

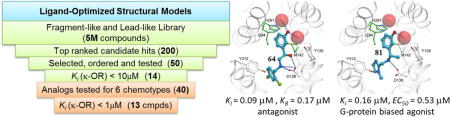
Abstract
The ongoing epidemics of opioid overdose raises an urgent need for effective anti-addiction therapies and addiction-free painkillers. The κ–opioid receptor (KOR) has emerged as a promising target for both indications, raising demand for new chemotypes of KOR antagonists, as well as G-protein-biased agonists. We employed the crystal structure of the KOR-JDTic complex and ligand-optimized structural templates to perform virtual screening of available compound libraries for new KOR ligands. The prospective virtual screening campaign yielded a high 32% hit rate, identifying novel fragment-like and lead-like chemotypes of KOR ligands. A round of optimization resulted in eleven new sub-micromolar KOR binders (best Ki = 90 nM). Functional assessment confirmed at least two compounds as potent KOR antagonists, while compound 81 was identified as a potent Gi biased agonist for KOR with minimal β-arrestin recruitment. These results support virtual screening as an effective tool for discovery of new lead chemotypes with therapeutically relevant functional profiles.
TOC image
INTRODUCTION
The classical μ, δ, and κ opioid receptors are G protein-coupled receptors (GPCRs) that play critical roles in pain, addiction and mood disorders.1–3 As such, opioid receptors are the targets for widely prescribed pain medications that often carry a major drug abuse liability.4, 5 Among the three opioid receptors, the κ-opioid receptor (KOR), is widely expressed throughout the central and peripheral nervous systems and is activated by the endogenous peptide dynorphin. KOR has a special physiological profile, associated with pain relief on one hand, and stress exacerbation and mood suppression on the other.6, 7 Moreover, there is accumulating evidence that dynorphin may serve as a body’s natural addiction control mechanism.6, 8 Exogenous KOR agonists, like the plant derived salvinorin A9 and the synthetic compound U-25048810 (Chart 1) are known to be effective in pain relief, but can also induce hallucinations and dysphoria,11 and therefore are deemed unsuitable as CNS analgesics. At the same time, KOR antagonists, such as NorBNI and JDTic have been in active development as potential antidepressants, anxiolytics and anti-addiction drugs.12, 13 In particular, JDTic showed robust response in rodent models of depression, anxiety, stress-induced cocaine relapse, and nicotine withdrawal.14–17 Although clinical development of JDTic was abandoned due to transient cardiovascular effects18 and exceptionally long lasting action,19 several other KOR selective antagonists are in various stages of clinical or preclinical studies.20, 21
Chart 1.
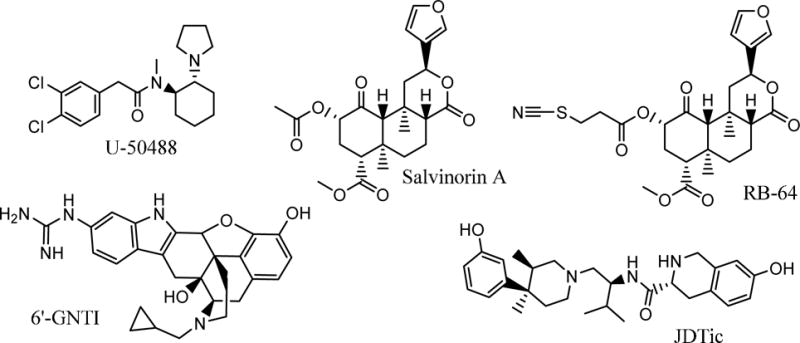
Examples of commonly used KOR small molecule agonists (U-50488, Salvinorin A), antagonist (JDTic), and G-protein biased ligands (RB-64 and 6′-GNTI).
In addition, it was recently shown that the KOR induced dysphoric and sedative effects are mediated by arrestin recruitment to KOR and corresponding signaling via p38 MAPK pathway activation.11, 22 Therefore, biased KOR agonists that activate the Gi/o pathway, but have reduced β-arrestin mediated responses, can possess the desired pain relief effects without the unwanted side effects. Indeed, several G-protein biased ligands, including 6′-guanidinonaltrindole (6′-GNTI),23 and the Salvinorin A derivative RB64,22 (Chart 1) have shown analgesia with significantly reduced addiction risk in animals, and several new series of biased KOR agonists are being actively developed.24, 25 However, the KOR ligand chemotypes are still largely limited to morphinans, diterpenes and JDTic analogues, so expanding their repertoire for both KOR antagonists and biased agonists would be highly beneficial for drug discovery.
A unique opportunity to effectively screen for new KOR ligand chemotypes is presented by the recently solved crystal structures of opioid receptors, including the high resolution structures of the KOR-JDTic complex26 (PDB ID 4DJH), morphinan bound δ-opioid receptor27 (DOR, PDB ID 4N6H) and μ-opioid receptor28 (MOR, PDB ID 4DLK). Structure-based virtual ligand screening (VLS) campaigns have been previously successful in discovery of novel ligand chemotypes for many GPCRs activated by small-molecule ligands, including new antagonists for β2 adrenergic,29 A2A adenosine,30, 31 H1 histamine,32 D3 dopamine,33 as well as agonists for β2 adrenergic,34 allosteric modulators for D3 dopamine,35 and more. Although opioid and other peptide- and protein-binding GPCRs present special challenges for virtual screening, usually limiting affinity of initial hits to the micromolar range,36–38 such screening has already demonstrated great utility, e.g. in discovery of a new class of MOR Gi-biased agonists with analgesic activity and reduced side effects.39
In this study we employed a multi-template screening strategy using the crystal structure of KOR and corresponding ligand-optimized atomistic models40 to discover several new KOR chemotypes with sub-micromolar activities and distinct functional features. The initial prospective screening achieved a 32% hit rate and identified six new promising lead-like scaffolds, while the follow up round of structure-activity relationship (SAR) yielded eleven additional hits in sub-micromolar affinity range. Functional characterization shows KOR antagonist or agonist activity for a number of new ligands, while compound 81 was identified as an agonist with a G-protein biased profile, relevant for therapeutic applications.
RESULTS
Benchmark docking and optimization of receptor models
To evaluate the performance of the structural models in correctly separating KOR binding compounds from inactives, we first performed a benchmark VLS using known KOR ligands and a decoy compound library (Figure 1). Docking into the model directly derived from the KOR-JDTic crystal structure (Model 1) showed high binding scores and conformations of JDTic analogues that were consistent with the crystal structure (PDB ID 4DJH).26 Docking poses for most morphinans in Model 1 were also consistent with the morphine core conformations found in crystal structures of other opioid receptors,27, 41 forming an anchor salt bridge between protonated amine and the carboxyl of Asp138. However, the docking scores calculated for KOR morphinan ligands and other non-JDTic chemotypes were suboptimal, resulting in the overall poor VLS benchmark performance of Model 1 (Figure 1c). This comes as no surprise, because the JDTic-bound conformation of the KOR orthosteric pocket (PDB ID 4DJH)26 is quite distinct from the pocket conformation in morphinan-bound opioid receptors, e.g. DOR (PDB ID 4N6H)27 or MOR (PDB ID 4DKL),28 despite almost exact residue conservation in the interaction sites.42 Thus, JDTic binds deeper in the pocket than the morphine scaffold, breaking a hydrogen bond interaction between Asp138 and Tyr320, which is otherwise conserved in opioid receptor structures, as well as in majority of aminergic receptors.43 This distinct JDTic-bound conformation in the KOR structure is apparently suboptimal for binding of morphinans and some other chemotypes, and requires separate treatment.
Figure 1. Optimization and validation of the models with known KOR ligands.
