

We demonstrate the use of selective electrocatalysts to run a direct methanol fuel cell at high concentrations of methanol.
Keywords: Direct methanol fuel cell, Nanocomposite, nanoparticle, Electrocatalyst, High-concentration methanol
Abstract
Owing to the serious crossover of methanol from the anode to the cathode through the polymer electrolyte membrane, direct methanol fuel cells (DMFCs) usually use dilute methanol solutions as fuel. However, the use of high-concentration methanol is highly demanded to improve the energy density of a DMFC system. Instead of the conventional strategies (for example, improving the fuel-feed system, membrane development, modification of electrode, and water management), we demonstrate the use of selective electrocatalysts to run a DMFC at high concentrations of methanol. In particular, at an operating temperature of 80°C, the as-fabricated DMFC with core-shell-shell Au@Ag2S@Pt nanocomposites at the anode and core-shell Au@Pd nanoparticles at the cathode produces a maximum power density of 89.7 mW cm−2 at a methanol feed concentration of 10 M and maintains good performance at a methanol concentration of up to 15 M. The high selectivity of the electrocatalysts achieved through structural construction accounts for the successful operation of the DMFC at high concentrations of methanol.
INTRODUCTION
Direct methanol fuel cell (DMFC) technology has the potential to prevail as a leader in the booming market for portable electronic devices because of its advantages of high energy density and quick refueling, which are crucial characteristics of portable power systems (1–4). In general, dilute methanol solutions, for example, 1 to 2 M for active DMFCs or about 3 M for passive DMFCs, are often used as fuel in DMFCs to inhibit the crossover of methanol from the anode to the cathode to achieve high performance (5, 6). However, to compete with lithium-based rechargeable batteries that currently dominate the portable power market, the use of high-concentration methanol as fuel is highly demanded to capitalize the high energy density of DMFCs. It has been reported that the specific energy of a DMFC system can be comparable with that of conventional Li-ion batteries only when a methanol solution with a concentration of 9 M or higher is fed as fuel (assuming that the overall efficiency of a DMFC system is 20%) (7).
Here, instead of the commonly used strategies [for example, improving the fuel-feed system (8–12), membrane development (13–17), modification of electrode (18–22), and water management (23–25)], we demonstrate the use of selective electrocatalysts to run a DMFC at high concentrations of methanol. In brief, at an operating temperature of 80°C, the as-fabricated DMFC with core-shell-shell Au@Ag2S@Pt nanocomposites at the anode for methanol oxidation reaction (MOR) and core-shell Au@Pd nanoparticles at the cathode for oxygen reduction reaction (ORR) produces a maximum power density of 89.7 mW cm−2 at a methanol feed concentration of 10 M and maintains good performance at a methanol concentration of up to 15 M. The good catalytic selectivity of the electrocatalysts achieved through structural construction accounts for the successful operation of the DMFC at high concentrations of methanol, and this concept may shed some light on the design of more cost-effective and efficient DMFC systems.
RESULTS AND DISCUSSION
The assembled DMFC with a total size of 8.9 cm × 9.9 cm × 10 cm is illustrated in Fig. 1 (scheme and practical photo). The membrane electrode assembly (MEA) in the assembled DMFC has an active area of 10 cm2 and a catalyst loading of 2 mg cm−2. The DMFC was tested on an electrochemical interface (Bio-Logic VMP3) at room temperature and ambient pressure. Methanol solutions with different concentrations (0.5 to 15 M) were supplied by a peristaltic pump with high precision (Kamoer LIs Plus), whereas sufficient pure oxygen gas was provided at a flow rate of 300 ml min−1. A constant current was applied, and the output voltage was monitored for a while until the final steady-state value was recorded. No leakage occurred during the long-term test.
Fig. 1. DMFC assemblies.
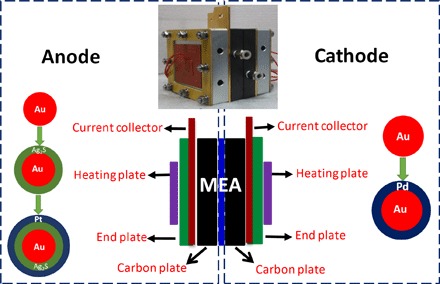
Schematic showing the DMFC fabricated with selective electrocatalysts at the anode and cathode chamber. Inset: Photograph of a practical cell.
