

Abstract
Covalently bound protein kinase inhibitors have been frequently designed to target non-catalytic cysteines at the ATP binding site. Thus, it is important to know if a given cysteine can form a covalent bond. Here we combine a function-site interaction fingerprint method and DFT calculations to determine the potential of cysteines to form a covalent interaction with an inhibitor. By harnessing the human structural kinome, a comprehensive structure-based binding site cysteine dataset was assembled. The orientation of the cysteine thiol group indicates which cysteines can potentially form covalent bonds. These covalent inhibitor accessible cysteines are located within five regions: P-loop, roof of pocket, front pocket, catalytic-2 of the catalytic loop and DFG-3 close to the DFG peptide. In an independent test set, these cysteines covered 95% of covalent kinase inhibitors. This study provides new insights into cysteine reactivity and preference which is important for the prospective development of covalent kinase inhibitors.
Graphical Abstract
Introduction
Abnormal kinase regulation is responsible for more than 200 diseases, notably various cancers.1,2 Consequently, protein kinases have been very important drug targets for anticancer drug discovery.3,4 However, to design a targeted kinase inhibitor with the desired selectivity is a daunting task as all kinases share a similar catalytic domain that binds ATP.5 Through July 2016, 30 kinase-targeted drugs had been approved by U.S. Food and Drug Administration (FDA) (http://www.fda.gov/),6 These drugs have proved successful7 in reducing patient suffering and prolonging patient survival in treating multiple diseases, especially cancers.8 However, reported off-target toxicities and acquired-mutation resistance dictate that prospective kinase-targeted inhibitors act at a lower dose and with higher specificity.4
Covalent targeting-kinase inhibitors have received increasing attention because of their high binding affinity and selectivity.9–11 Diverse covalent kinase inhibitors have been developed (see recent reviews9,12,13 and herein). More notably, the FDA approved three irreversible inhibitors recently. First is Afatinib for patients with metastatic non-small cell lung cancer (NSCLC), which is driven by epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution mutations.14 Second is the BTK inhibitor, Ibrutinib, for B-cell malignancies.15,16 Third is Osimertinib used to treat patients with EGFR T790M mutation-positive metastatic NSCLC17. All three drugs were designed by combining a reversible-inhibitor scaffold possessing competency against their primary targets and an electrophilic functional group capable of covalent Michael addition to cysteine.14,18 Recent studies9,19,20 have indicated that combining a reversible inhibitor with an electrophilic center is a practicable strategy for developing irreversible kinase inhibitors.3,9 The reversible-inhibitor scaffolds are important to assure targeting the desired kinase and the electrophilic functional group improves the specificity and binding affinity.3,19,21 Currently, diverse electrophilic functional groups,9 such as acrylamide and its derivatives,22 have been used to achieve covalent binding to the non-catalytic cysteines. Meanwhile, the abundant cysteine residues as nucleophilic groups, located within different parts of the binding site for various kinases, provides a potential opportunity for developing specific irreversible inhibitors by harnessing the different cysteine locations.6 Gray et al6,9 reviewed the locations of cysteines for the whole human kinome using primary sequence alignment and classified accessible cysteines into different regions including Gatekeeper, Roof region, Hinge region, DFG region, and P-loop, respectively. The known arrangement of cysteines provides useful information for designing a covalent inhibitor that can access the targeted cysteine by merging an appropriate electrophilic group with the given reversible inhibitor scaffold. However, having an electrophilic group and a spatially accessible cysteine is not necessarily sufficient. The Michael addition reaction requires both a favorable steric and electronic interaction between the nucleophilic and electrophilic groups.23 Thus, it is vital to learn which cysteines can indeed react with the electrophilic center; this requires structure-based information.
In this paper we address this question by combining a function-site interaction fingerprint (Fs-IFP) method and density functional theory (DFT) calculations across the whole human structural kinome, which is increasing rapidly.24–26 As of July 2016, there were277 4 kinase-ligand complex structures (235 kinases) in the PDB (http://www.rcsb.org/).27 Previously we have developed and applied the Fs-IFP approach to study binding modes across the human kinome and hence only a synopsis is given here.28,29 The Fs-IFP approach encodes the 3D binding modes of any given kinase-ligand complex structure into 1D interaction fingerprints for further large-scale analysis. The interaction fingerprint provides the interaction details of every atom at the interface of the binding pocket. In particular, the encoded fingerprint of the cysteine sulfur atom will directly indicate if the direction of the thiol group is favorable for covalent interaction. Thus the potential covalent reaction for every cysteinecan be analyzed
To further quantify the effect of microenvironments on cysteine reactivity within the binding site, DFT calculations are performed systematically. Cysteine-involved Michael addition has been widely studied (http://www.organic-chemistry.org/).30,31 Cronin et al32,33 suggest a simultaneous mechanism as shown in Figure 1, Scheme 1. Johannes et al34 and David J. et al35 reported a stepwise mechanism with the loose transition state in weak-base buffer solution for a set of α, β-unsaturated aldehydes, ketones or esters as the electrophilic groups, as shown in Figure 1, Scheme 2. In this paper, our purpose is to gain insights into solvent effects and steric effects for given cysteine covalent reactions within the binding site. Such an analysis is necessary to better interpret the reactivity of each cysteine-electrophile pair in the protein. To achieve this we built the representative computational model for the cysteine-involved addition reaction in the binding site. A series of multi-dimension potential energy surfaces with different dielectric constants including ε=0.0, ε=7.43 and ε=78.36 and the multi-dimension potential energy surface for the conformation space of the cysteine dihedral χ1 are calculated to qualitatively characterize the reactivity of any given cysteine.
Figure 1.

Model system and reaction schemes for cysteine-involved addition. di is the interatomic distance between two atoms.
Results
Cysteine distribution
Using the Fs-IFP method, we obtained the distribution of cysteines across the human kinome. In 2774 complex structures, 1599 structures (belong to 169 kinases) have at least one cysteine located at their respective binding sites. These cysteines are distributed at 63 different amino acid locations as indicated in Figure S1 and Table S1, with all positions marked.36 Furthermore, 17 amino acid locations have at least one contact between the cysteine and the corresponding ligand. The 17 amino acid locations are marked in different colors (Figure 2a) following the distribution at different regions within the binding pocket and labeled as listed in Table S1. We then counted the number of accessible cysteines at every positions and the number of cysteines involved in interactions with the ligands from the 1599 structures. Figure 2b illustrates the distribution of cysteines across all structure which are given in Table S2. The top two interacting positions(12% of kinases) are located at Hinge-2 and DFG-3 (Figure 2b). We also assembled the distribution of all 43 PDB structures with cysteine-ligand covalent contacts (green in Figure 2b; red in Table S2), which are distributed at P-loop-5, P-loop-9, Hinge-1, Front-2, Front-3 and DFG-3. Notably, 29 of the 43 cysteine-ligand covalent-contact structures occur at the sub-region of Front-2. What follows are details of the interactions between the cysteines and the corresponding ligands.
