

Abstract
Increasing concentration of counterions (salt) is known to reduce the bending persistence length of DNA. Here we use atomistic molecular dynamics simulations to predict that multivalent counterions have the opposite effect on double-stranded RNA, increasing its bending rigidity by at least 30%. This counter-intuitive effect is observed for various tri- and tetravalent ions alike, and is robust to methodological details and RNA sequence. In contrast to DNA, multivalent counterions bind inside the RNA major groove, causing significant contraction of the molecule along its helical axis — as a result, its further deformation due to bending becomes energetically more expensive compared to bending without bound multivalent ions. Thus, the relationship between mechanical properties of a charged polymer and its ionic atmosphere may be richer than previously thought.
Mechanical properties of nucleic acids (NA) are known to be strongly affected by ionic conditions of the solutions they are in [1–7]. Experiments show that increasing the concentration of monovalent salt reduces DNA bending persistence length [1, 3] from its classical value of ~ 500 Å at physiological NaCl concentration of ~ 0.145 M. An even more dramatic decrease of DNA persistence length with salt concentration has been observed when monovalent cations were replaced by multivalent ones [1]. The common explanation is that counterion atmosphere around a NA molecule screens the repulsion between the negatively charged phosphates along the polymer, thus increasing its bending flexibility [8–12].
In contrast to DNA, mechanical properties of double-stranded RNA (referred to simply as RNA for the remainder of the text) have not been investigated as extensively. From the similar charge density and overall structure of DNA and RNA double helices, one might expect that they should exhibit similar elastic responses under applied forces. Indeed, recent single-molecule force and torque measurements of RNA bending flexibilities [13–15] have shown analogous to DNA decrease of bending persistence length of RNA with increasing monovalent salt concentration [14].
Here we compare the effects of multivalent counterions on RNA bending flexibility using atomistic molecular dynamics simulations; the study was motivated by a recent finding that binding patterns of multivalent counterions to DNA and RNA are drastically different [16–18]. We investigate how this difference in multivalent ion binding affects RNA bending persistence length when monovalent counterions are replaced by ions with charges +3e and +4e, and propose an explanation for the opposing effects on bending flexibilities of DNA and RNA.
DNA and RNA duplexes of 25 base pairs (bp) with the same mixed sequence described in [16] and a homopolymeric poly(rA)·poly(rU) RNA fragment of the same length were generated in canonical B-form for DNA and A-form for RNA using Nucleic Acid Builder [19]. The NA molecules were then neutralized with three different combinations of monovalent Na+ and trivalent Cobalt(III) Hexammine (Co-Hex): (1) 48 Na+ (“no CoHex”); (2) 8 CoHex3+ and 24 Na+ (0.17 CoHex3+ per phosphate, indicated by P−); and (3) 16 CoHex3+ (0.33 CoHex3+/P−), the latter corresponding to estimated bulk concentrations (measured 32 Å from the helical axis of the duplexes) of 4 mM for RNA and 6 mM for DNA. In addition, mixed sequence RNA molecules were also neutralized with two other counterion types: 16 hypothetical “Na3+” ions and 12 Spermine4+ ions. “Na3+” was simulated by increasing threefold the Na+ charge in Amber topology file. Each system was then solvated with ~16800 TIP3P water molecules in a periodic box. To account for monovalent salt background roughly equivalent to physiological conditions, 24 Na+ Cl− ion pairs were added to all systems (low Na+ concentration regimes were not explored for technical reasons). All MD simulations were carried out using AMBER 12 [20] and ff99bsc0 force field [21, 22] at T = 300 K. Each system was first simulated for at least 100 ns while holding the duplex restrained to allow the ionic atmosphere to equilibrate around the molecule. Then, positional restraints were removed for production MD runs: 300 ns for CoHex3+ and “no CoHex” systems, and 400 ns for Spermine4+ and “Na3+” systems.