As schematically indicated in Fig. 1, we chose nanocomposites composed of Au, Ag2S, and Pt as the anode catalysts in the assembled DMFC, which have been verified to have high selectivity for the MOR because of the strong electronic coupling effect among different domains in the Au-Ag2S-Pt nanocomposites (26–28). The strategies used to prepare Au-Ag2S-Pt nanocomposites were usually carried out either by successive reduction of Au and Pt ions in the presence of Ag2S seeds in aqueous solution (29) or by a structural conversion process using core-shell Ag@Pt nanoparticles as precursors (30, 31). Unfortunately, deficiencies in current synthetic routes are apparent. For the seed-mediated growth, the expensive nature of BSPP [bis(p-sulfonatophenyl)phenylphosphane], which was used to direct the synthesis of Ag2S seeds, and the aqueous phase render that the synthesis of Au-Ag2S-Pt nanocomposites could only be performed at very low concentrations (29), whereas for the structural conversion route, the formed Au-Ag2S-Pt nanocomposites have very low surface areas exposed to the electrochemical reactions due to the high content of Ag in the core-shell Ag-Pt precursors (30). Instead, we developed a seed-mediated growth in an organic medium to produce Au-Ag2S-Pt nanocomposites. The strategy starts with preparing Au particles in oleylamine at an elevated temperature, which are then coated by Ag shells, followed by reacting with element sulfur to convert the Ag shells into Ag2S on the Au particle surface. Finally, the core-shell Au@Ag2S nanoparticles are used as seeds for the deposition of Pt metal, resulting in the formation of Au-Ag2S-Pt nanocomposites. The microscopic analyses (Fig. 2, A and B) indicate that the as-prepared Au-Ag2S-Pt nanocomposites are uniform in size (ca. 14.4 nm). In particular, in contrast to those synthesized in the aqueous phase, in which separated Pt dots are decorated on multiple sites on the surface of Ag2S nanocrystals (29), the nanoscale mapping (Fig. 2, C to H) and elemental profile analyses (Fig. 2I) confirm that the as-prepared Au-Ag2S-Pt nanocomposites in this study have core-shell-shell constructions. As exhibited in fig. S1 (A and B), the core-shell-shell Au@Ag2S@Pt nanocomposites were loaded on carbon substrates to examine their catalytic performance for electrochemical reactions. The electrochemically active surface area (ECSA) determined by cyclic voltammetry (fig. S2A) is 96.9 m2 gPt−1 for the core-shell-shell Au@Ag2S@Pt nanocomposites, slightly lower than that for commercial Pt/C catalysts (117.0 m2 gPt−1) but much higher than that for the Au-Ag2S-Pt nanocomposites obtained by aqueous synthesis (84.5 m2 gPt−1) or by structural conversion (24.8 m2 gPt−1). As expected, the core-shell-shell Au@Ag2S@Pt nanocomposites display superior activity for MOR, low activity for ORR, and better durability for MOR compared with commercial Pt/C catalysts, as shown in fig. S2 (B to D, respectively). The high MOR activity due to the electronic coupling effect renders the core-shell-shell Au@Ag2S@Pt nanocomposites a good candidate as selective catalysts at the anode of DMFCs.
Fig. 2. Core-shell-shell Au@Ag2S@Pt nanocomposites.
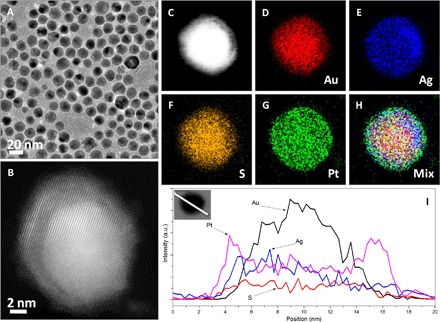
Transmission electron microscopy (TEM) image (A), aberration-corrected high-angle dark-field scanning TEM (STEM) image (B), nanoscale element mappings (C to H), and elemental profiles in STEM mode (I) of core-shell-shell Au@Ag2S@Pt nanocomposites prepared in oleylamine at elevated temperatures. a.u., arbitrary units.
We use core-shell Au@Pd nanoparticles with thin Pd shells as selective electrocatalysts for ORR at the cathode of the DMFC. As has been well documented, the thin Pd layers deposited on the surface of Au seeds or AuPd alloy nanoparticles have superior ORR activity because of the synergistic effects between Au and Pd components, for example, the lateral strain effect due to the lattice mismatch and the electronic coupling effect due to the difference in electronegativities between the two metals (32–35). Here, we extended the Au-catalyzed strategy, which could be used to produce core-shell Au@Pd nanoparticles with Pd shells up to three atomic layers in aqueous solution (36), to the synthesis of core-shell Au@Pd nanoparticles in an organic phase. In brief, Au nanoparticles served as seeds were firstly synthesized in oleylamine, followed by the growth of thin Pd shells, which were controlled by a low Pd/Au molar ratio (1:2) and a relatively low temperature (150°C). Owing to the catalysis of Au seeds, the reduction of Pd ions only occurs on the surface of Au cores, preventing the newly formed Pd atoms from self-nucleation. Figure 3A shows the TEM image of the as-prepared core-shell Au@Pd nanoparticles, indicating that the core-shell products are spherical with an average size of ca. 11.2 nm. The deposited Pd layer is not easily distinguished from the Au core in the TEM image, possibly because of their thin thickness and epitaxial growth on the surface of Au seeds. However, the aberration-corrected high-angle dark-field STEM image (Fig. 3B) reveals the core-shell construction with an Au core of ca. 9.3 nm and a surrounding Pd shell of three or four atomic layers in an arbitrary single particle, which is consistent with nanoscale mapping analyses (Fig. 3, C to F) and elemental profiles (Fig. 3G). The as-prepared core-shell Au@Pd nanocomposites could also be well dispersed on carbon substrates, as displayed in fig. S1 (C and D) as TEM and high-resolution TEM (HRTEM) images. The ECSA based on the unit weight of Pd and calculated from CO stripping voltammograms (fig. S3A) is 60.5 m2 gPd−1 for the core-shell Au@Pd nanoparticles, very close to that for commercial Pd/C catalysts (61.3 m2 gPd−1). Furthermore, as shown in fig. S3B, the half-wave potential for the core-shell Au@Pd nanoparticles is 556 mV, 41 mV more positive than that for the commercial Pd/C catalyst, indicating that the Pd shells in core-shell Au@Pd nanoparticles have higher ORR activity than that in the commercial Pd/C catalyst. Specifically, although the ORR activity of core-shell Au@Pd nanoparticles is lower than that of commercial Pt/C catalysts, which have a half-wave potential of 610 mV, the nondetectable MOR activity for core-shell Au@Pd nanoparticles (fig. S3C) makes them competitive as selective electrocatalysts at the cathode of the fabricated DMFCs. Also, the core-shell Au@Pd nanoparticles have comparable durability with commercial Pt/C catalysts, as evinced in fig. S3D with the chronoamperograms.