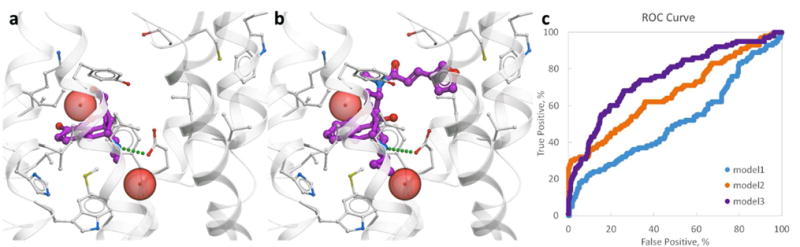
Docking poses of morpine (a) and nalfurafine (b) in the binding pocket of the optimized Model 3. Ligands are shown as sticks with carbon atoms colored magenta. Receptor is shown by grey cartoon and sticks, and tightly bound water molecules directly involved in ligand binding as red spheres. (c) ROC curves calculated for the KOR crystal structure (Model 1) and the two ligand-optimized models (Model 2 and Model 3).
To develop a more accurate model for docking of non-JDTic chemotypes, we employed ligand-guided optimization of the binding site using morphinans as seed compounds. The first optimization approach used the same JDTic bound crystal structure of KOR as a starting point (4DJH),26 and performed extensive refinement of the pocket conformation in presence of morphinans. The second approach used a model of KOR, built by homology from the DOR crystal structure template (PDB code 4N6H)27 as a starting point for ligand-guided optimization. The best performing models from these two approaches, referred to as Model 2 and Model 3, had better binding scores for morphinan ligands, which is reflected in their improved overall performance in the VLS benchmark (Figure 1c). The optimized Models 2 and 3 were selected for use in prospective VLS alongside with the original KOR crystal structure (Model 1).
Prospective VLS and identification of novel KOR binders
A commercial library of 4.5 million compounds was docked into all three different receptor models as described in Methods, and the binding scores were predicted for the best docking conformations. For each VLS run, the top 1000 compounds with the highest docking score were selected, and clustered based on their chemical structures. The Tanimoto distances were calculated for these compounds to evaluate their similarity with known KOR ligands, and the close analogues were removed. A final group of compounds was selected for purchase based on their chemical diversity, structural novelty and predicted binding affinity (Chart S1). Following mass spectrometry quality and purity verification, 43 compounds (18, 20 and 5 compounds for Models 1, 2 and 3, respectively) were tested for activity.
The candidate hits predicted by the virtual screen were tested for inhibition of 6H-U69593 binding to cloned human KOR at 50 μM concentration (Table S1). The compounds that showed at least 30% inhibition were further tested in concentration-response binding assays to determine KOR binding affinities; binding curves of six of the most potent compounds are shown in Figure 2. The best compounds identified by the virtual screen are listed in Table 1 and Chart 2. Out of the 43 tested compounds, binding affinity better than Ki <10 μM was measured for 14 compounds, which amounts to a 32% overall hit rate. Five of the hit compounds came from the KOR crystal structure-based Model 1 (27% hit rate), seven come from the ligand-optimized Model 2 (35% hit rate), and two from homology-based Model 3 (40% hit rate). The two most potent compounds discovered in the initial VLS round (28 and 11) had binding affinities of 0.2 and 0.5 μM, respectively.
Figure 2. Radioligand competition binding assays for the compounds predicted by structure based VLS.
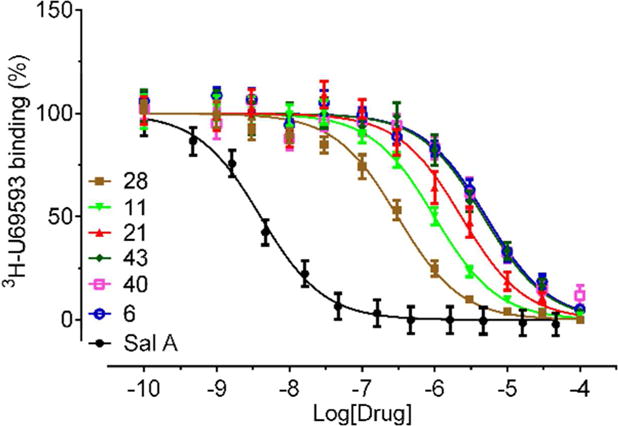
Concentration response curves for the top 6 hit compounds with KOR binding affinity (Ki) better that 3 μM. Normalized results (mean ± SEM) from three independent assays (each in triplicate) were pooled and analyzed in GraphPad Prism.
Table 1.
Compounds predicted by VLS with measured KOR binding affinity Ki < 10 μM
| # | Vendor ID | Vendor | Modela | %Inhibb ± SEMc | pKi ± SEM | Ki(μM) | LEd | Tanimotoe |
|---|---|---|---|---|---|---|---|---|
| 28 | Z1176486275 | Enamine | 1 | 96.9 ± 1.2 | 6.82 ± 0.16 | 0.2 | 0.43 | 0.42 |
| 11 | 42843630 | Chembridge | 1 | 95.3 ± 2.5 | 6.30 ± 0.12 | 0.5 | 0.40 | 0.19 |
| 21 | D202–0095 | ChemDiv | 3 | 77.5 ± 6.8 | 5.95 ± 0.26 | 1.1 | 0.31 | 0.29 |
| 43 | Z432878782 | Enamine | 2 | 76.5 ± 4.7 | 5.64 ± 0.16 | 2.3 | 0.32 | 0.39 |
| 40 | Z244392816 | Enamine | 3 | 77.5 ± 5.6 | 5.59 ± 0.22 | 2.6 | 0.36 | 0.40 |
| 6 | 9288549 | Chembridge | 1 | 80.1 ± 4.2 | 5.58 ± 0.17 | 2.6 | 0.39 | 0.39 |
| 17 | 72490033 | Chembridge | 2 | 53.0 ± 7.8 | 5.52 ± 0.12 | 3.0 | 0.32 | 0.52 |
| 16 | 70054354 | Chembridge | 2 | 60.7 ± 14.7 | 5.52 ± 0.17 | 3.0 | 0.37 | 0.38 |
| 20 | 99182372 | Chembridge | 2 | 49.1 ± 16.4 | 5.21 ± 0.14 | 6.2 | 0.30 | 0.42 |
| 35 | Z1582993358 | Enamine | 2 | 49.8 ± 8.4 | 5.19 ± 0.17 | 6.4 | 0.36 | 0.51 |
| 3 | 5460400 | Chembridge | 1 | 70.2 ± 3.9 | 5.18 ± 0.18 | 6.6 | 0.36 | 0.26 |
| 8 | 29033730 | Chembridge | 1 | 73.7 ± 4.0 | 5.09 ± 0.22 | 8.1 | 0.29 | 0.34 |
| 29 | Z1198176647 | Enamine | 2 | 48.6 ± 9.6 | 5.08 ± 0.12 | 8.3 | 0.59 | 0.15 |
| 22 | E557–0152 | ChemDiv | 2 | 47.3 ± 7.7 | 5.01 ± 0.13 | 9.8 | 0.28 | 0.39 |
VLS model used in compound selection
Inhibition of 3H-U69593 radioligand binding
Standard Error of the Mean, N=3
Ligand Efficiency
Tanimoto distance to the closest KOR binding ligand in ChEMBL database
Chart 2.
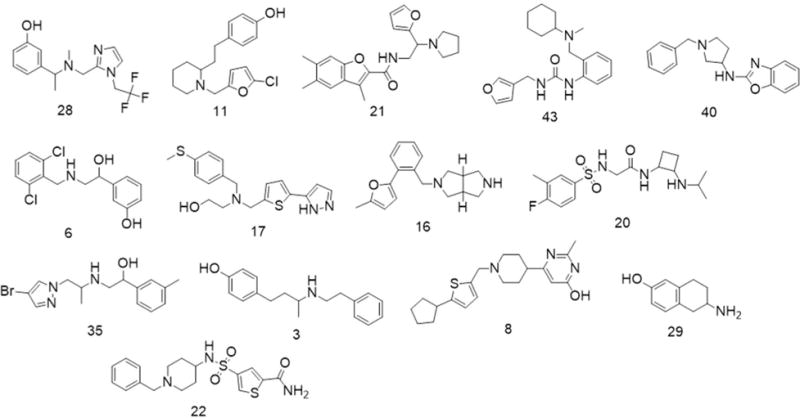
Chemical structures of VLS hit compounds in Table 1 with measured KOR Ki < 10 μM.