Figure 2.

(a) The locations of cysteines and the structure template from pdb id 3byu. (b) Statistics for the accessible cysteines, the details of the interactions between cysteines and ligands, and the existed covalent interactions between cysteines and ligands.
Details of the cysteine interactions
Figure 3a shows a cysteine composed of 6 non-hydrogen atoms: four (CA, C, O and N) as part of the backbone and the other two (CB and SG) as part of the side chain. Figure 3b indicates that two types of cysteine-ligand interactions dominate, hydrophobic interactions and hydrogen-bond interactions. 78.6% are hydrophobic contacts from atoms C(0.3%), CA(8.6%), CB(24.9%) and SG(44.7%). 5.1% are hydrogen-bond contacts with oxygen as an acceptor and 16.3% are hydrogen-bond contacts with nitrogen as a donor (Figure 3b). This distribution indicates that SG is structurally accessible in many kinases and provides abundant opportunities for developing covalent inhibitors. Figure 3c shows the detailed distribution of the interactions to which the atoms in cysteines contribute. In the following sections, we detail the possible covalent reactions at all locations described in Figures 2a and 3c.
Figure 3.

The detailed interactions of cysteines. (a) The six atoms of cysteine. (b) The distribution of different types of interactions for different atoms. (c) The rate of contributed interactions for different atoms at eachlocation.
P-loop
As shown in Figure 2a, there are three cysteine-involved positions P-loop-5, P-loop-7 and P-loop-9 located within the P-loop. Here the cysteine contributes mainly to the hydrophobic interactions via the side chain atoms CB and SG. The hydrophobic interaction from SG is the primary contribution (green in Figure 3c at locations P-loop-5, P-loop-7 and P-loop-9). Notably, at location P -loop-5, the cysteine contributes to not only the hydrophobic interaction from SG but also the hydrogen-bond interaction from N as a donor. In the binding pocket of the kinase domain, the P-loop is located at the front cleft,37 which is solvent-accessible38. Several released PDB structures exhibit covalent interactions with the P-loop. An example is shown in Figure 4a (pdb id 4r6v),21 where the cysteine at P-loop-5 donates the covalent contact with the ligand. Other structures with covalent interactions include pdb id4d9t, 4d9u, 4jg6, 4jg7 and 4jg8. 22,39 Thus, the P-loop can be utilized to design potential covalent inhibitor s.
Figure 4.

Cysteines at different locations; sulfur atom in yellow, cysteine in green; and ligand in blue. (a) Cysteine at P-loop (pdb id 4r6v). (b) Cysteine at roof of the binding pocket (pdb id 4riy). (c) Cysteines at location Hinge-1 of the hinge region (representative pdb id 3m2w). (d) Cysteines at location Hinge-2 of the hinge region (representative pdb id 2ywp). (e) Cysteines at location Hinge-3 of the hinge region (representative pdb id 3lco). (f) Cysteines at location Catalytic-2 of the catalytic loop (representative pdb id 1t46). (g) Cysteines at location Catalytic-9 of the catalytic loop (representative pdb id 4a0j). (h) Cysteine at location DFG-4 close to the DFG peptide (representative pdb id 3wf7).
Roof of pocket
Here the cysteine is located at Roof-3, which is on the roof of the binding pocket (Figures 2a and 4b). All cysteines are involved in the hydrophobic interaction from CB (4%) and SG (96%) (Figure 3c). Generally, the amino acid in this position is conserved to facilitate ATP binding.40 Previous reports28 have showed that this position is mainly occupied by an alanine, which provides a conserved interaction with the adenine ring of ATP (Figure 4b). Currently, most type-I and type-II inhibitors are also designed to utilize an adenine-like group that forms at least a hydrogen-bond with the kinase hinge,41 so the position is available to interact with a given electrophile. In designing a covalent inhibitor using the cysteine at the roof of the pocket, it is important to consider not only the covalent interaction at the roof, but also the hydrogen-bond interactions with the hinge.6
Hydrophobic subpocket
There are four sites Helix-9, Helix-11, Beta4-2 and Beta4-4, which contribute to possible cysteine interactions (Figures 2a and 3c). These cysteines provide hydrophobic interactions from the side chain atoms SG and CB. Notably, these sites are located at the deeper back pocket that forms the typical hydrophobic subpocket of the kinase domain.42 DFT calculations show that the cysteine-involved addition reaction is more favorable in polar solvent, which has high dielectric constant43, than in a hydrophobic environment( Figure 5).
Figure 5.

The potential energy surfaces(PESs). (a) For scheme 1 in different dielectric constants. (b) For scheme 2 in different dielectric constants. ΔE for energy barrier from the reactant to the transition state (TS) and the energy unit is kcal mol−1. ε is the dielectric constant.
Figure 5 shows the potential energy surfaces (PESs) of cysteine-ligand Michael addition using a DFT calculation. The PESs for two reaction mechanistic possibilities (Scheme 1 and 2) are shown in Figure 5a and 5b, respectively. In Figure 5a, there is an energy barrier 46.2 kcal mol−1 (ε=0), which is consistent with experiment23. Importantly, there is a lower energy barrier in higher dielectric constants (40.1 kcal mol−1 in ε=78.35 and 42.3 kcal mol−1 in ε=7.43). In weak basic solvent (an NH3 molecule as a micro-solvation molecule44) (Figure 5b), the cysteine-involved addition reaction shows much lower energy barriers (19.7 kcal mol−1 in ε=0, 17.3 kcal mol−1 in ε=7.43 and 16.6 kcal mol−1 in ε=78.35) than for Scheme 1. Comparatively speaking, our investigation indicates Scheme 2 is the energetically favored mechanism, again consistent with experiment45. In Scheme 2, the proton atom of the thiol group was transferred to the NH3 molecule leading to charge localization of the thiol group and a stabilized transition state. More notably, in either scheme, the addition reaction is more favorable in a polar environment (high dielectric constants)45, especially in basic buffer that facilitates producing the thiolate group.35 This makes sense as the high dielectric constant increases the dielectric screen between the general acid and the general base leading to the charge localization of the reaction groups. Thus, the transition state is further stabilized and the energy barrier reduced.