Fig. 3. Core-shell Au@Pd nanoparticles.
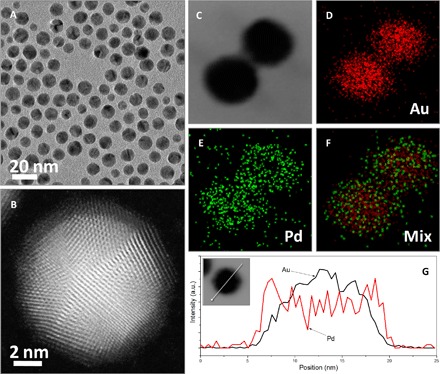
TEM image (A), aberration-corrected high-angle dark-field STEM image (B), nanoscale element mappings (C to F), and elemental profiles in STEM mode (G) of core-shell Au@Pd nanoparticles prepared in oleylamine using an Au-catalyzed strategy.
As demonstrated in a previous study (28), the selectivity of the catalysts for the electrochemical reactions could be evaluated by a prototype of the membraneless DMFC, which consists of a single compartment vessel with two electrodes in a common electrolyte of 1 M methanol in 0.1 M HClO4, as exhibited by the scheme and practical photo in fig. S4A and its inlet. The electrolyte is saturated with dissolved oxygen by continuously bubbling O2 into the solution. The performance of the membraneless DMFC was evaluated in terms of the open circuit voltage (OCV). Figure S4B shows that the membraneless DMFC with the core-shell-shell Au@Ag2S@Pt nanocomposites at the anode and the core-shell Au@Pd nanoparticles at the cathode maintains a stable OCV of ca. 0.30 V for 60 min, completely comparable with that of the same DMFC with a conventional Nafion 117 membrane, whereas the membraneless DMFC operating with commercial Pt/C catalysts at both the anode and the cathode has an OCV close to 0. The prototype does suggest the high selectivity of the core-shell-shell Au@Ag2S@Pt nanocomposites and core-shell Au@Pd nanoparticles for MOR and ORR, which is the essential prerequisite for a DMFC constructed to run at high concentrations of methanol.
Figure 4 shows the performance of the DMFC with selective catalysts, that is, core-shell-shell Au@Ag2S@Pt nanocomposites at the anode and core-shell Au@Pd nanoparticles at the cathode or with commercial Pt/C catalysts at both the anode and the cathode. These tests were performed with methanol concentrations of 0.5, 1, 2, 4, 6, 8, 10, and 15 M at 80°C. The polarization curves exhibit that the performance of the DMFC with selective catalysts at its electrodes (Fig. 4A) was improved compared to that of the DMFC with commercial Pt/C catalysts (Fig. 4C). As indicated in Fig. 4E and fig. S5A, the OCVs of the DMFC with selective electrocatalysts reach 0.59 V, despite the methanol solution concentration reaching up to 15 M. This high OCV manifests the selectivity of the cathode catalysts for ORR, which could overcome the mixed potentials at the cathode, as caused by serious methanol crossover. In case of commercial Pt/C catalysts, the OCVs drop rapidly from 0.58 to 0.49 V as the concentration of methanol increases from 0.5 to 15 M, as shown in Fig. 4E and fig. S5B, suggesting the negative effect induced by methanol crossover. With an improvement in the OCV, the performance of the DMFC with selective electrocatalysts at its electrodes is improved significantly. The DMFC has a maximum power density of 89.7 mW cm−2 under operation with 10 M methanol (Fig. 4, B and F). As summarized in table S1, this value is much higher than those reported in the last 5 years for the DMFCs using various strategies to emphasize high-concentration solutions of methanol (16, 18, 19, 22–24, 37–55). Although a slight decrease in power density is observed when further increasing the methanol concentration due to the complicated fundamentals involved in a DMFC using high concentrations of methanol, the power density of the DMFC with selective catalysts remains to be 82.4 mW cm−2 at the methanol concentration of 15 M. In contrast, a maximum power density of 69.8 mW cm−2 is achieved for the DMFC with commercial catalysts at the methanol concentration of 4 to 6 M, and the power densities decrease to 61.5 and 40.5 mW cm−2 at methanol concentrations of 10 and 15 M, respectively (Fig. 4, D and F).
Fig. 4. Performance of the assembled DMFC with selective or commercial catalysts.
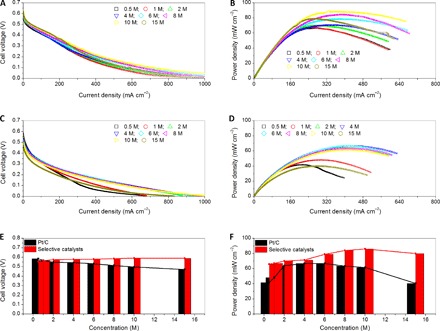
Polarization (A and C) and power density curves (B and D) of the assembled DMFC at different methanol feed concentrations with selective catalysts (A and B) and commercial Pt/C catalysts (C and D); comparison of the assembled DMFC with selective and commercial Pt/C catalysts in terms of open circuit cell voltage (E) and power density (F).
Figure S6 shows the transient discharging voltages at a constant current density (200 mA cm−2), with the cell to be fueled with 1 M methanol solution. Apparently, the discharging voltage remains higher and decreases with a slower rate for the DMFC with selective catalysts compared with that with commercial Pt/C catalysts at its anode and cathode electrodes.
Upon the success in using selective electrocatalysts in a single DMFC, we assembled a 20-W DMFC stack consisting of 15 MEAs connected in series, with a total active area of 225 cm2, and applied it to drive an experimental bulb. As illustrated in fig. S7, we noticed that the7-W white bulb remained glowing, provided that the methanol solution of 10 M is fed, manifesting the potentials for applying the selective catalysts in practical DMFCs. Unfortunately, owing to the lack of facilities, the detailed performance of the DMFC stack is yet to be evaluated.