The ligand efficiencies (LE) were found in the lead-like range for the vast majority of the hit compounds.44 Four of the hit compounds (16, 3, 40 and 29) had molecular weight less than 300 Da and satisfied the “rule of three” as fragment-like compounds. A fragment-sized compound 29 with a molecular weight of 163 Da reached the best LE = 0.59 kcal/mol per heavy atom. Overall, 11 hits had LE better than 0.3 kcal/mol per heavy atom. The small molecular weight and high LE can provide ample room for the further optimization of these fragment-like and lead-like hits.
All the discovered ligand chemotypes are novel to KOR, with 10 out of 14 compounds showing a Tanimoto distance > 0.3 to the known KOR ligands. The fragment-like compound 29 had the lowest Tanimoto distance (0.15), as its tetrahydronaphthalene ring structure has similar (but still distinct) chemotype as the tetrahydroisoquinoline core substructure of JDTic. Identification of this fragment by VLS confirms the established role of JDTic-like scaffolds as KOR drug-discovery leads.
Chemical and conformational diversity of the hit compounds
The newly identified ligands by VLS encompass a diverse set of chemical scaffolds, the top six of which are exemplified by compounds in Chart 2. The first scaffold, represented by compound 28 contains an imidazole ring and a phenol group. The second scaffold, represented by compound 11 has a phenol-ethyl-piperidine motif. The other two top chemotypes, represented by compounds 21 and 40 contain fused rings, benzofuran and benzooxazol, respectively. Compound 43 has a urea core, while compound 6 is based on amino-phenyl-ethanol scaffold, which can be found in some β2-adrenergic agonists, both not in previously documented KOR ligands.
Figure 3 shows the predicted binding for the six top chemotypes in their corresponding models of KOR. It illustrates the common salt bridge interactions between amino groups of the ligands and the carboxyl group of Asp138, but also the variety of secondary interactions. Thus, compounds 28, 11 and 6 are predicted to place a hydroxyphenyl group in the hydrophibic pocket flanked by transmembrane helices III, V, and VI, where it also forms a hydrogen bond to a conserved water molecule similar to water-mediated interactions of known ligands in opioid receptor structures. The same conserved subpocket in all opioid receptors is known to accommodate the phenoxy ring of the N-terminal Tyrosine residue of endogenous peptides, as well as in morphinans.27, 28 This interaction involves hydrophobic contact with Met142 and Ile294 side chains, but also a hydrogen bond network mediated by a highly conserved water molecule (Figure 3 a,b,f). Another three compounds, 21, 43 and 40 were predicted to extend more towards helix I and/or the extracellular side of the pocket, expanding the repertoire of contacts employed by the KOR ligands. Although the sidechain of Asp138 was given full flexibility in the docking models, the conformational changes in this sidechain are minor, and the residue retains a similar rotamer in most ligand complexes.
Figure 3. Binding poses for the top six novel KOR ligand chemotypes.
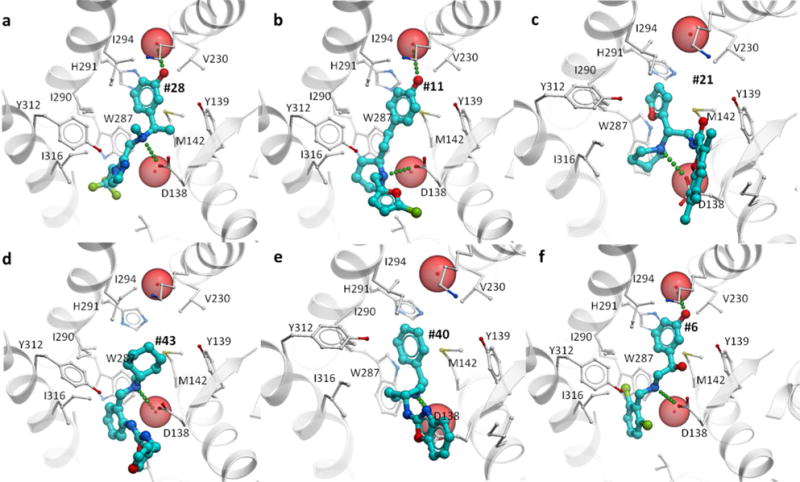
Hit compounds identified in primary round VLS and experimental evaluation are shown in stick representation with carbon atoms colored cyan. Receptor is shown by grey cartoon and sticks, and tightly bound water molecules directly involved in ligand binding as red spheres.
Initial optimization of the hit scaffolds
In the first round of compound optimization, we proceeded to explore the analogues of the top six chemical scaffolds identified by structure-based VLS, and assess their binding and functional properties. The search was performed for the top compounds based on chemical fingerprint similarity, followed by docking of the analogues into the structural models of KOR. Ten analogues each were selected for 28 and 11 chemotypes, and five analogues each were selected for 21, 43, 40 and 6 chemotypes (Chart S2), allowing us to explore available derivatives and to establish initial SAR for lead optimization.
These 40 analogue compounds were acquired from chemical vendors and tested in KOR binding assays. All 40 compounds displayed at least 30% inhibition at 50μM concentration, and therefore all were tested in the concentration-response binding assay to obtain binding affinities (Table S2). As many as 33 out of the 40 tested compounds showed Ki values of 10 μM or lower, and 11 compounds had sub-micromolar affinities up to Ki = 0.09 μM (Figure 4, Table 2 and Chart 3). The ligand efficiencies had also improved, with all 11 top hits having LE > 0.32 kcal/mol per heavy atom and six of them with LE> 0.4 kcal/mol per heavy atom. Compound 81, which is the second most potent ligand identified, had the best LE > 0.53 kcal/mol per heavy atom. All 11 of the most potent compounds represent scaffolds 28, 11, 21 and 6 (Chart 3). In particular, all five compounds from scaffold 21 display very good binding affinities with a Ki < 1μM. Scaffold 6 looks promising as well, as it yielded the first and the third best affinity compounds in the whole set. Three of the compounds in scaffold 28, were identified as potent binders, including 81. These results suggest selection of leads for further optimization, with scaffolds based on compounds 28, 21 and 6 looking the most encouraging.
Figure 4. Radioligand 6H-U69593 competition binding assays for the best hit analogs.
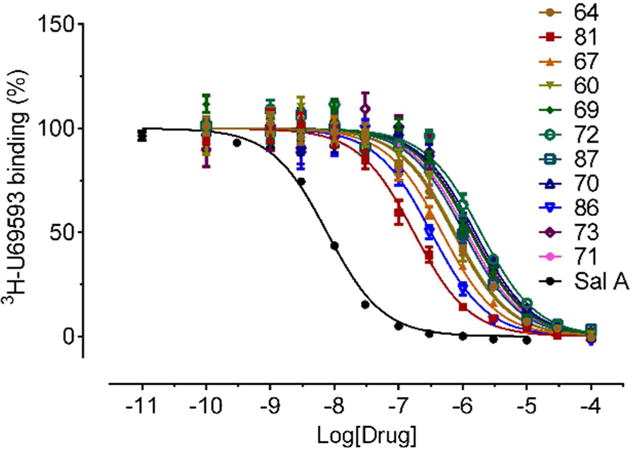
Binding curves shown for the best sub-micromolar compounds resulted from initial scaffold optimization (see also Table 2 and Chart 3). Normalized results (mean ± SEM) from three independent assays (each in triplicate) were pooled and analyzed in GraphPad Prism.
Table 2.