NA helical axis representations for each frame were generated from MD trajectories using Curves+ [23]. We then calculate, for each MD snapshot, the bending angle θ between average vectors of consecutive helical repeats (10 bp in our case), which should reduce the possibility of error from the non-symmetric nature of the double-helix[24]. The result of the calculation is the angular probability distribution P(θ, l), where l is the average contour length of the 10 bp helical repeat (calculated for each NA/ion system individually by summing all base-pair rise parameters). For l ≪ Lp we can use the inextensible worm-like chain [25–28] approximation for the bending energy:
| (1) |
For spherically isotropic distribution in 3D space[27]
| (2) |
which leads to
| (3) |
used to estimate Lp, Fig. 1.
FIG. 1.
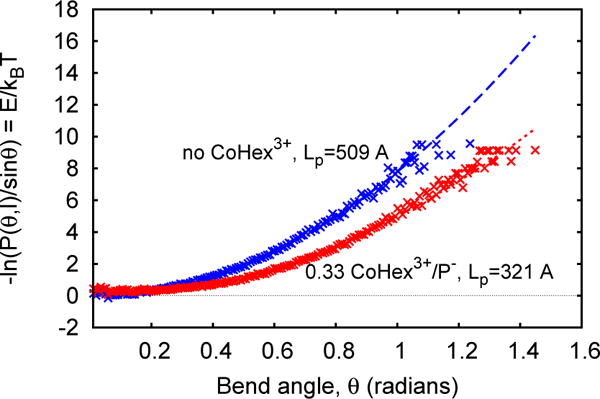
Estimation of bending persistence length Lp of DNA duplex with (red) and without (blue) CoHex3+ counterions. Lp is estimated by fitting (dashed lines) the data points to Eq. 3; each point represents a value of averaged over ~0.008 interval of θ.
For all calculated values of Lp in this work the relative statistical error never exceeded 2%, and is not reported below. For mixed sequence DNA the addition of neutralizing amount of CoHex yields the predicted relative change ΔLp/Lp = −37% (Table I, bottom row) which is within the reported error margin of the corresponding experimental estimate[1] of −44 ± 8%. Thus, despite unavoidable methodological limitations, including known imperfections of modern force-fields [29], our calculations predict the main quantity of interest — the relative change of persistence length ΔLp/Lp upon addition of multivalent ions — in acceptable agreement with experiment. Furthermore, recent WAXS [30] and CD spectra [16] measurements have not detected any significant changes in DNA structure upon addition of CoHex, consistent with our results that the molecule contracts by no more than 2%, Table I. Previous all-atom [31] and even coarse-grained models derived from the same atomistic potentials [32, 33] also reproduce correct quantitative dependence of DNA Lp on ionic strength.
TABLE I.
Effect of CoHex3+ counterions on DNA flexibility. Shown are relative changes in bending persistence length (Lp) and length of helical repeat (10 bp) segment (l), relative to the same system with no CoHex3+ ions (multivalent ions replaced by Na+ to maintain over-all neutrality).
| DNA w/CoHex3+ | ΔLP/LP (%) | Δl/l (%) |
|---|---|---|
| 0.17 CoHex3+/P− | −31 | −1.8 |
| 0.33 CoHex3+/P− | −37 | −1.8 |
Unexpectedly, the effect of CoHex on RNA is the opposite of its effect on the DNA: the addition of CoHex3+ results in a significantly higher Lp of the RNA, Table II. The counter-intuitive increase in RNA bending rigidity caused by CoHex does not appear to be sequence-dependent: the same strong effect of similar magnitude is seen when we replace the mixed sequence RNA with a homopolymeric poly(rA) ·poly(rU) fragment (first row of Table III). This effect on RNA persistence length is not limited to CoHex3+ counterions: tetravalent Spermine4+ (a linear polyamine ion) and hypothetical trivalent “Na3+” also stiffen the RNA, Table III.
TABLE II.
