clinical, histological, and molecular pathogenesis of oral cancer. (A) Clinical features of the progression of oral cancer. (B) The histological progression from normal-appearing mucosa to invasive cancer. (C) The progression of oral cancer is concomitant with the genetic alterations of multiple tumor suppressor genes and oncogenes. LOH: loss of heterozygosity. (Courtesy of Jeoseh A. Califano, M.D.).
With the advent of next-generation sequencing technologies, understanding of the molecular and genetic pathogenesis of HNSCC has never been clearer. Large-scale sequencing analyses of HNSCCs performed by our group and others have identified frequent alterations of genes in this tumor, including TP53, CDKN2A, CASP8, FAT1, NOTCH1, HRAS, PIK3CA, MLL2, and FBXW7 (Agrawal et al., 2011; Lawrence et al., 2015; Pickering et al., 2013; Stransky et al., 2011). These studies provided an important clue as to the key signaling molecules that contribute to HNSCC tumorigenesis. More importantly, they identified potential pharmacological targets for HNSCC-directed therapies.
Based on the recently identified mutations in HNSCCs the recent mutational landscape of HNSCC, the major pathological pathways implicated in the tumorigenesis of HNSCC include dysregulation of four processes: 1) Cellular survival and proliferation (e.g., TP53, HRAS, EGFR, and PIK3CA): recent genomic-scale mapping of HNSCC mutations confirmed that the incidences of TP53, HRAS, and PIK3CA mutations were high in this disease. 2) Cell-cycle control (e.g., CDKN2A and CCND1): homozygous deletion of CDKN2A (encoding cyclin-dependent kinase inhibitor 2A) occurs in about 30% of HNSCC cases. Also, amplification of CCND1, which encodes cyclin D1, occurs in about one third of HNSCCs. Taken together, the status of CDKN2A and CCND1 can contribute distinctively to prediction of survival of tongue cancer patients. 3) Cellular differentiation (e.g., NOTCH1): NOTCH1 is a transmembrane receptor and controls cell differentiation and embryonic development. In HNSCC patients, it may be a tumor suppressor gene, as significant inactivating mutations of NOTCH1 are present in HNSCC NOTCH1 (19%) and linked with squamous differentiation of tumors, although the exact mechanism of its tumor-suppressive function remains to be elucidated. 4) Adhesion and invasion signaling (e.g., FAT1): FAT1 encodes a protein that belongs to the cadherin superfamily. FAT1 suppresses cancer cell growth by binding to β-catenin and inhibiting its nuclear localization (Agrawal et al., 2011; Lawrence et al., 2015; Pickering et al., 2013; Stransky et al., 2011)
TP53 MUTATIONS IN HNSCCs
Among the genes mentioned above, mutation of the TP53 gene is the most common genomic alteration identified in HNSCC cases. Disruption of p53 functions can be caused by mutation or other mechanisms, such as HPV infection. In such cases, expression of HPV E6 protein targets p53 for proteasomal degradation. Because TP53 is the gene most frequently mutated in HNSCCs, investigators are focusing on the functional impact of these mutations on HNSCC progression and treatment response and, thus, their utility as prognostic or predictive biomarkers for HNSCC.
THE PREVALENCE AND ETIOLOGY OF TP53 MUTATIONS IN HNSCCs
Because of HNSCC heterogeneity (e.g., tumors at different anatomical sites) and different technologies used to detect TP53 mutations in HNSCCs (e.g., immunohistochemistry, mutation screening), the prevalence of TP53 mutations in HNSCC patients varies quite widely in studies in the literature, ranging from 30% to 70%. More recent next-generation sequencing studies confirmed that TP53 is the most commonly mutated tumor suppressor gene in HNSCCs, with a mutation frequency in non-HPV-associated HNSCC cases ranging from 75% to 85% (Agrawal et al., 2011; Lawrence et al., 2015; Pickering et al., 2013; Stransky et al., 2011). Updated TCGA (http://gdac.broadinstitute.org/) analysis results regarding 510 HNSCC cases demonstrated that 70.4% of tumors have TP53 mutations (Table 1). Moreover, tumors of the larynx and hypopharynx have the highest TP53 mutation rate (83.5%), tumors of the tongue and oral cavity have a TP53 mutation rate of 75.6%, and tumors of the oropharynx, including the tonsils, and base of the tongue have the lowest TP53 mutation rate (28.6%). The finding that the lowest TP53 mutation rate is for tumors of the oropharynx and tonsils is not surprising given that the vast majority of oropharyngeal cancers are HPV-associated.
Table 1.
TP53 mutation prevalence in HNSCC TCGA (n=510)
| Anatomical sites | Cases (n) | HPV+ (%) | HPV+ & WT TP53 (%) | TP53 Mutation (%) | TP53 Mutation/HPV− (%) |
|---|---|---|---|---|---|
| Larynx & Hypopharynx | 121 | 1.5 (2/121) | 1.5 (2/121) | 83.5 (101/121) | 84.5 (101/119) |
| Oral Cavity & Tongue | 312 | 3.8 (12/312) | 3.5 (11/312) | 75.6 (236/312) | 78.9 (236/300) |
| Oropharynx, Tonsil & Base of Tongue | 77 | 28.6 (22/77) | 28.6 (22/77) | 28.6 (22/77) | 40 (22/55) |
| Total | 510 | 7.1 (36/510) | 6.9 (35/510) | 70.4 (359/510) | 75.6 (236/312) |
Consistent with observations regarding many other cancers, most TP53 mutations in HNSCCs are missense mutations, frequently occurring within the central region of the protein that serves as the p53 DNA-binding domain. Updated HNSCC TCGA results demonstrated that although the spectrum of TP53 missense mutations is wide (Figure 2), several TP53 missense mutations, including those at codons R248, R273, G245, R175, R282, and H179, are the most prevalent hotspot mutations In HNSCCs (Table 2).
Figure 2.
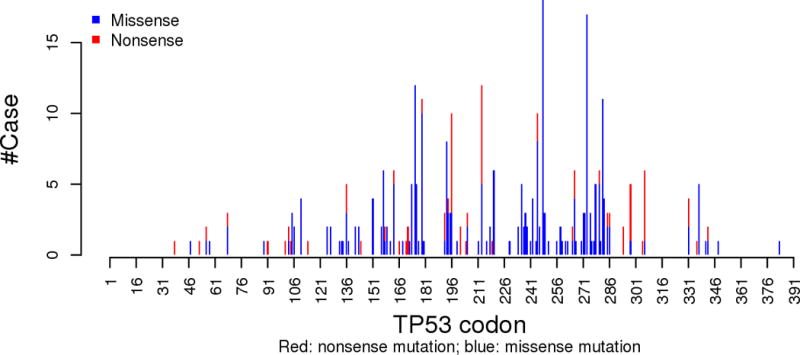
Histogram of number of cases with missense and nonsense TP53 mutations in HNSCC TCGA (n=510) by p53 codon position.