In summary, we have demonstrated the use of selective electrocatalysts for running a DMFC at high concentrations of methanol of up to 15 M. According to the experimental results, the assembled DMFC with core-shell-shell Au@Ag2S@Pt nanocomposites as selective anode catalyst for MOR and the core-shell Au@Pd nanoparticles as selective cathode catalyst for ORR generates a maximum power density of 89.7 mW cm−2 at a methanol feed concentration of 10 M and maintains acceptable performance at a methanol concentration of up to 15 M. The good catalytic selectivity of the electrocatalysts, which accounts for the successful operation of a DMFC at high concentrations of methanol, will be of benefit to reducing the complexity and increasing the flexibility in the design of DMFCs and hence enhancing their advantages as portable power sources. Additionally, by optimizing the overall size or composition of the core-shell system, for example, using Au nanoclusters with fine diameters as starting materials or a design of a semiconductor-metal system with energy level alignment more favorable for the electronic coupling effect, further enhancement in activity or selectivity for DMFC reactions might be expected.
MATERIALS AND METHODS
General materials
The following materials were used as received: potassium tetrachloroplatinate(II) (K2PtCl4; 98%), silver nitrate (AgNO3; 99%), gold(III) chloride trihydrate [HAuCl4·3H2O; ACS reagent; 49.0% Au basis), palladium(II) acetylacetonate [Pd(acac)2; 99%], acetic acid (CH3COOH; 98%), sulfur powder (S; chemical grade), and oleylamine (primary amine; 95.4%) from J&K Scientific; aqueous HClO4 solution (ACS reagent; 70%) and Nafion 117 solution (5% in a mixture of lower aliphatic alcohols and water) from Aladdin Reagents; ethanol (99%), methanol (99%), and toluene (99.5%) from Beijing Chemical Works; and Vulcan XC 72 carbon powders (XC 72C) with a BET (Brunauer–Emmett–Teller) surface area of ca. 250 m2 g−1 and average particle sizes of ca. 40 to 50 nm from Cabot Corporation. Commercial Pd/C and Pt/C catalysts were purchased from Johnson Matthey. All glassware and Teflon-coated magnetic stir bars were cleaned with aqua regia, followed by copious washing with deionized water before drying in an oven.
Synthesis of core-shell-shell Au@Ag2S@Pt nanocomposites
The seed-mediated growth method was used for the synthesis of the core-shell-shell Au@Ag2S@Pt nanocomposites. Typically, 41 mg of HAuCl4·3H2O was added to 20 ml of oleylamine in a three-necked flask equipped with a condenser and a stir bar. The solution was brought to 220°C and kept at this temperature under flowing N2 for 1 hour for the reduction of Au3+ ions by oleylamine. Then, 17 mg of AgNO3 was added swiftly, and the temperature of the reaction mixture was lowered to 160°C and maintained at this temperature under flowing N2 for 3 hours for the growth of Ag shells on the existing Au seeds. Subsequently, 15 mg of the element sulfur was added to the synthesized core-shell Au@Ag colloidal solution in oleylamine. The molar ratio of S/Ag in the mixture was calculated to be ca. 5:1. The mixture was heated for 3 hours at 80°C to convert the Ag into Ag2S on the surface of the Au core. Finally, 41 mg of K2PtCl4 was added swiftly, followed by heating and keeping the reaction mixture for 2 hours at 195°C under flowing N2 for the reduction of the Pt metal precursor in the presence of previously formed core-shell Au@Ag2S nanoparticles. After the reactions, the core-shell-shell Au@Ag2S@Pt nanocomposites were recovered by precipitation with methanol, centrifugation, and washing with methanol and were redispersed in 20 ml of toluene.
Synthesis of core-shell Au@Pd nanoparticles
A gold-catalyzed strategy was used for the synthesis of the core-shell nanoparticles with an Au core and a thin Pd shell. Typically, 41 mg of HAuCl4·3H2O was added to 20 ml of oleylamine in a three-necked flask equipped with a condenser and a stir bar. The solution was heated to 220°C and kept at this temperature under flowing N2 for 2 hours for the reduction of Au3+ ions by oleylamine. Then, after cooling the Au colloidal solution in oleylamine down to 150°C, 15 mg of Pd(acac)2 was added swiftly, followed by keeping the temperature at 150°C for 2 hours under flowing N2 for the reduction of the Pd shell precursors. Afterward, the core-shell Au@Pd nanoparticles were recovered by precipitation with methanol, centrifugation, and washing with methanol and were redispersed in 20 ml of toluene.
Yields of the core-shell products
To evaluate the yields of the core-shell-shell Au@Ag2S@Pt nanocomposites and the core-shell Au@Pd nanoparticles, the Au@Ag2S@Pt or Au@Pd colloidal solution in toluene was concentrated to 1 ml using flowing Ar. Then, 10 ml of methanol was added to precipitate the core-shell products, which were recovered by centrifugation, washed twice with methanol, and dried at room temperature under vacuum. The yields for the core-shell-shell Au@Ag2S@Pt nanocomposites and the core-shell Au@Pd nanoparticles were estimated to be ca. 90 and 92%, respectively. The losses were likely caused by centrifugation and adhesion to the container walls. On the other hand, the actual yield might also be lower because of surface oxidation of the Pt or Pd components during drying and the residual presence of oleylamine; both of these would add to the weights of the measured products.