Derivatives of the VLS hit compounds with measured sub-micromolar binding affinity to KOR.
| Comp# | Vendor ID | Vendor | %Inhiba | ± | SEMb | pKi | ± | SEM | Ki(μM) | LEc |
|---|---|---|---|---|---|---|---|---|---|---|
| 64 | 9040216 | Chembridge | 97.90 | ± | 0.63 | 7.08 | ± | 0.10 | 0.09 | 0.47 |
| 81 | Z1176485991 | Enamine | 78.81 | ± | 4.10 | 6.81 | ± | 0.03 | 0.16 | 0.53 |
| 67 | 9288333 | Chembridge | 98.25 | ± | 0.19 | 6.64 | ± | 0.07 | 0.23 | 0.44 |
| 60 | 36660379 | Chembridge | 96.87 | ± | 0.76 | 6.46 | ± | 0.06 | 0.35 | 0.41 |
| 69 | 9315946 | Chembridge | 69.10 | ± | 0.00 | 6.44 | ± | 0.07 | 0.38 | 0.35 |
| 72 | Z88446096 | Enamine | 86.45 | ± | 1.58 | 6.29 | ± | 0.06 | 0.52 | 0.34 |
| 87 | Z1347292377 | Enamine | 74.05 | ± | 3.05 | 6.25 | ± | 0.14 | 0.63 | 0.46 |
| 70 | 9323243 | Chembridge | 75.81 | ± | 2.55 | 6.19 | ± | 0.07 | 0.66 | 0.32 |
| 86 | Z973099512 | Enamine | 82.43 | ± | 1.09 | 6.17 | ± | 0.10 | 0.71 | 0.45 |
| 73 | Z27622033 | Enamine | 73.96 | ± | 2.82 | 6.15 | ± | 0.07 | 0.72 | 0.32 |
| 71 | Z32277632 | Enamine | 64.03 | ± | 0.95 | 6.04 | ± | 0.09 | 0.95 | 0.35 |
Chart 3.
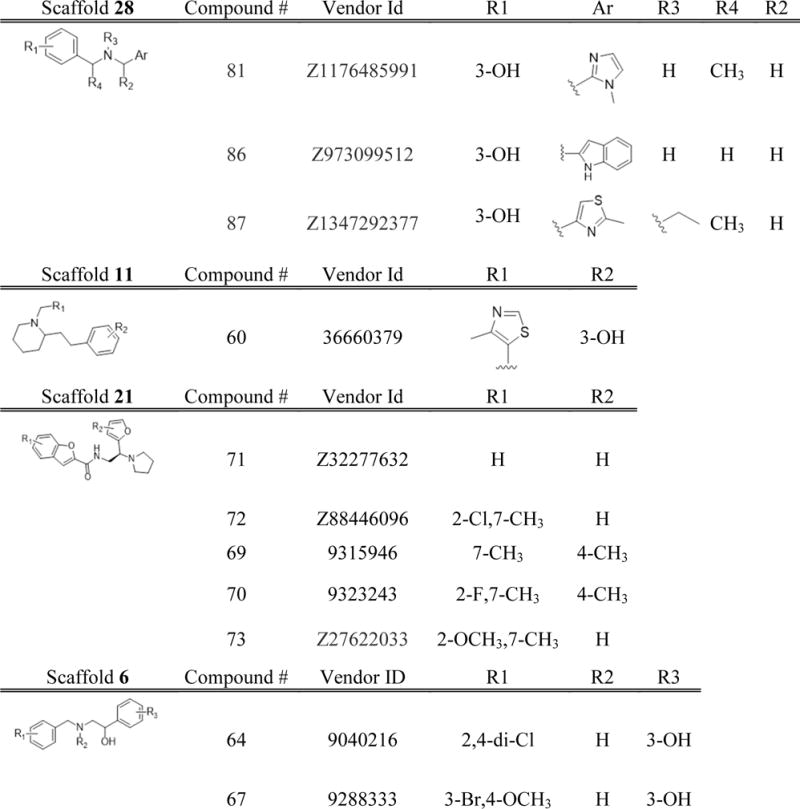
Derivatives of the VLS hit compounds in Table 2 with sub-micromolar binding affinity to KOR. The six scaffolds in this chart and Chart S2 and are based on the initial VLS hits 28, 11, 21, 43, 40, 6, respectively.
Effect of mutations on ligand binding
The affinities of the seven most potent KOR ligands identified in this study were retested in wild type (WT) KOR and 6 single point mutants (Table S3). Expression levels and 6H-U69593 radioligand binding were comparable with those of WT in most cases, except for mutants D138A and D138N, which had greatly reduced binding affinity for 6H-U69593 and were excluded from further assessment. The affinity of Sal A in WT KOR and mutants were found comparable with previously reported values.42 The results in Table S3 show distinct effect of mutations on binding different ligands. Thus, M142A mutant reduced binding of 64, 67 and Sal A at least 10 fold, at the same time enhancing binding of 69, 70 and 72. Mutations H291A and I294A resulted in more than 10 fold reduction of 64, 67, 81 and 87 affinity, while not affecting binding of the other three newly identified compounds or salvinorin A. This is in agreement with the predicted binding poses of the compounds (Figure 5), where the hydroxy phenyl moiety of compounds 64, 67, 81 and 87 are stacked between the hydrophobic side chains of Met142 and Ile294, while the hydroxy group can form hydrogen bonds via bridging waters to His291. In case of compounds 69 and 72, their hydrophobic benzofuran moieties do not reach His291 and Ile294 in helix VI, and also lack suitable polar groups capable of forming hydrogen bonds in this subpocket. Interestingly, for both 69 and 72, an increase in ligand binding was observed for M142A mutations, reflecting plausible steric strain between their large subsituted benzofuran group and Met142.
Figure 5. Docking poses of the top VLS hit derivatives.
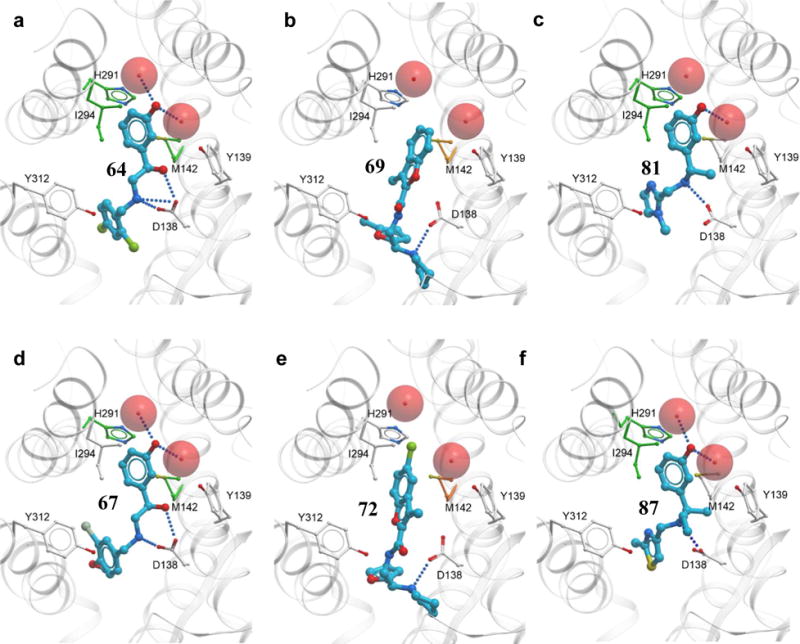
Compounds 64, 69, 81, 67, 72 and 87 are shown in sticks representaion. KOR is shown in cartoon representation with mutated residues as thin sticks, where white carbon atoms show side chains with no significant effect on ligand binding, green colored sticks show decrease in ligand binding upon mutation, while orange colored sticks show residues whose mutation led to increase in ligand binding. D138 side chain is shown for reference, though affinity data for D138A and D138N mutations were not obtained due to low affinity of 6H-U69593 radioligand. Tightly bound water molecules directly involved in ligand binding are shown as red spheres.
