

Summary
Synaptic refinement is a critical step in nervous system maturation, requiring a carefully timed reorganization and refinement of neuronal connections. We have identified myrf-1, a homologue of Myrf family transcription factors, as a key regulator of synaptic rewiring in C. elegans. MYRF-1 and its paralog MYRF-2 are functionally redundant specifically in synaptic rewiring. They co-exist in the same protein complex, and act cooperatively to regulate synaptic rewiring. We find that the MYRF proteins localize to the endoplasmic reticulum membrane, and that they are cleaved into active N-terminal fragments, which then translocate into the nucleus to drive synaptic rewiring. Over-expression of active forms of MYRF is sufficient to accelerate synaptic rewiring. MYRF-1 and MYRF-2 are the first genes identified to be indispensable for promoting synaptic rewiring in C. elegans. These findings reveal a molecular mechanism underlying synaptic rewiring and developmental circuit plasticity.
Keywords: Synaptic rewiring, GABAergic motor neuron, ER localization, proteolytic cleavage, ER to nuclear translocation, Intramolecular Chaperone of Endosialidase, larval development, critical period, synapse refinement
eTOC Blurb
Synaptic rewiring of DD neurons in C. elegans provides a powerful in vivo model to study developmental plasticity of the nervous system. Meng et al. identified myrf-1 and myrf-2, members of Myelin Regulatory Factor family transcription factors, as essential regulators that drive synaptic rewiring via a cleavage-dependent mechanism.
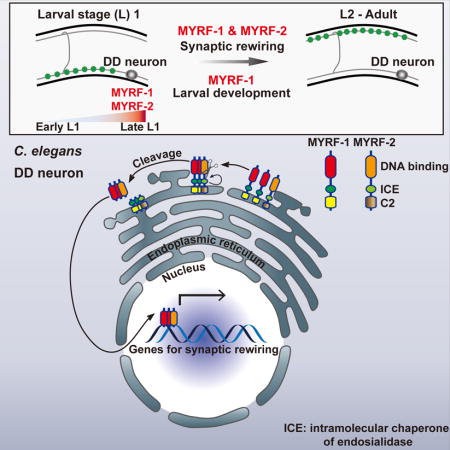
Introduction
One of the enigmatic characteristics of the developing nervous system is its ability to remodel and refine neuronal connections during its transition into a functional, mature nervous system. During development, neural circuits add or eliminate synapses dynamically, refining and pruning their connections until a mature state is reached. While the cellular processes that drive neuronal remodeling can lead to changes in synaptic connections at various scales, many forms of neuronal plasticity must be tightly regulated, involving well-defined cascades of events that occur during a critical developmental window.
Decades of studies have yielded insights into the principles underlying synaptic refinement in different neural circuits (O’Leary and Koester, 1993; Purves and Lichtman, 1980). In developing visual systems, for example, synaptic refinement is required for final positioning of axonal arbors, a process that involves extensive synapse elimination and axon pruning. The segregation of axonal arbors occurs in two phases, and a combination of genetic and environmental inputs influence many steps of synaptic refinement (Espinosa and Stryker, 2012). During the initial phase, axon refinement is controlled by both innate molecular signals and spontaneous neuronal activity. During the secondary phase, segregation and pruning predominantly depend on external visual experiences. While a number of genes regulating refinement processes of the visual system have been identified (Espinosa and Stryker, 2012), it remains to be understood how neurons initiate this form of plasticity and instruct refinement processes, regardless of whether these processes are completely innate or whether they incorporate spontaneous activities or experiences.
A striking and informative example of synaptic refinement occurs during the development of the C. elegans nervous system, in which six GABAergic motor neurons, known as dorsal D (DD) neurons, reverse their axonal and dendritic domains, with no overt changes in morphology (White et al., 1978). Born during embryogenesis, DD neurons form ventral and dorsal processes connected by commissures. Their ventral processes synapse onto ventral muscles, acting as axonal processes, while dorsal processes function as dendrites, receiving cholinergic inputs from the dorsal A and B (DA, DB) motor neurons. At the end of the first larval (L1) stage, however, a rewiring event occurs, in which the dorsal processes of DD neurons begin forming new synapses onto dorsal muscles while the existing ventral synapses are disassembled. Concurrently, new ventral (V) classes of motor neurons, including ventral A, B, and D (VA, VB, VD), are generated and incorporated into the motor circuitry. By late L2 stage, DD neurons complete rewiring, with their dorsal processes being axonal, and ventral processes being dendrites, receiving inputs from cholinergic VA and VB. Thus, DD rewiring is integral to maturation of the motor circuit. This dramatic synaptic rewiring offers a powerful model in which we can identify genes that are essential for the developmental plasticity of neurons (Kurup and Jin, 2016).
Initial insights into the mechanisms regulating DD rewiring suggested a key role for the transcription factor LIN-14 in controlling the timing of DD rewiring (Hallam and Jin, 1998). Loss of lin-14 causes DD neurons to rewire prematurely, suggesting that lin-14 represses the DD rewiring program. A second, Ig-domain containing, gene, oig-1, was recently found to be a transcriptional target of LIN-14. OIG-1 functions as a synaptic organizer to maintain DD’s early synaptic pattern (He et al., 2015; Howell et al., 2015). Loss of oig-1 results in premature rewiring of DD neurons, partially resembling lin-14 null. While lin-14 and oig-1 act as negative regulators of DD rewiring, no essential factors that promote rewiring at late L1 have been identified.
Here, in a genetic screen for genes specifically required for DD synaptic rewiring, we identified myrf-1, a C. elegans homolog of Myelin Regulatory Factor (Myrf). Myrf family genes are essential for organism development in mice (Emery et al., 2009), Dictyostelium (Senoo et al., 2012), and C. elegans (Russel et al., 2011). In mice, Myrf is specifically expressed in oligodendrocytes of the CNS (Emery et al., 2009) and critical for myelin development and maintenance (Emery et al., 2009; Koenning et al., 2012; Xiao et al., 2016). Whether Myrf genes are involved in neuronal plasticity remains unknown.
We show that myrf-1 and its paralog myrf-2 are essential for synaptic rewiring in C. elegans. N-terminal MYRF-1 fragments are released from the ER after being cleaved and translocate into the nucleus to drive rewiring. MYRF-1 and MYRF-2 physically interact and act cooperatively to promote synaptic rewiring. Collectively, our findings open up an avenue for dissecting mechanisms of synaptic rewiring.
Results
A Newly Discovered Mutation of myrf-1, a Homolog of Myelin Regulatory Factor (Myrf), Blocks DD Rewiring
To identify genes regulating DD rewiring (Figure 1A), we designed a forward genetic screen using two markers: an unc-25pro-mCherry::RAB-3 to visualize presynaptic sites in DD and VD neurons; and an acr-2pro-GFP reporter, which labels ventral cord cholinergic motor neurons, for staging ventral nerve cord development. In wild type L2 animals expressing these transgenes, the rewiring of the DDs is readily discernible by the fluorescent clusters of mCherry::RAB-3 along the dorsal cord (Figure S1A, S1B).
Figure 1. A novel mutation in myrf-1 blocks DD rewiring.
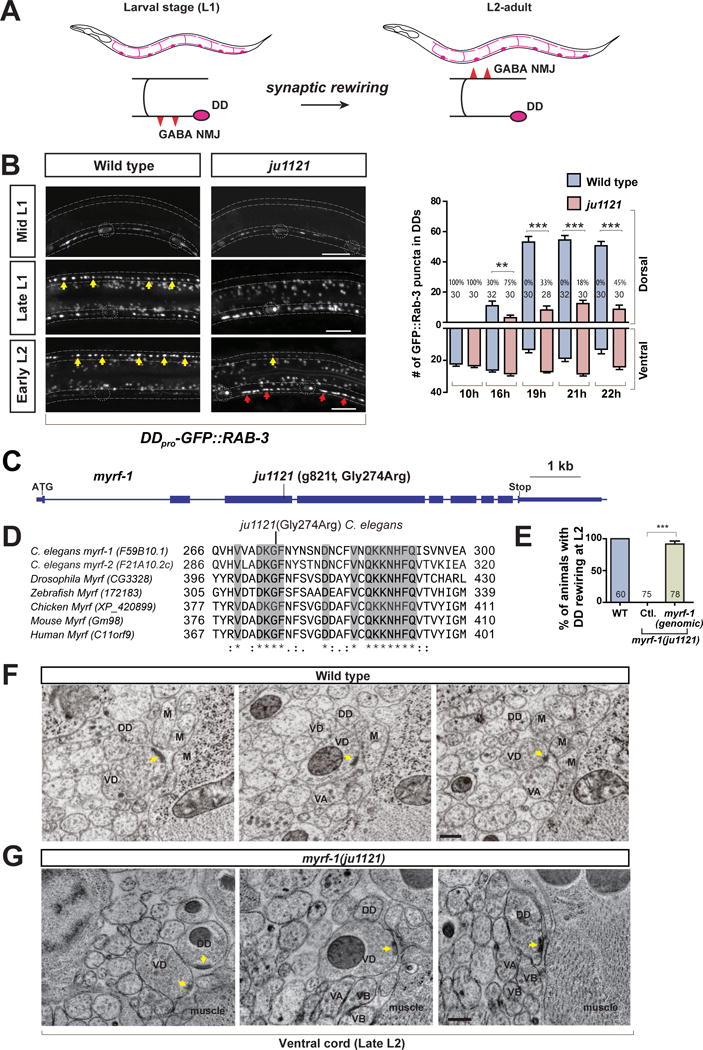
A. Illustration of DD synaptic rewiring during L1-L2 transition.
B. Dorsal cord in myrf-1(ju1121) mutants lack clear synaptic clusters at either late L1 or early L2, labeled by flp-13pro-GFP∷RAB-3 (ybqIs47). Dotted lines, ventral and dorsal cords; Dotted circles, DD soma; Yellow arrows, new dorsal synapses; Red arrows, retained ventral synapses. Scale, 10 μm. Number of synapses is shown as mean ± SEM; t-test (**P<0.01, ***P < 0.001); the number of animals analyzed is shown on each bar; %, penetrance for animals with no dorsal synapse.
C. Gene structure for myrf-1(F59B10.1) showing ju1121 mutation.
D. Alignment between MYRF-1 protein and its orthologs, showing the segment with conserved Glycine 274 altered in ju1121
E. Rewiring defect in ju1121 is rescued by expressing genomic fragment of myrf-1 (ybqEx55). Percentage of animals with rewired DDs is shown as mean ± SEM; t-test (***P<0.001); the number of animals analyzed is shown on each bar.
F. EM images showing mature VD synapses and no DD synapses at discrete cross positions along wild type ventral cord of late L2, Yellow arrow, presynaptic density; Scale, 200 nm (also in G).
G. EM images showing prominent DD synapses and mature VD synapses at discrete cross positions along the ventral cord of myrf-1(ju1121) at late L2. (See also Figure S1, S2.)
Following visual screening, we isolated a mutant, ju1121, displaying few clusters of unc-25pro-mCherry::RAB-3 in dorsal cords at the L2 stage (Figure S1B), indicative of defective DD rewiring. To confirm this, we examined additional presynaptic markers, flp-13pro-GFP::RAB-3 and flp-13pro-GFP::UNC-57 (endophilin), which only label DDs but not VDs in the ventral cord. Using these markers, we observed a lack of synaptic clusters in the dorsa cords of ju1121 mutants at the L2 stage (Figure 1B; Figure S1C).
Next, using linkage analyses, we mapped ju1121 to myrf-1 (F59B10.1, previously named pqn-47) (Russel et al., 2011) and determined that ju1121 is a G-to-T change at nucleotide 821 of the myrf-1 open reading frame (Figure 1C). MYRF-1 is a C. elegans ortholog of the recently identified Myrf (myelin regulatory factor) family (Emery et al., 2009; Li and Richardson, 2016; Senoo et al., 2012). The ju1121 mutation causes a highly conserved Glycine 274 to be changed to Arginine (Figure 1D). Transgenes expressing full-length genomic DNA of myrf-1, injected at a low concentration (0.1 ng/μl), rescued DD rewiring defects in ju1121 mutants (Figure 1E). At higher concentrations, however, transgenic expression of myrf-1 caused larval lethality, precluding the establishment of transgenic lines, suggesting that tightly regulated expression of myrf-1 is critical for larval development.
myrf-1 has a Specific Role in DD Synaptic Rewiring
We next examined the progression of synaptic rewiring in wild type and myrf-1(ju1121) mutants. Prior to DD rewiring, in early and mid-L1 stages, wild type animals exhibit ventral synaptic clusters, as evidenced by the presence of GFP::RAB-3 and GFP::UNC-57 fluorescent puncta. During a short time between late L1 and early L2 (approximately three hours), fluorescent clusters started to emerge in the dorsal cord, as rewiring began. At this stage, the clusters rapidly increased in number, the fluorescence of individual clusters intensified, and clusters became evenly distributed along the dorsal cord. During the critical window for rewiring, the presence of ventral synaptic clusters also declined steadily (Figure 1B; Figure S1B, S1C).
Importantly, during early and mid-L1 stages, myrf-1(ju1121) mutants also formed ventral synaptic clusters; the pattern and number of synapses were similar to wild type animals (Figure 1B; Figure S1C). These results suggest that embryonic neurogenesis and synaptogenesis were unaffected in myrf-1(ju1121) mutants.