Loading core-shell-shell Au@Ag2S@Pt nanocomposites and core-shell Au@Pd nanoparticles on Vulcan carbon substrates
The core-shell-shell Au@Ag2S@Pt nanocomposites and core-shell Au@Pd nanoparticles dispersed into toluene were loaded on XC 72 carbon for electrochemical examinations. In detail, a calculated amount of carbon powders was firstly added to the toluene solution of Au@Ag2S@Pt and Au@Pd particle solutions. After stirring the mixture for 12 hours, carbon-supported core-shell-shell Au@Ag2S@Pt nanocomposites and core-shell Au@Pd nanoparticles (labeled as Au@Ag2S@Pt/C and Au@Pd/C; 20 weight % Pt and Pd on carbon support) were collected by centrifugation and washed thrice with ethanol. Then, the Au@Ag2S@Pt/C and Au@Pd/C catalysts were redispersed in 30 ml of acetic acid by ultrasonication, and the resulting mixtures were refluxed for 3 hours at 120°C to remove the organic surfactants from the particle surface. Finally, the Au@Ag2S@Pt/C and Au@Pd/C catalysts were collected by centrifugation, washed thrice with water, and then dried at room temperature under vacuum.
Particle characterizations
TEM and HRTEM were performed on a JEOL JEM-2100F electron microscope operating at 200 kV, with supplied software for automated electron tomography. For the TEM measurements, a drop of the nanoparticle solution was dispensed onto a 3-mm carbon-coated copper grid. Excessive solution was removed by an absorbent paper, and the sample was dried under vacuum at room temperature. STEM and high-angle, annular dark-field imaging were performed on an aberration-corrected JEM-ARM200F TEM operated at 200 kV, providing a nominal image resolution of 0.08 nm. An energy dispersive x-ray spectroscopy analyzer attached to the TEM operating in the STEM mode was used to analyze the chemical compositions of the synthesized nanocomposites or nanoparticles.
Electrochemical measurements
Electrochemical measurements were carried out in a standard three-electrode cell connected to a Bio-Logic VMP3 (with EC-Lab software version 9.56) potentiostat. A leak-free calomel electrode (saturated with KCl) was used as the reference electrode. The counter electrode was a platinum mesh (1 × 1 cm2) attached to a platinum wire. The working electrode was a thin layer of Nafion-impregnated catalyst cast on a vitreous carbon disk. This electrode was prepared by ultrasonically dispersing 5 mg of the catalyst in 10 ml of aqueous solution containing 4 ml of ethanol and 0.1 ml of Nafion solution. A calculated volume of the ink was dispensed onto the 5-mm glassy carbon disc electrode to produce a nominal catalyst loading of 20 μg cm−2 (Pt/Pd basis). The carbon electrode was then dried in a stream of warm air for 1 hour at 70°C. The room temperature cyclic voltammograms of Au@Ag2S@Pt/C and commercial Pt/C catalysts in argon-purged HClO4 (0.1 M) were recorded between −0.2 and 1 V at 50 mV s−1 and used for the determination of the ECSAs of Au@Ag2S@Pt/C and commercial Pt/C catalysts.
The catalytic performance of Au@Ag2S@Pt/C and commercial Pt/C catalysts at room temperature MOR was also measured by cyclic voltammetry. For these measurements, the potential window of 0.0 to 1 V was scanned at 20 mV s−1 until a stable response was obtained with the electrolyte of methanol (1 M) in perchloric acid (0.1 M). The ORR performance of Au@Ag2S@Pt/C and commercial Pt/C catalysts at room temperature was evaluated in 0.1 M HClO4 electrolyte solution using a glassy carbon rotating disc electrode (RDE) at a rotation rate of 1600 rpm. Negative-going linear sweep voltammograms were recorded from 1.0 to 0 V at 10 mV s−1 in the presence of bubbling ultrapure oxygen to maintain a saturated oxygen atmosphere near the working electrode. For each carbon-supported catalyst (Au@Ag2S@Pt and commercial Pt), the current density was normalized in reference to the ECSAs to obtain the specific activities.
Electrochemical CO stripping voltammograms used to determine the ECSA of the Au@Pd/C and commercial Pd/C catalysts were obtained by the oxidation of preadsorbed CO (COad) in 0.1 M HClO4 at a scan rate of 50 mV s−1. CO was introduced into 0.1 M HClO4 for 20 min to allow for complete adsorption of CO onto the catalyst. Excessive CO in the electrolyte was then purged out using N2 with high purity. The amount of COad was measured by integration of the COad stripping peak and corrected for electric double-layer capacitance. The specific ECSA was calculated on the basis of the equation ECSA = Q/(420G), where Q is the charge of CO desorption-electrooxidation in microcoulomb (μC), which is calculated by dividing the scan rate by the integral area of the CO desorption peak. G represents the total amount of Pd [in micrograms (μg)] on the electrode, and the number (420) is the charge [in microcoulomb per centimeter squared (μC cm−2)] required to oxidize a monolayer of CO on the catalyst.
The performance of Au@Pd/C and commercial Pd/C catalysts at room temperature ORR was evaluated in 0.1 M HClO4 electrolyte solution using a glassy carbon RDE at a rotation rate of 1600 rpm. Negative-going linear sweep voltammograms were recorded from 1.0 to 0 V at 10 mV s−1 at room temperature in the presence of bubbling ultrapure oxygen to maintain a saturated oxygen atmosphere near the working electrode. In the ORR polarization curves, for each catalyst (Au@Pd/C and commercial Pd/C), the measured current density was normalized with reference to its ECSAs to obtain the specific activity.
