

Abstract
We report on an infant with a previously un-described chromosome 15 deletion (q26.1→ qter) and compare the clinical findings with those of 7 reported patients with deletions of distal 15q, as well as ring chromosome 15 syndrome patients. Most of the patients with deletions of distal 15q, including our patient, have intrauterine growth retardation (IUGR), microcephaly, abnormal face and ears, micrognathia, highly arched palate, renal abnormalities, lung hypoplasia, failure to thrive, and developmental delay/mental retardation. Several genes have been assigned to the 15q25→qter region, including insulin-like growth factor 1 receptor (IGF1R). DNA analysis from our patient documented the loss of one IGF1R gene copy. Our study further localizes the IGF1R gene distal to the 15q26.1 band. It is interesting to speculate that the severe IUGR and postnatal growth deficiency of our patient and other patients with similar chromosome 15 deletions are related to the loss of an IGF1R gene copy which may lead to an abnormal number and/or structure of the receptors.
Keywords: chromosome 15 deletions, intrauterine growth retardation (IUGR), IGF1R, ring chromosome 15 syndrome
INTRODUCTION
There is a paucity of clinical reports of patients with deletions of the distal long arm of chromosome 15. Excluding ring chromosome 15 syndrome, only 7 patients with such abnormalities have been described [Clark, 1984; Formiga et al., 1988; Fryns et al., 1982; Kristofferson et al., 1987; Pasquali et al., 1973]. Here, we report on an infant with a previously undescribed deletion of chromosome 15 (15q26.1→qter), summarize the clinical findings of the 7 reported patients with 15q deletions and compare the clinical findings of these patients with those having the ring chromosome 15 syndrome. Because insulin-like growth factor 1 is important in normal growth, and the insulin-like growth factor 1 receptor gene (IGF1R) maps to the distal end of 15q, DNA analysis was undertaken on our patient.
CLINICAL REPORT
K.S. is a 12-month-old white girl born to a 38-year-old G4P3→4 mother and a nonconsanguineous 46-year-old father. The pregnancy was complicated by intrauterine growth retardation (IUGR) and oligohydramnios. These findings prompted percutaneous umbilical blood sampling (PUBS) at 34 weeks gestation, which documented a deletion of 15q(15q26.1→qter).
At 37 weeks of gestation, persistent fetal heart rate decelerations necessitated an emergency C-section. The Apgar scores were 8 and 9 at one and 5 minutes, respectively. The birth length was 36.5 cm (<3rd centile), birth weight 1,263 g (<3rd centile) and the head circumference (OFC) was 27.5 cm (<3rd centile). Multiple anomalies were noted at birth, including relative hypertelorism, relatively large, well-formed ears, small palpebral fissures, micrognathia, highly arched palate, diastasis recti, small hands, rocker-bottom feet with proximally placed fifth toes bilaterally, hypoplastic finger and toe nails, one flexion crease of the second finger bilaterally, sacral dimple, and hypotonia (Fig. 1). The chromosome abnormality [del(15)(q26.1→qter)] detected in the fetal blood cells was confirmed during the neonatal period from lymphocyte cultures.
Fig. 1.

Several views of our patient at one week of age showing full body, facial appearance, and hands and feet.
Shortly after delivery, respiratory distress developed and the infant was intubated. Chest film showed a left pneumothorax, hypoplastic lungs, and an enlarged cardiac silhouette. A chest tube was inserted and the infant was placed on a ventilator; however, she breathed on her own by the second day of life and mechanical ventilation was discontinued. The infant had a weak cry, poor suck reflex, and slightly decreased muscle tone.
A soft systolic murmur (grade II/VI) was also noted at birth but electro- and echocardiograms were normal and she was diagnosed with a clinically insignificant peripheral pulmonary flow murmur. Renal ultrasound showed small kidneys, measuring 1.3 cm in length on the right and 1.2 cm on the left. Serum creatinine was 2.6 mg/dl on the second day of life and increased to a maximum of 3.6 mg/dl on the fifth day of life.
The patient was discharged on the 28th day of life on nasogastric tube feedings. Her creatinine was 2.4 mg/dl and BUN was 37 mg/dl.