Strikingly, however, myrf-1(ju1121) mutants became progressively abnormal during late L1 stage. In contrast to wild type animals, myrf-1(ju1121) mutants exhibited no increase in expression, or intensification of synaptic clusters in dorsal cords (Figure 1B; Figure S1B, S1C). Occasionally, we detected small and weak fluorescent clusters that were minimal in comparison to wild type animals. During late L1 and onward the ventral synaptic clusters in myrf-1(ju1121) mutants either remained unchanged or became brighter. The lack of dorsal synaptic clusters was not the consequence of aberrant axon outgrowth, as DD axon morphology was normal in myrf-1(ju1121) animals (Figure S1D).
Electron microscopy (EM) analyses of the myrf-1(ju1121) mutants confirmed our observations. In wild type animal at late L2, synapses of VD, but not DD, were observed in the ventral cord (Figure 1F). In myrf-1(ju1121) mutant at late L2, EM images showed that DD neurons had prominent presynaptic densities surrounded with a cluster of vesicles in the ventral cord (Figure 1G), indicating a failure in rewiring. Moreover, EM analyses revealed mature VD synapses in the ventral cord, marked by distinctive presynaptic density, abundant synaptic vesicles, and close contact with muscles (Figure 1G). Thus, VD synaptogenesis is normal in myrf-1(ju1121). Taken together, these data strongly suggest that the progression of DD rewiring in myrf-1(ju1121) mutants is severely and specifically blocked, while the synapse formation in VDs is normal.
We also examined whether myrf-1(ju1121) might affect the development of other neuronal classes within the motor circuit. The development of cholinergic motor neurons in the ventral cord was largely normal, based on acr-2pro-GFP expression (Figure S2A). Embryonic cholinergic DA/DB neurons developed normally, as did postembryonic VA/VB neurons. The synapses from these neurons, and particularly the ventral synapses formed by VA/VBs in late L1, showed no discernible defects in myrf-1(ju1121) mutants (Figure S2B). As a further control, we took advantage of the mig-13pro-GFP∷SNB-1 marker, which selectively labels synapses of DA9 neuron at L1. We found no difference in DA9 synapse formation between wild type and myrf-1(ju1121) mutant (Figure S2C). Additionally, VA12, which is born in late L1 and forms ventral synapses, can also be labeled by mig-13pro-GFP∷SNB-1. Again, we found no abnormalities in VA12 synapses in myrf-1(ju1121) mutants. Collectively, the normal cholinergic synaptogenesis suggests that myrf-1(ju1121) specifically affects DD synaptic rewiring.
Rewiring Defect in myrf-1 Mutants is Not Due to Developmental Arrest
myrf-1(ju1121) mutants never grow to L4 or fertile adults. To determine at which developmental stage the mutants were arrested, we tracked their growth by measuring the body length of the animals. We found that throughout the L1 stage the body length for myrf-1(ju1121) mutants increased at a similar rate to that for control animals (Figure 2A, B), but the increase for the mutants stopped before mid L2, whereas the increase for the control animals continued (Figure 2B). While the mutants were arrested in body size, their developmental progression appeared to have continued, as all mutants all gradually reached molting by the end of L2, but they invariably failed to shed the cuticle and eventually perished within the unshed cuticles (Figure 2A, C).
Figure 2. Rewiring defect in myrf-1(ju1121) is independent of the mutant’s larva arrest.
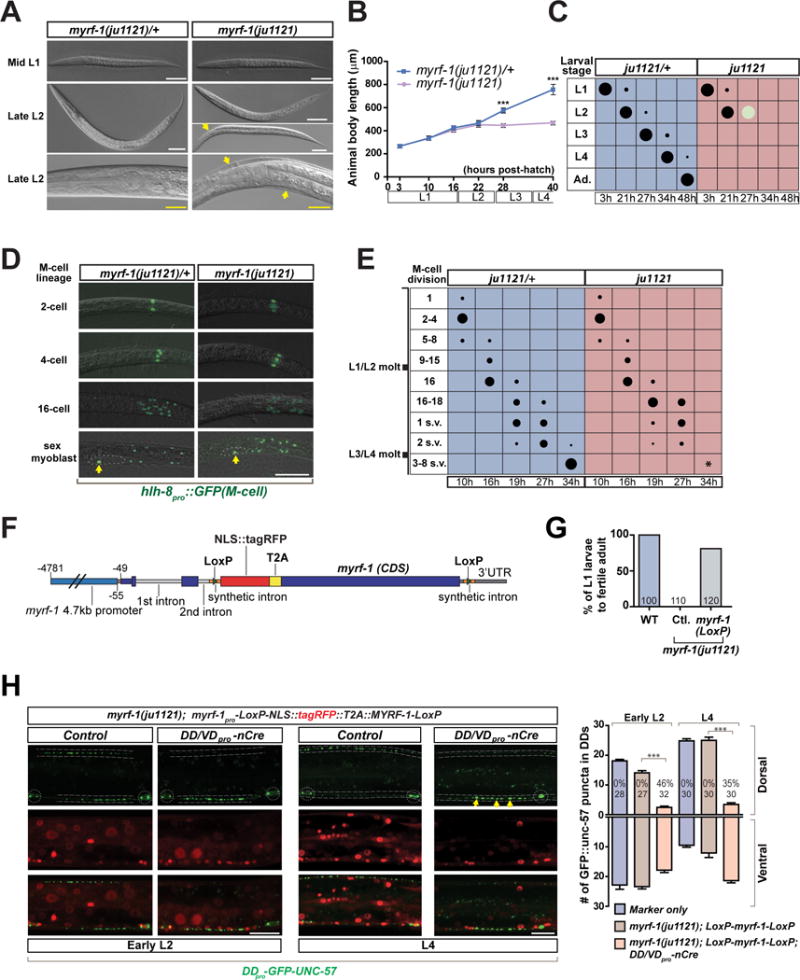
A. DIC images showing similar body size for myrf-1(ju1121) and control animal at mid L1 (a, b); control animal (c) being bigger than myrf-1(ju1121) mutants (d, e) at late L2. d, a late L2 mutant before molting. e, a late L2 mutant that failed to shed cuticle. f and g, close view of head regions. Arrows, unshed cuticles. Scale, 50 μm (white); 20 μm (yellow).
B. Body length for myrf-1(ju1121) and control animals is quantified and shown as mean ± SEM; t-test (***P<0.001); n=30.
C. Assessment of larval development for myrf-1(ju1121) and control animals. Size of black dots corresponds to percentage of animals at specific larval stages. Light color dots represents arrested animals. Ad., adult. n=300.
D. Images of DIC and hlh-8pro∷GFP (ayIs6), labeling progenitors of M-cell lineage, which exhibits stage-characteristic division. After 4 divisions (16 cell), two of 16 cells divide one more time, producing two sex myoblasts, which later migrate to prospective vulva (dotted line) (s.v. in E), and divide again at the end of L3 (3–8 s.v. in E). Scale, 50 μm.
E. Quantification of M-cell lineage progression in myrf-1(ju1121) and control animals. Size of dots corresponds to percentage of animals with M-cell lineage pattern. Asterisk indicates lack of division of sex myoblasts in myrf-1(ju1121). n=200.
F. Illustration of myrf-1 conditional rescue transgene MYRF-1LoxP.
G. Percentage of myrf-1(ju1121) mutants carrying MYRF-1LoxP transgene (ybqEx401) (picked at L1) that developed into fertile adults.
H. MYRF-1LoxP transgene (ybqEx401) rescued DD synaptic rewiring in myrf-1(ju1121) mutant, labeled by flp-13pro-GFP∷UNC-57 (ybqIs49). In the presence of unc-25pro-nCre(ybqEx322), DDs lose the rescuing myrf-1 while the rest of mutant retains transgene expression and exhibit sustained rewiring block. Yellow arrows, sustained DD ventral synapses. Dotted lines, ventral and dorsal cords; Dotted circles, DD neuron soma. Scale, 10 μm. Number of synapses per DD neuron is shown as mean ± SEM; t-test (***P < 0.001); the number of animals analyzed is shown on each bar.
To further define the developmental arresting stage for myrf-1(ju1121) mutants, we analyzed the M-cell division, which undergoes larval stage-specific lineage progression (Harfe et al., 1998). Using hlh-8pro∷GFP to label progenitors of M-cell lineage, we determined that myrf-1(ju1121) mutants and control animals were similar in M-cell lineage progression from the 1st (2-cell) through 4th cell division (16-cell) (Figure 2D, E). However, the 5th cell division, from which the sex myoblasts arise, was delayed in a fraction of myrf-1(ju1121) mutants (Figure 2E). The migration of sex myoblasts to prospective vulva was also delayed in some myrf-1(ju1121) mutants (Figure 2E). Further division of the sex myoblasts was never observed in myrf-1(ju1121) mutants, indicating the mutants do not progress beyond L3-L4 molt. Together, our analysis on M-cell division indicates that myrf-1(ju1121) mutants have developed into L2 stage, consistent with normal development of VD, VA and VB motor neurons in the mutants.
To address whether blocked DD rewiring was caused by developmental arrest in myrf-1(ju1121), we designed a tissue-specific deletion scheme (Figure 2F), in which rescuing myrf-1 is expressed broadly to restore organismal growth, but is deleted specifically in DDs. We created an ORF cassette encoding NLS∷tagRFP and MYRF-1, connected by a T2A sequence such that NLS∷tagRFP and MYRF-1 can be simultaneously expressed as two separate proteins. The whole ORF cassette was flanked by two LoxP sites, and was driven under myrf-1 promoter. This MYRF-1LoxP transgene rescued the developmental arrest in myrf-1(ju1121) because the mutants carrying the transgene could grow to fertile adults (Figure 2G). The MYRF-1LoxP transgene also rescued blocked DD rewiring in myrf-1(ju1121) (Figure 2H). We observed that red fluorescence from NLS∷tagRFP was present in DD neurons (Figure 2H), suggesting endogenous MYRF-1 is expressed in DDs. To test whether myrf-1(ju1121) specifically acts in DDs for synaptic rewiring, we constructed a second transgene expressing unc-25pro-nCre, which causes excision of rescuing MYRF-1LoxP cassette in DDs and VDs specifically. Importantly, myrf-1(ju1121) mutants that carry MYRF-1LoxP and nCre transgenes developed normally into L4 and adults. We observed a sustained block in DD rewiring by L4 in these mutants, such that they showed prominent ventral synapses but fewer fully developed synapses in the dorsal cord (Figure 2H). These data indicate that the rewiring phenotype in myrf-1(ju1121) is not secondary to developmental arrest of the mutant animals, and suggest a cell-autonomous function of myrf-1.
MYRF-1 Expression is Temporally Regulated at Late L1
A previous study reported that myrf-1 is widely expressed in many tissues, including neuronal, muscle, and epidermal seam cells (Russel et al., 2011). However, these initial analyses did not determine myrf-1 expression within specific neuronal subsets. To address whether myrf-1 was expressed in DD neurons, we generated a transcriptional reporter using the 4.7 kb of myrf-1 promoter, but failed to detect GFP signal in ventral cord neurons. We reasoned that the first two introns of myrf-1 may contain additional cis-regulatory information. We then constructed a new transgene of the 4.7kb myrf-1 promoter and myrf-1 CDS that also includes the first two introns (Figure 3A). We inserted GFP in frame after Alanine 171 within a less conserved region, as described previously (Russel et al., 2011), and a Flag at the C-terminus immediately before the stop codon. Thus, GFP from this new transgene, referred as the myrf-1 minigene, could act as a reporter for both tissue expression patterns of myrf-1 and for MYRF-1 protein’s subcellular localization. Expression of these transgene restored DD rewiring, and also rescued the larval arrest of myrf-1(ju1121) mutants (Figure 3B, C), suggesting the transgene recapitulates endogenous myrf-1 expression. Importantly, GFP∷MYRF-1 was barely detectable at early L1, but its intensity significantly increased at late L1 (Figure 3D) and co-localized with unc-25pro-mCherry∷RAB-3 (Figure 3E). We confirmed that myrf-1 was expressed in DDs at late L1 and continued to express in DDs in the L2 stage (Figure 3E). The upregulation of MYRF-1 in ventral cord neurons at late L1 supports our hypothesis that myrf-1 plays a specific role in DD rewiring.
Figure 3. Dual localization of MYRF-1 in cytoplasm and nucleus.