Performance of the assembled DMFC
The MEA in the assembled DMFC was composed of a single anode layer with Au@Ag2S@Pt/C or commercial Pt/C catalysts, an Nafion 117 membrane, and a cathode layer with Au@Pd/C or commercial Pt/C catalysts. The MEA with an active area of 10.0 cm2 and a loading of 2 mgmetal cm−2 was sandwiched between two carbon plates, in which crossed channels were adopted for methanol or oxygen flow. A copper plate coated with gold was used for collecting current. Electrical heaters and thermocouples were embedded in the plates to control operating temperature. A pump was used to supply aqueous methanol solution from a reservoir without back pressure. Oxygen was fed from a cylinder, and the pressure was controlled by a pressure regulator.
The performance of the DMFC was examined at 80°C. In all experiments, 0.5 to 15 M methanol solution was pumped through the DMFC anode flow field at a flow rate of 1 ml min−1 for 8 hours to activate the MEA before collecting the data. The cathode was fed with oxygen under pressure of 0.1 MPa and at a flow rate of 300 ml min−1. Current-voltage and power density curves were obtained stepwise using a Bio-Logic VMP3 (with EC-Lab software version 9.56) potentiostat.
Supplementary Material
Acknowledgments
Funding: This work was financially supported by the National Natural Science Foundation of China (grant nos. 21376247, 21506225, and 21573240) and the Center for Mesoscience, Institute of Process Engineering, Chinese Academy of Sciences (COM2015A001). Author contributions: Y.F. and H.L. performed the materials synthesis, characterization, and electrochemical measurements. J.Y. supervised the project and wrote the main manuscript text, and all authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700580/DC1
fig. S1. Core-shell-shell Au@Ag2S@Pt nanocomposites and core-shell Au@Pd nanoparticles supported on carbon substrates.
fig. S2. Electrochemical measurements of core-shell-shell Au@Ag2S@Pt nanocomposites.
fig. S3. Electrochemical measurements of core-shell Au@Pd nanoparticles.
fig. S4. A prototype of the membraneless DMFC.
fig. S5. Open circuit voltage.
fig. S6. Transient voltages.
fig. S7. DMFC stack with 15 selective catalyst–based MEAs connected in series.
table S1. Comparison between this study and literatures in the recent 5 years on DMFCs, with emphasis on the high-concentration solutions of methanol.
REFERENCES AND NOTES
- 1.Kirubakaran A., Jain S., Nema R. K., A review on fuel cell technologies and power electronic interface. Renew. Sustain. Energy Rev. 13, 2430–2440 (2009). [Google Scholar]
- 2.Mekhilef S., Saidur R., Safari A., Comparative study of different fuel cell technologies. Renew. Sustain. Energy Rev. 16, 981–989 (2012). [Google Scholar]
- 3.Joghee P., Malik J. N., Pylypenko S., O’Hayre R., A review on direct methanol fuel cells—In the perspective of energy and sustainability. MRS Energy Sustain. 2, E3 (2015). [Google Scholar]
- 4.Mehmood A., Scibioh M. A., Prabhuram J., An M.-G., Ha H. Y., A review on durability issues and restoration techniques in long-term operations of direct methanol fuel cells. J. Power Sources 297, 224–241 (2015). [Google Scholar]
- 5.Zhao T. S., Chen R., Yang W. W., Xu C., Small direct methanol fuel cells with passive supply of reactants. J. Power Sources 191, 185–202 (2009). [Google Scholar]
- 6.Li X., Faghri A., Review and advances of direct methanol fuel cells (DMFCs) part I: Design, fabrication, and testing with high concentration methanol solutions. J. Power Sources 226, 223–240 (2013). [Google Scholar]
- 7.Zhao T. S., Yang W. W., Chen R., Wu Q. X., Towards operating direct methanol fuel cells with highly concentrated fuel. J. Power Sources 195, 3451–3462 (2010). [Google Scholar]
- 8.Eccarius S., Krause F., Beard K., Agert C., Passively operated vapor-fed direct methanol fuel cells for portable applications. J. Power Sources 182, 565–579 (2008). [Google Scholar]
- 9.Xu C., Faghri A., Li X., Development of a high performance passive vapor-feed DMFC fed with neat methanol. J. Electrochem. Soc. 157, B1109−B1117 (2010). [Google Scholar]
- 10.Wang L., Zhang Y., Zhao Y., An Z., Zhou Z., Liu X., Design, fabrication and testing of an air-breathing micro direct methanol fuel cell with compound anode flow field. J. Micromech. Microeng. 21, 104012 (2011). [Google Scholar]
- 11.Yuan W., Zhang Z., Hu J., Zhou B., Tang Y., Passive vapor-feed direct methanol fuel cell using sintered porous metals to realize high-concentration operation. Appl. Energy 136, 143–149 (2014). [Google Scholar]
- 12.Mallick R. K., Thombre S. B., Shrivastava N. K., Vapor feed direct methanol fuel cells (DMFCs): A review. Renew. Sustain. Energy Rev. 56, 51–74 (2016). [Google Scholar]
- 13.Zhang Y., Cai W., Si F., Ge J., Liang L., Liu C., Xing W., A modified Nafion membrane with extremely low methanol permeability via surface coating of sulfonated organic silica. Chem. Commun. 48, 2870–2872 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Beauger C., Lainé G., Burr A., Taguet A., Otazaghine B., Rigacci A., Nafion®–sepiolite composite membranes for improved proton exchange membrane fuel cell performance. J. Membr. Sci. 430, 167–179 (2013). [Google Scholar]