Table 2.
Top 8 most frequently mutated p53 codon in HNSCC TCGA (n=510)
| Codon | Original Amino Acid | # Mutation | # Missense Mutation | # Nonsense Mutation |
|---|---|---|---|---|
| 248 | R | 18 | 18 | 0 |
| 273 | R | 18 | 18 | 0 |
| 175 | R | 12 | 12 | 0 |
| 213 | R | 12 | 3 | 9 |
| 179 | H | 12 | 12 | 0 |
| 282 | R | 11 | 11 | 0 |
| 245 | G | 10 | 10 | 0 |
| 196 | R | 10 | 3 | 7 |
While TCGA results confirmed that HNSCC arises via the multistep process of carcinogenesis, it appears that alterations of TP53 often occur early in this progression, as they can be detected in premalignant lesions (Boyle et al., 1993). Moreover, a study using direct sequencing demonstrated no difference of TP53 mutations between primary HNSCCs and matched nodal metastatic lesions (Tjebbes et al., 1999). In another study of 20 patients with invasive HNSCC, the researchers found no TP53 mutations in clinically normal-appearing mucosa (taken at least 3 cm from the carcinoma) but detected TP53 mutations in seven dysplastic lesions, with five of these lesions having the same mutations as the corresponding carcinoma samples (el-Naggar et al., 1995). Taken together, these results strongly indicate that early occurrence of TP53 mutations contributes to stepwise clonal expansion and progression down the multistep pathway to HNSCC development.
Whether risk factors such as use of tobacco or alcohol and exposure to chemicals can affect the frequency of TP53 mutations in patients with HNSCC is still controversial. Whereas several previous studies demonstrated that the frequency of TP53 mutations is positively associated with tobacco exposure, others failed to demonstrate such an association (Poeta et al., 2007). These inconsistent results may have resulted from use of different methods to detect TP53 mutations. In the recently updated TCGA data on the 510 HNSCC cases, the association between smoking and TP53 mutations was not significant. Similarly, investigators showed that young age was associated with high rates of TP53 mutations (De Paula et al., 2009). However, our recent study on squamous cell carcinoma of the oral tongue demonstrated that tumors in younger never-smoking patients (<45 years old) appeared genomically similar to those in older smoking patients, including similarly high TP53 mutation rates (Pickering et al., 2014).
It was well documented that there is an inverse relationship between the presence of HPV DNA in squamous cell carcinoma of the oropharynx and the presence of TP53 mutations in this tumor. In support of this, nearly one third (28.6%) of oropharyngeal and tonsil tumors in the TCGA HNSCCs are HPV+ and wild-type (WT) for TP53, whereas tumors at other sites are less prone to be HPV+ and have higher rates of TP53 mutations (Table 1). This result strongly suggests that in patients with HPV infection, in whom WT p53 is inactivated by binding to the E6 viral proteins rather than by mutation, HPV infection plays an important role in the development of oropharyngeal and tonsil tumors, whereas TP53 mutations, which result in loss-of-function and/or gain-of-function (GOF), are vital for HNSCC tumorigenesis and progression in HPV− patients.
P53’S BIOLOGICAL ACTIVITIES IN TUMOR SUPPRESSION AND THE CONSEQUENCES OF TP53 MUTATIONS
The TP3 gene consists of 11 exons, the first of which is noncoding. The p53 protein consists of 393 amino acids and comprises four regions with different functions: the N-terminal transactivational domain (amino acids 20–42), the central DNA-binding domain (amino acids 103–292), the C-terminal tetramerization domain (amino acids 319–360), and the C-terminal regulatory domain (amino acids 364–393). WT p53 is a pivotal tumor suppressor—termed the “guardian of the genome” — because it ensures genomic stability and thus prevents cancer onset. P53 exerts its tumor-suppressive function by regulating the transcription of numerous downstream target genes involved in cell-cycle arrest, apoptosis, senescence, DNA repair, and metabolism as a transcription factor (Lane and Levine, 2010).
Under normal unstressed physiological conditions, p53 protein has a very short half-life and is maintained at a low level, mainly by being degraded by its E3 ubiquitin ligase, MDM2, pirh2, and COP1 (Pant and Lozano, 2014). Once cells are exposed to genotoxic stresses, p53 is posttranslationally modified via phosphorylation, acetylation, and other modifications and stabilized, and its level rises dramatically, resulting in activation and transcription of hundreds of genes with important roles in cell-cycle arrest, senescence, apoptosis, metabolism, and differentiation (Vousden and Prives, 2009). Together, these activities ensure that cells with DNA damage do not propagate this damage via proliferation. Thus, when p53 activity is lost owing to gene deletion or mutation, normal cells lose the ability to control and address DNA damage via repair or death, leading to genomic instability and, ultimately, cancer (Muller and Vousden, 2013). The observation that more than 70% of HNSCCs have mutations of the TP53 gene and that many HNSCC patients with WT p53 are infected with HPV (Table 1), which also inactivates WT p53’s function, indicates the indispensability of intact p53 activity for the suppression of HNSCC development.
An intriguing question is what is the functional mechanism of p53-mediated tumor suppression under physiological conditions? Although regulation of cell-cycle arrest, apoptosis, and senescence is widely accepted as the main function of p53 that contributes to its tumor-suppressive role, recent studies with animal models have challenged this paradigm. For instance, p53 efficiently suppresses tumor development in the complete absence of its cell-cycle-inhibitory and proapoptotic effectors p21, Puma, and Noxa. Moreover, mice with expression of mutated p53 at three acetylation sites (K117, K161, and K162), which abolished p53-mediated cell-cycle arrest, apoptosis, and senescence, were not prone to early onset of spontaneous tumorigenesis, suggesting that cell-cycle arrest, apoptosis, and senescence are dispensable for p53’s tumor-suppressive role. In addition to cell-cycle arrest, apoptosis, and senescence under genotoxic stress, p53 regulates many aspects of cell metabolism. For example, it inhibits aerobic glycolysis, lipid synthesis, and the pentose phosphate pathway and promotes mitochondrial respiration via multiple mechanisms (Berkers et al., 2013). More importantly, in the mice described above, acetylation-site-mutated p53 retained the ability to inhibit glucose uptake, glycolysis, and reactive oxygen species production by regulating expression of the metabolic p53 target genes GLS2, TIGAR, and SLC7A11, strongly suggesting that p53-mediated regulation of cell metabolism (e.g., cysteine metabolism, ferroptosis) plays an important role in p53’s tumor-suppressive function (Jiang et al., 2015).