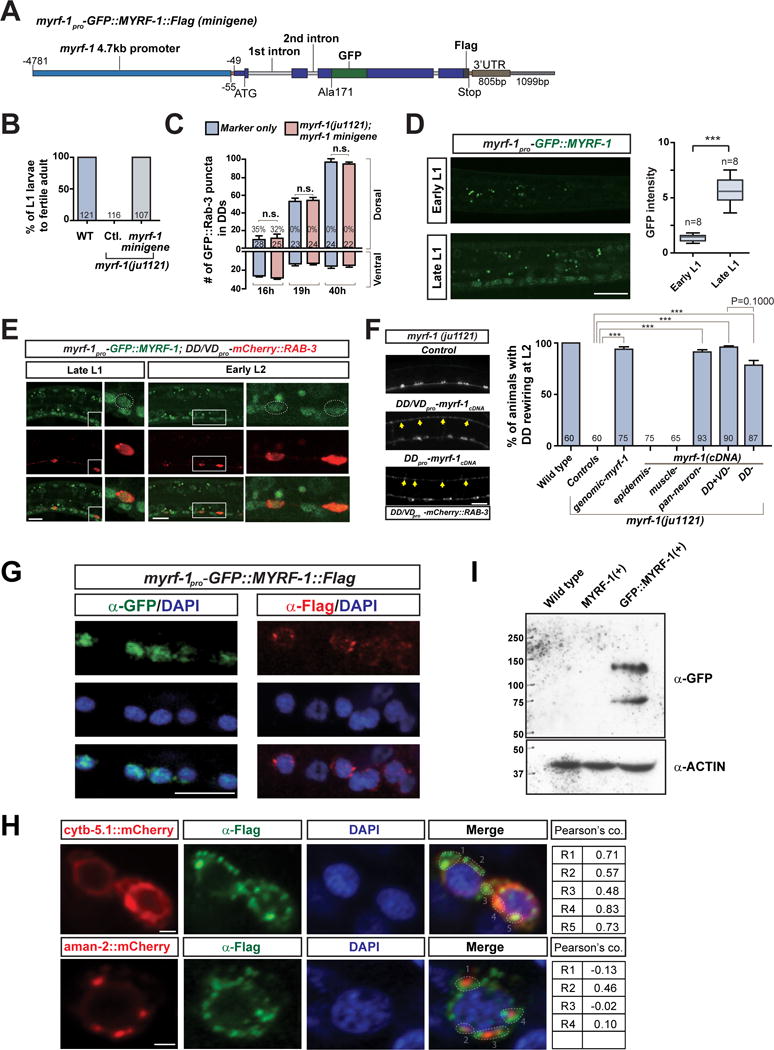
A. Illustration of myrf-1pro-GFP∷MYRF-1∷Flag minigene transgene.
B. Percentage of myrf-1(ju1121) mutants carrying myrf-1 minigene (ybqIs13) (picked at L1) that developed into fertile adults.
C. Number of synapses is quantified for myrf-1(ju1121) carrying myrf-1 minigene transgene (ybqIs13), labeled by flp-13pro-GFP∷Rab-3 (ybqIs47), and shown as mean ± SEM; t-test (n.s.); the number of animals analyzed is shown on each bar.
D. GFP signal from myrf-1 minigene (ybqIs13) increased at late L1. Arrows, ventral cord neurons with GFP signal. GFP signal in soma normalized by background signal is shown as mean ± SEM (box bars); t-test (***P<0.001); Line bars, min/max values; n, number of images analyzed.
E. Co-localization of native GFP signal from GFP∷MYRF-1 (ybqEx164) and unc-25pro-mCherry∷RAB-3 (juIs236).
F. Tissue specific expression of myrf-1 cDNA in myrf-1(ju1121) mutants. Genomic myrf-1 (ybqEx55), 11.5 kb amplified from genomic DNA; DD flp-13 (ybqEx94); DD/VD unc-25 (ybqEx85); epidermis dpy-7 (ybqEx16); body wall muscle myo-3 (ybqEx93); pan-neuron rgef-1 (ybqEx86). Percentage of animals with rewired DDs is shown as mean ± SEM; t-test (***P < 0.001); the number of animals analyzed is shown on each bar. Arrows, dorsal DD synapses.
G. Immunostaining of myrf-1 minigene transgene (ybqIs13) using anti-GFP and anti-Flag. DAPI, a DNA-binding dye. Scale, 10 μm.
H. Immunostaining using anti-Flag on dual-transgene animals with myrf-1 minigene (ybqIs13) and pan-neuronally (rgef-1pro) expressed ER marker cytb-5.1∷mCherry transgenes (ybqEx595); or Golgi marker aman-2∷mCherry (ybqEx597). Red and green signals in each ROIs are test for co-localization; R, Pearson’s coefficiency. Scale, 1 μm.
I. Protein extracts from N2, myrf-1(+), and GFP-myrf-1(+) transgene animals (ybqEx55, ybqIs13) are analyzed by Western Blot. Two bands (140 kDa, 75 kDa) are detected by anti-GFP. (See also Figure S3, S4)
MYRF-1 Acts Cell-Autonomously in DD Rewiring
To determine the spatial requirement for myrf-1 in the regulation of synaptic rewiring, we next expressed the myrf-1 cDNA under tissue specific promoters (Figure 3F). We found that, when expressed under epidermal (dpy-7) or body wall muscle (myo-3) promoters, the myrf-1 transgene could not rescue the rewiring defect of the myrf-1(ju1121) mutant. In contrast, when expressed under pan-neuronal (rgef-1), DD/VD-specific (unc-25), or DD-specific (flp-13) promoters, myrf-1 transgene effectively restored DD rewiring in myrf-1(ju1121) mutant (Figure 3F). These data are consistent with our analyses of DD-specific deletion of myrf-1 (see above), and collectively, indicate that myrf-1 regulates DD rewiring cell-autonomously.
MYRF-1 Shows Cytoplasmic and Nuclear Dual Localization
In our initial analyses, we observed that GFP∷MYRF-1∷Flag minigene showed some GFP expression in nuclei. Because native GFP∷MYRF-1 fluorescence was weak, we performed immunostaining using anti-GFP to enhance the detection of GFP∷MYRF-1. We observed immunostaining signals in the nuclei of many cells, including ventral cord neurons (Figure 3G). The anti-GFP signal was not present in cytoplasm of any stained cells. We also immunostained the same transgenic animals using anti-Flag to detect the C-terminally inserted Flag tag, and remarkably, observed anti-Flag signals predominantly in the cytoplasm (Figure 3G).
To further confirm this observation, we generated a CRISPR-engineered allele of myrf-1(ybq14) by knocking in GFP at the N-terminal alanine 171 and 3xFlag at the C-terminus (Figure S3A). Immunostaining using anti-GFP showed consistent nuclear signals (Figure S3B). An earlier report on myrf-1 in C. elegans has suggested MYRF-1 predominantly co-localizes with ER (Russel et al., 2011). To determine the localization of cytoplasmic MYRF-1, we generated animals co-expressing the myrf-1 minigene transgene and neuronally expressed marker lines, including cytb-5.1∷mCherry for general ER and aman-2∷mCherry for Golgi (Rolls et al., 2002), respectively. We immunostained such animals using anti-Flag for MYRF-1 and analyzed the co-localization. We found that Flag immunostaining signals exhibited significant co-localization with ER-targeted CYTB-5.1∷mCherry, but not with Golgi AMAN-2∷mCherry (Figure 3H). The C. elegans neurons are typically small in size (2–3 μm), which gives limited resolution for observing cellular organelles. To seek further evidence for MYRF-1’s ER localization, we expressed C-terminal fragment of MYRF-1 in cultured HEK293 cells. By immunostaining, we found that C-terminal fragment of MYRF-1 co-localized with ER marker but not with Golgi marker (Figure S3C). Thus, N-terminal MYRF-1 (GFP tagged) localizes to the nucleus, whereas C-terminal MYRF-1 (Flag tagged) is present predominantly in the ER.
The dual localization of tagged MYRF-1 in C. elegans is consistent with the findings on mammalian MYRF, which have suggested that MYRF may undergo self-cleavage in the ER membrane, releasing its N-terminal fragment to the nucleus (Bujalka et al., 2013; Li et al., 2013). To test whether MYRF-1 in C. elegans could be cleaved into N and C-terminal fragments, we performed Western Blot analysis on protein extracts from myrf-1 minigene animals. We detected two bands of size 140 kDa and 75 kDa using anti-GFP (Figure 3I). The large band was of the size expected for the full-length GFP∷MYRF-1, while the small band, for the N-terminal fragment after cleavage. We obtained similar results for protein samples from GFP knock-in myrf-1 animals (Figure S3D). Collectively, these data suggest that MYRF-1 is cleaved into N- and C-terminal fragments.
N-terminal Cleavage and Nuclear Localization of MYRF-1 are Necessary for DD Rewiring
MYRF-1 has multiple functional domains and signaling motifs (Figure 4A), including the NDT80/PhoG-like DNA binding domain (pfam05224), an Intramolecular Chaperone of Endosialidase (pfam13884), two nuclear-localization signals (NLSs), one transmembrane domain (TM), a coiled coil domain (also designated C1), and a functionally unknown MYRF conserved domain 2 (C2). These domains are highly conserved between C. elegans MYRF-1 and its mammalian orthologs. The most notable feature of these domains is the intramolecular chaperone of endosialidase, which is found in bacteriophage endosialidases, proteins that constitute the end-tail-spikes of many bacteriophages (Stummeyer et al., 2005). This domain allows protein trimerization and catalyzes cleavage at the serine-lysine dyad (Schulz et al., 2010). When we deleted the chaperone domain from MYRF-1 or mutated the serine of the cleavage site (S483) to Alanine, the mutant MYRF-1 failed to restore DD rewiring in myrf-1(ju1121) (Figure 4B, 4C). The result supports the model that MYRF-1 is cleaved into N-terminal (1-482aa) and C-terminal (483-931aa) fragments.
Figure 4. Cleavage and nuclear localization of MYRF-1 are necessary for DD rewiring.
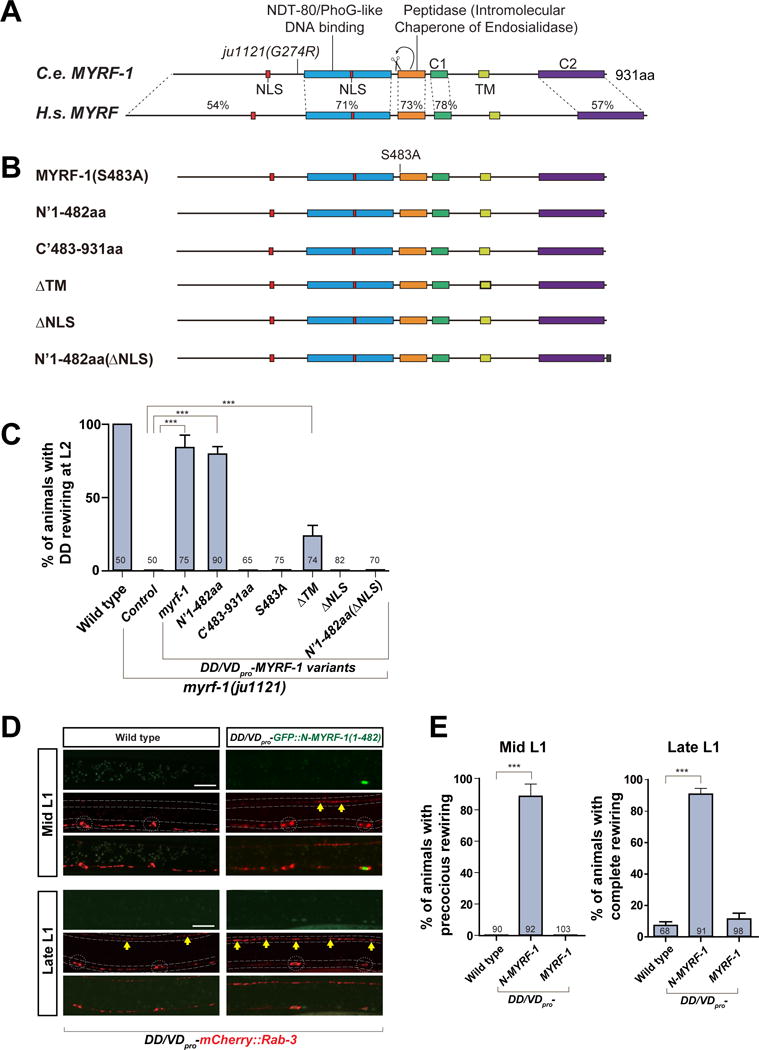
A. Comparison of protein sequence and domain similarity between C. elegans MYRF-1 and human Myrf.
B. Constructs of MYRF-1 proteins with truncation, deletion, and point mutation.
C. MYRF-1 variants (also see STAR methods) were expressed under unc-25pro. Percentage of animals with rewired DD is shown as mean ± SEM; t-test (***P < 0.001); the number of animals analyzed is shown on each bar.
D. Over-expression of unc-25pro-N-MYRF-1 (ybqEx102) drives early DD rewiring, labeled by unc-25pro-mCherry∷RAB-3 (juIs236). In DDs with adequate transgene expression marked by GFP signals (white arrow), dorsal synapses (yellow arrows) appear at mid L1. Synaptic rewiring is complete in DDs with transgene at late L1 while the rewiring is just initiated in controls. Dotted lines, ventral and dorsal cords; Dotted circles, DD neuron soma. Scale, 10 μm.
E. Percentage of animals with over-expression of N-MYRF-1 (ybqEx102) (or full length MYRF-1, ybqEx64) showing precocious rewiring at mid L1 (left graph) and animals showing complete rewiring at late L1 (right graph) are shown as mean ± SEM; t-test (***P < 0.001); the number of animals analyzed is shown on each bar.
Our results using the myrf-1 minigene suggest that the N-terminal MYRF-1 is localized in the nucleus. To confirm that the cleavage at Ser483 indeed resulted in the release of a functional N-terminal fragment, we tested whether expression of the N- or C-fragment alone was sufficient for synaptic rewiring. Consistent with our observation above, the N-fragment (1-482aa) transgene rescued the rewiring defect in myrf-1(ju1121), whereas the C-fragment (483-931aa) did not (Figure 4B, 4C). When GFP was inserted into the N-terminal fragment of MYRF-1, the GFP signal localized to the nucleus (Figure 4D). The GFP signal became diffuse in both neuron soma and nucleus, only when the two NLS sequences were deleted (not shown). Without its NLSs, expression of the N-terminal fragment of MYRF-1 also failed to rescue DD rewiring in the mutant (Figure 4C). Finally, the full-length MYRF-1 also lost its activity to rescue myrf-1(ju1121) activity when the two NLSs were removed. These results indicate that nuclear localization of the N-terminal MYRF-1 is required for DD rewiring.