Effect of CoHex3+ on RNA flexibility. Persistence length (Lp) of RNA increases with CoHex3+ concentration — an effect that is opposite to the DNA response to CoHex3+. Bound CoHex3+ also significantly shrinks RNA duplex along its helical axis.
| RNA w/CoHex3+ | ΔLP/LP (%) | Δl/l (%) |
|---|---|---|
| 0.17 CoHex3+/P− | +68 | −12 |
| 0.33 CoHex3+/P− | +90 | −14 |
TABLE III.
CoHex3+ increases persistence length (Lp) of homopolymeric poly(rA) ·poly(rU) RNA molecules. Other tri- and tetravalent ions have the same qualitative effect: Spermine4+ and hypothetical “Na3+” increase Lp and decrease the fragment length (l) of RNA duplexes.
| RNA w/+3 and +4-valent ions | ΔLP/LP (%) | Δl/l (%) |
|---|---|---|
| 0.33 CoHex3+/p− (poly(rA) ·poly(rU)) | +56 | −14 |
| 0.33 “Na3+”/P− (mixed RNA) | +85 | −16 |
| 0.25 Spermine4+/P− (mixed RNA) | +29 | −14 |
We propose the following qualitative explanation for this novel effect. The persistence length of a charged polymer in the presence of counterions can be conceptually decomposed into two distinct contributions: Lp ≃ Lint + ΔLscr [34]. The “base” contribution Lint depends on the internal structure (short-range interactions) and charge distribution of the polymer, while ΔLscr represents a correction to Lint due to the screening by the counterions of the long-range charge-charge interactions along the polymer. Both of these contributions can significantly affect the total persistence length Lp [35]. In the case of NA, ΔLscr is always negative because the screening results in a reduced effective electrostatic repulsion between the negatively charged phosphate groups. In general, DNA and RNA double helices have very similar overall structure and electrostatic properties, including linear charge density and other parameters relevant to Lp according to existing models [34, 36]. Thus, given the same amount of reduction in the charge-charge repulsion along the NA polymer due to partial neutralization by the counterions, the ΔLscr term is expected to be roughly equal for DNA and RNA surrounded by the same type and amount of counterions. Indeed, increasing the concentration of monovalent ions (which are known to form loose distributions around NA molecules [37–39]) leads to similar changes in observed Lp of both DNA and RNA. However, the situation is very different with multivalent counterions. For DNA, these ions mostly bind to the externally exposed surface of the phosphate groups [16], Fig. 2 (left panel), leading to their partial neutralization, which mainly affects ΔLscr in the expected way. In contrast, CoHex binds preferentially inside the RNA molecule, closer to the helical axis [16, 17], Fig. 2 (right panel). This striking difference in the binding pattern can be rationionalized as follows. Structural differences between DNA and RNA helices (B- vs. A-form) lead to a much more negative electrostatic potential in the major groove of RNA[16, 40], causing the preferential Co-Hex binding. While qualitative, this picture is consistent with experimental differences in the CoHex binding constants: the ion binds much stronger to the RNA (binding constant ~ 104 M−1)[41] compared to the DNA (~ 102 M−1)[42] at near physiological NaCl concentrations. This specific binding preference of trivalent CoHex is expected to be robust: dsRNA is always in A-form, and the fraction of bound ions is insensitive to water model and force-field choice; simulations with TIP4P water and the latest chiOL3 modifications for RNA to ff99bsc0 force-field[43] result in a negligible 3% difference in the number of CoHex ions bound to the RNA. The internally and strongly bound CoHex ions can be considered as part of the internal structure of RNA itself, mainly affecting Lint. As sufficient number of CoHex counterions bind into the major grove of RNA, they pull the oppositely charged phosphate groups closer together. The strong pull results in the contraction of the duplex along its helical axis (Tables II, III). Changes in experimental WAXS profile of dsRNA (which correlate well with simulated profiles from MD) [30], specifically the “sharpening” of the features observed in the WAXS regime, are consistent with better defined structures when Co-Hex is present. This suggests a stiffer (less flexible) RNA duplex. In contrast, virtually no structural change occurs in DNA upon binding of the same multivalent counterions (see Table I). Note that even though “Na3+” is sterically about 3 times smaller than CoHex3+, the relative contraction of the RNA double helix caused by “Na3+” remains almost the same, Tables II and III, suggesting that any further contraction of the RNA double helix becomes highly unfavorable energetically. The strong electrostatic pull between phosphates and buried counterions is balanced by the short-range interactions that maintain the internal structure of the double helix. Bending of a polymer implies some combination of contraction and stretching (of the opposite side of the chain), and both of these deformations require more energy in the already contracted state of the RNA compared to the original “realaxed” conformation, which ultimately results in higher bending modulus of the polymer. Thus, the binding of multivalent counterions to the RNA helix increases Lint significantly, resulting in a net increase of its Lp, while in the DNA the ions mostly reduce Lp via negative ΔLscr.