At age 2 months her weight was 1.8 kg, length was 41.5 cm, and OFC was 36 cm (all <3rd centile). She had good head control but had no social smile. Serum creatinine was 1.9 mg/dl and BUN was 56 mg/dl.
At age 4 months, she continued to have poor growth and development. Her length was 47.5 cm and weight 2.6 kg (both <3rd centile). Renal ultrasonography demonstrated a right kidney of 1.8 cm and a left kidney of 2.2 cm (both <3rd centile). Her serum creatinine was 1.5 mg/dl and BUN was 25 mg/dl. She developed hyperchloremic metabolic acidosis with a serum chloride of 109 mEq/L and carbon dioxide of 16 nmol/L. In the evaluation of growth retardation, her insulin-like growth factor 1 level was found to be normal and DNA analysis of the insulin-like growth factor 1 receptor gene (IGF1R) was undertaken.
MATERIALS AND METHODS DNA Analysis
Genomic DNA was extracted from lymphoblastoid cell lines using previously described methods [Aldridge et al., 1984]. The cell lines from both parents and the patient were established by transforming peripheral lymphocytes with Epstein Barr virus [Neitzel, 1986].
Genomic DNA (5 μg) digested with HindIII was fractionated by electrophoresis on agarose gel and transferred to a nylon membrane (Hybond N +, Amersham). The membrane was pre-hybridized and probed with radioactive DNA, washed, and exposed to x-ray film as previously described [Mbikay et al., 1988].
A 0.7-kb EcoRI fragment of the cDNA for the human insulin-like growth factor 1 receptor (IGF1R, obtained from American Tissue Culture Collection, Rockville, Maryland) was used to probe the loci. The IGF1R gene has been localized to 15q25→qter [Ullrich et al., 1986]. A 1.3-kilobase (kb) HindIII fragment derived from an intron of the human 7B2 gene, which codes for a neuroendocrine protein and localized to proximal 15q [Mattei et al., 1990], was used as an internal control probe. The DNA fragments were uniformly labeled with 32P using the multi-priming method of Feinberg and Vogelstein [1984]. The 2 probes were concomitantly hybridized to Southern blot membranes in order to determine the presence or absence of the IGF1R gene copies.
The autoradiographic bands were scanned using a Bio-Rad video densitometer, model 620, and peaks were analyzed using the DPACK computer program. To obtain a linear range of band intensities, an autoradiogram of varying amounts of radioactive DNA spotted onto a nylon membrane was subjected to a similar densitometric analysis. The relative intensities of bands on genomic Southern blots were computed as described by Tantravahi et al. [1983].
RESULTS AND DISCUSSION
The rare del(15q) detected in fetal blood from our patient was also identified from lymphocytes obtained after delivery. High resolution chromosome analysis documented a distal deletion of chromosome 15 (q26.1→qter) (Fig. 2).
Fig. 2.

A prometaphase chromosome 15 (850 band level) idiogram [Harnden and Klinger, 1985] and representative metaphase and prometaphase chromosome 15s are shown. The chromosome with 15q26.1→qter deletion is on the left of each chromosome pair.
Parental chromosome were normal, although chromosome 15 polymorphisms indicated that the chromosome 15 donated by the father led to the deletion in the child (Fig. 3). Our results on the parental origin of de novo deleted chromosome 15s are consistent with preferential paternal origin of the deleted chromosome 15 in Prader-Willi syndrome patients [Butler et al., 1986] and in other non-Robertsonian structural rearrangements [Chamberlin and Magenis, 1980].
Fig. 3.