We next asked whether over-expression of the N-fragment of MYRF-1 could induce a premature initiation of synaptic rewiring. When GFP-tagged N-terminal MYRF-1 was expressed in DDs, synapses were observed in the dorsal cord at mid L1 (Figure 4D). Due to mosaic expression of transgene, not all DDs contained adequate expression of GFP∷N-MYRF-1. Intriguingly, DDs that did not exhibit a GFP signal rarely initiated early rewiring at mid L1 (Figure 4D). By late L1, when DD rewiring was just initiating in wild type animals, the rewiring was complete in DDs showing expression of GFP-N-MYRF-1 (Figure 4D, E). Surprisingly, overexpressing full-length MYRF-1 failed to promote early rewiring even though the same transgene rescued the rewiring defects in myrf-1(ju1121) (Figure 4E). One possible explanation is that the processing of full-length MYRF-1 may be rate-limiting, hence an over-expression of full-length MYRF-1 might not have produced sufficient amount of N-fragment of MYRF-1 to drive early rewiring.
MYRF-1 and its Paralog MYRF-2 Function Redundantly to Control Rewiring
myrf-1(ju1121) behaves as genetic loss of function by larval arrest phenotype, resembling myrf-1 deletion mutants (gk3366, ok3445, and tm2707), all of which exhibit larval arrest. Although the deletion mutants show more severe larval arrest phenotype than myrf-1(ju1121), we found that none of the deletion mutants had obvious effects on DD rewiring. Because the three deletions affected coding sequence for the C-terminal half of MYRF-1, we were concerned that the N-fragment of MYRF-1 protein might be present in those deletion mutants, conferring partial function. To obtain a null allele of myrf-1, we used CRISPR editing (Dickinson et al., 2013) to generate indel mutations close to the 5′-end of myrf-1 ORF such that the N-terminal portion of MYRF-1 was ensured to be disrupted (Figure 5A). We analyzed multiple newly acquired alleles of myrf-1. Surprisingly, none of them exhibited any severe rewiring defects seen with myrf-1(ju1121), although all showed penetrant larval arrest phenotypes similar to other myrf-1 deletion alleles (Figure 5B–D).
Figure 5. myrf-1 and myrf-2 cooperatively controls DD rewiring.
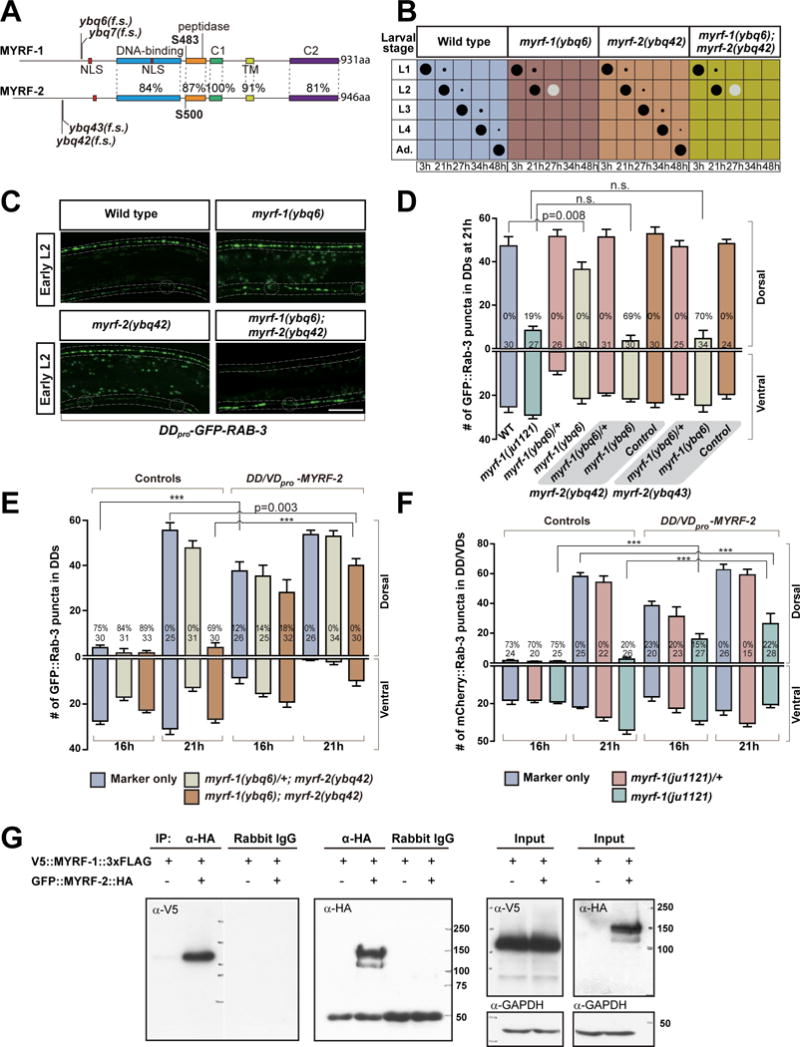
A. Comparison shows protein sequence and domain similarity between MYRF-1 and MYRF-2 (isoform c). Cleavage sites, S483 in MYRF-1 and S500 in MYRF-2, are conserved. The indel mutations cause frame shift (f.s.).
B. Assessment of larval development for myrf-1 and myrf-2 mutants. Size of black dots corresponds to percentage of the animals at specific larval stage. Light color dots represents arrested animals. n=300.
C. DD rewiring in myrf-1(ybq6), myrf-2(ybq42), and myrf-1(ybq6); myrf-2(ybq42) double mutants, labeled by flp-13pro-GFP∷RAB-3 (ybqIs47). Note the lack of dorsal synapses in double mutant. Dotted lines, ventral and dorsal cords; Dotted circles, DD neuron soma. Scale, 10 μm.
D. Number of synapses in myrf-1 and myrf-2 mutants labeled by flp-13pro-GFP∷RAB-3 (ybqIs47) is shown as mean ± SEM; t-test; the number of animals analyzed is shown on each bar. %, penetrance for animals with no dorsal synapse.
E. Number of synapses in animals over-expressing unc-25pro-myrf-2 (ybqEx529) labeled by flp-13pro-GFP∷RAB-3 (ybqIs47) is shown as mean ± SEM; t-test (***P<0.001); the number of animals analyzed is shown on each bar. %, penetrance for animals with no dorsal synapse. myrf-2 transgene rescues blocked DD rewiring in myrf-1; myrf-2 double mutants, and caused precocious DD rewiring at 16h.
F. Number of synapses in animals over-expressing unc-25pro-myrf-2 (ybqEx528) labeled by unc-25pro-mCherry∷RAB-3 (juIs236) is shown as mean ± SEM; t-test (***P<0.001); the number of animals analyzed is shown on each bar. %, penetrance for animals with no dorsal synapse. myrf-2 transgene partially suppresses the blocked rewiring in myrf-1(ju1121) mutant.
G. Co-immunoprecipitation of V5∷MYRF-1∷3xFlag and GFP∷MYRF-2∷HA, expressed in HEK293 cells.
We considered the possibility that other factors may function redundantly with myrf-1’s role in rewiring. C. elegans MYRF-2 shows over 80% similarity to MYRF-1 in protein sequence (Figure 5A). We generated a GFP knock-in allele myrf-2(ybq46), in which GFP was inserted at Ile190, in the N-terminal part of MYRF-2. We immunostained the knock-in animals using anti-GFP and observed the immunostaining GFP signals are localized to the nucleus (Figure S5A, B). We further generated a myrf-2pro-GFP∷myrf-2∷HA minigene transgene (Figure S5C), in which GFP was inserted in the N-terminal part of MYRF-2, and HA tagged at the C-terminus. We immunostained the transgene animals using anti-HA and analyzed its co-colocalization with neuronally expressed ER and Golgi transgene markers (Figure S5D). We found that the immunostaining HA signals had significant co-localization with ER marker but not with Golgi (Figure S5D). The nuclear localization of N-terminal GFP tag and the cytoplasmic localization of C-terminal HA tag suggests the dual localization of MYRF-2. Furthermore, when expressed in HEK293 cells, N-MYRF-2 was localized to the nucleus, while C-MYRF-2 to the ER (Figure S5E), supporting the dual localization of MYRF-2 in vivo.
By Western Blot analyses on protein extracts from myrf-2pro-GFP∷myrf-2∷HA minigene animals, we detected two protein fragments of 140 kDa and 75 kDa in size (Figure S5F), expected for full-length GFP∷MYRF-2 and GFP∷N-MYRF-2 after cleavage, respectively. These data strongly support that MYRF-2 can be cleaved and exhibits subcellular localization similar to MYRF-1. To test whether myrf-2 is expressed in DD neurons, we generated myrf-2pro-nls∷tagRFP as a transcription reporter, and determined that myrf-2 was expressed in DDs (Figure S5G–I). Furthermore, we confirmed that MYRF-2 protein was present in DDs and observed a significant upregulation of MYRF-2 protein in DDs at late L1 by analyzing the GFP∷myrf-2 knock-in animals (Figure S5K and legend). Thus, the close resemblance borne by MYRF-2 to MYRF-1 strongly suggests a functional role for MYRF-2 in DD rewiring.
To test whether MYRF-2 may act redundantly with MYRF-1, we generated indel mutations in myrf-2 using CRISPR editing (Figure 5A, S6A). The indel mutations of myrf-2 we obtained are expected to affect all the transcripts of myrf-2, therefore likely acting as nulls. myrf-2 mutants grew like wild type animals and exhibited normal DD rewiring (Figure 5B–D). However, in myrf-1; myrf-2 double CRISPR-mediated indel alleles, DD rewiring was blocked, and the severity of the defect was comparable to that of myrf-1(ju1121) (Figure 5C, D). At least two independent indel mutations for myrf-2 were tested, resulting in similar observations. The indel mutations of myrf-2 we obtained are expected to affect all the transcripts, therefore more likely acting as nulls. These observations suggest myrf-1 and myrf-2 have redundant roles in promoting DD rewiring.
The rewiring defect in myrf-1; myrf-2 double mutants could be rescued by over-expressing myrf-2 in DDs (Figure 5E), indicating that myrf-2 mutation is responsible for the rewiring defect. Moreover, over-expressing myrf-2 in DDs dramatically advanced the timing of DD rewiring (Figure 5E), supporting a direct role for myrf-2 in promoting DD rewiring. Over-expressing myrf-2 in DDs also partially rescued the blocked rewiring in myrf-1(ju1121) (Figure 5F), again supporting that MYRF-1 and MYRF-2 function in common pathways.
The intramolecular chaperone of endosialidase domain enables the bacteriophage endosialidase to trimerize (Schulz et al., 2010). If this domain in MYRF-1 functions in a similar manner, it would likely catalyze the trimerization of MYRF-1 in ER. Because MYRF-1 and MYRF-2 share similar domain features, we hypothesized that they may form a hetero-oligomer in ER through their chaperone of endosialidase domains. To test this, we asked whether MYRF-1 and MYRF-2 could bind to each other in cultured human cell lines. We found that the MYRF-1 could be co immuno-precipitated with MYRF-2, and vice versa (Figure 5E; Figure S6B). The binding of MYRF-1 and MYRF-2 in cultured cells suggests that they may also bind with each other in DD neurons; thus, overexpressing non-cleavable form of MYRF-1 in DDs may sequester MYRF-2, suppressing MYRF-2’s function in promoting DD rewiring. Indeed, overexpression of MYRF-1(S483A) in DDs, a mutation that abolishes protein cleavage, caused severely blocked DD rewiring (Figure S6C). Together with their biochemical interaction, this result supports our hypothesis that MYRF-1 and MYRF-2 function in a same protein complex.
MYRF-1(G274R) Interferes with MYRFs in a Dosage-Dependent Manner
Since the myrf-1(ju1121) single mutant mimics the myrf-1(null); myrf-2(null) double mutant animals in DD rewiring, we reasoned that the mutant form MYRF-1(G274R) protein must display two characteristics: loss of rewiring-promoting activity, and interference with wild type MYRF-2.
To understand how G274R may affect the MYRF-1 protein, we further compared MYRF-1 sequence with that of yeast protein Ndt80. Ndt80 belongs to a family of transcription factors with S-type Ig-fold DNA binding domain (Rudolph and Gergen, 2001). The structure of the Ndt80-MSE complex has been previously solved, detailing the features that enable its DNA binding (Lamoureux et al., 2002). Among these features, one of the Ndt80-DNA contacts is made by a segment called a-b loop. The Glycine 274 in MYRF-1 marks the beginning of a-b loop (Figure 6A), and the change to Arginine likely breaks the contact between a-b loop and DNA such that MYRF-1(G274R) may no longer bind to DNA. To test the hypothesis, we constructed the equivalent mutation in mouse MYRF, G384R, and found that when expressed in HEK293 cells, N-MYRF(G384R) proteins were localized to the nucleus (Figure S7A–C). The result indicates that the Glycine-to-Arginine mutation does not affect the cleavage of MYRF, nor the translocation of N-MYRF into nucleus. Despite its localization in nucleus, however, MYRF(G384R) mutant proteins failed to bind to DNA in Gel Shift Assays (EMSA), while wild type MYRF could. (Figure 6B).
Figure 6. MYRF-1(G274R) mutant interferes with MYRF’s normal function.