Mutations of the TP53 gene occur mainly in the DNA-binding domain, and the majority of them are missense mutations. Mutant p53 can be categorized roughly into two types: 1) DNA contact mutants, which are present on the amino acids that bind directly to the p53-responsive element in DNA (e.g., p53R273H, p53R280K), and 2) conformational mutants, which alter the structure of p53 protein to abolish its DNA-binding ability (e.g., p53R175H, p53V143A). Both mutant types not only lose WT p53’s transcriptional function but also have dominant-negative activity by heteroligomerization with WT p53.
Over the course of tumor evolution, mutant TP53 derived from the mutated allele co-exists with WT TP53 from the other allele for varying periods until the WT TP53 allele is generally lost owing to loss of heterozygozity, leaving only the mutant TP53 allele. Interestingly, during the co-existence phase of both WT and mutant p53 proteins, haploinsufficiency of TP53 leads to the propensity for increased tumor development, as demonstrated in Li-Fraumeni syndrome patients as well as animal models. However, dominant-negative effects of mutant p53 over the functions of WT p53 are often observed with co-overexpression of mutant TP53 and WT TP53. In such cases, mutant p53 inactivates WT p53 via oligomerization and sequesters WT p53 away from its target gene promoters and/or quenches co-factors required for transactivation by the WT p53 bound to the promoters, thereby inhibiting WT p53’s ability to suppress cellular transformation and transactivate target gene expression.
Finally, in addition to the loss-of-function and dominant-negative forms of p53, some p53 mutations are associated with GOF activities that can enhance tumor progression, metastatic potential, and/or drug resistance when overexpressed, even in cells lacking WT TP53 (Freed-Pastor and Prives, 2012). This explains why TP53 was originally described as an oncogene, as researchers unknowingly used plasmids encoding mutations of TP53. Thus, single-amino-acid changes in the TP53 gene may result in profound changes to its function, converting p53 from a tumor suppressor to an oncogene that greatly contributes to the malignant properties of cancer cells.
GOF ACTIVITIES OF MUTANT P53
Thus far, researchers have gained understanding of GOF activities of mutant p53 proteins primarily using cell culture studies of TP53-deficient isogenic cell lines to examine the effects of different overexpressed TP53 mutants. For example, introduction of various tumor-derived human TP53 mutants into p53-null tumor cells increased the cells’ tumorigenic and metastatic potential and resistance to chemotherapeutic drugs and DNA-damaging agents (Muller and Vousden, 2013). Similarly, we observed that GOF p53 mutants promoted invasive growth of (Neskey et al., 2015; Zhou et al., 2014), resistance to treatment with cisplatin of (Osman et al., 2015c), and anoikis in (Xie et al., 2013) HNSCC cells. Furthermore, GOF p53 mutants mediate genomic instability; drive epithelial-to-mesenchymal transition; regulate angiogenesis, nucleotide metabolism, lipid metabolism, and glycolysis; and promote inflammation and cellular programming in cancer stem cells (Haupt et al., 2016).
In addition, generation and studies of TP53–knock-in mice provided direct in vivo evidence of the GOF activities of mutant p53. Knock-in mouse models with expression of mutant p53R172H (equivalent to human R175H) and p53R270H (equivalent to human R273H) protein develop tumors that exhibit a GOF phenotype in vivo with higher metastatic capacity than that of tumors in mice with TP53−/− alleles (Lang et al., 2004). Similarly, in a murine model of HNSCC, mutant p53R172H, cooperating with oncogenic K-ras, accelerated oral tumor growth and promoted tumor progression to carcinoma (Acin et al., 2011).
Whereas p53 mutants are often considered to be equivalent to each other, accumulating evidence indicates that different mutants exhibit distinct profiles with respect to loss-of-function, dominant-negative, and GOF activity. Comparative studies of the R270H (equivalent to human R273H) and R172H (equivalent to human R175H) mutants in mutant p53 knock-in mice demonstrated different tumor spectra, suggesting that the GOF activities of different p53 mutants vary. Consistently, the p53 mutant R248Q had a strong GOF phenotype in contrast with the p53 mutant G245S in mutant TP53–knock-in mice (Hanel et al., 2013), indicating that GOF may not be a universal phenomenon resulting from p53 mutations. Moreover, not only the position of the mutation but also the nature of the substitution of p53 may influence the activity of a mutant protein, resulting in different GOF potential. For example, ectopic expression of R248Q but not the substitution mutant R248W resulted in enhanced tumor invasion in H1299 xenografts in vivo (Yoshikawa et al., 2010). In agreement with this, researchers observed strong in vivo GOF of R248Q (Hanel et al., 2013) but weaker in vivo GOF of R248W (Song et al., 2007) in mutant TP53–knock-in animal models. A possible explanation for these intracodon-specific differences in GOF activities is that these two mutations may have different structural consequences, leading to differential functions that affect their GOF properties. Consistent with this, investigators demonstrated that structural changes in p53 mediated by certain TP53 mutations, including R248Q, result in co-aggregation of mutant p53 proteins into higher order structures with other tumor-suppressive transcription factors, whereas R248W is not able to co-aggregate (Xu et al., 2011). Therefore, the altered structural properties of p53 resulting from these mutations (e.g., aggregation propensity) may be among the important features accounting for the GOF properties of mutant p53. Given the complexity of varied natures of different p53 mutants, mutant p53 actually refers to many different proteins.