Our survey has shown that the thiol group of cysteine in the hydrophobic sub-pocket is not easily polarized. Therefore it is difficult to form a covalent interaction between any given ligand and the cysteine in the hydrophobic sub-pocket because of the lack of the polarization step in Michael addition.23
Hinge region
Here the cysteine has the most contacts since the majority of inhibitors bind at the anchor site, e.g., interacting with ATP through 2~3 contacts (Figure 2b).6 For Hinge positions, Hinge-1, Hinge-2 and Hinge-3, the major contacts are from the backbone atoms (Figure 3c). At location Hinge-1, the majority of interactions are from two different atoms: SG (56%) and CA (40%). This suggests that both backbone and side chain atoms contribute to ligand interactions in the hinge region. This agrees with the observation of at least two or three contacts between the hinge and the corresponding ligand.6,24,42 The side chain of cysteine points outside the pocket (Figure 4c), thus it is difficult to design an irreversible inhibitor by utilizing the cysteine at Hinge-146. At location Hinge-2, the main contacts are from the main-chain atoms CA (40%) and N (30%), consistent with the observation of more than one contact point between the hinge and the corresponding ligand, as was true for Hinge-1. We further inspect the directions of the side chain of cysteine at Hinge-2 (Figure 2b). 360 structures have this protein-ligand interaction and the distribution of the χ1 dihedral (N-CA-CB-SG) is shown in Figure 6a, in which the distribution of χ1 is from −76.0 to −34.0 degree with the peak at about −62.0 degrees. Based on the distribution and the premise of the backbone atom N pointing to the Gatekeeper (Figure 4d), the side chains always point towards the bottom of the pocket, and all SG atoms point toward the deeper hydrophobic sub-pocket (Figure 4d). In this orientation the cysteine side chain is not easily accessed because of an unfavorable direction and unfavorable polarization of the thiol group, similar to that found in the hydrophobic sub-pocket. Moreover, there is a strong steric clash at location Hinge-2 when linking the given electrophile. If the reaction were to occur, the linked electrophilic group should locate at the bottom of binding pocket along the direction of the cysteine side chain, but there is no space for this interaction, as shown in Figure 4d. On the other hand, if the dihedral χ1 of cysteine is rotated and adjusted to accommodate the linked electrophilic group, the rotation action needs to overcome a high energy barrier, as shown on the calculated potential energy surface (Figure 6b), in which the low energy points are located around χ1 = −175.0°, that correspond to the bonded distance between electrophile and nucleophile (d1 = 1.83 Å). The calculated potential energy surface also quantitatively confirms the energy barrier along the rotation of the dihedral χ1. An energy barrier of at least 20.0 kcal mol−1needs to be overcome following the rotation pathway via (χ1 = −175.0°, d1 = 1.83 Å) as shown in Figure 6b. Due to the steric effect, the cysteine located at Hinge-2 is not easily accessible to the electrophilic group. At location Hinge-3 the main interactions are from the backbone atom CA. Here the side chains point outside the binding pocket, as shown in Figure 4e. The thiol group on the side chain of cysteine is not accessible for covalent reaction. In summary, although many contacts occur at the hinge region, it is difficult to design covalent inhibitors.
Figure 6.

(a) Distribution of the cysteine dihedralangle χ1at location Hinge -2. (b) PES for the conformation space of the cysteine dihedralangle χ1;Y -axis for the interatomic distance d1.
Front pocket
The interaction at Front-2 and Front-3 primarily involves the side-chain atoms (CB and SG) (Figure 3c). Further, the front pocket is located at the edge of the binding site, which not only exposes the favorable polar environment for covalent reaction,38 but also provides sufficient open space to tolerate a diverse set of electrophilic groups.9,47 This region is capable of forming a covalent bond to cysteine, as also validated by binding of irreversible inhibitors including the aforementioned three FDA-approved irreversible drugs, Afatinib, Ibrutinib and Osimertinib3,15,17,19 and 31 covalent-interaction kinase-ligand PDB structures (Table S1).
Catalytic loop
There are two cysteine-involved positions within the catalytic loop, Catalytic-2 and Catalytic-9 (Figure 2a). Catalytic-2 is located at the bottom of the allosteric site between the C-Helix and the DFG peptide. The tails of several type-II inhibitors frequently touch this position, for example, Imatinib in binding c-Kit (pdb 1t46) (Figure 4f).48 Here the cysteine side-chain atoms provide the contacts. It is possible to design an irreversible inhibitor by bonding with the cysteine at this site. At location Catalytic-9, the interactions are from the backbone atom CA. 125 structures have the cysteine at this position (Figure 2b). We aligned these binding sites and confirmed that all cysteine side chains at location Catalytic-9 have the same conformation, and point toward the outside of the binding pocket (Figure 4g). This is in agreement with the fact that only the atom CA provides the contact, as shown in Figure 3c (an example from pdb id 4aoj (Figure 4g)). Both positions, Catalytic-2 and Catalytic-9, are located at the flexible catalytic loop between the N-lobe and C-lobe, which may change the conformations of the cysteine. Thus, in using this position to design a covalent ligand, it is necessary to have the cysteine in a suitable conformational state.49
DFG peptide
Close to the DFG peptide, there are two cysteine-accessible locations at DFG-3 and DFG-4 (Figure 2a). For DFG-3, approximately 88% of the interactions are from side chain atoms. Moreover, the position is close to two polar residues, Asp from the DFG peptide and Lys, a catalytic residue from the roof50. This suggests the position can be used to design irreversible inhibitors. Within the structural kinome, there are several covalent kinase-ligand complexes such as pdb ids 4zzm, 4zzo, 5lcj and 5lck.51,52 For DFG-4, the majority of interactions come from the backbone oxygen atom and the side chains of all cysteines point outside the binding pocket (Figure 4h). It is less likely for the cysteine at this position will form a covalent bond with the ATP competitive inhibitor. However, the cysteine is available if, when designing a covalent inhibitor, the binding mode is that of a type-III inhibitor, which resides in the allosteric binding site. DFG-4 is at the location of the activation loop, which is flexible. It is necessary to model the orientation of the side chain of any cysteine to determine if it points towards the binding pocket.
In summary, we inspected the orientation of every cysteine located at the binding site using the metric of regional hydrophilicity, the side chain conformation, and the interaction details of the thiol for every cysteine. We found that the five regions, including P-loop, roof of pocket, front pocket, Catalytic-2 of catalytic loop and DFG-3 close to the DFG peptide, are the most accessible for covalent inhibitor design (Figure 7a).
Figure 7.

Reactive cysteines across the human kinome. (a) The reactive cysteines distributed at the five regions of binding sites marked in different colors. (b) The kinases with released 3D kinase structures. (c) The kinases without released kinase structures. (b) and (c) were generated using KinMap (http://kinhub.org/) and the high resolution figures are available in supporting information Figure S2–3.