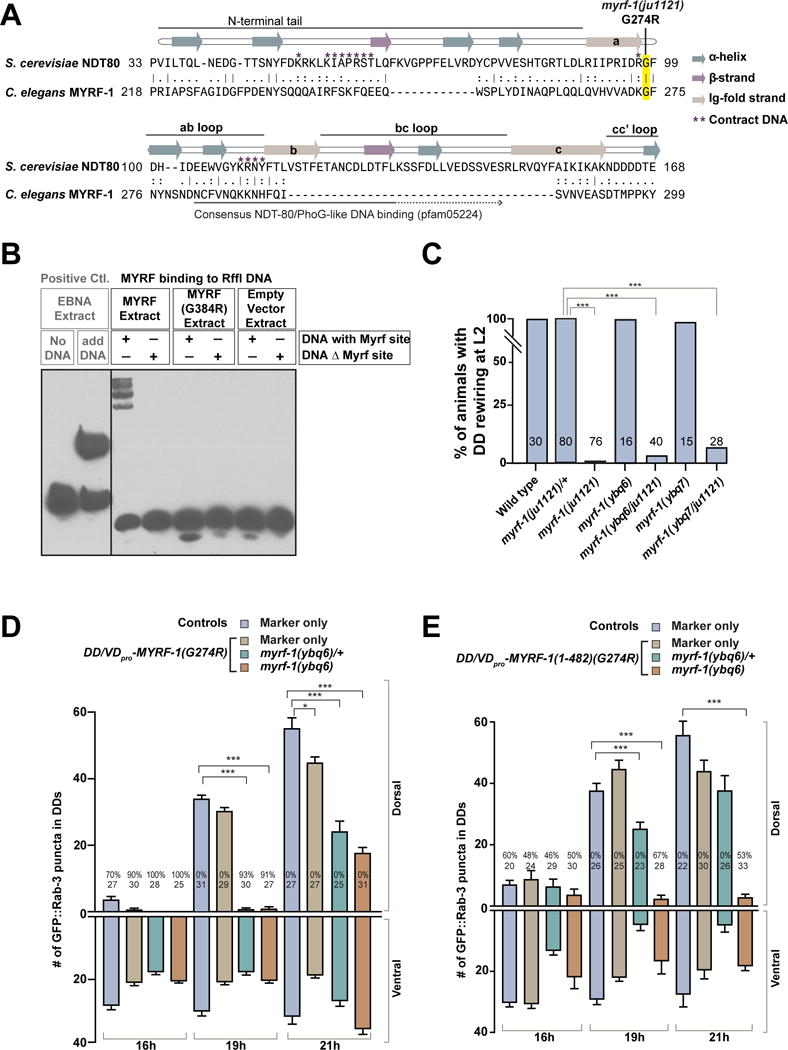
A. Alignment between segments of DNA-binding domains of MYRF-1 and yeast Ndt80. Annotation of Ndt80 strands are redrawn based on Lamoureux et al. 2002.
B. Electrophoretic mobility shift assay (EMSA) testing mouse MYRF binding on Rffl DNA. MYRF(R384R) mutation is equivalent to MYRF-1(G274R) in C. elegans.
C. Percentage of animals with normal DD rewiring. Animals of myrf-1(ybq6/ju1121) are cross progenies of myrf-1(ybq6)/mIn1 and myrf-1(ju1121)/mIn1. myrf-1(ybq7/ju1121) was generated similarly. Pearson’s chi-squared test (***P<0.001).
D. Number of synapses in animals expressing unc-25pro-MYRF-1(G274R) (ybqIs27) labeled by flp-13pro-GFP∷RAB-3 (ybqIs47) is shown as mean ± SEM; t-test (*P<0.05, ***P<0.001); the number of animals analyzed is shown on each bar; %, penetrance for animals with no dorsal synapse.
E. Number of synapses labeled by flp-13pro-GFP∷RAB-3 (ybqIs47) in animals expressing unc-25pro-N-MYRF-1(G274R) (ybqEx517, ybqEx541) is shown as mean ± SEM; t-test (***P<0.001); the number of animals analyzed is shown on each bar; %, penetrance for animals with no dorsal synapse.
(See also Figure S7.)
By co-immunoprecipitation from cultured cells, we observed that MYRF-1(G274R) mutant could still bind with MYRF-2 (Figure S7D), suggesting MYRF-1(G274R) may act in a dominant negative manner. Yet the myrf-1(ju1121) mutation is genetically recessive, because heterozygous myrf-1(ju1121)/+ animals show normal rewiring (Figure 6C). When wild type myrf-1 was removed, as in myrf-1(ju1121/null), DD rewiring was blocked (Figure 6C). To test whether the presence of wild type MYRF-1 might counter balance MYRF-1(G274R), we over-expressed MYRF-1(G274R) protein in DDs wild type background, as well as in myrf-1(0). We found that the severity of DD rewiring defects depended on how many wild type myrf-1 gene copies were present (Figure 6D). This result supports a conclusion that MYRF-1(G274R) can act as a dosage-dependent negative form.
If MYRF-1(G274R) disrupts DNA-binding, we may expect that its interfering activity takes place in the nucleus but not in ER. Indeed, we observed rewiring defects in transgenic animals over-expressing the nuclear GFP∷N-MYRF-1(G274R) in DDs (Figure 6E). The severity of defects also depended on the presence of wild type myrf-1 (Figure 6E). Thus, these data support the model that MYRF-1(G274R) obstructs the function of MYRF-1 and MYRF-2 in the nucleus (Figure 7)
Figure 7. Model for MYRF-1 and MYRF-2 regulation of synaptic rewiring.
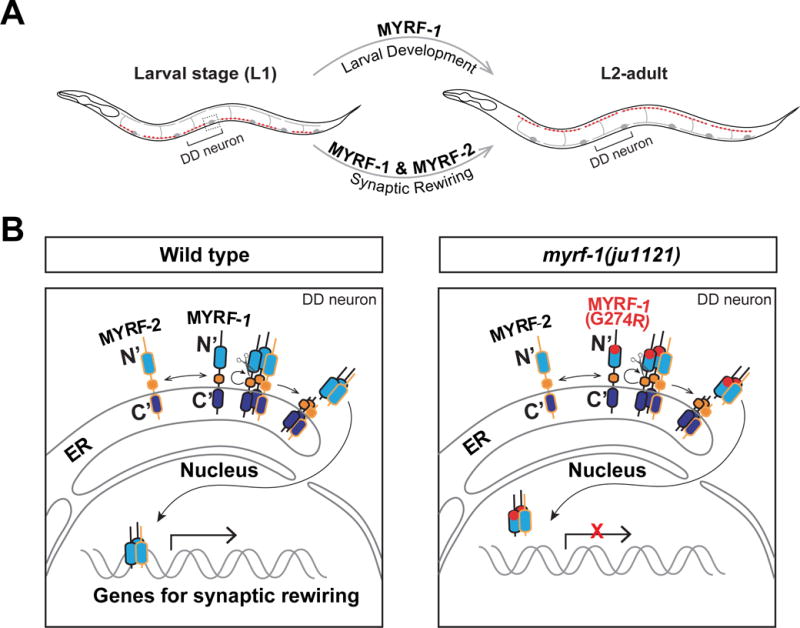
A. MYRF-1 and MYRF-2 are specifically required for synaptic rewiring, whereas MYRF-1 alone is required for larval development.
B. MYRF-1 and MYRF-2 are processed on ER. Their N-terminal fragments translocate into the nucleus and regulate DD synaptic rewiring. In myrf-1(ju1121) mutants, MYRF-1(G274R) still interact with MYRF-2 but fails to bind to DNA. Light blue, DNA binding domain; Orange, chaperone of endosialidase domain; purple blue, C2 domain.
Discussion
Synaptic refinement is a critical step in neural development, but as yet we have limited knowledge on how neurons are instructed to carry out refinement processes. We explored such genetic programs using DD synaptic rewiring in C. elegans. Through studies of a newly discovered mutation, myrf-1(G274R), which blocks DD synaptic rewiring, we find that myrf-1 and its paralog myrf-2 cooperatively regulate DD rewiring, and that myrf-1(G274R) acts in a dosage-dependent negatively interfering manner. Over-expression of either MYRF in DD neurons is sufficient to accelerate synaptic rewiring. To our knowledge, myrf-1 and myrf-2 are the first genes identified to be indispensable for promoting DD synaptic rewiring. These findings build a framework for dissecting core mechanisms underlying DD developmental synaptic plasticity. Our analyses also provide insights into the functions of the conserved Myrf family proteins.
Genetic control of developmental synaptic plasticity
The identification of lin-14 provided the first insight into genetic control of synaptic rewiring in C. elegans. lin-14 functions to maintain early synaptic connectivity and to suppress DD rewiring throughout L1 stage until the end of L1 (Hallam and Jin, 1998). While the timing of LIN-14 downregulation at late L1 coincides with the onset of DD rewiring, it is unlikely that the downregulation of LIN-14 triggers DD rewiring at late L1, because the rewiring still occurs in mutants in which LIN-14 protein remains high at late L1 (YBQ and YJ, unpublished data). These observations suggest that other factors may play positive roles in the initiation of rewiring at the end of L1.
Several other genes have been associated with preventing ectopic remodeling in VD neurons, a subclass GABAergic neurons that have analogous functions to DDs except that they synapse onto ventral muscles in adults. Loss of the COUP nuclear hormone receptor unc-55 causes VD neurons to form dorsal synapses instead of making ventral synapse, an ectopic rewiring in VDs (Zhou and Walthall, 1998). Several genes were identified as downstream of unc-55 based on their ability to suppress the ectopic rewiring in unc-55 mutants (Miller-Fleming et al., 2016; Petersen et al., 2011; Thompson-Peer et al., 2012). However, mutants for these genes show only mild defects in DD rewiring. Furthermore, unc-55 itself is specifically expressed in VDs but not in DDs (Zhou and Walthall, 1998). Collectively, these data suggest that the pathways regulating ectopic rewiring in VD neurons may be distinct from those regulating the rewiring of DD neurons.
Here, we have demonstrated that myrf-1 and myrf-2 are essential positive regulators of DD rewiring. lin-14 keeps DDs in an immature and plastic state, while myrf-1 and myrf-2 promote DD rewiring and possibly other aspects of maturation. LIN-14 and MYRFs thus act at temporally separate stages to instruct the developmental plasticity in DDs.
Our findings provide strong support for the concept that innate genetic programs have major roles in refining connectivity, in resonance with studies that have linked transcriptional factors to circuit refinement in mammals. For example, loss of homeodomain transcription factor Otx1 causes incomplete elimination of subcortical projections from layer 5 cortical neurons (Weimann et al., 1999). Overexpressing orthodenticle homeobox 2 (Otx2) in parvalbumin (PV)-expressing-cell induces precocious visual plasticity, while reducing Otx2 uptake in PV cell reopens plasticity in adulthood (Sugiyama et al., 2008). When circadian clock gene Clock (Bmal1) is deleted, the maturation of PV cell network is delayed, and the loss of visual acuity in response to brief monocular deprivation is concomitantly delayed (Kobayashi et al., 2015). Moreover, in retina, transforming growth factor (TGF)-β regulates neuronal C1q expression in synaptic pruning (Bialas and Stevens, 2013). Collectively, these findings support the idea that circuit refinement is instructed in part by developmental programs.
Many questions remain about how myrf-1 and myrf-2 promote synaptic rewiring. At least two lines of future work should shed light on their mechanisms. First, our data demonstrate that an up-regulation of myrf-1 expression at the late L1 stage is necessary to promote rewiring. The upregulation of myrf-1 is not triggered by down-regulation of LIN-14 (JM and YBQ, unpublished data); and we speculate that it may involve other developmental signals regulating late L1 development. The second line remaining is the identification of the downstream targets of myrf-1 and myrf-2. Our data indicate both myrf-1 and myrf-2 can promote early rewiring when over-expressed in DDs, suggesting they can directly control rewiring. Knowing their transcriptional targets will yield mechanistic insights into synapse formation, synapse elimination, neural polarity, and plasticity.
Our analyses suggest myrf-1 and myrf-2 have distinct roles in larval development and synaptic rewiring. While both myrf-1 and myrf-2 are broadly expressed across similar tissue types, the myrf-1 single mutants exhibit larval arrest, while the myrf-2 single mutants grow normally. Yet, myrf-1 and myrf-2 need to be deleted simultaneously to block DD rewiring in DDs. It is interesting to speculate that MYRF-1 and MYRF-2 must have both common and distinct functions in specific tissues. Our co-immunoprecipitation analyses suggest that MYRF-1 and MYRF-2 co-exist in the same protein complex. However, it remains unclear whether their interaction is regulated temporally or spatially. Future work will delineate how MYRF-1 and MYRF-2 function differentially in distinct cellular contexts, which may yield insights into the question why specific subsets of neurons exhibit plasticity while others do not.
Conserved proteolytic processing for MYRF proteins
The most prominent domain features for Myrf are an Ndt80-like DNA-binding domain and a Chaperone of Endosialidase domain. All members of Myrf family show high sequence similarity for these two domains. Additionally, extensive evidence indicate that Myrf family proteins may bind to DNA. Mouse Myrf can directly bind DNA, and mutating critical residues within the DNA-binding domain abolishes its DNA-binding activity (Bujalka et al., 2013; Li et al., 2013). MrfA in Dictyostelium was identified in a screen for factors that have affinity to a GC-rich regulatory DNA sequence, and mutations on DNA-binding motifs in MrfA disrupted its DNA-binding (Senoo et al., 2012). There is also strong evidence suggesting the proteolytic processing of Myrf from two studies on mouse and human Myrf (Bujalka et al., 2013; Li et al., 2013) and one study on MrfA in Dictyostelium (Senoo et al., 2013). After cleavage by the chaperone of endosialidase domain, N-Myrf enters into the nucleus while the C-Myrf remains on the ER. Biochemical analysis of human Myrf in vitro has confirmed the formation of the Myrf trimer (Li et al., 2013), consistent with the trimerization of bacteriophage endosialidases.