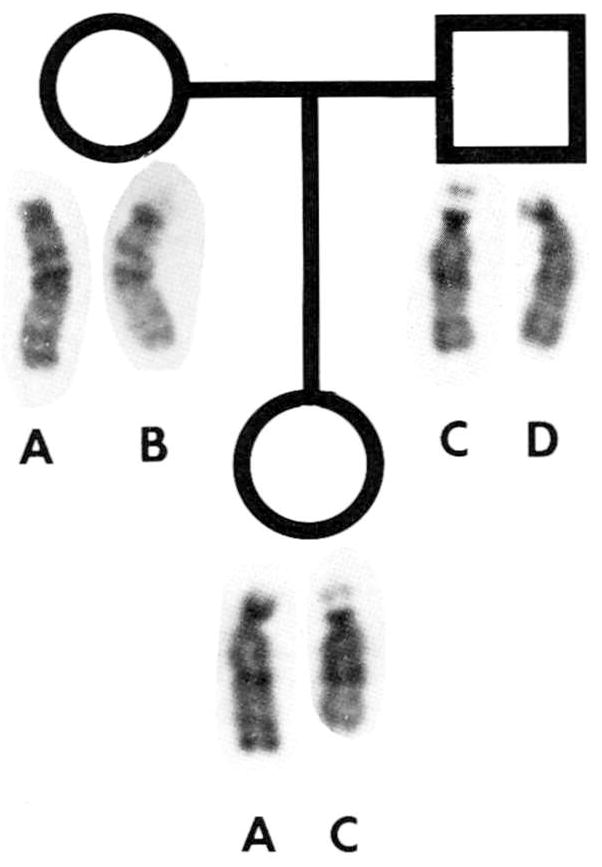
GTG-banded chromosome 15s from the mother, father, and child. Chromosome C was identified in the father and recognized as the deletion in the child by satellite size and stalk length polymorphisms.
Several genes have been assigned to the distal region of 15q including FES (feline sarcoma viral [v-fes] oncogene, 15q25→qter), FUR (furin, membrane associated receptor protein, 15q25-q26), CD13 antigen (15q25→qter), CTSH (cathepsin H, 15q24-q25), and IGFIR (insulin-like growth factor 1 receptor, 15q25→qter) [Cox and Donlon, 1989].
Quantitative Southern blot analyses of the IGF1R gene locus were performed on DNA from K.S. and her parents, using the 7B2 locus as an internal control probe. The densitometric data from the Southern blot analysis are shown in Table I. A representative Southern blot autoradiogram of HindIII digests of lymphoblastoid DNA is shown in Figure 4. The 7B2 probe hybridizes to a 1.3-kb fragment; the IGF1R probe to a 1.6-kb fragment. Variations among samples in the intensity of the 7B2 band would indicate differences in amounts of DNA loaded on the gel. It is apparent that the amounts were equivalent in all 3 lanes (mother, father, and child). The IGF1R band in the proposita was about half the intensity of that of the 2 parents, indicating that the chromosome deletion observed in this patient resulted in the loss of an IGF1R gene copy.
TABLE I.
Densitometric Analysis of IGF1R Gene Copy Number
| DNA source | |||
|---|---|---|---|
|
| |||
| Mother | Father | Daughter | |
| CPM ratio (IGF1R/SGNE-1)a | 0.78 | 0.74 | 0.43 |
| IGF1R Copy No. per genomeb | 2 | 1.90(2) | 1.10(1) |
The cpm values for the IGF1R and the SGNE-1 bands were derived as described by Tantravahi et al. [1983], after densitometric scanning of autoradiograms of Southern blots and of radioactivity standards. They represent averages from two separate blots.
Assuming the mother’s DNA carried two IGF1R copies per genome, its cpm ratio was used to deduce the copy No. for the other two DNA samples. Rounded values are given in parentheses.
Fig. 4.

Southern blot autoradiogram of HindIII digests of lymphoblastoid DNA from the mother, father, and proposita with the 15q26.1→qter deletion detected by two probes [IGF1R and SGNE-1(7B2)], both localized to different regions of chromosome 15. The intensity of the IGF1R band is approximately one-half that of the SGNE-1 (7B2) band in the proposita, which indicates that an IGF1R gene copy is missing.