Finally, although researchers have accumulated extensive evidence highlighting the existence of GOF activities of mutant p53 using cell culture and animal models, the degree of GOF activities may depend on different contextual factors. For example, R246S mutant (equivalent to human R249S) mice did not exhibit clear indication of GOF activities over those in control mice (Lee et al., 2012), whereas this mutant had GOF activities in cell line-based studies (Yan and Chen, 2010; Zhu et al., 2015). Consistent with these findings, GOF activities of mutant p53 are generally not observed in untransformed cellular contexts but are primarily observed in the cancer cell context. In numerous tissue samples derived from mouse models with mutant TP53 alleles, mutant p53 proteins were not stabilized due to the presence of Mdm2 and p16INK4a, which modify p53 protein stability. Only upon loss of Mdm2 or p16INK4a, which leads to stabilization of mutant p53, have mice had lower survival rates and more metastases than control mice with intact Mdm2 or p16INK4 (Terzian et al., 2008). Therefore, in addition to the difference in each p53 mutant, the GOF activity of mutant p53 depends largely on multiple signals that control the stabilization and modification of mutant p53, which may vary in normal cells as well as tumor cells.
MECHANISMS OF MUTANT P53 GOF ACTIVITY
Researchers have gone to extensive efforts to understand the mechanistic basis for GOF activity of mutant p53. Thus far, at least three categories of actions have been proposed to understand the possible role of mutant p53 GOF in regulation of novel gene transcription: 1) direct DNA binding of mutant p53, 2) Binding of mutant p53 to and cooperating with transcription factors or co-factors (Muller and Vousden, 2013), and 3) epigenetic regulation of chromatin remodeling (Pfister et al., 2015; Zhu et al., 2015).
In the first instance, whereas mutant p53 usually fails to regulate the expression of p53’s target genes due to loss of its specific ability to bind to canonical p53-responsive elements on the promoters of p53’s target genes, studies also have suggested that mutant p53s have the ability to bind to novel target gene promoters in a DNA structure-specific manner to activate them. Alternatively, mutant p53 can bind to and cooperate with other transcription factors, such as p63, p73, nuclear factor-κB, NF-Y, E2F1, VDR, SP1, SREBP, and ETS, to enhance or repress target gene expression (Muller and Vousden, 2013). More recently, investigators found that mutant p53 engaged in epigenetic regulation of chromatin remodeling via the SWI/SNF complex (Pfister et al., 2015), and chromatin regulatory proteins methyltransferase MLLs and acetyltransferase MOZ (Zhu et al., 2015).
In addition to its nuclear transcriptional activities, mutant p53 is localized in the cytoplasm as well where it can exert its GOF activity. Our recent work demonstrated that mutant p53-mediated cytoplasmic inhibition of 5′ AMP-activated protein kinase contributed to the oncogenic function of GOF p53 mutants in HNSCC cells (Zhou et al., 2014), suggesting that a transcription-independent mechanism is also involved in the GOF activities of mutant p53. In support of this, it was recently reported that the cytoplasmic mutant p53 regulated tumor necrosis factor signaling and metastatic programming in a transcription-independent manner (Di Minin et al., 2014).
Given that mutant p53 gains the ability to modulate gene expression in transcription-dependent and independent manners, many downstream genes and signaling pathways are regulated by GOF mutant p53, including coding and noncoding genes involved in promotion of proliferation, inhibition of cell death, DNA repair, promotion of migration, inflammation, metastasis, and lipid, glucose, and nucleotide metabolism (Haupt et al., 2016; Muller and Vousden, 2013).
Finally, even though studies using different in vitro and in vivo models have led to the discovery of many pathways controlled by mutant p53, whether these pathways have the same central roles in different cellular contexts is unclear. In fact, the diversity of the mechanisms of p53’s GOF properties strongly suggests that GOF activities may depend on and vary according to mutation type, cell type, and even stimuli. Therefore, efforts aimed at targeting mutant p53 as described below must take into account these factors affecting mutant p53’s properties and functions.
CLINICO-PATHOLOGICAL IMPLICATIONS OF P53 MUTATIONS IN HNSCCs
A prognostic biomarker can provide information about a cancer patient’s potential outcome, whereas a predictive biomarker can indicate the likelihood of a patient or tumor expressing the biomarker in response to a specific treatment. Given the high prevalence of TP53 mutations in HNSCC patients, translational researchers have been trying to determine the potential utility of TP53 mutational status as both a prognostic and predictive biomarker for HNSCC. Some of the studies holding promise for HNSCC patients are here summarized below.
P53 MUTATION AS A MARKER OF RISK AND PROGNOSIS FOR HNSCC
Studies have suggested that functional categories of TP53 mutations are correlated with prognosis for HNSCC. In one study, Poeta et al. (2007) analyzed the TP53 mutation locations in exons 2 through 11 and the nature of amino acid substitutions (i.e., conservative vs nonconservative) in the mutated codons. They then classified the mutations as disruptive or non-disruptive mutations, with disruptive mutations being those yielding truncated p53 protein or a change in amino acids in the DNA-binding domain. Using this functional classification, a prospective 7-year study of 420 HNSCC patients suggested that decreased overall survival rates in patients who underwent surgery were significantly associated with disruptive TP53 mutations (Poeta et al., 2007). We applied this classification scheme to evaluation of the impact of disruptive mutations on HNSCC cells. Our results demonstrated that expression of disruptive TP53 mutants in p53-null HNSCC cells promoted faster tumor growth and more aggressive lymph node metastases with shorter animal survival rates than that of non-disruptive TP53 mutants did in an orthotopic murine model of tongue cancer (Sano et al., 2011).
Recently, we developed a computational strategy for mutant p53 classification called evolutionary action or EAp53, in which each p53 missense mutation is assigned an evolutionary action score (http://mammoth.bcm.tmc.edu/EAp53/) and classified as either high- or low-risk (Neskey et al., 2015). Evolutionary action scores take into account the functional sensitivity of p53 to sequence variations, with sequence positions with high scores giving rise to larger phylogenetic divergences than those for sequence positions with lower scores, and the least conservative amino acid changes. Therefore, the functional impact of mutations with high evolutionary action scores is significant. We demonstrated that in both in vitro and in vivo models of HNSCC, expression of high-risk TP53 mutations conferred tumor cells with more invasive and tumorigenic characteristics than did expression of low-risk mutations (Neskey et al., 2015). Furthermore, in contrast with previous functional classification systems based on the nature of sequence substitutions in p53, our new strategy integrates functional sensitivity to evolution, and we further refined it according to clinical sample validation to reflect the clinical impact of TP53 mutations. Therefore, it can effectively stratify HNSCC patients into different prognostic groups. Using 168 TCGA HNSCC samples as a training set and 96 HNSCC samples as a validation set, we demonstrated that patients with high-risk TP53 mutations were prone to suffer worse survival outcome and more metastasis than were patients with low-risk mutations (Neskey et al., 2015). Such results were further validated in recently-updated TCGA cohort with 507 HNSCC patients (Figure 3).