Previous observations on C. elegans MYRF-1 led to the question whether it functions as a transcription factor (Russel et al., 2011). The authors did not observe nuclear localization GFP in their GFP∷MYRF-1 transgenic animals. In our GFP∷MYRF-1 construct, we inserted GFP at the identical position and confirmed cytoplasmic localization of GFP∷MYRF-1 in DD/VD neurons expressing multi-copy array transgene. However, in our myrf-1 minigene transgene or GFP knock-in animals, we detected GFP consistently in the nucleus, but not in the cytoplasm. When GFP was knocked into the myrf-2 gene, GFP signals also localized in the nucleus (Figure S5B, K). We reckon that the discrepancy is likely due to the expression level of MYRF proteins. We examined multiple transgene lines of unc-25pro-GFP∷MYRF-1 as well as unc-25pro-GFP∷MYRF-2, and our imaging data suggest when the expression is lowered to a barely detectable level, GFP exhibits a nuclear localization. Our results collectively support a model in which MYRF-1 and MYRF-2 undergoes Serine 483-dependent cleavage in ER through the chaperone of endosialidase domain, suggesting that proteolytic cleavage is a conserved feature in C. elegans MYRF.
Previous studies on mammalian Myrf suggest the cleavage is auto-processed upon trimerization of Myrf. However, two pieces of data in our study suggest the cleavage of C. elegans MYRF-1 may need additional factors. First, MYRF-1 variants cannot be adequately processed when they are overexpressed in DD neurons, resulting in the release of low amount of N-fragments. Second, when MYRF-1 is expressed in human cell lines, MYRF-1 is localized on ER membrane but cannot be cleaved (Figure S3C, S4). Because the over-expression of MYRF-1 using endogenous myrf-1 promoter results in lethality, the quantity and processing of MYRF-1 may be under tight developmental regulation. It will be interesting to examine whether such regulatory mechanisms apply to other Myrf family proteins in specific developmental contexts.
Conserved and derived functions of MYRF
The appearance of Myrf during evolution is intriguing. Myrf proteins show significant homology to yeast Ndt80 in their DNA binding domains, but Ndt80 does not have the chaperone of endosialidase domain. Myrf proteins also exhibit strong homology with bacteriophage endosialidase in the intramolecular chaperone domain, but bacteriophage endosialidase does not have DNA binding domain. Thus, the emergence of Myrf ancestor is likely a result of invasion event, during which a phage DNA integrated into the genome of an early protist, producing a fusion protein with both DNA binding domain and the chaperone domain (Li and Richardson, 2016).
Evidence across studies from different model organisms collectively and strongly suggest an essential role for Myrf in development. In Dictyostelium, MrfA was originally identified as a regulator of prestalk (PstA) cell differentiation, (Senoo et al., 2012). However, MrfA-deficient strains exhibit pleiotropic defects including a developmental delay not readily explainable by aberrant PstA cell differentiation. The loss of myrf-1 in C. elegans results in larval arrest (Russel et al., 2011); this study), indicating that myrf-1 is indispensable for the growth of vital organs. Similarly, constitutive deletion of Myrf in mice led to early embryonic lethality (Emery et al., 2009), suggesting that Myrf is necessary for embryonic development in mice independent of its later role in myelination. The role of Myrf in these developmental contexts has not been studied in depth. The best characterized role for Myrf is in myelin development. It is interesting to note that the myelination of central nervous system is an evolutionary invention for vertebrates. As invertebrates do not have central myelin but clearly have Myrf orthologs, it is possible that the ancestral function of Myrf was to regulate developmental progression, with Myrf later becoming co-opted for the myelin program in vertebrates. To further understand these possibilities, it will be informative to determine whether any of the transcriptional targets of MYRF are conserved.
The role of Myrf in myelination has been extensively characterized. While Myrf is expressed in non-neural cells of mouse, within the CNS its expression is restricted to pre-myelinating oligodendrocyte (Emery et al., 2009). When Myrf is specifically deleted in the oligodendrocyte lineage of mice, the specification and proliferation of oligodendrocyte progenitors are normal, but the expression of myelin genes is mostly absent, and the progenitors fail to differentiate, undergoing apoptosis (Emery et al., 2009). Therefore, the late stage of oligodendrocyte differentiation towards myelination is specifically blocked by the loss of Myrf. When Myrf is deleted in oligodendrocytes in adult mouse, it causes the progressive loss of myelin (Koenning et al., 2012). Transcriptional target analyses by ChIP-seq for Myrf using rat oligodendrocyte progenitor cell cultures revealed that putative Myrf targets include not only myelin component genes but also a broader spectrum of other genes, which may coordinate the differentiation of oligodendrocytes (Bujalka et al., 2013). C. elegans does not contain structures or genes homologous to myelin, which is mostly found in vertebrates; therefore, it seems unlikely that MYRF-1 regulates the homologs of myelin genes in C. elegans. However, considering MYRFs constituting a highly conserved protein family throughout animals and in slime molds, we speculate some functions of C. elegans MYRF may also be conserved in vertebrates. Elucidation of MYRF-1 pathways in C. elegans and comparative analysis of Myrf homologs may open ways for understanding the biological functions controlled by Myrf family proteins.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yingchuan B. Qi (ybq@hznu.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Wild-type C. elegans were Bristol N2 strain. For SNP mapping the polymorphic wild-type Hawaiian strain, CB4856 was used. Strains were cultured on NGM plates using standard procedures (Brenner, 1974) at 20–23 °C. Animals analyzed in this paper were hermaphrodite.
Cell lines
Human embryonic kidney cells 293 (HEK 293) were cultured in DMEM supplied with 10% FBS and antibiotics.
METHOD DETAILS
Transgene alleles
All alleles generated in YBQ lab are designated as “ybq” alleles, and all strains, as “BLW” strains. The origination of other alleles/strains are noted individually. “Ex” denotes exchromosomal array transgene alleles. “Is” denotes integrated transgene array alleles. The exchromosomal array transgenes were generated by injecting mix of DNA constructs (of 20 ng/μl concentration, unless noted differently) into gonads of young adult hermaphrodites, and screening for F1 lines that produce transgene-carrying F2 progenies. The integrated transgene alleles were generated by a process of UV-irradiation induced mutagenesis. In brief, the exchromosomal array transgene animals of L4 or young adult were irradiated in UV crosslinker (UVP HL-2000 HybriLinker) under setting of “Energy” (35–50 mj). The F1s of irradiated P0 animals were clonally cultured and screened for integration. The isolated integrated transgene are outcrossed with N2 animals at least four times to rid of background mutations.
Fluorescence markers
Localization of presynaptic sites in DDs and VDs: unc-25pro-mCherry∷Rab-3 (juIs236) (YJ lab); Localization of presynaptic sites in DDs: flp-13pro-GFP∷Rab-3 (ybqIs46) and flp-13pro-UNC-57∷GFP (ybqIs49); Analysis of axonal morphology in DD and VDs: unc-47pro-GFP (oxIs12) (Erik Jorgensen lab, Univ. of Utah); Labeling of A and B motor neurons: acr-2pro-GFP (juIs14) (YJ lab); Localization of presynaptic sites in DA, DB, VA, VB: acr-2pro-GFP∷SNB-1 (nuIs94) (Josh Kaplan lab, Harvard Univ.); Localization of presynaptic sites in DA9 and VA12: mig-13pro-SNB-1∷GFP (wyIs109) (Kang Shen lab, Stanford Univ.); M-cell lineage: hlh-8pro∷GFP (ayIs6) (Andrew Fire lab, Stanford Univ.)
Genomic myrf-1 rescue transgene
Forward primer 5′-TATTTGAACAATTTGACAATCTGG and reverse primer 5′-GAGAGGGTCTTCGTTACTAT were used to amplify 11.5 kb myrf-1 genomic sequence that includes 4.8 kb of promoter region, complete coding region, and 2 kb of downstream region of myrf-1. The rescuing transgene (ybqEx55) was made by injecting PCR product at 0.1 ng/μl into myrf-1(ju1121)/mIn1.
Tissue specific rescues transgenes
DD and VD: unc-25pro- myrf-1 (ybqEx85); DD: flp-13pro- myrf-1∷FLAG (ybqEx94); Pan-neuron: rgef-1pro- myrf-1 (ybqEx86); Epidermis: dpy-7pro- myrf-1 (ybqEx16); Body wall muscles: myo-3pro- myrf-1 (ybqEx93).
Truncated myrf-1 proteins and other myrf-1 mutations
unc-25pro-MYRF-1∷Flag (ybqEx17); unc-25pro-GFP∷MYRF-1(N’1-482aa) (ybqEx102); unc-25pro-MYRF-1(C’485-931aa)∷Flag (ybqEx199); unc-25pro-GFP∷MYRF-1(∆TM)∷Flag (ybqEx183), deleted TM sequence is “NSCHLGFHYGRLSSRHVSSLCP”(AACAGTTGTCACCTTGGTTTCCATTATGG CCGCTTGTCTTCTCGCCATGTCAGCTCTTTATGTCCTT); unc-25pro-GFP∷MYRF-1(∆NLS)∷FLAG (ybqEx278), deleted NLS sequences are “RKRSRL” (CGTAAGAGAAGCCGACTTGA) and “KQKN” (AAACAAAAGAATCG); unc-25pro-GFP∷MYRF-1(N’1-482aa)(∆NLS) (ybqEx280); unc-25pro-GFP∷MYRF-1(S483A)∷Flag (ybqEx139); unc-25pro-GFP∷MYRF-1(G274R) (ybqIs27); unc-25pro-GFP∷MYRF-1(N’1-482aa)(G274R), Pflp-13-GFP∷Rab-3 (ybqEx517, ybqEx541); unc-25pro-MYRF-1∷Flag, unc-25pro-mCherry∷Rab-3 (ybqEx609); unc-25pro-MYRF-1(S483A)∷Flag, unc-25pro-mCherry∷Rab-3 (ybqEx607).
myrf-1 expression analyses
myrf-1 minigene (also see Figure 3A) transgene (ex-chromosomal array): myrf-1pro-GFP∷MYRF-1∷Flag-myrf-13′UTR (ybqEx164) (injected at 0.1 ng/μl); myrf-1 minigene transgene (integrated): myrf-1pro-GFP∷MYRF-1∷Flag-myrf-13′UTR (ybqIs13) (integrated ybqEx164); (GFP is inserted after the Ala171 of MYRF-1, and Flag is inserted at the C-terminus.)
myrf-2 rescue and expression analyses
myrf-2 over-expression: unc-25pro-myrf-2 (ybqEx528), unc-25pro-gfp∷myrf-2∷HA (ybqEx529); myrf-2 minigene (also see Figure S5C) transgene: myrf-2pro-GFP∷MYRF-2∷HA (ybqIs107), (GFP is inserted after Ile190 of MYRF-2, and HA is inserted before the stop codon); myrf-2 transcription reporter (also see Figure S5G): myrf-2pro-NLS∷tagRFP (ybqEx560).
Localization of MYRF-1 and MYRF-2 to subcellular compartments
MYRF-1 and ER: myrf-1pro-GFP∷MYRF-1∷FLAG (ybqIs13); rgef-1pro-cytb-5.1∷mCherry (ybqEx595); MYRF-1 and Golgi: myrf-1pro-GFP∷MYRF-1∷FLAG (ybqIs13); rgef-1pro-aman-2∷mCherry (ybqEx597); MYRF-2 and ER: myrf-2pro-GFP∷MYRF-2∷HA (ybqIs107); rgef-1pro-cytb-5.1∷mCherry (ybqEx599); MYRF-2 and Golgi: myrf-2pro-GFP∷MYRF-2∷HA (ybqIs107); rgef-1pro-aman-2∷mCherry (ybqEx601).
Conditional rescue-transgene myrf-1LoxP
myrf-1LoxP (also see Figure 2F) transgene: myrf-1pro-LoxP-NLS∷tagRFP∷T2A∷MYRF-1-LoxP-myrf-13′UTR (ybqEx401); DD/VD expressed nCre transgene: unc-25pro-nCre (ybqEx322).