Focusing on these five regions, we extracted all reactive cysteines in each region across the whole human structural kinome as shown in Figure 7b. 69 kinases were predicted to accommodate a covalent inhibitor (the complete list is found in Table S3 and Figure S2).Moreover, for all other kinases without PDB structures, we extracted the reactive cysteines based on a multiple sequence alignment (Figure 7c). Here 75 kinases were predicted to be accessible to a covalent inhibitor (complete data are listed in Table S4 and Figure S3). Thus, given any kinase, it is possible to determine if a covalent inhibitor can likely be designed based on these reactive cysteines. It is interesting that only Her3 (ErbB3)53, a well-known anticancer target,54 has a cysteine at the region of Roof across the whole human kinome (Figure 7b–c). Thus, it would be a potential strategy to utilize the roof cysteine to design the covalent inhibitor to achieve the desired selectivity for Her3.55,56 Another example is the TEC family kinase57, which comprises five members in mammals: BMX, BTK, ITK, TEC and RLK. All of them have a reactive cysteine located at the front pocket as shown in Figure 7a–c. Currently, there is more than one irreversible inhibitor for the members BMX, BTK and ITK58. This implies that the covalent inhibitors of BMX, BTK, and ITK can be repurposed to treat diseases associated with TEC and RLK 59.
Further, we found that the kinase MAP2K7, an essential component of MAP kinase signal pathway,60 has three reactive cysteines distributed within the P-loop, front pocket, and close to the DFG region. This provides new opportunities to design highly selective covalent inhibitors by taking advantage of the reactive cysteines at different positions. This provides a strategy to overcome the resistance of covalent inhibitors61 by selectively reacting with cysteines at different positions on the same target.
Evaluation of the cysteine accessibility rule
We collected available covalent kinase inhibitors (CKIs) as a CKI dataset (details shown in Table S5). The CKI dataset includes 124 CKIs that bind to 44 kinases, as visualized using the kinase profiling visualization tool, TREEspot (https://www.discoverx.com/) (Figure 8a). The lipid kinase family62 and every major group of protein kinase63 with the exception of CK1 have released CKIs. The number of CKIs varies from kinase to kinase. For example, EGFR has 36 CKIs. More importantly, the cysteines, which are used to form covalent interactions, are from different regions as shown in Figure 8b (the complete cysteine-site and compounds information is available in supporting information Table S5 for all CKIs ). We note that there are 119 CKIs covalently bound to cysteines located near the ATP binding site. Among them, 74 CKIs are at Front pocket, 31 CKIs at P-loop, 10 CKIs at DFG-3, 2 CKIs at Roof, 1 CKI at Catalytic-2 and 1 CKI at Hinge-1, respectively. Thus, a total of 118 CKIs are located at our five predicted favorable regions (Front pocket, P-loop, DFG-3, Roof and Catalytic-2). Additionally, one CKI (BLU9931) is located in the hinge region, which we predicted to be a very challenging design location. The binding mode obtained by Kohl et al46 at Hinge-146 is shown in Figure 9a, and proved selectivity for FGFR4 from FGFR1-3.
Figure 8.

(a) Distribution of the kinases with the released CKIs. (b) Distribution of cysteines of contributing to form covalent adducts.
Figure 9.
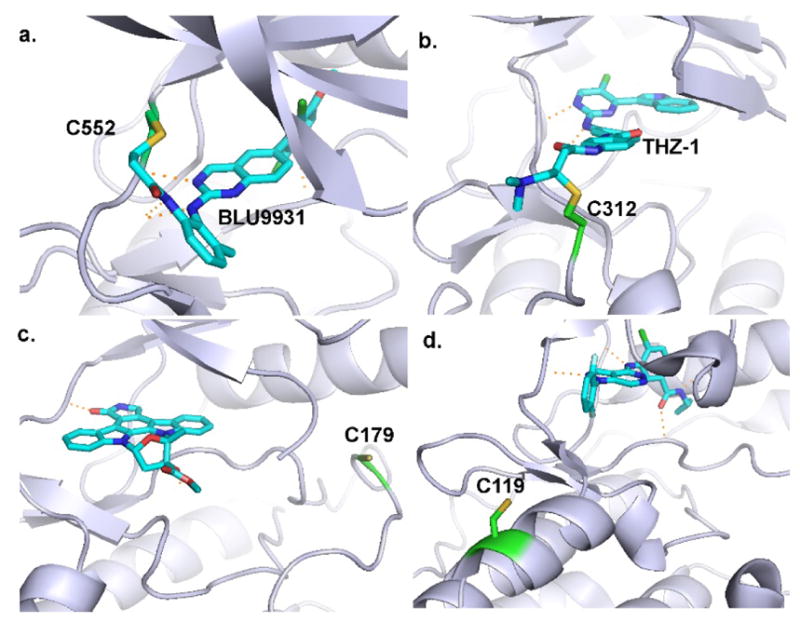
Cysteines located at different locations. (a) Cysteine at Hinge-1 with the covalent ligand (pdb id 4xcu). (b) Remote cysteine from the binding site (pdb id 1ua2). (c) Cysteine at the activation loop (pdb id 4e3c). (d) Cysteine at the extended front pocket (pdb id 3itz).
Five other CKIs are bound to cysteines located at Remote cysteine, Extended front pocket and Activation loop, which are a little far away from the ATP binding site (Figure 9b–d). Figure 9b shows that the two covalent inhibitors, THZ1 and Bio-THZ164, covalently bind Cys312 in CDK7, which is at the tail region and remote from the ATP binding site. Gray et al 64 found the unanticipated binding mode where the Cys312 traverses the ATP cleft to locate near the front pocket and binds with the acrylamide moiety of THZ1. Recently, Gray et al published another similar CKI (THZ531) which targets Cys1039 in CDK12 and Cys1017 in CDK13 with the same binding mode.47 The CKIs CDDO-Me65 and Nimbolide66 form covalent interactions with the cysteine located at the activation loop (Figure 9c), which was identified by a residue mutation experiment that showed the two CKIs could form adducts with the Cys179 of the kinase IKKβ65,66. Figure 9d shows the Cys119 forming a covalent interaction with one CKI (23225637-2) of p38α67,68. It is worth noting that the position of Cys119 is adjacent to the front pocket, named the extended front pocket67,68. Li et al used a D-recruitment site probe to explore the covalent interaction with Cys11967. It showed that the scaffold did not bind into the ATP binding site68. It would be interesting to develop new CKIs by designing an ATP competitive scaffold with a long tail electrophilic moiety that can form a covalent bond with Cys119. Finally, all of the cysteines bound to the five CKIs are located on the surface of the kinase domain and thus are important in understanding protein-protein interactions, protein-substrate interactions and signal pathways64,65.