Functional characterization of the novel KOR ligands
The top 11 sub-micromolar compounds from the analog optimization set and the top 6 original VLS hits were further characterized in Gi-cAMP and β-arrestin recruitment (Tango) assays to probe their functional profiles at KOR (Figures 6 and 7). Most of the new KOR ligands exhibited pronounced antagonist activity in Tango assays (Figure 6a), with the ligand 64 completely inhibiting Sal A-induced β-arrestin recruitment. In the agonist mode of the Tango assays, only compound 28 showed measurable β-arrestin recruitment activity at 10 μM (Figure 6b and 7a). In contrast, in the Gi-mediated inhibition of cAMP production assays, a number of compounds showed agonism at 10 μM, with compounds 28 and 81 inducing as much activity as Sal A (95% and 80% of Sal A activity, respectively) (Figure 6c). Several other compounds (40, 69, 72, 70) showed partial (40%–60%) activation of the receptor in the cAMP assays.
Figure 6. Functional characterization of the most potent ligands at KOR receptors.
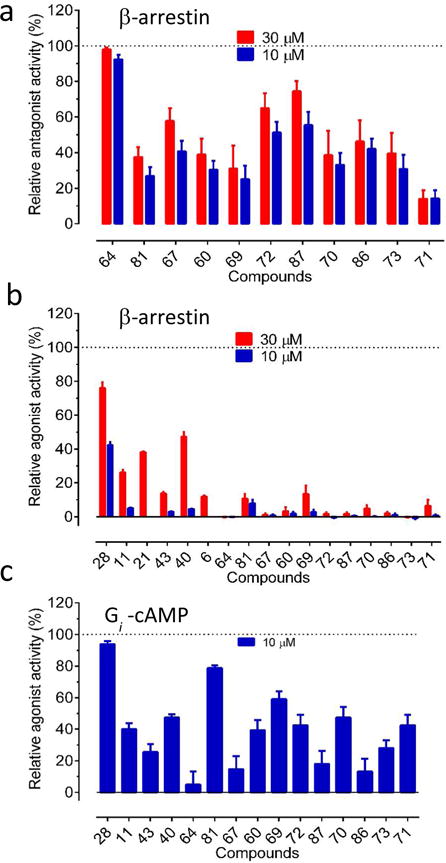
Selected compounds were tested in β-arrestin recruitment (a and b) and Gi-mediated inhibition of cAMP production (c). Values represented mean ± SEM from a minimum of 4 measurements. Results were normalized and analyzed in Prism. (a) Relative antagonist and (b) agonist activity in recruiting β-arrestin at two ligand concentrations. Agonist activity was normalized to that of Sal A (100%) and antagonist activity was normalized to that of 5-GNTI (100%). (c) Relative agonist activity in Gi-mediated inhibition of cAMP production. Results were normalized to that of Sal A (100%).
Figure 7. Functional characterization of selected compounds.
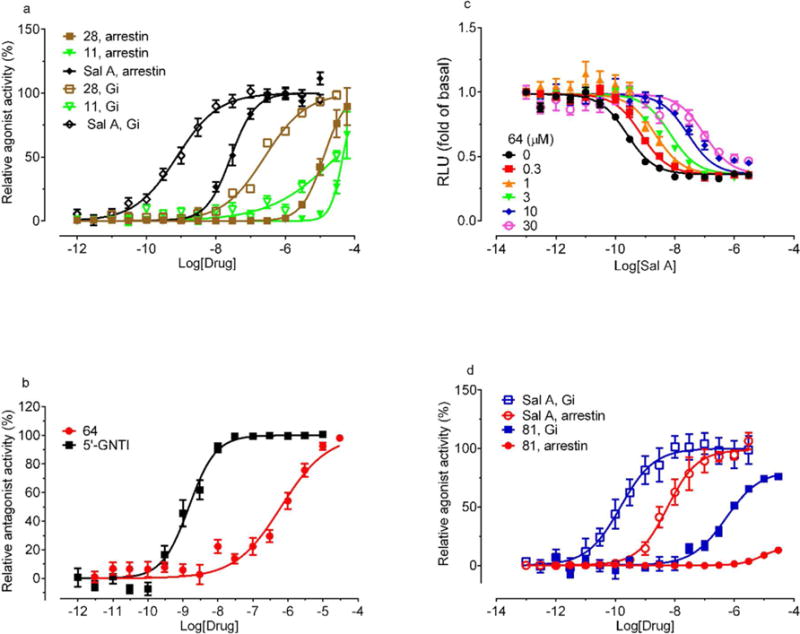
(a) KOR agonist activity of compounds 28 and 11 in β-arrestin recruitment and Gi-mediated inhibition of cAMP production, with Sal A as positive control. (b) Compound 64 concentration-dependent inhibition of Sal A in β-arrestin recruitment assays with 5-GNTI as a positive control. (c) Compound 64 concentration-dependent inhibition of Sal A mediated Gi activation. Schild analysis resulted in pA2 of 6.78 ± 0.11 and Schild slope of 1.15 ± 0.06, and average KB = 0.017 μM. Results were normalized to fold of basal. (d) Comparison of compound 81 activity at both β-arrestin and Gi pathways. Values represented mean ± SEM from a minimum of 3 independent assays, each in triplicate or quadruplicate. Results were normalized, corrected with its nonspecific activity at control cells if necessary, and pooled for analysis in Prism.
The highest affinity compounds 64 and 81 were then further tested for their antagonist and agonist activity in a concentration-dependent manner. In the β-arrestin recruitment assays (Figure 7b), compound 64 inhibited Sal A activity with an IC50 value of 1.1 μM (pIC50 = 5.96 ± 0.33). According to Cheng-Prusoff equation,45 the estimated functional affinity of 64 was 0.23 μM (pKi = 6.63 ± 0.20), while in the Gi-mediated inhibition of cAMP production assays, Schild analysis indicated KB = 0.17 μM (Figure 7c). The values from these two functional assays were comparable with each other and with the compound 64 binding affinity (Ki = 0.09 μM), suggesting consistent antagonism of 64 in the sub-micromolar range.
Most interesting though, agonist 81 showed much stronger agonist activity in the Gi-pathway (EC50 = 0.53 μM) than in the arrestin pathway (EC50 = 8.1 μM) and analysis of the pooled results46 indicated that compound 81 has a Gi-bias factor of 6.0 over arrestin recruitment (Figure 7d). Compounds 28 and 81, the two with the strongest KOR agonist activity, were also tested at DOR and MOR for Gi-mediated inhibition of cAMP production. Results show at least 50% and 70% reduced agonist activity at either DOR or MOR (Figure S1), suggesting KOR selectivity of these compounds.
DISCUSSION
Development of novel antagonists and G-protein biased agonists for KOR is a highly promising strategy for effective and potentially liability-free treatment of pain, anxiety and addictive disorders.20, 21 This study employed crystal structure-derived models of KOR26 and a multi-template VLS approach to discover new fragment-like and lead-like chemotypes for KOR. The initial round of structure-based prospective VLS resulted in a high hit rate (32%) and the discovery of at least six novel KOR chemotypes, with best ligand affinity Ki = 0.2 μM. High prospective hit rate, diversity and potency of the best hits in this study are on par with most successful structure-based VLS campaigns for small-ligand-binding GPCRs including adrenergic β2-adrenergic,29 adenosine A2A,30, 31 histamine H1,32 and dopamine D3,35,33 receptors. This outcome is very encouraging for KOR, because its large orthosteric site is optimal for binding of endogenous peptides, but represents a substantial challenge for structure-based discovery of small molecule ligands.36, 37
Our initial benchmarking of the structural VLS models and prospective screening results suggest several key strategies helping to improve VLS performance and discover chemically and functionally diverse high affinity chemotypes. One of them is inclusion of relevant tightly bound water molecules in the screening models. Thus, the initial benchmarks of the KOR structure-based models with a set of known ligands suggested poor performance of the structural models that have all water molecules removed. Inclusion of the tightly bound waters, conserved between high-resolution opioid receptor structures yielded Model 1. This model improved docking poses and scores for JDTic and similar compounds, due to formation of the water mediated interactions with the receptor. Water interactions play important role in ligand binding, and accounting for water molecules is essential in ligand docking for many GPCRs,47 including opioid receptors.26,48, 49 Therefore, we believe that the current trend for explicit inclusion of relevant waters in VLS models30, 50, 51 is an important step for improved virtual screening efficiency in GPCRs.