Generation of myrf-1 and myrf-2 null alleles and knock-in alleles via CRISPR editing
We designed sgRNA sequences using web-based program Optimized CRISPR Design (http://crispr.mit.edu/), then cloned in Peft-3-cas9-NLS-pU6-sgRNA empty vector (Dickinson et al., 2013). For generating myrf-1 CRISPR indel alleles (see Figure 5A, S6A), four Peft-3-cas9-NLS-pU6-sgRNA plasmids were constructed and their sgRNA target on sequences AGACAGCTCCCTGTGTATAC AGG; GAGCCACAAGATACGGACAT TGG; AAAGAATTGCAACGGCCCAC TGG; AGAAAAGACATTTCTGGGCG AGG (the last three nucleotide of each sequence is PAM site), respectively. The sgRNA targets were selected to generate indel mutations close to the 5′-end of myrf-1 ORF such that the N-terminal portion of MYRF-1 was ensured to be disrupted. The four mixed constructs together with plasmids expressing fluorescent co-injection marker (myo-3pro-mCherry) were injected into N2 hermaphrodites. Over 200 F1 animals carrying transgene was singly picked and cultured. The F2 animals descended from individual F1 plates were screened visually for larval arrest phenotype, and further by PCR-sequencing. Two of the generated myrf-1 indel alleles are the following:
-
wt: …aacggatacgctggagccaatGAtaacggaaatcaaacaatga…
myrf-1(ybq6): …aacggatacgctggagccaatTtaacggaaatcaaacaatga…
-
wt: …ctgcagtcaatcaacctacaAACACcctggctcaactcaaccttt…
myrf-1(ybq7): …ctgcagtcaatcaacctacaGcctggctcaactcaaccttt…
For generating myrf-2 CRISPR indel alleles (see Figure 5A, S6A), two Peft-3-cas9-NLS-pU6-sgRNA plasmids were constructed and their sgRNA target on sequences TTAAGACCTTCTGGTGAGTA TGG; GGAACACAACGTCCAAACCA CGG (the last three nucleotide of each sequence is PAM site), respectively. The sgRNA targets were selected to generate indel mutations close to the 5′-end of myrf-2 ORF such that the N-terminal portion of MYRF-2 was ensured to be disrupted. The two mixed constructs together with plasmids expressing fluorescent co-injection marker (myo-3pro-mCherry) were injected into N2 hermaphrodites. Over 200 F1 animals carrying transgene was singly picked and cultured. The F2 animals descended from individual F1 plates were screened by PCR-sequencing. Two of the isolated myrf-2 indel alleles are the following:
-
wt: …cggactaggaacacaacgtccaa-accacggagtgattccaaacga…
myrf-2(ybq42): …cggactaggaacacaacgtccaaCGGAGTGATGGAGTGCAACGGACCAAGGGAGTGAaccacggagtgattccaaacga…
-
wt: …ggactaggaacacaacgtccaAACCAcggagtgattccaaacgata…
myrf-2(ybq43): …ggactaggaacacaacgtcca-cggagtgattccaaacgata…
For generating GFP and 3xFlag knock-in alleles of myrf-1 (See Figure S3A), besides the plasmids expressing Cas9 and sgRNA, additional plasmid that serves as DNA repair template is required for homology-dependent DNA repair. The sgRNA site for GFP insertion into MYRF-1 is TGACTGCAGCAGTTCCCGGT TGG (TGG is PAM site). The repair template plasmid was generated by assembling the 5′, 3′ homologous arm (about 500–700bp) and the GFP fragments into pCR8 vector. We then introduced synonymous mutation into the PAM domain of related sgRNA by using site-directed mutagenesis of repair template plasmid. The Cas9-sgRNA plasmid and repair template plasmid were co-injected into N2 hermaphrodites. About 150 F1 animals carrying transgene was singly picked and cultured. The F2 animals descended from individual F1 plates were screened by PCR-sequencing. The resulted GFP∷myrf-1(ybq11) knock-in allele is the following:
- GFP is inserted after Ala171 (See Figure S3A):
- WT: …ccaACCGGGAACTGCT-GCAGTCA…(cca is PAM site)
- GFP insertion: …tcaACCGGGAACTGCT-GFP-GCAGTCA… (tca is PAM site being mutated)
(Underline denotes sgRNA sequence.)
For knocking in 3xFlag at the C-terminus of myrf-1, a Cas9-sgRNA plasmid was constructed with sgRNA site GATCAGATTGTACTCCTCGT AGG (AGG is PAM site). A repair template plasmid was generated by assembling the 5′, 3′ homologous arm (about 500–700bp) and the 3xFlag fragments into pCR8 vector. The PAM site was mutated subsequently. The Cas9-sgRNA plasmid and repair template plasmid were co-injected into GFP∷myrf-1(ybq11) hermaphrodites. About 200 F1 animals carrying transgene was singly picked and cultured. The F2 animals descended from individual F1 plates were screened by PCR-sequencing. The resulted GFP∷myrf-1∷3xFlag (ybq14) knock-in allele is the following:
- 3xFlag is inserted at C-terminus right before stop codon (see Figure S3A):
- WT: …GGATTCAAATGGATCCTACGaggAGTACAATCTGATCTTTTATCGCATGTgt aagtgacttcatgaatttttaaacatgaatttaataatgtatttcagGCACCCTATCATCTTCA-TAA (agg is PAM site.)
- 3xFlag insertion: …GGATTCAAATGGATCCTACGaagAGTACAATCTGATCTTTTATCGCATGTgt aagtgacttcatgaatttttaaacatgaatttaataatgtatttcagGCACCCTATCATCTTCA-3xFlag-TAA (aag is PAM site being mutated.)
(Underline denotes sgRNA sequence.)
For knocking in GFP into myrf-2 (see Figure S5A), a Cas9-sgRNA plasmid was constructed with sgRNA site CATAACTTCCTGGCGTTGGA TGG (TGG is PAM site). A repair template plasmid was generated by assembling the 5′, 3′ homologous arm (about 500–700bp) and the GFP fragments into pCR8 vector. The PAM site was mutated subsequently. The Cas9-sgRNA plasmid and repair template plasmid were co-injected into N2 hermaphrodites. About 200 F1 animals carrying transgene was singly picked and cultured. The F2 animals descended from individual F1 plates were screened by PCR-sequencing. The resulted GFP∷myrf-2 (ybq46) knock-in allele is the following:
- GFP is inserted after Ile190 (see Figure S5A):
- WT: …ccaTCCAACGCCAGGAAGTTATGTTCAGgtataaggttaaacaaaagtttatttttcaaaaacgttgttttcagTCAATT-ATGAGTAAAGGAGA… (cca is PAM site)
- GFP knock-in: …tcaTCCAACGCCAGGAAGTTATGTTCAGgtataaggttaaacaaaagtttatttttcaaaaac gttgttttcagTCAATT-GFP-ATGAGTAAAGGAGA… (tca is PAM site being mutated)
(Underline denotes sgRNA sequence.)
Mutagenesis and screening
Strains carrying two fluorescent makers were used for mutagenesis. One is unc-25pro-mCherry∷RAB-3(juIs236) to label synapses in DD/VDs. The second is acr-2pro-GFP(juIs14), which marks the developmental transition from L1 to L2 because this reporter labels VA/VBs that are generated at the end of L1. Moreover, acr-2pro-GFP reporter is upregulated in DA/DBs at L2 stage. L4 animals were treated in 0.5 mM Ethyl methanesulfonate (Sigma M0880) for 4 hours, and then allowed to lay eggs/F1s for a couple days. Gravid adult F1s were collected and treated briefly in a 1:1 mixture of 1N NaOH and household bleach so that the bleach did not disintegrate fully the adult cuticles. The F2 embryos hatched inside the dead hermaphrodite body, fed on the carcass, and some may develop into L2 or even older larvae while remaining inside the body of dead parent. The larvae inside the carcass developed at various paces and were allowed to grow for at least 24 hours before such “wormbags” were examined under fluorescence microscope for up-regulation of acr-2pro-GFP or synaptic patterns of DD neurons. The “wormbags” that showed aberrant DD synapse pattern were retrieved for further examination.
Mapping of myrf-1(ju1121)
To outcross ju1121 we followed DD synaptic rewiring phenotype. We found the allele was linked to the transgene unc-25pro-mCherry∷RAB-3(juIs236) on chromosome II. The ju1121 allele was mapped using genetic markers including unc-4 (Chr II: +1.77) and dpy-10 (Chr II: +0.00). Further mapping used SNP markers between Hawaiian strain CB4858 and Bristol N2 strain. We determined that ju1121 was between Chr II +2.5461 and +2.585. We surveyed the genes in this genomic region and found F59B10.1 (pka. pqn-47, here renamed myrf-1) to be a good candidate based on the reported developmental arrest phenotype in pqn-47 (Russel et al., 2011). We sequenced myrf-1 in ju1121 mutants and found a change of nucleotide G-to-T at the position 821 of myrf-1 open reading frame, which caused an amino acid change of Glycine on 274 to Arginine.
Stage-synchronization of C. elegans cultures
Gravid adults were treated with a 1:1 mixture of 1N NaOH and household bleach to allow worm cuticles to disintegrate. Eggs were washed, dumped on unseeded NGM plates, and incubated at 20°C overnight. Hatched worms were collected, seeded on fresh NGM plates grown with OP50 lawn, and incubated at 20°C. The hour counting started from the seeding point. In cases where a specific hour not noted, worms that were newly hatched or on food for 0–4 hours were considered as Early L1; on food for 8–12h, as Mid L1; on food for 16–20h, as Late L1; on food for 21h, as Early L2.
Electron microscopy analyses
Late L2 stage myrf-1(ju1121) and N2 animals were individually picked and frozen (Leica HPM100) and processed as described with modifications (Lim et al., 2016).
For ju1121, about 1000 continuous 50nm serial ultra-thin cross sections that covered the third and fourth DD motor neurons were sectioned, imaged, stitched and aligned using an in-house developed pipeline (Hung et al., 2013; Lim et al., 2016). For wild type, the whole animal was sectioned at 30nm resolution, and the entire nervous system was reconstructed. The identity of all motor neuron processes at the ventral and dorsal nerve cords were identified based on their relative positions, trajectory and other anatomic features derived from C. elegans larva connectomics projects (Witvliet, Mitchel, Mucalhy, et al., unpublished). Example NMJs by VDs and DDs are shown in this study.
Immunostaining
Stage-synchronized animals were collected and washed in 1xPBS. They were then fixed in 4% PFA on ice for 20 min. The worms are briefly sonicated to allow the cuticles to crack open. The worms were then fixed in 4% PFA for an additional 20 min. The worms were then treated in cold acetone, which had been kept at −20°C, for 15 min on ice. The samples were washed extensively in PBST and incubated in 5% goat serum in PBST at 20°C for 1h. The samples were then incubated with the primary antibody diluted in PBST with 5% goat serum at 4°C for overnight (anti-GFP, rabbit polyclonal IgG, Invitrogen A11122, 1:500; anti-FLAG, mouse monoclonal IgG, Sigma A2220, 1:2000; anti-HA, rabbit polyclonal IgG, Abcam ab9110, 1:500). The next day, the samples were washed extensively in PBST and incubated in secondary antibody diluted in PBST with 5% serum (Alexa Fluor 488-conjugated goat anti-rabbit IgG, Invitrogen, 1:2000, Alexa Fluor 594-conjugated goat anti-mouse IgG, Invitrogen, 1:2000) plus DAPI (1:10000 diluted) at 20°C for 1–2h. The samples were washed in PBST and mounted on slides in Mowiol mounting medium.
Microscopic analysis and quantification
Live animals were anesthetized using 0.5 % 1-Phenoxy-2-propanol in M9 and mounted on 5 % Agar gel pad, or immobilized using 11 % Agarose gel pad. The animals were examined under Zeiss Imager M2 microscope. The DIC images or fluorescence images of Z-focal stacks were taken by a Zeiss MR3 camera on Zeiss M2 wild field fluorescent microscope, which is driven by AxioVision software. Images of live animals or immunostained samples were also acquired on Zeiss LSM710.
Among the images presented in the figures, those for rescued synaptic rewiring in myrf-1(ju1121) (Figure 3F), myrf-2pro-nls∷tagRFP reporter (Figure S5H, I), and axonal pattern of DDs (Figure S1D) were captured by a Zeiss MR3 camera on Zeiss M2 wild field fluorescent microscope. All other images were captured on Zeiss LSM710. Among these confocal microcopy images, those for synaptic rewiring phenotypes, M-cell division, and A/B motor neuron development are projected Z-stack slices using maximum intensity. Others including images for the upregulation of GFP∷MYRF-1, whole mount immunostaining, co-colocalization, and immunostaining of cultured cells are acquired in single confocal slice at 0.8μm thickness.
For quantification of synapse number, images of the whole body of animals were acquired in Zeiss MR3, a wide field camera. The fluorescent images were projected on Z-axis by maximum intensity in ImageJ. The number of fluorescent puncta on dorsal and ventral cords were manually counted. The values and statistics are then processed in GraphPad Prism. The mean number of puncta for individual group was presented in the bar graphs. The error bars denotes SEM. The percentage of animals with zero dorsal puncta was noted in the graphs. The number of analyzed animals was noted on each bar. The difference between two groups was tested by Student’s t-test (given Gaussian distribution).
For analysis of percentages of animals that show rewiring, the animals were examined for evidence of dorsal synapse using fluorescent microscope. The scoring examined binary phenotype, non-rewired state as in myrf-1(ju1121), and rewired state as in wild type or rescued mutants. The rewired group included both complete rewiring and incomplete rewiring, as long as the rewiring was evident judged by the presence of strong dorsal synaptic clusters. Independent scorings on three cohorts of animals were conducted, and the mean percentage from three scorings was quantified and presented in the bar graphs. The total number of analyzed animals was noted on each bar. The difference between two groups was tested by Student’s t-test (given Gaussian distribution).
For analysis of percentages of animals with rewiring for myrf-1(ju1121/ybq6) and myrf-1(ju1121/ybq7) (Figure 6C), the animals analyzed are cross progenies of myrf-1(ju1121) and myrf-1(ybq6), and cross progenies of myrf-1(ju1121) and myrf-1(ybq7), respectively (also see Supplemental Strain List). The quantification was performed similarly to the above, i.e., the scoring examined binary phenotypes, rewired and non-rewired. The percentage of animals showing rewiring for individual group was presented in the bar graph. The difference between each group was tested by Pearson’s chi-squared test (chi-square test for goodness of fit in GraphPad Prism).
For analysis of DD axonal pattern in myrf-1(ju1121) (Figure S1D), the myrf-1(ju1121) animals at late L1 stage were examined on Zeiss fluorescent microscope and scored for axonal pattern. Axon branching, Commissure branching, and dorsal and ventral gaps were scored. The difference between myrf-1(ju1121/+) and myrf-1(ju1121) was tested by Pearson’s chi-squared test (chi-square test for goodness of fit in GraphPad Prism).