- 15.Abouzari-Lotf E., Nasef M. M., Ghassemi H., Zakeri M., Ahmad A., Abdollahi Y., Improved methanol barrier property of Nafion hybrid membrane by incorporating nanofibrous inter layer self-immobilized with high level of phosphotungstic acid. ACS Appl. Mater. Interfaces 7, 17008–17015 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Yan X. H., Wu R., Xu J. B., Luo Z., Zhao T. S., A monolayer graphene–Nafion sandwich membrane for direct methanol fuel cells. J. Power Sources 311, 188–194 (2016). [Google Scholar]
- 17.Lee J.-S., Hwang I.-T., Jung C.-H., Choi J.-H., Surface modification of Nafion membranes by ion implantation to reduce methanol crossover in direct methanol fuel cells. RSC Adv. 6, 62467–62470 (2016). [Google Scholar]
- 18.Park Y.-C., Kim D.-H., Lim S., Kim S.-K., Peck D.-H., Jung D.-H., Design of a MEA with multi-layer electrodes for high concentration methanol DMFCs. Int. J. Hydrogen Energy 37, 4717–4727 (2012). [Google Scholar]
- 19.Kang K., Lee G., Gwak G., Choi Y., Ju H., Development of an advanced MEA to use high-concentration methanol fuel in a direct methanol fuel cell system. Int. J. Hydrogen Energy 37, 6285–6291 (2012). [Google Scholar]
- 20.Shrivastava N. K., Thombre S. B., Mallick R. K., Effect of diffusion layer compression on passive DMFC performance. Electrochim. Acta 149, 167–175 (2014). [Google Scholar]
- 21.Chai J., Zhou Y., Fan J., Jiang J., Yuan T., Zhang H., Zou Z., Qian H., Yang H., Fabrication of nano-network structure anode by zinc oxide nanorods template for passive direct methanol fuel cells. Int. J. Hydrogen Energy 40, 6647–6654 (2015). [Google Scholar]
- 22.Yuan W., Yan Z., Tan Z., Wang A., Li Z., Tang Y., Anode optimization based on gradient porous control medium for passive liquid-feed direct methanol fuel cells. Renewable Energy 89, 71–79 (2016). [Google Scholar]
- 23.He Y.-L., Miao Z., Yang W.-W., Characteristics of heat and mass transport in a passive direct methanol fuel cell operated with concentrated methanol. J. Power Sources 208, 180–186 (2012). [Google Scholar]
- 24.Deng H. C., Zhang Y. F., Zheng X., Li Y., Zhang X. L., Liu X. W., A CNT (carbon nanotube) paper as cathode gas diffusion electrode for water management of passive mu-DMFC (micro-direct methanol fuel cell) with highly concentrated methanol. Energy 82, 236–241 (2015). [Google Scholar]
- 25.Yan X. H., Zhao T. S., Zhao G., An L., Zhou X. L., A hydrophilic-hydrophobic dual-layer microporous layer enabling the improved water management of direct methanol fuel cells operating with neat methanol. J. Power Sources 294, 232–238 (2015). [Google Scholar]
- 26.Liu H., Feng Y., Chen D., Li C., Cui P., Yang J., Noble metal-based composite nanomaterials fabricated via solution-based approaches. J. Mater. Chem. A 3, 3182–3223 (2015). [Google Scholar]
- 27.Qu J., Ye F., Chen D., Feng Y., Yao Q., Liu H., Xie J., Yang J., Platinum-based heterogeneous nanomaterials via wet-chemistry approaches toward electrocatalytic applications. Adv. Colloid Interface Sci. 230, 29–53 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Feng Y., Yang J. H., Liu H., Ye F., Yang J., Selective electrocatalysts toward a prototype of the membraneless direct methanol fuel cell. Sci. Rep. 4, 3813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J., Ying J. Y., Nanocomposites of Ag2S and noble metals. Angew. Chem. Int. Ed. 50, 4637–4643 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Liu H., Ye F., Cao H., Ji G., Lee J. Y., Yang J., A core–shell templated approach to the nanocomposites of silver sulfide and noble metal nanoparticles with hollow/cage-bell structures. Nanoscale 5, 6901–6907 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Feng Y., Liu H., Wang P., Ye F., Tan Q., Yang J., Enhancing the electrocatalytic property of hollow structured platinum nanoparticles for methanol oxidation through a hybrid construction. Sci. Rep. 4, 6204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kibler L. A., El-Aziz A. M., Hoyer R., Kolb D. M., Tuning reaction rates by lateral strain in a palladium monolayer.Angew. Chem. Int. Ed. 44, 2080–2084 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Shim J. H., Kim J., Lee C., Lee Y., Porous Pd layer-coated Au nanoparticles supported on carbon: Synthesis and electrocatalytic activity for oxygen reduction in acid media. Chem. Mater. 23, 4694–4700 (2011). [Google Scholar]
- 34.Kuai L., Yu X., Wang S., Sang Y., Geng B., Au–Pd alloy and core–shell nanostructures: One-pot coreduction preparation, formation mechanism, and electrochemical properties. Langmuir 28, 7168–7173 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Deming C. P., Zhao A., Song Y., Liu K., Khan M. M., Yates V. M., Chen S., Alkyne-protected AuPd alloy nanoparticles for electrocatalytic reduction of oxygen. ChemElectroChem 2, 1719–1727 (2015). [Google Scholar]
- 36.Chen D., Li J., Cui P., Liu H., Yang J., Gold-catalyzed formation of core–shell gold–palladium nanoparticles with palladium shells up to three atomic layers. J. Mater. Chem. A 4, 3813–3821 (2016). [Google Scholar]