Discussion and Conclusions
Despite recent advances in kinase-targeted covalent inhibitor discovery, most covalent inhibitors seem to have been found serendipitously. In this work, we systematically explore which cysteines can react covalently with the electrophilic group of the inhibitor ligand. We introduce a new approach to characterize the reactive cysteines across the human kinome. Our approach integrates the structural Fs-IFP method with quantum chemistry calculations thereby studying the interaction between cysteine and ligand at atomic detail. DFT calculations further quantify the environment and the preference for the cysteine to be involved in a covalent reaction.
With a detailed analysis of the cysteine-involved interactions at every location, we provided new insights into potential covalent-reaction positions. As shown in Figure 2a, the positions, including the P-loop, roof of the pocket, front pocket, Catalytic-2 of catalytic loop and DFG-3 close to the DFG peptide, have the potential to form covalent inhibitors. In contrast, within the Hinge region it is difficult for the cysteine to undertake a covalent reaction with the ligand, although there are many contacts between cysteine and ligand. These insights provide guidelines for the design of irreversible kinase inhibitors with the desired affinity20,22 or residence time20,22.
Our analysis benefits from the rapidly growing number of PDB structures. Besides the marked positions in Figure 2a, we also indexed all kinases with released PDB structures (Table S1), which includes 63 locations around the binding site( Figure S1), and can be used for further exploration. Table S1 contains the cysteine-involved positions, details of the interaction, the kinases involved, the UniProt entry and the corresponding PDB structures, respectively. As more PDB structures are added, we will update Table S1 periodically.
In summary, we analyzed the potential covalent-reaction sites for every cysteine in the binding pocket of the human kinome with the aim of determining which cysteines are available for developing irreversible kinase inhibitors. Associated with this work is a dataset of all protein kinase structures and their associated cysteines where each cysteine-ligand interaction is described. The overall aim is to aid in the design of covalent protein kinase inhibitors.
Experimental Section
Fs-IFP encoding
The Fs-IFP method is an efficient means of delineating the binding-site on a proteome scale, as detailed in our previous paper.28 In brief, the Fs-IFP method includes three steps. Step 1 is to prepare the kinase-ligand dataset. In this study, we download all kinase structures released through July, 2016 from the RCSB Protein Data Bank (http://www.rcsb.org/)27. Then the homology models, apo structures and the structures without kinase domains were excluded, resulting in 2774 kinase-ligand complex structures from 235 independent kinases and 2084 unique ligands. Step 2 is to align all binding sites using SMAP 2.1.69–71 For every complex structure, the residues at less than 6.5 Å72 from any heavy atom of the ligand were used to define the binding site; all other parameters were set to their defaults. This results in a matrix, where each row represents the amino acid residues that constitute the binding site for every kinase-ligand complex structure and every column represents the accessible amino acid residues located at the same spatial location. Step 3 is to encode the binding site-ligand interaction using Fs-IFP. Every binding site is described using 80 amino acids, and every amino acid is encoded using a 7-bit array that represents 7 types of interactions.73 Thus, every binding site of every kinase-ligand complex structure was encoded using a length of 560 bits (7 bits × 80 residues). In this paper, we performed the encode by using IChem software,74,75 which is a toolkit for detecting the protein-ligand interaction and which not only outputs the interaction types between binding site and ligand, but also the contribution of every atom to the interaction.
DFT calculation
Guided by the cysteine-involved reaction mechanism in Figure 1, the solvent effects were explored by calculating the multi-dimension potential energy surfaces (PESs). Due to multiple bonds forming and breaking in both schemes, a multi-dimensional reaction coordinate driving method76,77 was used to obtain a series of PESs with different dielectric constants. Starting from the optimized cysteine-electrophile-addition product, every energy point on a two-dimensional PESs was obtained by geometry optimization along the restrained reaction coordinates. Here the reaction coordinate was restrained to a designated value by using a harmonic restraining potential . The reaction coordinate RCj is defined as a combination of interatomic Euclidean distances involved with forming/breaking bonds in the j-th dimension, namely, RCj = Σ cidi, where di are the distances, with ci = 1 if the bond is to be broken, or ci = −1 if the bond is to be formed. The restraining force constant krestrained is 1000 kcal mol−1 Å−2. The j-th dimension of reference reaction coordinate value RCj(ref) starts from a starting conformation and is changed by 0.1 Å after one point has been optimized to optimize the next point. The actual reaction coordinate values after restrained optimization are always within 0.01 Å from the respective reference value. In Scheme 1, we used the first dimension to designate the nucleophilic attack on the thiol group of cysteine. Thus, the first dimension of the reaction coordinate (j = 1) was defined as RC1 = d1, and d1 described the distance between the sulfur atom (S) of the thiol group and the carbon atom (C2) of α,β unsaturated carbonyls. The RC1(ref) value means the designed distance between the sulfur atom and the carbon atom of α,β unsaturated carbonyls. The second dimension of the reaction coordinate is for the transfer of the proton and the reaction coordinate is RC2 = d3 − d2, where the interatomic distances d2[S-H] and d3[H-C1], mark the distance between the sulfur atom and the hydrogen atom, the distance between the hydrogen atom and the carbon atom of the αβ, unsaturated carbonyls, respectively. In Scheme 2, the proton is transferred from the NH3 molecule to the carbon atom (C2) of α,β unsaturated carbonyls as the first dimension reaction coordinate: RC1 = d4 − d5. The proton transfer from the thiol group to the NH3 molecule and the thiolate group attacking the carbon atom of the α,β unsaturated carbonyls as nucleophile are used as the second reaction coordinate: RC2 = d1 + d3 − d2 as shown in Scheme 2. In different polarizable continuum models (ε=0.0, ε=7.43 and ε=78.36 respectively), every energy point on the PESs was recalculated using a single-point energy calculation at the level of B3LYP/ 6-31G*/PCM78–81.
To indicate the steric hindrance effect for the addition reaction, the thiol group conformational space was explored by calculating the PES of the rotation of dihedral χ1. The optimized product conformation P as starting point is shown in Scheme 1. The dihedral χ1 is chosen and every 10° one conformation is calculated with the restrained distance d1. The interatomic distance d1 is restrained82 from 2.00 to 1.83 Å at 0.01 Å intervals to show the process of the nucleophile attacking of the thiol group by the electrophilic group and following the d1, the energy change was obtained.
The computational model, which retained all the important elements of the Michael addition reaction in the binding site, was built. The electrophilic group was the representative acryloyl group, which is also part of the three FDA-approved covalent drugs14,15,17. The protein part and the reversible scaffold part were also modeled as shown in Figure 1. Using the micro solvation approach83, a base molecule NH3 is added to provide a weak-base-buffer experiment ensemble for Scheme 2. All DFT calculations were performed by Gaussian0984 on the NIH high-performance Biowulf cluster (https://hpc.nih.gov/). All structures were optimized at the B3LYP/6-31G* level78–81 with the Gaussian09 default convergence threshold84.