With the exception of the ring chromosome syndrome, patients with deletions of the terminal bands of 15q are rare, with only 7 cases found in the literature (Table II). Of these patients, only those of Kristofferson et al. [1987] and Pasquali et al. [1973] had a chromosome deletion comparable to the deletion in our patient, but the chromosome abnormalities in their patients were due to unbalanced chromosome translocations. Coincidentally, their patients as well as our patient had similar manifestations, including IUGR, abnormal face and ears, developmental delay, failure to thrive, and skeletal abnormalities. Of the other chromosome 15 deletion patients reported in the literature, only those with more distal deletions had IUGR. The patient of Fryns et al. [1982] who had a 15q21 deletion did not have IUGR or postnatal growth retardation at age one year, although severe failure to thrive was reported subsequently. The patient reported by Formiga et al. [1988] with a 15q21-q24 deletion, which was more proximal than in our patient, did not have IUGR or postnatal growth retardation. Two of the 3 patients reported by Kristofferson et al. [1987] were monosomic for 15q24→qter and had IUGR, while their sib, who was trisomic for this region, was of normal size.
TABLE II.
Comparison of Our Patient and Other Patients With Deletions of Distal 15q
| del 15q26→qter (Pasquali et al. [1973])a | del 15q21 (Fryns et al. [1982]) | del 15q22–q24 (Clark [1984]) | del 15q24→qter (Kristofferson et al. [1987]—two siblings with the same deletion)b | del 15q21–q24 del 15q22–q25 (Formiga et al. [1988]—two de novo cases) | Del 15q26.1→qter (present case) | |||
|---|---|---|---|---|---|---|---|---|
| Pregnancy and family data | ||||||||
| Intrauterine growth retardation | + | − | + | + | + | − | + | + |
| Oligohydramnios | NRc | + | + | NR | NR | NR | NR | + |
| Maternal age (yr) | 29 | 25 | 27 | 26 | 27 | 31 | NR | 38 |
| Maternal chromosomes | NR | Normal | Normal | t(6;15) (p25;q24) | Normal | Normal | Normal | |
| Paternal age (yr) | 33 | 29 | NR | 29 | 30 | 33 | NR | 46 |
| Paternal chromosomes | NR | Normal | NR | NR | NR | Normal | Normal | Normal |
| Craniofacial | ||||||||
| Microcephaly | + | + | NR | + | NR | − | + | + |
| Hypertelorism | NR | + | NR | NR | NR | + | + | + |
| High-arched palate | + | + | NR | NR | − | + | + | + |
| Large/abnormal ears | + | + | NR | + | NR | + | + | + |
| Micrognathia | + | − | NR | − | NR | + | + | + |
| Eyes | ||||||||
| Microcornea/microphthalmia | − | − | NR | − | NR | + | + | − |
| Coloboma | − | + | NR | − | NR | + | − | − |
| Hypopigmented iris | − | + | NR | − | NR | + | + | − |
| Cardiopulmonary | ||||||||
| Cardiac abnormality | − | − | + | − | NR | + | − | − |
| Lung hypoplasia | − | − | + | + | + | − | − | + |
| Diaphragmatic hernia | − | − | − | + | + | − | − | − |
| Renal | ||||||||
| Small/hypoplastic kidneys | − | − | + | − | − | − | − | + |
| Polycystic kidneys | − | − | + | + | + | − | − | − |
| Skeletal | ||||||||
| Clinodactyly | + | + | − | − | NR | + | − | + |
| Proximal placement of thumb or toe | − | + | − | − | NR | + | + | + |
| Club feet | − | + | − | + | NR | − | − | − |
| Kyphosis or scoliosis | + | + | − | − | NR | − | − | − |
| Neurodevelopment | ||||||||
| Developmental delay/MR | + | + | NAd | NA | NA | + | + | + |
| Hypotonia | NR | NR | NR | NR | NR | + | + | + |
| Seizures/EEG abnormality | + | − | NA | − | NA | + | + | − |
| Growth | ||||||||
| Failure to thrive | + | + | NA | NA | NA | − | + | + |
| Truncal obesity | + | + | NR | NR | NR | − | − | − |
Patient with 45,XY t(15;13)(15pter→15q26::13qll→13qter), therefore monosomic for 15q26 and for the short arm and centromeric region of chromosome 13.
Due to an unbalanced chromosome translocation between chromosomes 6 and 15 inherited from a balanced translocation mother.
NR = not reported.
NA = not applicable.