FIG. 2.
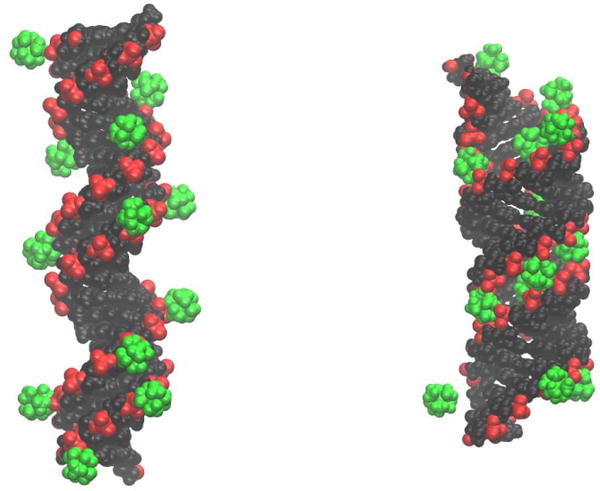
In DNA (left), multivalent counterions (illustrated for CoHex3+, green) bind mostly externally [16] onto the negatively charged phosphates (red), with little effect on the DNA structure. The binding reduces the effective electrostatic repulsion along the helix, which in turn decreases the helix bending rigidity. In contrast, the same ions bind deep inside the RNA major groove (right) [16] causing the double helix to contract and significantly stiffen its internal structure. This leads to an over-all increase of the RNA bending rigidity — the pull of the ions works as taut bicycle spokes that tighten the wheel. The distributions (not shown) of bound Spermine4+ around DNA and RNA are similar to the above.
Our main result — that multivalent counterions can significantly increase RNA bending persistence length — is unexpected from the perspective of the completely opposite and well-known effect of increasing salt concentration on RNA’s closest cousin, the DNA. The physics behind the salt effect on bending rigidity of long charged polymers appeared well understood: the charge-charge repulsion along the polymer is screened out by the counterions, making it easier to bend. But apparently, this is only one side of the story: small differences in structure between RNA and DNA can make dominant a previously unexplored consequence of counterion binding, changing the sign of the over-all salt effect on the polymer’s persistence length. While the magnitude of the predicted effect might have some dependence on details of the methodology, it is clear that the effect is strong, persists over a range of ion concentrations, and is robust to ion type and sequence details, all of which should facilitate direct experimental verification. We believe that single-molecule measurements such as magnetic or optical tweezers experiments[1, 14] are best suited for studying this phenomenon. AFM studies[26] are another possible way to observe this effect, provided the interactions of the surface with the NA duplexes have minimal influence on the binding of ions to the molecule. The new effect may manifest itself in other scenarios: mechanical properties of DNA with sequences and/or structure that have relatively stronger affinity for multivalent ions compared to canonical B-form DNA considered here may also show unexpected, or even counter-intuitive response. For example, it is known that under low hydration[44, 45], as well as with increasing concentrations of CoHex[46, 47], DNA can spontaneously transition from B- to A-form, which in turn may alter mechanical properties of the double helix, either directly (A-form is expected to be stiffer) or via the mechanism proposed here for the RNA. In reality, the net change in the siffness maybe a combination of both effects; dehydration may become particularly important in living cells. Given the importance of nucleic acid stiffness in genome packing, these effects may have significant biological consequences. The influence of divalent ions such as Mg2+, known to bind to nucleic acids, is also worth exploring. Perhaps most importantly, the physics of the relationship between mechanical properties of charged polymers and counterion binding is worth revisiting.