Sequence alignment
All 532 human kinase-domain sequences were downloaded from kinase.com, and online Clustal Omega (v1.2.4) (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used for multiple sequence alignment. The default parameters were set for the alignment. For every region of accessible cysteines, the sequence slices of the corresponding kinases with the involved cysteines were extracted (Table S6).
CKI dataset
We collected all potential CKIs from two databases with the quantitative cysteine-inhibitor interaction information. One database is the IUPHAR/BPS Guide to Pharmacology database (version 2017.1; http://www.guidetopharmacology.org), which provides expert-curated quantitative intermolecular interactions between the successful and potential drugs and their targets across the human genome85. First, we extracted all human kinase drugs and potential drugs from every kinase target represented by UniProtKB ID (http://www.uniprot.org/docs/pkinfam). Then the gene name (i.e. geneID) of every kinase was obtained by UniProtKB ID from UniProt database (http://www.uniprot.org),86 641 kinase-targeted molecules were directly downloaded from PubChem (http://pubchem.ncbi.nlm.nih.gov) 87 using the keywords “geneID and section=curated ligands”. Finally the 27 CKIs were obtained by screening the quantitative texts of 641 molecules in IUPHAR/BPS Guide to Pharmacology database and meanwhile the cysteine site information was also confirmed manually. The second database, Cyteinome (version 2016; http://www.cysteinome.org/)88, collects proteins with targetable cysteine and their covalent inhibitors from public scientific literatures and database resources27,86,87,89. Similar to the former procedure, we also use UniProtKB ID86 as the reference to download every kinase web page. Then we extracted 122 covalent modulators from the web pages. Molecule probes and inhibitors were eliminated if they were bound to the non-kinase domain. Finally, 106 active covalent inhibitors were collected from the Cyteinome database. 9 CKIs were common to the two databases. In total there were 124 covalent small-molecule inhibitors in the CKI dataset.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Library of Medicine, National Institutes of Health(Z.Z., S.B and P.E.B.), the National Library of Medicine, National Institutes of Health under Award R01LM011986 (L.X.), and the National Institute on Minority Health and Health Disparities, National Institutes of Health, under Award G12MD007599 (L.X.).
Abbreviations Used
- IFP
interaction fingerprint
- Fs-IFP
functional site interaction fingerprint
- DFT
density functional theory
- DFG
Asp-Phe-Gly peptide
- CKI
covalent kinase inhibitor
- BTK
Bruton’s tyrosine kinase
- ITK
interleukin-2 (IL-2)-inducible T-cell kinase
- BMX
bone-marrow tyrosine kinase gene on chromosome X
- RLK
resting lymphocyte kinase; also called TXK
Footnotes
Ancillary Information
Table S1, Structure-based cysteine dataset across the human structural kinome (xlsx). Table S2–6 and Figure S1–3, Additional list of PDB ids with accessible cysteines for Figure 2b and additional list of kinases with reactive cysteines following Figure 7b–c; Covalent kinase inhibitor dataset; The sequence alignment result for the cysteine available regions; Accessible cysteines at the different locations presented on a reference binding site; The high resolution figures for Figure 7b–c.( PDF)
References
- 1.Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat Rev Genet. 2010;11:60–74. doi: 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller S, Chaikuad A, Gray NS, Knapp S. The ins and outs of selective kinase inhibitor development. Nat Chem Biol. 2015;11:818–821. doi: 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- 4.Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat Rev Clin Oncol. 2016;13:209–227. doi: 10.1038/nrclinonc.2015.213. [DOI] [PubMed] [Google Scholar]
- 5.Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev. 2001;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:8–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barouch-Bentov R, Sauer K. Mechanisms of drug resistance in kinases. Expert Opin Investig Drugs. 2011;20:153–208. doi: 10.1517/13543784.2011.546344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rask-Andersen M, Zhang J, Fabbro D, Schioth HB. Advances in kinase targeting: Current clinical use and clinical trials. Trends Pharmacol Sci. 2014;35:604–620. doi: 10.1016/j.tips.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 11.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert AM. Recent advances in irreversible kinase inhibitors. Pharm Pat Anal. 2014;3:375–386. doi: 10.4155/ppa.14.24. [DOI] [PubMed] [Google Scholar]
- 13.Barf T, Kaptein A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J Med Chem. 2012;55:6243–6262. doi: 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]
- 14.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. Bibw2992, an irreversible egfr/her2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The bruton tyrosine kinase inhibitor pci-32765 blocks b-cell activation and is efficacious in models of autoimmune disease and b-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong KK, Janne PA, Eck MJ. Overcoming egfr(t790m) and egfr(c797s) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–132. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. Azd9291, an irreversible egfr tki, overcomes t790m-mediated resistance to egfr inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Discovery of selective irreversible inhibitors for bruton's tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, Kath JC, Xu M, Wani R, Murray BW. Covalent egfr inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Natl Acad Sci U S A. 2014;111:173–178. doi: 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol. 2015;11:525–531. doi: 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, Zhu SJ, Zhang Y, Ercan D, Liao RG, Capelletti M, Zhou W, Hur W, Kim N, Sim T, Gaudet S, Barbie DA, Yeh JR, Yun CH, Hammerman PS, Mohammadi M, Janne PA, Gray NS. Development of covalent inhibitors that can overcome resistance to first-generation fgfr kinase inhibitors. Proc Natl Acad Sci U S A. 2014;111:E4869–4877. doi: 10.1073/pnas.1403438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little RD, Masjedizadeh MR, Wallquist O, McLoughlin JI. The intramolecular michael reaction. Org React. 2004;47:315–552. [Google Scholar]
- 24.Zhao Z, Martin C, Fan R, Bourne PE, Xie L. Drug repurposing to target ebola virus replication and virulence using structural systems pharmacology. BMC Bioinf. 2016;17:90. doi: 10.1186/s12859-016-0941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boran ADW, Iyengar R. Systems approaches to polypharmacology and drug discovery. Curr Opin Drug Discovery Dev. 2010;13:297–309. [PMC free article] [PubMed] [Google Scholar]