Another major factor for effective VLS in this study is ligand-guided optimization strategy to generate multiple screening models for KOR.40, 52 Thus, our assessment showed that even though water inclusion in Model 1 improved binding of JDTic-like compounds, the model was still suboptimal in VLS benchmarks of other compounds, such as morphinans. Ligand-guided optimization with morphinan 6′-GNTI as a seed compound led to Models 2 and 3 with dramatically improved performance for morphinan-based compounds and overall (Figure 1). While Model 2 was limited to side-chain optimization of the KOR pocket, Model 3 was generated using homology modeling, starting with the high-resolution crystal structure of morphinan-bound DOR.27 The rationale for using a homology model is very high sequence and structural conservation between the core binding pockets of the two opioid receptors, and a hypothesis that morphinan scaffolds make similar interactions in the two receptors, shaping the pocket conformations in the two receptors similarly. All three models were employed in perspective VLS using the multiple template approach,53 which resulted in a 26% hit rate for Model 1, and even higher 35% and 40% hit rates for optimized Models 2 and 3 respectively. Though the number of compounds tested for each model is not sufficient to establish statistical significance of improved hit rates for Models 2 and 3, this result suggests that ligand-optimized VLS models, including homology-based, can be beneficial for overall performance of screening and yield more chemically and functionally diverse chemotypes.
We also noted that reproducibility of the KOR docking models critically depends on extended ligand sampling in docking procedure, requiring at least 5 times more sampling as compared to GPCRs with small molecule cognate ligands. As mentioned above, the large size and relatively open shape of the KOR binding pocket is more suitable for peptide interactions than for small molecule binding. It creates a substantial challenge that should be taken into account in VLS attempts not only for opioid receptors, but for other peptide and protein-binding receptors.
Based on experimental validation, the high ligand efficiency (LE) for many of the discovered compounds (LE > 0.40 kcal/mol per heavy atom) suggests the potential utility of these KOR ligands as leads for optimization. Indeed, the analogue search performed for the best six chemotypes resulted in identification of 33 additional hits with Ki < 10 μM, of which 11 had sub-micromolar affinities in the binding assays. Scaffolds 28, 21 and 6 have multiple sub-micromolar analogues and, therefore, present promising candidates for further optimization for their affinity, selectivity and functional properties.
Functional assessments performed for the new sub-micromolar ligands shows KOR antagonism for most of these compounds in both β-arrestin recruitment (Tango assay) and G-protein activation (Gi-cAMP assay). The most potent antagonist identified is compound 64 (Scaffold 6), which has the highest affinity (Ki = 0.09 μM), and comparable potencies Ki = 0.23 μM and KB = 0.17 μM in β-arrestin and G-protein assays respectively (Figure 7b,c), suggesting that 64 is a balanced antagonist. The two most potent agonists discovered here belong to the same chemotype (scaffold 28) and have similar affinities (Ki = 0.16 μM). However, while 28 has balanced activity in both arrestin and G-protein assays, compound 81 apparently shows characteristics of G-protein biased agonist with G-protein KB= 0.17 μM, and Gi-bias factor of 6.0 over arrestin recruitment (Figure 7d). Interestingly, compound 81 represents a substructure of 28, after removal of the methyl moiety from its amino group and 3-fluoromethyl from diazole ring. The first modifications can increase flexibility of the ligands at the amine located in the middle of the compound and anchored at Asp138 side chain conserved in opioid receptors. The other modification removes the 3-fluoromethyl group that points towards conserved Trp287 side chain, which is implicated in activation mechanism in opioid receptors.54 Both changes can easily impact the activation profile, and lead to biased signaling of 81, though further SAR analysis will be required to fully understand the mechanisms involved in biased signaling of KOR.
CONCLUSIONS
Prospective VLS for novel chemotypes of KOR ligands shows high hit rate for fragment-like and hit like compound libraries (32%), combined with high ligand affinities (Ki= 0.09 μM) and ligand efficiencies (LE > 0.40 kcal/mol per heavy atom) for the best hit compounds suggesting their amenability for optimization. The initial round of optimization resulted in 11 sub-micromolar hits based on four distinct novel chemotypes, providing further insights for lead optimization of these scaffolds. Functional characterization reveals a number of antagonists with the best KB = 0.17 μM, and a few agonists, most potent of which, 81, has a strong preference to G-protein pathway (bias factor 6.0, calculated with Sal A as a reference compound as described in ref.46), which has been associated lower addiction liability and fewer side effectsfor KOR-targeted analgesics.22
EXPERIMENTAL SECTION
Binding assays
Radioligand binding assays with 6H-U69593 were performed using cloned, human KOR receptors expressed in HEK293 cells as previously detailed in ref.55 and PDSP assay protocols (https://pdspdb.unc.edu/pdspWeb/?site=assays). Initial primary screening assays were carried out at the single concentration of 50 μM in quadruplicate. Compounds with a minimum of 30% inhibition were subjected to concentration-response binding assays (12 points ranging from 100 μM to 10 nM in triplicate) to determine their binding affinities, Ki. Quality and purity of all tested compounds was verified using mass spectrometry; 5 out of 48 initially obtained compounds (1, 14, 23, 32, 47) were excluded from further consideration due to inadequate quality. With the exception of compounds 6, 20 and 30, which had >90% purity, all the remaining compounds were certified at >95% purity (Table S1).
Ligand efficiency (LE) was calculated for the hits as the ratio of experimental affinity and number of heavy atoms in the molecule: LE = 1.4(−logKi)/N (ref44, 56). High value of LE (usually > 0.3 kcal/mol per heavy atom) is considered one of the key properties of a compound that reflects its propensity for successful lead optimization.
KOR-arrestin translocation assays
KOR Tango assays were performed as described.57 Briefly, HTLA cells were transfected overnight and then plated in DMEM supplemented with 1% dialyzed FBS in Poly-L-Lys (PLL) coated 384-well white clear bottom cell culture plates at a density of 15,000 cells per well in total of 40 μl. The cells were incubated for at least 6 h before receiving drug stimulation. Drug solutions were prepared in DMEM with 1% dFBS at 5× and added to cells (10 μl per well) for overnight incubation. For antagonist assays, 30 nM of reference agonist (Sal A) was added 30 min after compounds. The next day, media and drug solutions were removed and 20 μl per well of BrightGlo reagents (purchased from Promega, after 1:20 dilution) were added. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted and analyzed in GraphPad Prism 5.0.
cAMP assays
HEK293T cells were co-transfected with human KOR receptor along with a split luciferase-based cAMP biosensor (GloSensor; Promega) as described57 with modifications. In brief, transfected cells were plated in Poly-L-Lys coated 384-well white clear bottom cell culture plates with DMEM + 1% dialyzed FBS at a density of 15,000 cells per 40 μl per well overnight. On the day of assay, cells were removed of culture medium and loaded with 20 μl/well assay buffer (20 mM HEPES, 1× HBSS, pH 7.4) supplemented with 4 mM luciferin for 1 hour. To measure agonist activity for Gi coupled receptors, we added 10 μl of 3× of drug solutions for 15 min, followed by 10 μl of Isoproterenol at a final concentration of 200 nM (to activate Gs via endogenous β2-adrenergic receptors), and luminescence intensity was quantified 15 min later. To measure antagonist activity at Gi coupled receptors, cells were preincubated with drugs for 15 min before the addition of 20 nM reference agonist (Sal A) for another 15 min, then 10 μl of isoproterenol at final concentration of 200 nM was added and luminescence was quantified 15 min later. Results (relative luminescence units) were plotted and analyzed in GraphPad Prism 5.0. Transduction coefficients log(τ/KA) and ligand bias ΔΔlog(τ/KA) were calculated as described by Kenakin et al.46
Mass spectrometry analysis
LCMS data for all compounds were acquired using an Agilent 6110 series system with a UV detector set to 220 nm. To each well of a Greiner 96 well plate with 2 μL of each sample (10 mM solution in DMSO) we added 38 μL of MeOH. After mixing, 15 μL of each sample was injected onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μm, C18 column at room temperature (rt) at a flow rate of 1.0 mL/min. A linear gradient from 10% to 100% (vol/vol) B over 5.0 min followed by 2.0 min at 100% B with a mobile phase of (A) H2O + 0.1% acetic acid and (B) MeOH + 0.1% acetic acid was used. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 series single quadrupole mass spectrometer with an electrospray ionization (ESI) source.