For co-localization analysis of C-terminal MYRF-1 and ER/Golgi markers, as well as C-terminal MYRF-2 and ER/Golgi markers, the animals carrying dual transgenes expressing MYRF and ER/Golgi markers were immunostained, and images of the samples in single confocal slice of 0.8μm thickness were acquired on Zeiss LSM710. The images were processed in ImageJ. The regions of interest on the two color images were selected and the pixel intensity from the two color channels were analyzed using “Cocol 2”, a plug-in program in ImageJ.
Transient transfection in cell line and co-Immunoprecipitation
HEK293 cells were cultured in DMEM supplied with 10% FBS. Transient transfection was performed using Lipofectamine 2000 (Thermo Fisher). For co-IP assays HEK293 cells were grown on 30 mm culture dishes and were transfected with the V5∷myrf-1∷3xFlag (pQA1228), or V5∷myrf-1(G274R)∷3xFlag (pQA1261) and GFP∷myrf-2∷HA (pQA1225) plasmids. The transfected cells were incubated at 37°C for 24hrs. The cells were then lysed in 1 mL of Cell Lysis Buffer (50 mM Tris PH7.4, 150 mM NaCl, 1% NP40, 0.25% Sodium taurodeoxycholate hydrate, 1 mM EDTA, protease inhibitor cocktail). Cell lysates were gently sonicated and centrifuged at 14,000Xg for 15 min at 4°C. The supernatant was collected and incubated with addition of primary antibodies, including anti-HA (ab9110, Abcam, 1:500) and anti-FLAG (F1804, Sigma, 1:500), as well as rabbit IgG (12–370, Millipore) and mouse IgG (A7028, Sangon Biotech) controls at the same molar concentration as primary antibodies, respectively, at 4°C for overnight. The next day, protein A beads (16–157, Millipore) were added to the reactions and incubated at 4°C for 4 hours to precipitate the IgGs. Beads were washed and treated in SDS sample loading buffer. The samples were denatured at 70°C for 10 min and analyzed by SDS-PAGE and Western Blot. The antibody used in Western Blot includes: mouse anti-V5 (ab27671, Abcam, 1:5000); rabbit anti-HA (ab9110, Abcam, 1:5000); mouse anti-GAPDH (ABS16, Millipore, 1:5000); goat anti-mouse HRP-conjugated (Maibio, Shanghai, 1:5000), goat anti-rabbit HRP-conjugated (HuaAn, Hangzhou, 1:3000).
Preparation of C. elegans protein extract and Western Blot analysis
Synchronized animals of L2-L3 stages were washed in PBS and snap frozen in liquid N2. The frozen pellet was ground in liquid N2 using a mortar and pestle. The ground worm powder was thawed and incubated in extraction buffer (50 mM Tris PH7.4, 150 mM NaCl, 1% SDS, 1% Triton, 0.5% Sodium deoxycholate, protease inhibitor cocktails) on ice for 1 hour. The samples were centrifuged at 4°C for 20 min. The supernatant was then collected, mixed with SDS sample loading buffer, and heated at 70°C for 10 min. The denatured samples were analyzed by SDS-PAGE (8 % gel) and Western Blot. Additional antibodies used were monoclonal antibody against GFP (sc-9996, Santa Cruz, 1:200) and mouse anti-beta-Actin (ab3280, Abcam, 1:5000).
EMSA
The DNA sequence chosen for the Myrf binding assay was from Rat Rffl gene, and the binding of mouse Myrf onto the sequence was reported previously (Bujalka et al., 2013). We used the same sequences to synthesize biotin labeled DNA probes: WT, biotin 5′-TGACTACCCCACAAGCTGGCACTGCCTGGCGCGGCCA; mutated (Myrf site gone), biotin 5′-TGACTACCCCACAAGtcaacatTGCCTGGCGCGGCCA (shown are forward strands). Both forward and reverse oligos are synthesized carrying biotin label at the 5′-end. The oligos were annealed to generate DNA probes for gel shit assay. Plasmids expressing CMV promoter-driven mouse Myrf, Myrf(G384R), and empty vector were transfected in HEK293 cells. The nuclear extract of the cell lysis using NE-PER Nuclear Extraction (ThermoFisher) were tested for EMSA, which was carried out using LightShift Chemiluminescent EMSA Kit (ThermoFisher).
Protein domain analysis
The protein sequences were analyzed using multiple web based programs including Pfam (EMBO-EBI), PSORT II, InterPro (EMBO-EBI), Conserved Domains (NCBI), and PROSITE (ExPASy). Sequence alignment was analyzed using Clustal 2.1 (EMBO-EBI).
QUANTIFICATION AND STATISTICAL ANALYSIS
Details on quantification and statistics were described in the section of Method details - Microscopic analysis and quantification. Summaries of quantification and statistics were also noted in figure legends.
DATA AND SOFTWARE AVAILABILITY
N/A.
ADDITIONAL RESOURCES
N/A.
Supplementary Material
Table S3. C. elegans strains list. Related to STAR methods.
Highlights.
C. elegans myrf-1 and myrf-2 are indispensable for developmental synaptic rewiring
MYRF proteins localize on ER and are cleaved into active N-terminal fragments
Nuclear translocation of N-terminal MYRFs drives synaptic rewiring
MYRF-1 and MYRF-2 function cooperatively in the same protein complex
Acknowledgments
We thank Andrew Chisholm for critical reading, comments, and editing of the manuscript. We thank Douglas Holmyard and Ben Mulcahy for EM sample processing, section and image processing. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by the National Natural Science Foundation of China (NFSC) general programs #31571272 and #31171197 to Y.B.Q.; CIHR MOP-93619, -123250, and NSERC RGPIN 262112-07 to M.Z. (U. Toronto); Chinese Ministry of Science and Technology 973 grant 2014CB849800 to M.-Q.D. Y.B.Q. was partly supported by NIH R01 035546 and HHMI to Y.J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.M. carried out most experiments with significant assistance from X.M. and H.T.; X.J. carried out of the characterization of myrf-1 and myrf-2 nulls; J.M., D.W., J.M. (Harvard), and M.Z. (U. Toronto) performed the EM analysis; M.Z. (NIBS) and M.-Q.D. performed the protein interaction analysis; Y.J. and Y.B.Q. designed the genetic screen and Y.B.Q. carried out the screen. Y.B.Q. designed and interpreted other main experiments and wrote the paper. Y.J. co-wrote the paper, and M.Z. (U. Toronto) edited the paper.
References
- Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16:1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujalka H, Koenning M, Jackson S, Perreau VM, Pope B, Hay CM, Mitew S, Hill AF, Lu QR, Wegner M, et al. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS biology. 2013;11:e1001625. doi: 10.1371/journal.pbio.1001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–1034. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, Barres BA. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JS, Stryker MP. Development and plasticity of the primary visual cortex. Neuron. 2012;75:230–249. doi: 10.1016/j.neuron.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam SJ, Jin Y. lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature. 1998;395:78–82. doi: 10.1038/25757. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Vaz Gomes A, Kenyon C, Liu J, Krause M, Fire A. Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes & development. 1998;12:2623–2635. doi: 10.1101/gad.12.16.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Philbrook A, McWhirter R, Gabel CV, Taub DG, Carter MH, Hanna IM, Francis MM, Miller DM., 3rd Transcriptional Control of Synaptic Remodeling through Regulated Expression of an Immunoglobulin Superfamily Protein. Curr Biol. 2015;25:2541–2548. doi: 10.1016/j.cub.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell K, White JG, Hobert O. Spatiotemporal control of a novel synaptic organizer molecule. Nature. 2015;523:83–87. doi: 10.1038/nature14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung WL, Hwang C, Gao S, Liao EH, Chitturi J, Wang Y, Li H, Stigloher C, Bessereau JL, Zhen M. Attenuation of insulin signalling contributes to FSN-1-mediated regulation of synapse development. Embo J. 2013;32:1745–1760. doi: 10.1038/emboj.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Ye Z, Hensch TK. Clock genes control cortical critical period timing. Neuron. 2015;86:264–275. doi: 10.1016/j.neuron.2015.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenning M, Jackson S, Hay CM, Faux C, Kilpatrick TJ, Willingham M, Emery B. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J Neurosci. 2012;32:12528–12542. doi: 10.1523/JNEUROSCI.1069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup N, Jin Y. Neural circuit rewiring: insights from DD synapse remodeling. Worm. 2016;5:e1129486. doi: 10.1080/21624054.2015.1129486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoureux JS, Stuart D, Tsang R, Wu C, Glover JN. Structure of the sporulation-specific transcription factor Ndt80 bound to DNA. Embo J. 2002;21:5721–5732. doi: 10.1093/emboj/cdf572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Richardson WD. Evolution of the CNS myelin gene regulatory program. Brain Res. 2016;1641:111–121. doi: 10.1016/j.brainres.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Park Y, Marcotte EM. A Bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLoS biology. 2013;11:e1001624. doi: 10.1371/journal.pbio.1001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MA, Chitturi J, Laskova V, Meng J, Findeis D, Wiekenberg A, Mulcahy B, Luo L, Li Y, Lu Y, et al. Neuroendocrine modulation sustains the C. elegans forward motor state. Elife. 2016;5 doi: 10.7554/eLife.19887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Fleming TW, Petersen SC, Manning L, Matthewman C, Gornet M, Beers A, Hori S, Mitani S, Bianchi L, Richmond J, et al. The DEG/ENaC cation channel protein UNC-8 drives activity-dependent synapse removal in remodeling GABAergic neurons. Elife. 2016;5 doi: 10.7554/eLife.14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DD, Koester SE. Development of projection neuron types, axon pathways, and patterned connections of the mammalian cortex. Neuron. 1993;10:991–1006. doi: 10.1016/0896-6273(93)90049-w. [DOI] [PubMed] [Google Scholar]
- Petersen SC, Watson JD, Richmond JE, Sarov M, Walthall WW, Miller DM., 3rd A transcriptional program promotes remodeling of GABAergic synapses in Caenorhabditis elegans. J Neurosci. 2011;31:15362–15375. doi: 10.1523/JNEUROSCI.3181-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D, Lichtman JW. Elimination of synapses in the developing nervous system. Science. 1980;210:153–157. doi: 10.1126/science.7414326. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Hall DH, Victor M, Stelzer EH, Rapoport TA. Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol Biol Cell. 2002;13:1778–1791. doi: 10.1091/mbc.01-10-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MJ, Gergen JP. DNA-binding by Ig-fold proteins. Nat Struct Biol. 2001;8:384–386. doi: 10.1038/87531. [DOI] [PubMed] [Google Scholar]
- Russel S, Frand AR, Ruvkun G. Regulation of the C. elegans molt by pqn-47. Dev Biol. 2011;360:297–309. doi: 10.1016/j.ydbio.2011.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz EC, Dickmanns A, Urlaub H, Schmitt A, Muhlenhoff M, Stummeyer K, Schwarzer D, Gerardy-Schahn R, Ficner R. Crystal structure of an intramolecular chaperone mediating triple-beta-helix folding. Nat Struct Mol Biol. 2010;17:210–215. doi: 10.1038/nsmb.1746. [DOI] [PubMed] [Google Scholar]
- Senoo H, Araki T, Fukuzawa M, Williams JG. A new kind of membrane-tethered eukaryotic transcription factor that shares an auto-proteolytic processing mechanism with bacteriophage tail-spike proteins. J Cell Sci. 2013;126:5247–5258. doi: 10.1242/jcs.133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo H, Wang HY, Araki T, Williams JG, Fukuzawa M. An orthologue of the Myelin-gene Regulatory Transcription Factor regulates Dictyostelium prestalk differentiation. Int J Dev Biol. 2012;56:325–332. doi: 10.1387/ijdb.120030jw. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stummeyer K, Dickmanns A, Muhlenhoff M, Gerardy-Schahn R, Ficner R. Crystal structure of the polysialic acid-degrading endosialidase of bacteriophage K1F. Nat Struct Mol Biol. 2005;12:90–96. doi: 10.1038/nsmb874. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Di Nardo AA, Aizawa S, Matsuo I, Volovitch M, Prochiantz A, Hensch TK. Experience-dependent transfer of Otx2 homeoprotein into the visual cortex activates postnatal plasticity. Cell. 2008;134:508–520. doi: 10.1016/j.cell.2008.05.054. [DOI] [PubMed] [Google Scholar]
- Thompson-Peer KL, Bai J, Hu Z, Kaplan JM. HBL-1 patterns synaptic remodeling in C. elegans. Neuron. 2012;73:453–465. doi: 10.1016/j.neuron.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimann JM, Zhang YA, Levin ME, Devine WP, Brulet P, McConnell SK. Cortical neurons require Otx1 for the refinement of exuberant axonal projections to subcortical targets. Neuron. 1999;24:819–831. doi: 10.1016/s0896-6273(00)81030-2. [DOI] [PubMed] [Google Scholar]
- White JG, Albertson DG, Anness MA. Connectivity changes in a class of motoneurone during the development of a nematode. Nature. 1978;271:764–766. doi: 10.1038/271764a0. [DOI] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, Richardson WD. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci. 2016;19:1210–1217. doi: 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HM, Walthall WW. UNC-55, an orphan nuclear hormone receptor, orchestrates synaptic specificity among two classes of motor neurons in Caenorhabditis elegans. J Neurosci. 1998;18:10438–10444. doi: 10.1523/JNEUROSCI.18-24-10438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. C. elegans strains list. Related to STAR methods.