- 26.Vidovic D, Muskal SM, Schurer SC. Novel kinase inhibitors by reshuffling ligand functionalities across the human kinome. J Chem Inf Model. 2012;52:3107–3115. doi: 10.1021/ci3003842. [DOI] [PubMed] [Google Scholar]
- 27.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Z, Xie L, Xie L, Bourne PE. Delineation of polypharmacology across the human structural kinome using a functional site interaction fingerprint approach. J Med Chem. 2016;59:4326–4341. doi: 10.1021/acs.jmedchem.5b02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, Xie L, Bourne PE. Insights into the binding mode of mek type-iii inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. bioRxiv. 2016 doi: 10.1371/journal.pone.0179936. http://dx.doi.org/10.1101/076711. [DOI] [PMC free article] [PubMed]
- 30.Ranu BC, Banerjee S. Ionic liquid as catalyst and reaction medium. The dramatic influence of a task-specific ionic liquid, [bmim]oh, in michael addition of active methylene compounds to conjugated ketones, carboxylic esters, and nitriles. Org Lett. 2005;7:3049–3052. doi: 10.1021/ol051004h. [DOI] [PubMed] [Google Scholar]
- 31.Chalker JM, Bernardes GJ, Lin YA, Davis BG. Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem Asian J. 2009;4:630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- 32.Schwobel JA, Madden JC, Cronin MT. Examination of michael addition reactivity towards glutathione by transition-state calculations. SAR QSAR Environ Res. 2010;21:693–710. doi: 10.1080/1062936X.2010.528943. [DOI] [PubMed] [Google Scholar]
- 33.Mulliner D, Wondrousch D, Schuurmann G. Predicting michael-acceptor reactivity and toxicity through quantum chemical transition-state calculations. Org Biomol Chem. 2011;9:8400–8412. doi: 10.1039/c1ob06065a. [DOI] [PubMed] [Google Scholar]
- 34.Schwobel JA, Wondrousch D, Koleva YK, Madden JC, Cronin MT, Schuurmann G. Prediction of michael-type acceptor reactivity toward glutathione. Chem Res Toxicol. 2010;23:1576–1585. doi: 10.1021/tx100172x. [DOI] [PubMed] [Google Scholar]
- 35.Ebbrell DJ, Madden JC, Cronin MT, Schultz TW, Enoch SJ. Development of a fragment-based in silico profiler for michael addition thiol reactivity. Chem Res Toxicol. 2016;29:1073–1081. doi: 10.1021/acs.chemrestox.6b00099. [DOI] [PubMed] [Google Scholar]
- 36.Liao JJ. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem. 2007;50:409–424. doi: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- 37.van Linden OP, Kooistra AJ, Leurs R, de Esch IJ, de Graaf C. Klifs: A knowledge-based structural database to navigate kinase-ligand interaction space. J Med Chem. 2014;57:249–277. doi: 10.1021/jm400378w. [DOI] [PubMed] [Google Scholar]
- 38.Patel RY, Doerksen RJ. Protein kinase-inhibitor database: Structural variability of and inhibitor interactions with the protein kinase p-loop. J Proteome Res. 2010;9:4433–4442. doi: 10.1021/pr100662s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J Am Chem Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor SS, Kornev AP. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brooijmans N, Chang YW, Mobilio D, Denny RA, Humblet C. An enriched structural kinase database to enable kinome-wide structure-based analyses and drug discovery. Protein Sci. 2010;19:763–774. doi: 10.1002/pro.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kooistra AJ, Kanev GK, van Linden OP, Leurs R, de Esch IJ, de Graaf C. Klifs: A structural kinase-ligand interaction database. Nucleic Acids Res. 2016;44:D365–371. doi: 10.1093/nar/gkv1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern HA, Feller SE. Calculation of the dielectric permittivity profile for a nonuniform system: Application to a lipid bilayer simulation. J Chem Phys. 2003;118:3401–3412. [Google Scholar]
- 44.Merrill GN, Fletcher GD. A microsolvation approach to the prediction of the relative enthalpies and free energies of hydration for ammonium ions. Theor Chem Acc. 2007;120:5–22. [Google Scholar]
- 45.Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN. The thiol-michael addition click reaction: A powerful and widely used tool in materials chemistry. Chem Mater. 2014;26:724–744. [Google Scholar]
- 46.Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, Bifulco N, Kim JL, Hodous B, Brooijmans N, Shutes A, Winter C, Lengauer C, Kohl NE, Guzi T. First selective small molecule inhibitor of fgfr4 for the treatment of hepatocellular carcinomas with an activated fgfr4 signaling pathway. Cancer Discov. 2015;5:424–437. doi: 10.1158/2159-8290.CD-14-1029. [DOI] [PubMed] [Google Scholar]
- 47.Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, Manz T, Hao M, Bartkowiak B, Greenleaf AL, Marto JA, Geyer M, Bullock AN, Young RA, Gray NS. Covalent targeting of remote cysteine residues to develop cdk12 and cdk13 inhibitors. Nat Chem Biol. 2016;12:876–884. doi: 10.1038/nchembio.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and sti-571 inhibition of c-kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 49.Rastelli G, Rosenfeld R, Reid R, Santi DV. Molecular modeling and crystal structure of erk2-hypothemycin complexes. J Struct Biol. 2008;164:18–23. doi: 10.1016/j.jsb.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Carrera AC, Alexandrov K, Roberts TM. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring atp. Proc Natl Acad Sci U S A. 1993;90:442–446. doi: 10.1073/pnas.90.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward RA, Colclough N, Challinor M, Debreczeni JE, Eckersley K, Fairley G, Feron L, Flemington V, Graham MA, Greenwood R, Hopcroft P, Howard TD, James M, Jones CD, Jones CR, Renshaw J, Roberts K, Snow L, Tonge M, Yeung K. Structure-guided design of highly selective and potent covalent inhibitors of erk1/2. J Med Chem. 2015;58:4790–4801. doi: 10.1021/acs.jmedchem.5b00466. [DOI] [PubMed] [Google Scholar]
- 52.Lebraud H, Wright DJ, East CE, Holding FP, O'Reilly M, Heightman TD. In-gel activity-based protein profiling of a clickable covalent erk1/2 inhibitor. Mol Biosyst. 2016;12:2867–2874. doi: 10.1039/c6mb00367b. [DOI] [PubMed] [Google Scholar]
- 53.Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. Erbb3/her3 intracellular domain is competent to bind atp and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. Her3 overexpression and survival in solid tumors: A meta-analysis. J Natl Cancer Inst. 2013;105:266–273. doi: 10.1093/jnci/djs501. [DOI] [PubMed] [Google Scholar]