- 37.Nam K., Lim S., Kim S.-K., Yoon S.-H., Jung D.-H., Application of silica as a catalyst support at high concentrations of methanol for direct methanol fuel cells. Int. J. Hydrogen Energy 37, 4619–4626 (2012). [Google Scholar]
- 38.Yuan W., Tang Y., Yang X., Liu B., Wan Z., Structural diversity and orientation dependence of a liquid-fed passive air-breathing direct methanol fuel cell. Int. J. Hydrogen Energy 37, 9298–9313 (2012). [Google Scholar]
- 39.Kim Y.-S., Peck D.-H., Kim S.-K., Jung D.-H., Lim S., Kim S.-H., Effects of the microstructure and powder compositions of a micro-porous layer for the anode on the performance of high concentration methanol fuel cell. Int. J. Hydrogen Energy 38, 7159–7168 (2013). [Google Scholar]
- 40.Yuan W., Tang Y., Yang X.-j., Liu B., Wan Z.-p., Manufacture, characterization and application of porous metal-fiber sintered felt used as mass-transfer-controlling medium for direct methanol fuel cells. Trans. Nonferrous Met. Soc. China 23, 2085–2093 (2013). [Google Scholar]
- 41.Lin C. W., Lu Y. S., Highly ordered graphene oxide paper laminated with a Nafion membrane for direct methanol fuel cells. J. Power Sources 237, 187–194 (2013). [Google Scholar]
- 42.Wu Q. X., Zhao T. S., Chen R., An L., A sandwich structured membrane for direct methanol fuel cells operating with neat methanol. Appl. Energy 106, 301–306 (2013). [Google Scholar]
- 43.Yan X. H., Zhao T. S., An L., Zhao G., Zeng L., A micro-porous current collector enabling passive direct methanol fuel cells to operate with highly concentrated fuel. Electrochim. Acta 139, 7–12 (2014). [Google Scholar]
- 44.Yuan W., Deng J., Zhang Z., Yang X., Tang Y., Study on operational aspects of a passive direct methanol fuel cell incorporating an anodic methanol barrier. Renewable Energy 62, 640–648 (2014). [Google Scholar]
- 45.Kang K., Park S., Gwak G., Jo A., Kim M., Lim Y.-D., Kim W.-G., Hong T., Kim D., Ju H., Effect of variation of hydrophobicity of anode diffusion media along the through-plane direction in direct methanol fuel cells. Int. J. Hydrogen Energy 39, 1564–1570 (2014). [Google Scholar]
- 46.Yao B., Yan X., Ding Y., Lu Z., Dong D., Ishida H., Litt M., Zhu L., Synthesis of sulfonic acid-containing polybenzoxazine for proton exchange membrane in direct methanol fuel cells. Macromolecules 47, 1039–1045 (2014). [Google Scholar]
- 47.Paneri A., Heo Y., Ehlert G., Cottrill A., Sodano H., Pintauro P., Moghaddam S., Proton selective ionic graphene-based membrane for high concentration direct methanol fuel cells. J. Memb. Sci. 467, 217–225 (2014). [Google Scholar]
- 48.Yuan W., Tang Y., Zhang Z., Zhou B., Deng J., Operational and structural aspects of a vapor-feed semi-passive direct methanol fuel cell supplied with concentrated methanol. Chin. Sci. Bull. 59, 3216–3221 (2014). [Google Scholar]
- 49.Zhang Y., Xue R., Zhang X., Song J., Liu X., rGO deposited in stainless steel fiber felt as mass transfer barrier layer for μ-DMFC. Energy 91, 1081–1086 (2015). [Google Scholar]
- 50.Nataraj S. K., Wang C.-H., Huang H.-C., Du H.-Y., Chen L.-C., Chen K.-H., Functionalizing biomaterials to be an efficient proton-exchange membrane and methanol barrier for DMFCs. ACS Sustain. Chem. Eng. 3, 302–308 (2015). [Google Scholar]
- 51.Gago A. S., Esquivel J.-P., Sabaté N., Santander J., Alonso-Vante N., Comprehensive characterization and understanding of micro-fuel cells operating at high methanol concentrations. Beilstein J. Nanotechnol. 6, 2000–2006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J., Cai W., Ma L., Zhang Y., Chen Z., Cheng H., Towards neat methanol operation of direct methanol fuel cells: A novel self-assembled proton exchange membrane. Chem. Commun. 51, 6556–6559 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Yuan W., Wang A., Yan Z., Tan Z., Tang Y., Xia H., Visualization of two-phase flow and temperature characteristics of an active liquid-feed direct methanol fuel cell with diverse flow fields. Appl. Energy 179, 85–98 (2016). [Google Scholar]
- 54.Sebastián D., Serov A., Artyushkova K., Atanassov P., Aricò A. S., Baglio V., Performance, methanol tolerance and stability of Fe-aminobenzimidazole derived catalyst for direct methanol fuel cells. J. Power Sources 319, 235–246 (2016). [Google Scholar]
- 55.Sebastián D., Baglio V., Aricò A. S., Serov A., Atanassov P., Performance analysis of a non-platinum group metal catalyst based on iron-aminoantipyrine for direct methanol fuel cells. Appl. Catal. B Environ. 182, 297–305 (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700580/DC1
fig. S1. Core-shell-shell Au@Ag2S@Pt nanocomposites and core-shell Au@Pd nanoparticles supported on carbon substrates.
fig. S2. Electrochemical measurements of core-shell-shell Au@Ag2S@Pt nanocomposites.
fig. S3. Electrochemical measurements of core-shell Au@Pd nanoparticles.
fig. S4. A prototype of the membraneless DMFC.
fig. S5. Open circuit voltage.
fig. S6. Transient voltages.
fig. S7. DMFC stack with 15 selective catalyst–based MEAs connected in series.
table S1. Comparison between this study and literatures in the recent 5 years on DMFCs, with emphasis on the high-concentration solutions of methanol.