Generation of Receptor Models
The virtual screening models of KOR were prepared from three different sources. The first model (Model 1) was built using the KOR crystal structure (PDB ID: 4DJH) with the molecular object conversion procedure implemented in ICM molecular modeling software (www.molsoft.com). The procedure includes the addition of hydrogen atoms, selection of the energetically favorable conformations of His, Asn, and Gln side chains, and local minimization of polar hydrogens using the internal coordinates space. Five water molecules in the orthosteric binding site, preserved in high resolution structures of opioid receptors were retained in the model. Hydrogen atoms of the structured water molecules were co-optimized together with KOR hydrogens, while coordinates of water oxygen atoms were retained as in the crystal structure. The second receptor model (Model 2) was generated starting with the same KOR crystal structure (PDB ID: 4DJH) by co-optimizing it with the morphine derivative 6′-GNTI. The compound 6′-GNTI was initially placed into the binding site by superimposing it with the naltrindole as in DOR complex structure27 (DOR, PDB ID 4N6H). The resulting KOR-6′-GNTI complex was optimized using energy-based global optimization with ICM Biased probability Monte Carlo procedure,58 where full flexible sidechains of residues within 5 Å of the ligand were extensively sampled to accommodate the compound.
The third receptor model (Model 3) was built using KOR homology model generated from the high resolution structure of DOR (PDB ID: 4N6H)27, which shares as much as 70% sequence identity to KOR. The homology modeling procedure in the ICM Model Builder58 inherits the conformation of the template backbone in the conserved residues regions, while re-optimizing conformation of the non-conserved sidechains and loop regions. Finally, the global energy optimization procedure was then performed for the KOR-6′-GNTI complex as describe for Model 2 above.
To validate the ICM VLS models, a small set of 100 known KOR ligands from the ChEMBL database59 were mixed with a decoy set of 900 compounds, and docked into all three receptor models. The docking models were analyzed for their ability to separate ligands from decoys by calculating its Receiver Operator Characteristic (ROC) curves, which were plotted with True Positive Rate on the Y axis vs False Positive Rate on the X axis. Based on the calculated area under the ROC curve (AUC) and normalized square root AUC (NSQ_AUC) (ref.40) the ligand-optimized Models 2 and 3 show improved VLS benchmark performance as compared to the original Model 1.
Virtual Ligand Libraries
Lead-like and fragment-like libraries were built using commercial compounds from five vendors (Chembridge, Enamine, Life Chemicals, Chemdiv and Maybridge). The libraries contain 4.5 million compounds in total. Duplicates and compounds with molecular weight more than 500 Da were removed. Compounds were also filtered to exclude reactive functional groups and promiscuous PAINS chemotypes.60 Compound formal charges were assigned at pH 7 using the pKa calculation module within ICM software.58
The Tanimoto chemical similarity distances between the VLS hits and known ligands of KOR were calculated using dynamic linear fingerprints in the ICM-pro suite.58 Compounds with a Tanimoto distance of less than 0.3 Å are considered similar in a chemical search. The search was performed in the ChEMBL database59 subset of ligands with affinity higher than 10 μM to any of the KOR orthologues.
Virtual Screen
Virtual screens of the 4.5 million compounds library for each model were carried out using the ICM VLS procedure with partial flexibility in the binding pocket.58 The receptor all-atom models were transformed into energy potential maps calculated using a fine 3D grid (0.5 Å). Energy terms including van der Waals, hydrogen-bonding, hydrophobic, and electrostatic interactions between the ligand and the receptor were taken into account. The anchor binding sidechain of Asp138 was excluded from this grid potentials, and its conformation and interactions with ligand were treated explicitly as in an all-atom model with flexible torsion angles. In virtual screen docking, ligands were given full torsion flexibility in internal coordinates. The docking was performed using a Monte Carlo global energy minimization procedure. The conformation of each compound was sampled using BPMC algorithm61 and scored to find the best binding energy. The initial benchmark virtual screens with the default thoroughness level 1 yielded only ~25% redundancy among the top ranked compounds predicted in two independent runs. The same test performed using thoroughness level of 5, however, achieved ~60% overlap between independent screening results. This benchmark suggests that the large size of the KOR binding pocket may require a higher thoroughness level of sampling to achieve robust docking convergence. The final virtual screen for the full compound library was performed with a thoroughness level of 5 to enhance convergence. No empirical constraints on ligand conformation were used in the docking. Overall, each virtual screen run on the 4.5 million compound library used 28,000 CPU core hours on Linux workstations totaling 128 CPUs cores.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants DA035764 (RCS, BLR and VK), and DA038858 (VK), as well as National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. The authors thank Angela Walker for assistance with manuscript preparation.
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- KOR
κ-opioid receptor
- DOR
δ-opioid receptor
- MOR
μ-opioid receptor
- VLS
Virtual Ligand Screening
- LE
Ligand Efficiency
- SAR
structure activity relationship
- AUC
Area Under Curve
Footnotes
Supporting Information. Supporting information includes chemical structures, purity, binding and functional data for all experimentally assessed compounds in this study. The Supporting Information is available free of charge on the ACS Publications website.
Author contributions: ZZ performed virtual screening, analyzed results and wrote the manuscript, XPH, TJM, RZ, XC performed experiments, analyzed results and helped to write the manuscript, SZ analyzed results, BLN, RCS analyzed results and helped to write the manuscript, VK conceived and coordinated the study, analyzed results and wrote the manuscript.
Notes: The authors declare no competing financial interest.