Acknowledgments
We thank Dr. Suzette Pabit for helpful discussions. This work was supported by the National Institutes of Health (NIH) R01 GM099450.
References
- 1.Baumann CG, Smith SB, Bloomfield VA, Bustamante C. Proceedings of the National Academy of Sciences. 1997;94:6185. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning GS. Quarterly reviews of biophysics. 1978;11:179. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 3.Wenner JR, Williams MC, Rouzina I, Bloomfield VA. Biophysical journal. 2002;82:3160. doi: 10.1016/S0006-3495(02)75658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan ZJ, Chen SJ. Biophysical Journal. 2006;90:1175. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazur AK. Physical Review E. 2011;84:021903. doi: 10.1103/PhysRevE.84.021903. [DOI] [PubMed] [Google Scholar]
- 6.Luan B, Aksimentiev A. Physical review letters. 2008;101:118101. doi: 10.1103/PhysRevLett.101.118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao L, Zhang X, Jin L, Tan ZJ. Chinese Physics B. 2016;25:18703. [Google Scholar]
- 8.Odijk T. Journal of Polymer Science: Polymer Physics Edition. 1977;15:477. [Google Scholar]
- 9.Skolnick J, Fixman M. Macromolecules. 1977;10:944. [Google Scholar]
- 10.Fenley MO, Manning GS, Olson WK. The Journal of Physical Chemistry. 1992;96:3963. [Google Scholar]
- 11.Schlick T, Li B, Olson WK. Biophysical journal. 1994;67:2146. doi: 10.1016/S0006-3495(94)80732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podgornik R, Hansen PL, Parsegian VA. The Journal of Chemical Physics. 2000;113:9343. [Google Scholar]
- 13.Abels J, Moreno-Herrero F, Van der Heijden T, Dekker C, Dekker N. Biophysical journal. 2005;88:2737. doi: 10.1529/biophysj.104.052811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herrero-Galán E, Fuentes-Perez ME, Car-rasco C, Valpuesta JM, Carrascosa JL, Moreno-Herrero F, Arias-Gonzalez JR. Journal of the American Chemical Society. 2012;135:122. doi: 10.1021/ja3054755. [DOI] [PubMed] [Google Scholar]
- 15.Lipfert J, Skinner GM, Keegstra JM, Hens-gens T, Jager T, Dulin D, Köber M, Yu Z, Donkers SP, Chou F-C, et al. Proceedings of the National Academy of Sciences. 2014;111:15408. doi: 10.1073/pnas.1407197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolokh IS, Pabit SA, Katz AM, Chen Y, Drozdetski A, Baker N, Pollack L, Onufriev AV. Nucleic Acids Research. 2014;42:10823. doi: 10.1093/nar/gku756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pabit SA, Qiu X, Lamb JS, Li L, Meisburger SP, Pollack L. Nucleic Acids Research. 2009;37:3887. doi: 10.1093/nar/gkp257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Pabit SA, Meisburger SP, Pollack L. Physical Review Letters. 2011;106:108101. doi: 10.1103/PhysRevLett.106.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macke T, Case D. Washington DC: American Chemcial Society. 1998:379. [Google Scholar]
- 20.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. Journal of computational chemistry. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheatham TE, Cieplak P, Kollman PA. Journal of Biomolecular Structure and Dynamics. 1999;16:845. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 22.Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Biophysical Journal. 2007;92:3817. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K. Nucleic Acids Research. 2009;37:5917. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mazur AK. Biophysical journal. 2006;91:4507. doi: 10.1529/biophysj.106.091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantor CR, Schimmel PR. Biophysical chemistry: Part III: the behavior of biological macro-molecules. Macmillan; 1980. [Google Scholar]