- 55.Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, Udayakumar D, Gurbani D, Tae HS, Riddle SM, Sim T, Marto JA, Janne PA, Crews CM, Gray NS. Pharmacological targeting of the pseudokinase her3. Nat Chem Biol. 2014;10:1006–1012. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim SM, Xie T, Westover KD, Ficarro SB, Tae HS, Gurbani D, Sim T, Marto JA, Janne PA, Crews CM, Gray NS. Development of small molecules targeting the pseudokinase her3. Bioorg Med Chem Lett. 2015;25:3382–3389. doi: 10.1016/j.bmcl.2015.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwartzberg PL, Finkelstein LD, Readinger JA. Tec-family kinases: Regulators of t-helper-cell differentiation. Nat Rev Immunol. 2005;5:284–295. doi: 10.1038/nri1591. [DOI] [PubMed] [Google Scholar]
- 58.Liu F, Zhang X, Weisberg E, Chen S, Hur W, Wu H, Zhao Z, Wang W, Mao M, Cai C, Simon NI, Sanda T, Wang J, Look AT, Griffin JD, Balk SP, Liu Q, Gray NS. Discovery of a selective irreversible bmx inhibitor for prostate cancer. ACS Chem Biol. 2013;8:1423–1428. doi: 10.1021/cb4000629. [DOI] [PubMed] [Google Scholar]
- 59.Gomez-Rodriguez J, Kraus ZJ, Schwartzberg PL. Tec family kinases itk and rlk / txk in t lymphocytes: Cross-regulation of cytokine production and t-cell fates. FEBS J. 2011;278:1980–1989. doi: 10.1111/j.1742-4658.2011.08072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foltz IN, Gerl RE, Wieler JS, Luckach M, Salmon RA, Schrader JW. Human mitogen-activated protein kinase kinase 7 (mkk7) is a highly conserved c-jun n-terminal kinase/stress-activated protein kinase (jnk/sapk) activated by environmental stresses and physiological stimuli. J Biol Chem. 1998;273:9344–9351. doi: 10.1074/jbc.273.15.9344. [DOI] [PubMed] [Google Scholar]
- 61.Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation egfr tyrosine kinase inhibitors. Front Med. 2016;10:383–388. doi: 10.1007/s11684-016-0488-1. [DOI] [PubMed] [Google Scholar]
- 62.Nacht M, Qiao L, Sheets MP, St Martin T, Labenski M, Mazdiyasni H, Karp R, Zhu Z, Chaturvedi P, Bhavsar D, Niu D, Westlin W, Petter RC, Medikonda AP, Singh J. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase pi3kalpha. J Med Chem. 2013;56:712–721. doi: 10.1021/jm3008745. [DOI] [PubMed] [Google Scholar]
- 63.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 64.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS. Targeting transcription regulation in cancer with a covalent cdk7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid cddo-me blocks the nf-kappab pathway by direct inhibition of ikkbeta on cys-179. J Biol Chem. 2006;281:35764–35769. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 66.Gupta SC, Prasad S, Reuter S, Kannappan R, Yadav VR, Ravindran J, Hema PS, Chaturvedi MM, Nair M, Aggarwal BB. Modification of cysteine 179 of ikappabalpha kinase by nimbolide leads to down-regulation of nf-kappab-regulated cell survival and proliferative proteins and sensitization of tumor cells to chemotherapeutic agents. J Biol Chem. 2010;285:35406–35417. doi: 10.1074/jbc.M110.161984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J, Kaoud TS, LeVieux J, Gilbreath B, Moharana S, Dalby KN, Kerwin SM. A fluorescence-based assay for p38alpha recruitment site binders: Identification of rooperol as a novel p38alpha kinase inhibitor. Chembiochem. 2013;14:66–71. doi: 10.1002/cbic.201200529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J, Kaoud TS, Laroche C, Dalby KN, Kerwin SM. Synthesis and biological evaluation of p38alpha kinase-targeting dialkynylimidazoles. Bioorg Med Chem Lett. 2009;19:6293–6297. doi: 10.1016/j.bmcl.2009.09.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie L, Xie L, Bourne PE. A unified statistical model to support local sequence order independent similarity searching for ligand-binding sites and its application to genome-based drug discovery. Bioinformatics. 2009;25:i305–312. doi: 10.1093/bioinformatics/btp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci U S A. 2008;105:5441–5446. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie L, Bourne PE. A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites. BMC Bioinf. 2007;8(Suppl 4):S9. doi: 10.1186/1471-2105-8-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Desaphy J, Bret G, Rognan D, Kellenberger E. Sc-pdb: A 3d-database of ligandable binding sites--10 years on. Nucleic Acids Res. 2015;43:D399–404. doi: 10.1093/nar/gku928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 74.Desaphy J, Raimbaud E, Ducrot P, Rognan D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J Chem Inf Model. 2013;53:623–637. doi: 10.1021/ci300566n. [DOI] [PubMed] [Google Scholar]
- 75.Da Silva F, Desaphy J, Bret G, Rognan D. Ichempic: A random forest classifier of biological and crystallographic protein-protein interfaces. J Chem Inf Model. 2015;55:2005–2014. doi: 10.1021/acs.jcim.5b00190. [DOI] [PubMed] [Google Scholar]
- 76.Zhang YK, Liu HY, Yang WT. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio qm/mm potential energy surface. J Chem Phys. 2000;112:3483–3492. [Google Scholar]
- 77.Zhang Y, Zhao Z, Liu H. Deriving chemically essential interactions based on active site alignments and quantum chemical calculations: A case study on glycoside hydrolases. ACS Catal. 2015;5:2559–2572. [Google Scholar]
- 78.Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can J Phys. 1980;58:1200–1211. [Google Scholar]
- 79.Becke AD. Density-functional thermochemistry. Iii. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 80.Lee C, Yang W, Parr RG. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 81.Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 82.Pulay P, Fogarasi G. Geometry optimization in redundant internal coordinates. J Chem Phys. 1992;96:2856–2860. [Google Scholar]
- 83.Parchment OG, Vincent MA, Hillier IH. Speciation in aqueous zinc chloride. Anab initiohybrid microsolvation/continuum approach. J Chem Phys. 1996;100:9689–9693. [Google Scholar]
- 84.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision E01. Gaussian, Inc; Wallingford, CT, USA: 2009. [Google Scholar]
- (:5) Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Spedding M, Catterall WA, Fabbro D, Davies JA, Nc I. The iuphar/bps guide to pharmacology in 2016: Towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 2016;44:D1054–1068. doi: 10.1093/nar/gkv1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.The UniProt C. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, Bryant SH. Pubchem substance and compound databases. Nucleic Acids Res. 2016;44:D1202–1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu S, Luo Howard H, Wang H, Zhao W, Hu Q, Yang Y. Cysteinome: The first comprehensive database for proteins with targetable cysteine and their covalent inhibitors. Biochem Biophys Res Commun. 2016;478:1268–1273. doi: 10.1016/j.bbrc.2016.08.109. [DOI] [PubMed] [Google Scholar]
- 89.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. Kegg for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.