Figure 3.
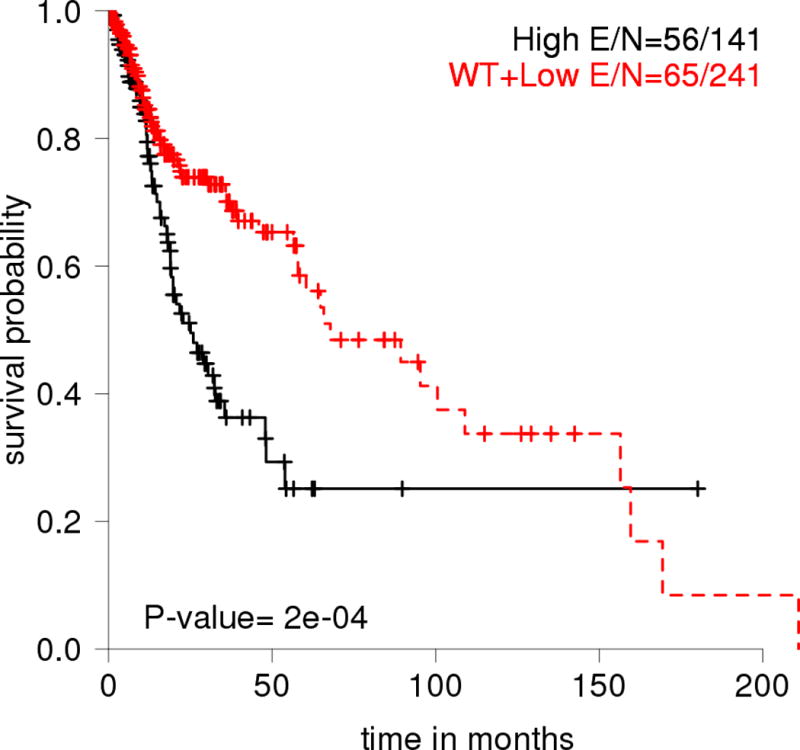
Overall survival probability for HNSCC patients in the TCGA data set (n=507) stratified by EAp53 scoring of missense TP53 mutations. High: high-risk missense mutation; Low: low-risk missense mutation; E: Event, which represents numbers of patients who passed away.
Finally, in support of the importance of TP53 mutations in HNSCCs, the TCGA analysis of indicated that TP53 mutations are significantly associated with decreased overall survival probability of HNSCC patients (Figure 4).
Figure 4.
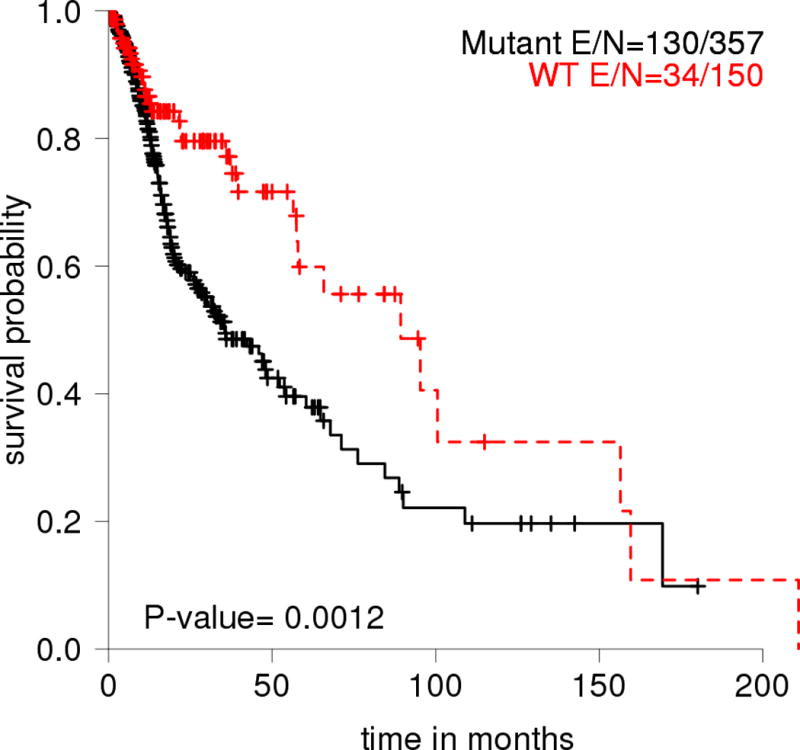
Overall survival probability for HNSCC patients in the TCGA data set (n=507) stratified by mutant and WT p53. E: Event, which represents numbers of patients who passed away.
TP53 MUTATION AS A PREDICTOR OF CLINICAL RESPONSE OF HNSCC TO TREATMENT
In the clinic, researchers have demonstrated that TP53 mutations have predictive significance in evaluating response of HNSCC to platinum-based therapy. In a recent study, Perrone et al. (2010) categorized TP53 mutations based on the transactivation activity of the mutated proteins as determined using information in the International Agency for Research on Cancer TP53 database and scored them as “functional,” “partially functional,” or “nonfunctional.” They found that nonfunctional p53 (with loss of transactivation activity) may be indicative of a low pathological complete remission rate and a low rate of response to cisplatin-based neoadjuvant chemotherapy (Perrone et al., 2010).
Despite the role of TP53 in predicting therapy response of HNSCC, an effective system of evaluating TP53 status for stratifying patients into different response groups is still lacking. Recently, by reanalyzing the TP53 mutational status with the EAp53 scoring system in the cohort of 68 patients who underwent cisplatin-based chemotherapy followed by surgical resection for locally advanced HNSCC, we identified a subset of patients harboring high-risk TP53 mutations (13 of 14) who had decreased sensitivity of HNSCC to a cisplatin-based regimen. Compared with the low-risk TP53 mutation group, the high-risk TP53 mutation group was 10-fold more likely to have residual disease following cisplatin-based treatment. In in vitro preclinical models of HNSCC cells expressing WT and mutant p53 with a range of evolutionary action scores, high-risk TP53 mutants conferred non-TP53-expressing HNSCC cells with resistance to cisplatin-based treatment. Interestingly, HNSCC cells bearing p53 mutations with low evolutionary action scores retained some of the cisplatin responsiveness seen in WT p53-expressing cells (Osman et al., 2015b). Similar results were observed in an orthotopic mouse model of tongue cancer, in which high-risk TP53 mutations were associated with decreased response to treatment with cisplatin and overall survival outcome (Osman et al., 2015b).