The 2 patients reported by Kristofferson et al. [1987] also had renal and pulmonary abnormalities including hypoplastic lungs, left diaphragmatic hernia and cystic kidneys. Our patient had hypoplastic lungs and kidneys. Thus, renal abnormalities were identified in 4 of 8 reported patients with distal 15q deletions. In a review of the cytogenetic literature, Barakat and Butler [1987] also documented that the frequency of renal anomalies was significantly higher in patients with chromosomal abnormalities than in the general population.
Patients with the ring chromosome 15 syndrome have deletions of the 15q26.2 or 15q26.3 bands as well as the distal short arm of chromosome 15 [Wilson et al., 1985; Butler et al., 1988]. Typically, these patients have IUGR, variable mental retardation, microcephaly, hypertelorism, triangular face, abnormal ears, cafe-au-lait spots, cryptorchidism, cardiac anomalies, and fifth finger clinodactyly [Butler et al., 1988]. Many of these traits are also present in our patient with the 15q26.1→qter deletion (Table III).
TABLE III.
Clinical Comparison of Our Patient With 15q26.1→qter Deletion and Ring Chromosome 15 Syndrome Patients*
| Present case | Ring chromosome 15 syndromea | |
|---|---|---|
| Growth | ||
| Birth weight (<3rd centile) | + | 19/26 |
| Birth length (<3rd centile) | + | 14/17 |
| Short stature | + | 27/27 |
| Cranio-facial | ||
| Triangular face | − | 10/24 |
| Microcephaly (<3rd centile) | + | 21/24 |
| High-arched palate | + | 6/27 |
| Abnormal ears | + | 8/27 |
| Micrognathia | + | 7/24 |
| Hypertelorism | + | 11/24 |
| Cardio-pulmonary | ||
| Cardiac abnormalities | − | 8/27 |
| Lung hypoplasia | + | NR |
| Uro-genital | ||
| Cryptorchidism | NA | 3/10 |
| Hypospadias | NA | 2/10 |
| Renal abnormality | + | 2/27 |
| Skeletal | ||
| Clinodactyly | + | 7/27 |
| Club feet | − | 4/27 |
| Small hands | + | 6/26 |
| Skin | ||
| Café-au-lait spots | − | 7/23 |
| CNS function | ||
| Developmental delay/mental retardation | + | 20/21 |
| Hypotonia | + | 7/27 |
NA = not applicable; NR = not recorded.
Fourteen of the 17 patients with ring chromosome 15 syndrome reviewed by Butler et al. [1988] had IUGR, while all patients had postnatal growth deficiency. Growth hormone and IGF1 levels were reported to be normal [Wilson et al., 1985; Butler et al., 1988]. Interestingly, Francke et al. [1988] found an IGF1R gene copy absent in 3 of 5 patients with r(15). Those missing one IGF1R gene copy had severe growth failure, while those with both IGF1R gene copies present had only mild growth failure. One could speculate that the r(15) syndrome patients with one IGF1R gene copy missing may have a larger deletion (e.g., 15q26.2→qter) and severe growth retardation. Those ring chromosome 15 syndrome patients with mild growth failure may have smaller deletions (e.g., 15q26.3) and both IGF1R gene copies present. DNA analysis from our patient also revealed that her deletion included the loss of one IGF1R gene copy (Fig. 4). Thus, the data from ring chromosome 15 syndrome patients as well as our patient would support the location of the IGF1R gene proximal to 15q26.3 or qterminus but distal to 15q26.1. Additional studies are needed to further localize the gene.
It is interesting to hypothesize that the severe IUGR and postnatal growth deficiency of our patient, and other patients with similar chromosome 15 deletions, may be related to the loss of one of the IGF1R gene copies leading to an abnormal number and/or structure of the receptors. Additional research is underway to further characterize the location of recognized genes on distal 15q and to identify the IGFlR receptor abnormality.