- 26.Wiggins PA, Van Der Heijden T, Moreno-Herrero F, Spakowitz A, Phillips R, Widom J, Dekker C, Nelson PC. Nature nanotechnology. 2006;1:137. doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- 27.Mazur AK. Physical review letters. 2007;98:218102. doi: 10.1103/PhysRevLett.98.218102. [DOI] [PubMed] [Google Scholar]
- 28.Wiggins PA, Nelson PC. Physical Review E. 2006;73:031906. doi: 10.1103/PhysRevE.73.031906. [DOI] [PubMed] [Google Scholar]
- 29.Fadrná E, Spačková N, Štefl R, Koča J, Cheatham TE, Šponer J. Biophysical Journal. 2004;87:227. doi: 10.1529/biophysj.103.034751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pabit SA, Katz AM, Tolokh IS, Drozdet-ski A, Baker N, Onufriev AV, Pollack L. The Journal of Chemical Physics. 2016;144:205102. doi: 10.1063/1.4950814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bomble YJ, Case DA. Biopolymers. 2008;89:722. doi: 10.1002/bip.21000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korolev N, Luo D, Lyubartsev AP, Nor-denskiöld L. Polymers. 2014;6:1655. [Google Scholar]
- 33.Savelyev A, Papoian GA. Proceedings of the National Academy of Sciences. 2010;107:20340. doi: 10.1073/pnas.1001163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manning GS. Biophysical journal. 2006;91:3607. doi: 10.1529/biophysj.106.089029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savelyev A, Materese CK, Papoian GA. Journal of the American Chemical Society. 2011;133:19290. doi: 10.1021/ja207984z. [DOI] [PubMed] [Google Scholar]
- 36.Sivolob A, Khrapunov SN. Biophysical chemistry. 1997;67:85. doi: 10.1016/s0301-4622(97)00022-7. [DOI] [PubMed] [Google Scholar]
- 37.Savelyev A, Papoian GA. Journal of the American Chemical Society. 2006;128:14506. doi: 10.1021/ja0629460. [DOI] [PubMed] [Google Scholar]
- 38.Kirmizialtin S, Silalahi ARJ, Elber R, Fenley MO. Biophysical Journal. 2012;102:829. doi: 10.1016/j.bpj.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirmizialtin S, Pabit SA, Meisburger SP, Pollack L, Elber R. Biophysical Journal. 2012;102:819. doi: 10.1016/j.bpj.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chin K, Sharp KA, Honig B, Pyle AM. Nature Structural & Molecular Biology. 1999;6:1055. doi: 10.1038/14940. [DOI] [PubMed] [Google Scholar]
- 41.Colmenarejo G, Tinoco I. Journal of molecular biology. 1999;290:119. doi: 10.1006/jmbi.1999.2867. [DOI] [PubMed] [Google Scholar]
- 42.Braunlin W, Anderson C, Record MT., Jr Biochemistry. 1987;26:7724. doi: 10.1021/bi00398a028. [DOI] [PubMed] [Google Scholar]
- 43.Zgarbová M, Otyepka M, Šponer J, Mládek A, Banáš P, Cheatham TE, III, Jurecka P. Journal of chemical theory and computation. 2011;7:2886. doi: 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mazur AK. Journal of the American Chemical Society. 2003;125:7849. doi: 10.1021/ja034550j. [DOI] [PubMed] [Google Scholar]
- 45.Mazur AK. ChemPhysChem. 2008;9:2691. doi: 10.1002/cphc.200800446. [DOI] [PubMed] [Google Scholar]
- 46.Arscott PG, Ma C, Wenner JR, Bloomfield VA. Biopolymers. 1995;36:345. doi: 10.1002/bip.360360309. [DOI] [PubMed] [Google Scholar]
- 47.Cheatham TE, Kollman PA. Structure. 1997;5:1297. doi: 10.1016/s0969-2126(97)00282-7. [DOI] [PubMed] [Google Scholar]