In addition to functionally and computationally defined TP53 mutations, TP53 polymorphism may also affect the clinical responses to chemoradiotherapy in advanced HNSCC patients. For example, TP53 codon 72 polymorphism encoding two amino acids (arginine versus proline) differentially modulates p73 functions, accounting for differing clinical outcomes in HNSCC patients, with 72R mutants conferring poorer response to cisplatin-based treatment than did 72P mutants (Bergamaschi et al., 2003).
As demonstrated in the aforementioned studies, TP53 is one of the most important prognostic predictors of disease state and treatment response for HNSCC patients. Although a TP53 mutational status has not been incorporated into clinical evaluation of HNSCC patients, we expect that further investigation of the TP53 mutational status and its association with outcome in HNSCC patients would contribute significantly to prediction of clinical outcomes and therefore is of significant interest to both patients and clinicians.
THERAPEUTIC STRATEGIES FOR HNSCC WITH P53 MUTATIONS
Given that the majority of tumors, including HPV− HNSCCs, have TP53 mutations and that many cancer cells depend on mutant TP53 for their survival and growth, reactivation of WT p53’s tumor-suppressive properties and eliminating mutant p53’s GOF activities are potentially effective strategies for treatment of HNSCC patients with p53 mutations. Moreover, in spite of the diversity of GOF mechanisms among different p53 mutants, the possibility of distinguishing mutant p53-specific processes from common mechanisms of GOF shared by some mutant p53 variants will aid the development of personalized strategies for targeting mutant p53 and/or downstream pathways for the treatment of HNSCC.
STRATEGIES TO RESTORE WT P53 ACTIVITY TO MUTANT P53
Because the activation of WT p53 in many cancer cells leads to p53-mediated apoptosis and senescence, researchers have explored introduction of exogenous WT TP53 into HNSCC cells using either viral or nonviral methods. In particular, because of its affinity for the cells of the upper aerodigestive tract, adenovirus was used to deliver WT TP53 to HNSCC patients 20 years ago. Since then, many attempts have been made to use adenoviral p53 to treat HNSCC in phase 1, 2, and 3 clinical trials. In many of those trials, adenoviral p53 alone or in combination with chemotherapy or radiotherapy produced tumor regression (Tassone et al., 2013). In spite of these encouraging results, the long-term effects of this adenovirus-mediated TP53 gene therapy still need to be tested in larger cohorts, and further improvements in gene delivery methods are required for safe, effective use of WT p53 gene therapy for HNSCC patients. This concern is also true for nonviral methods of WT p53 gene delivery in HNSCC patients (e.g., photochemical internalization, nanoparticle approaches).
In addition to introduction of WT p53 into tumors, reactivation of some level of WT function in mutant p53-bearing cells is an attractive therapeutic strategy for cancers with p53 mutations. Over the past few years, researchers have characterized a variety of compounds and peptides that may restore WT p53 function: 1) small molecules (e.g., PhiKan083, PK7088) that bind to a site in the p53 Y220C mutant by stabilizing its structure, increasing the level of p53 protein with WT conformation and activity; 2) compounds (e.g., CP-31398, STIMA-1, PRIMA-1, PRIMA-1met/APR-246, P53R3, SCH29074) that bind to multiple mutant p53 proteins and interact with the DNA-binding domain, thereby promoting proper folding of mutant proteins and restoration of WT p53 functions; 3) compounds that restore the WT p53 conformation and transcriptional activity to mutant p53 proteins (e.g., MIRA-1 and its analogs, chetomin, WR-1065, RITA); 4) compounds (e.g., NSC319726) that chelate the metal ion Zn(2+) and help mutant p53 R175H fold correctly to WT conformation (reviewed by (Parrales and Iwakuma, 2015); and 5) peptides (e.g., ReACp53) that prevent mutant p53 aggregation and restore WT p53 function (Soragni et al., 2016).
Of all the compounds that restore WT activity, the only drug directly targeting mutant p53 that has reached the clinical stage is PRIMA-1met/APR-246, which successfully completed phase 1/2 clinical trials in patients with hematological malignancies or prostate cancer that included mutant p53 patients (Lehmann et al., 2012). PRIMA-1 also can induce apoptosis of and enhance the cytotoxicity of standard chemotherapy in HNSCC cells (Roh et al., 2011).
Of note is that in many of the cases described above, the precise mechanisms of the p53 reactivation by compounds are largely unknown, and some off-target effects of the compounds are expected. For example, we observed that the p53-reactivating small molecule RITA induces senescence in HNSCC cells, partially in a p53-independent manner (Chuang et al., 2014). In addition, although many reactivating compounds seem to target more than one p53 mutant, whether these compounds can reactivate all p53 mutants or specific mutant types remains unclear, making this strategy quite challenging.
STRATEGIES TO PROMOTE MUTANT P53 DEGRADATION
Another approach to targeting oncogenic mutant p53 is to discover and administer treatment with compounds that specifically degrade mutant p53. Some of the first such approaches included treatment with inhibitors of heat shock protein 90, a chaperone molecule that contributes to accumulation of mutant p53 by inactivating p53 ubiquitin ligases MDM2 and CHIP. The heat shock protein 90 inhibitors, geldanamycin, 17-AAG, and ganetespib, destabilize mutant p53 and induce mutant p53 depletion, increasing apoptosis in tumors in in vivo mouse models (Alexandrova et al., 2015). Treatment with other compounds, such as the histone deacetylase inhibitor SAHA (vorinostat), arsenic compounds, gambogic acid, spautin-1, YK-3-237, NSC59984, and disulfiram, has downregulated the expression of mutant p53 variants in different kinds of cells (reviewed by (Parrales and Iwakuma, 2015). The U.S. Food and Drug Administration approved vorinostat for the treatment of cutaneous T-cell lymphoma, and ganetespib is currently under evaluation in a phase 3 clinical trial for lung cancer.