References
- Aldridge J, Kunkel L, Bruns G, Tantravahi U, Lalande M, Brewster T, Moreau E, Wilson M, Bromley W, Roderick T, Latt SA. A strategy to reveal high frequency RFLPs along the human X chromosome. Am J Hum Genet. 1984;36:546–564. [PMC free article] [PubMed] [Google Scholar]
- Barakat AY, Butler MG. Renal and urinary tract abnormalities associated with chromosome aberrations. Int J Pediatr Nephrol. 1987;8:215–226. [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Fogo AB, Fuchs DA, Collins FS, Dev VG, Phillips JA., III Brief clinical report and review: Two patients with ring chromosome 15 syndrome. Am J Med Genet. 1988;29:149–154. doi: 10.1002/ajmg.1320290119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Meaney FJ, Palmer CG. Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet. 1986;23:793–809. doi: 10.1002/ajmg.1320230307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin J, Magenis RE. Parental origin of de novo chromosome rearrangements. Hum Genet. 1980;53:343–347. doi: 10.1007/BF00287054. [DOI] [PubMed] [Google Scholar]
- Clark RD. Letter to the editor: del(15) (q22q24) syndrome with Potter sequence. Am J Med Genet. 1984;19:703–705. doi: 10.1002/ajmg.1320190409. [DOI] [PubMed] [Google Scholar]
- Cox DW, Donlon TA. Report of the committee on the genetic constitution of chromosome 14 and 15. Cytogenet Cell Genet. 1989;51:280–298. doi: 10.1159/000132795. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1984;137:266, 267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- Formiga LF, Poenaru L, Couronne F, Flori E, Eibel JL, Deminatti MM, Savary JB, Lai JL, Gilgenkrantz S, Pierson M. Interstitial deletion of chromosome 15: two cases. Hum Genet. 1988;80:401–404. doi: 10.1007/BF00273663. [DOI] [PubMed] [Google Scholar]
- Francke U, Darras BT, Foellmer BE. Loss of IGF-I receptor gene in patients with ring chromosome 15 is related to Russell-Silver like phenotype. Proceedings of the 9th Annual David W. Smith Workshop on Malformations and Morphogenesis; Oakland CA. August 1988.1988. [Google Scholar]
- Fryns JP, Muelenaere A, Berghe H. Interstitial deletion of the long arm of chromosome 15. Ann Genet (Paris) 1982;25:59–60. [PubMed] [Google Scholar]
- Harnden DG, Klinger HP. An International System for Human Cytogenetic Nomenclature. Basel: S. Karger; 1985. p. 55. [Google Scholar]
- Kristofferson V, Heim S, Mandahl N, Sundkvist L, Szelest J, Hagerstrand I. Monosomy and trisomy of 15q24→qter in a family with a translocation t(6;15)(p25;q24) Clin Genet. 1987;32:169–171. doi: 10.1111/j.1399-0004.1987.tb03348.x. [DOI] [PubMed] [Google Scholar]
- Mattei MG, Mbikay M, Sylla BS, Lenoir G, Mattei JF, Seidah NG, Chrétien M. Assignment of the gene for neuroendocrine protein 7B2 (SGNE1 locus) to mouse chromosome region 2[E3-F3] and to human chromosome region 15qll–ql5. Genomics. 1990;6:436–440. doi: 10.1016/0888-7543(90)90473-8. [DOI] [PubMed] [Google Scholar]
- Mbikay M, Linard CG, Sirois F, Lazure C, Seidah NG, Chrétien M. Tissue-specific expression of the prostatic secretory protein PSP94 in cyanomolgus monkey (Macaca fascicularis) Cell Mol Biol. 1988;34:387–398. [PubMed] [Google Scholar]
- Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73:320–326. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- Pasquali F, Zuffardi O, Severi F, Colombo A, Burgio GR. Tandem translocation 15/13. Ann Genet (Paris) 1973;16:47–50. [PubMed] [Google Scholar]
- Tantravahi U, Kirschner DA, Beauregard L, Page L, Kunkel L, Latt SA. Cytologic and molecular analysis of 46, XXq – cells to identify a DNA segment that might serve as probe for a putative human X chromosome inactivation center. Hum Genet. 1983;64:33–38. doi: 10.1007/BF00289475. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Hentzel W, Le Bon T, Kathuria S, Chen E, Jacobs S, Francke U, Ramanchandran J, Fugita-Hamagushi Y. Insulin-like growth factor receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986;5:2503–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GN, Sauder SE, Bush M, Beitins IZ. Phenotypic delineation of ring chromosome 15 and Russell-Silver syndromes. J Med Genet. 1985;22:233–236. doi: 10.1136/jmg.22.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]