References
- 1.Pasternak GW. Opioids and their receptors: Are we there yet? Neuropharmacology. 2014;76(Pt B):198–203. doi: 10.1016/j.neuropharm.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 3.Lutz PE, Kieffer BL. Opioid receptors: Distinct roles in mood disorders. Trends Neurosci. 2013;36:195–206. doi: 10.1016/j.tins.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dart RC, Surratt HL, Cicero TJ, Parrino MW, Severtson SG, Bucher-Bartelson B, Green JL. Trends in opioid analgesic abuse and mortality in the united states. N Engl J Med. 2015;372:241–248. doi: 10.1056/NEJMsa1406143. [DOI] [PubMed] [Google Scholar]
- 5.Kolodny A, Courtwright DT, Hwang CS, Kreiner P, Eadie JL, Clark TW, Alexander GC. The prescription opioid and heroin crisis: A public health approach to an epidemic of addiction. Annu Rev Public Health. 2015;36:559–574. doi: 10.1146/annurev-publhealth-031914-122957. [DOI] [PubMed] [Google Scholar]
- 6.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van’t Veer A, Carlezon WA., Jr Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frankel PS, Alburges ME, Bush L, Hanson GR, Kish SJ. Striatal and ventral pallidum dynorphin concentrations are markedly increased in human chronic cocaine users. Neuropharmacology. 2008;55:41–46. doi: 10.1016/j.neuropharm.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan F, Roth BL. Salvinorin a: A novel and highly selective kappa-opioid receptor agonist. Life Sci. 2004;75:2615–2619. doi: 10.1016/j.lfs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Piercey MF, Lahti RA, Schroeder LA, Einspahr FJ, Barsuhn C. U-50488h, a pure kappa receptor agonist with spinal analgesic loci in the mouse. Life Sci. 1982;31:1197–1200. doi: 10.1016/0024-3205(82)90341-1. [DOI] [PubMed] [Google Scholar]
- 11.Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll FI, Carlezon WA., Jr Development of kappa opioid receptor antagonists. J Med Chem. 2013;56:2178–2195. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrich JM, Phillips PE, Chavkin C. Kappa opioid receptor activation potentiates the cocaine-induced increase in evoked dopamine release recorded in vivo in the mouse nucleus accumbens. Neuropsychopharmacology. 2014;39:3036–3048. doi: 10.1038/npp.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson KJ, Carroll FI, Negus SS, Damaj MI. Effect of the selective kappa-opioid receptor antagonist JDTic on nicotine antinociception, reward, and withdrawal in the mouse. Psychopharmacology (Berl) 2010;210:285–294. doi: 10.1007/s00213-010-1803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll FI, Harris LS, Aceto MD. Effects of JDTic, a selective kappa-opioid receptor antagonist, on the development and expression of physical dependence on morphine using a rat continuous-infusion model. Eur J Pharmacol. 2005;524:89–94. doi: 10.1016/j.ejphar.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Carroll I, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Pharmacological properties of JDTic: A novel kappa-opioid receptor antagonist. Eur J Pharmacol. 2004;501:111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 17.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Identification of (3r)-7-hydroxy-n-((1s)-1-[[(3r,4r)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro- 3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 18.Chavkin C, Martinez D. Kappa antagonist JDTic in phase 1 clinical trial. Neuropsychopharmacology. 2015;40:2057–2058. doi: 10.1038/npp.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melief EJ, Miyatake M, Carroll FI, Beguin C, Carlezon WA, Jr, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. Duration of action of a broad range of selective kappa-opioid receptor antagonists is positively correlated with c-jun n-terminal kinase-1 activation. Mol Pharmacol. 2011;80:920–929. doi: 10.1124/mol.111.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urbano M, Guerrero M, Rosen H, Roberts E. Antagonists of the kappa opioid receptor. Bioorg Med Chem Lett. 2014;24:2021–2032. doi: 10.1016/j.bmcl.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 21.Carlezon WA, Jr, Krystal AD. Kappa-opioid antagonists for psychiatric disorders: From bench to clinical trials. Depression Anxiety. 2016;33:895–906. doi: 10.1002/da.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. The G protein-biased kappa-opioid receptor agonist rb-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rives ML, Rossillo M, Liu-Chen LY, Javitch JA. 6′-guanidinonaltrindole (6′-gnti) is a G protein-biased kappa-opioid receptor agonist that inhibits arrestin recruitment. J Biol Chem. 2012;287:27050–27054. doi: 10.1074/jbc.C112.387332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, Greenhill SD, Hale VA, Sharman JL, Bonner TI, Catterall WA, Davenport AP, Delagrange P, Dollery CT, Foord SM, Gutman GA, Laudet V, Neubig RR, Ohlstein EH, Olsen RW, Peters J, Pin JP, Ruffolo RR, Searls DB, Wright MW, Spedding M. IUPHAR-DB: The IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2009;37:D680–685. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dogra S, Yadav PN. Biased agonism at kappa opioid receptors: Implication in pain and mood disorders. Eur J Pharmacol. 2015;763:184–190. doi: 10.1016/j.ejphar.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of delta-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci USA. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katritch V, Jaakola VP, Lane JR, Lin J, Ijzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R. Structure-based discovery of novel chemotypes for adenosine a(2a) receptor antagonists. J Med Chem. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlsson J, Yoo L, Gao ZG, Irwin JJ, Shoichet BK, Jacobson KA. Structure-based discovery of a2a adenosine receptor ligands. J Med Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, Iwata S, Shimamura T, Stevens RC, de Esch IJ, Leurs R. Crystal structure-based virtual screening for fragment-like ligands of the human histamine h(1) receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine d(3) receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss DR, Ahn S, Sassano MF, Kleist A, Zhu X, Strachan R, Roth BL, Lefkowitz RJ, Shoichet BK. Conformation guides molecular efficacy in docking screens of activated beta-2 adrenergic G protein coupled receptor. ACS Chem Biol. 2013;8:1018–1026. doi: 10.1021/cb400103f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane JR, Chubukov P, Liu W, Canals M, Cherezov V, Abagyan R, Stevens RC, Katritch V. Structure-based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol Pharmacol. 2013;84:794–807. doi: 10.1124/mol.113.088054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daga PR, Polgar WE, Zaveri NT. Structure-based virtual screening of the nociceptin receptor: Hybrid docking and shape-based approaches for improved hit identification. J Chem Inf Model. 2014;54:2732–2743. doi: 10.1021/ci500291a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Negri A, Rives ML, Caspers MJ, Prisinzano TE, Javitch JA, Filizola M. Discovery of a novel selective kappa-opioid receptor agonist using crystal structure-based virtual screening. J Chem Inf Model. 2013;53:521–526. doi: 10.1021/ci400019t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. Structure-based ligand discovery for the protein-protein interface of chemokine receptor cxcr4. Proc Natl Acad Sci USA. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, Huang XP, Sassano MF, Giguere PM, Lober S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katritch V, Rueda M, Abagyan R. Ligand-guided receptor optimization. Methods Mol Biol. 2012;857:189–205. doi: 10.1007/978-1-61779-588-6_8. [DOI] [PubMed] [Google Scholar]
- 41.Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Structure and dynamics of the m3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vardy E, Mosier PD, Frankowski KJ, Wu H, Katritch V, Westkaemper RB, Aube J, Stevens RC, Roth BL. Chemotype-selective modes of action of kappa-opioid receptor agonists. J Biol Chem. 2013;288:34470–34483. doi: 10.1074/jbc.M113.515668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hopkins AL, Groom CR, Alex A. Ligand efficiency: A useful metric for lead selection. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 45.Cheng Y, Prusoff WH. Relationship between the inhibition constant (k1) and the concentration of inhibitor which causes 50 per cent inhibition (i50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 46.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Miller RL, Thompson AA, Trapella C, Guerrini R, Malfacini D, Patel N, Han GW, Cherezov V, Calo G, Katritch V, Stevens RC. The importance of ligand-receptor conformational pairs in stabilization: Spotlight on the n/ofq G protein-coupled receptor. Structure. 2015;23:2291–2299. doi: 10.1016/j.str.2015.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldfeld DA, Murphy R, Kim B, Wang L, Beuming T, Abel R, Friesner RA. Docking and free energy perturbation studies of ligand binding in the kappa opioid receptor. J Phys Chem B. 2015;119:824–835. doi: 10.1021/jp5053612. [DOI] [PubMed] [Google Scholar]
- 50.Langmead CJ, Andrews SP, Congreve M, Errey JC, Hurrell E, Marshall FH, Mason JS, Richardson CM, Robertson N, Zhukov A, Weir M. Identification of novel adenosine a(2a) receptor antagonists by virtual screening. J Med Chem. 2012;55:1904–1909. doi: 10.1021/jm201455y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beuming T, Lenselink B, Pala D, McRobb F, Repasky M, Sherman W. Docking and virtual screening strategies for gpcr drug discovery. Methods Mol Biol. 2015;1335:251–276. doi: 10.1007/978-1-4939-2914-6_17. [DOI] [PubMed] [Google Scholar]
- 52.Rueda M, Totrov M, Abagyan R. Alibero: Evolving a team of complementary pocket conformations rather than a single leader. J Chem Inf Model. 2012;52:2705–2714. doi: 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: A fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2009;52:397–406. doi: 10.1021/jm8009958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Demene H, Granier S. Propagation of conformational changes during mu-opioid receptor activation. Nature. 2015;524:375–378. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shultz MD. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg Med Chem Lett. 2013;23:5980–5991. doi: 10.1016/j.bmcl.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 57.Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, Sciaky N, Roth BL. Presto-tango as an open-source resource for interrogation of the druggable human gpcrome. Nat Struct Mol Biol. 2015;22:362–369. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abagyan RA, Orry A, Raush E, Budagyan L, Totrov M. ICM manual, 3.8. MolSoft LLC; La Jolla, CA: 2016. [Google Scholar]
- 59.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. Chembl: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 61.Abagyan R, Totrov M. Biased probability monte carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.