STRATEGIES FOR TARGETING MUTANT P53-REGULATED PATHWAYS AND INDUCING MUTANT P53-RELATED SYNTHETIC LETHALITY
In addition to targeting mutant p53 directly, investigators have used strategies targeting mutant p53-regulated downstream targets and signaling pathways. For example, many mutant p53 proteins are believed to achieve GOF activity by interacting with and inhibiting p63 and p73. The small molecule RETRA destabilizes p73 mutant-p53 interaction, leading to activation of p73 target genes and a concomitant decrease in tumor-cell survival and xenograft tumor growth suppression (Kravchenko et al., 2008). In addition, because GOF mutant p53 regulates sterol metabolism, pharmacological inhibition of the rate-limiting enzyme HMG CoA reductase during sterol biosynthesis by treatment with simvastatin, a lipophilic statin, resulted in a reduction in invasive growth as well as extensive death of GOF-mutant p53 cell lines in both three-dimensional culture and a xenograft breast tumor model (Freed-Pastor et al., 2012). Recently, researchers found that GOF-mutant p53 regulated the chromatin remodeling pathway through the methyltransferase MLL1, and pharmacological inhibition of the MLL1 methyltransferase complex with OICR-9429, an antagonist of interaction of WDR5 with MLL1 in the COMPASS complex (complex proteins associated with Set1), inhibited the proliferation of GOF-mutant p53-expressing cancer cells but not of WT p53-bearing cancer cells (Zhu et al., 2015).
Synthetic lethality is generally used to describe the condition in which a mutation of a gene is not lethal by itself but causes cell death in combination with drugs or other mutations. Because most HNSCC patients have mutations of the TP53 gene, targeting p53 mutations to induce synthetic lethality of tumors is an attractive strategy. Researchers showed that treatment with G2/M checkpoint blockers such as the protein kinase C inhibitor UCN01, the polo-like kinase inhibitor BI-2536, and the Wee-1 kinase inhibitor PD0166285 abrogated DNA damage-induced cell-cycle arrest at G2/M phase and increased cytotoxicity in cancer cells with TP53 mutations [reviewed by (Parrales and Iwakuma, 2015)]. Consistent with this, our work has demonstrated that treatment with the Wee-1 kinase inhibitor AZD-1775 can help overcome the cisplatin resistance of HNSCC associated with p53 mutations via mitotic arrest followed by senescence (Osman et al., 2015a). However, the observed synthetic lethality of inhibitors associated with loss of cell-cycle arrest at G2/M phase in mutant p53 cells likely is dependent not on oncogenic GOF activity of mutant p53 but rather on loss of WT p53 activity, leading to mitotic catastrophe in cancer cells lacking WT p53 activity.
CONCLUSIONS
The most comprehensive integrative genomic analysis of HNSCCs by TCGA confirmed that TP53 is the most frequently mutated gene in HNSCC. The mutually exclusive relationship between p53 mutations and HPV positivity and various p53 mutation rates in head and neck tumors at different anatomical sites provide great insight into the molecular pathogenesis of HNSCC. Clinically, TP53 mutations are significantly associated with short survival probability in HNSCC patients. This indicates that TP53 mutations may be useful prognostic biomarkers for HNSCC. Moreover, given that the standard treatments of HNSCC, including radiotherapy and chemotherapy, are usually effective against tumors with WT TP53 and that TP53 mutations are often associated with tumor radioresistance and/or chemoresistance, TP53 status also may be considered a useful predictive biomarker for determining the clinical response of HNSCC to treatment. Also, developing better therapeutic strategies for HNSCC in patients with tumors bearing TP53 mutations is imperative. Recent studies revealed that TP53 mutation can lead to not only loss of WT p53 function but also GOF activity. Furthermore, mutant p53 is not just one protein but actually a multitude of proteins that can contribute to an exceptionally vast network of tumor-promoting processes in cells. Discovering drug-based strategies to safely and efficiently target mutant p53 in HNSCC is therefore highly challenging and will require a better understanding of mutant p53’s biology, such as its interaction partners, degradation pathways, and downstream signaling pathways. Furthermore, because of the heterogeneity of mutant p53 forms, determining whether p53-activating and mutant p53-depleting drugs impact only specific mutant p53 types or all p53 mutants or other proteins or pathways in HNSCCs will be important. It is also important to investigate any synergistic or additive effects of these compounds with standard radiotherapy and chemotherapy for HNSCC. Therefore, much effort is needed to target mutant p53 for personalized therapy for HNSCC. However, given the high frequency of p53 mutations in patients with this tumor and their association with poor response to treatment, these types of efforts are needed to make an impact on this deadly disease.
Acknowledgments
We apologize to our colleagues for the exclusion of other important contributions to the field and their references due to space limitations.
Contract grant sponsor: National Institute of Health RO1 DE14613, 5 R01 DE024601-02, and UO1 DE025181
References
- Acin S, Li Z, Mejia O, Roop DR, El-Naggar AK, Caulin C. Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras. The Journal of pathology. 2011;225:479–489. doi: 10.1002/path.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, Lozano G, Dobbelstein M, Moll UM. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523:352–356. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, Yulug I, Merlano M, Numico G, Comino A. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer cell. 2003;3:387–402. doi: 10.1016/s1535-6108(03)00079-5. [DOI] [PubMed] [Google Scholar]
- Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell metabolism. 2013;18:617–633. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa RA, Correo R, Eby YJ, Ruppert JM, Sidransky D. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer research. 1993;53:4477–4480. [PubMed] [Google Scholar]
- Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer research. 1996;56:2488–2492. [PubMed] [Google Scholar]
- Chuang HC, Yang LP, Fitzgerald AL, Osman A, Woo SH, Myers JN, Skinner HD. The p53-reactivating small molecule RITA induces senescence in head and neck cancer cells. PloS one. 2014;9:e104821. doi: 10.1371/journal.pone.0104821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paula AM, Souza LR, Farias LC, Correa GT, Fraga CA, Eleuterio NB, Silveira AC, Santos FB, Haikal DS, Guimaraes AL, et al. Analysis of 724 cases of primary head and neck squamous cell carcinoma (HNSCC) with a focus on young patients and p53 immunolocalization. Oral oncology. 2009;45:777–782. doi: 10.1016/j.oraloncology.2008.11.015. [DOI] [PubMed] [Google Scholar]
- DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA: a cancer journal for clinicians. 2013;63:151–166. doi: 10.3322/caac.21173. [DOI] [PubMed] [Google Scholar]
- Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R, Sommaggio R, et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Molecular cell. 2014;56:617–629. doi: 10.1016/j.molcel.2014.10.013. [DOI] [PubMed] [Google Scholar]
- el-Naggar AK, Lai S, Luna MA, Zhou XD, Weber RS, Goepfert H, Batsakis JG. Sequential p53 mutation analysis of pre-invasive and invasive head and neck squamous carcinoma. International journal of cancer Journal international du cancer. 1995;64:196–201. doi: 10.1002/ijc.2910640309. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes & development. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell death and differentiation. 2013;20:898–909. doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Raghu D, Haupt Y. Mutant p53 Drives Cancer by Subverting Multiple Tumor Suppression Pathways. Frontiers in oncology. 2016;6:12. doi: 10.3389/fonc.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravchenko JE, Ilyinskaya GV, Komarov PG, Agapova LS, Kochetkov DV, Strom E, Frolova EI, Kovriga I, Gudkov AV, Feinstein E, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6302–6307. doi: 10.1073/pnas.0802091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harbor perspectives in biology. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Sougnez C, Lichtenstein L, Cibulskis K, Lander E, Gabriel SB, Getz G, Ally A, Balasundaram M, Birol I, et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, Sabapathy K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer cell. 2012;22:751–764. doi: 10.1016/j.ccr.2012.10.022. [DOI] [PubMed] [Google Scholar]
- Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, Uggla B, Yachnin J, Juliusson G, Moshfegh A, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:3633–3639. doi: 10.1200/JCO.2011.40.7783. [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. p53 mutations in cancer. Nature cell biology. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- Neskey DM, Osman AA, Ow TJ, Katsonis P, McDonald T, Hicks SC, Hsu TK, Pickering CR, Ward A, Patel A, et al. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer research. 2015;75:1527–1536. doi: 10.1158/0008-5472.CAN-14-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, Frederick MJ, Woo SH, Caulin C, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Molecular cancer therapeutics. 2015a;14:608–619. doi: 10.1158/1535-7163.MCT-14-0735-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman AA, Neskey DM, Katsonis P, Patel AA, Ward AM, Hsu TKK, Hicks SC, McDonald TO, Ow TJ, Alves MO, et al. Evolutionary Action Score of TP53 Coding Variants Is Predictive of Platinum Response in Head and Neck Cancer Patients. Cancer research. 2015b;75:1205–1215. doi: 10.1158/0008-5472.CAN-14-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman AA, Neskey DM, Katsonis P, Patel AA, Ward AM, Hsu TK, Hicks SC, McDonald TO, Ow TJ, Alves MO, et al. Evolutionary Action Score of TP53 Coding Variants Is Predictive of Platinum Response in Head and Neck Cancer Patients. Cancer research. 2015c;75:1205–1215. doi: 10.1158/0008-5472.CAN-14-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes & development. 2014;28:1739–1751. doi: 10.1101/gad.247452.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Frontiers in oncology. 2015;5:288. doi: 10.3389/fonc.2015.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone F, Bossi P, Cortelazzi B, Locati L, Quattrone P, Pierotti MA, Pilotti S, Licitra L. TP53 Mutations and Pathologic Complete Response to Neoadjuvant Cisplatin and Fluorouracil Chemotherapy in Resected Oral Cavity Squamous Cell Carcinoma. Journal of Clinical Oncology. 2010;28:761–766. doi: 10.1200/JCO.2009.22.4170. [DOI] [PubMed] [Google Scholar]
- Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, Freed-Pastor WA, Laptenko O, Neo SP, Bargonetti J, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes & development. 2015;29:1298–1315. doi: 10.1101/gad.263202.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering CR, Zhang J, Neskey DM, Zhao M, Jasser SA, Wang J, Ward A, Tsai CJ, Ortega Alves MV, Zhou JH, et al. Squamous cell carcinoma of the oral tongue in young non-smokers is genomically similar to tumors in older smokers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:3842–3848. doi: 10.1158/1078-0432.CCR-14-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer discovery. 2013;3:770–781. doi: 10.1158/2159-8290.CD-12-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2007;357:2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JL, Kang SK, Minn I, Califano JA, Sidransky D, Koch WM. p53-Reactivating small molecules induce apoptosis and enhance chemotherapeutic cytotoxicity in head and neck squamous cell carcinoma. Oral oncology. 2011;47:8–15. doi: 10.1016/j.oraloncology.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano D, Xie TX, Ow TJ, Zhao M, Pickering CR, Zhou G, Sandulache VC, Wheeler DA, Gibbs RA, Caulin C, et al. Disruptive TP53 Mutation Is Associated with Aggressive Disease Characteristics in an Orthotopic Murine Model of Oral Tongue Cancer. Clinical Cancer Research. 2011;17:6658–6670. doi: 10.1158/1078-0432.CCR-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature cell biology. 2007;9:573–580. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- Soragni A, Janzen DM, Johnson LM, Lindgren AG, Thai-Quynh Nguyen A, Tiourin E, Soriaga AB, Lu J, Jiang L, Faull KF, et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer cell. 2016;29:90–103. doi: 10.1016/j.ccell.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone P, Old M, Teknos TN, Pan Q. p53-based therapeutics for head and neck squamous cell carcinoma. Oral oncology. 2013;49:733–737. doi: 10.1016/j.oraloncology.2013.03.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes & development. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjebbes GW, Leppers vd Straat FG, Tilanus MG, Hordijk GJ, Slootweg PJ. p53 tumor suppressor gene as a clonal marker in head and neck squamous cell carcinoma: p53 mutations in primary tumor and matched lymph node metastases. Oral oncology. 1999;35:384–389. doi: 10.1016/s1368-8375(98)00127-4. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Xie TX, Zhou G, Zhao M, Sano D, Jasser SA, Brennan RG, Myers JN. Serine substitution of proline at codon 151 of TP53 confers gain of function activity leading to anoikis resistance and tumor progression of head and neck cancer cells. The Laryngoscope. 2013;123:1416–1423. doi: 10.1002/lary.23846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nature chemical biology. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- Yan W, Chen X. Characterization of functional domains necessary for mutant p53 gain of function. The Journal of biological chemistry. 2010;285:14229–14238. doi: 10.1074/jbc.M109.097253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Hamada J, Tada M, Kameyama T, Nakagawa K, Suzuki Y, Ikawa M, Hassan NM, Kitagawa Y, Moriuchi T. Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomedical research. 2010;31:401–411. doi: 10.2220/biomedres.31.401. [DOI] [PubMed] [Google Scholar]
- Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Molecular cell. 2014;54:960–974. doi: 10.1016/j.molcel.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–211. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]