

Abstract
Antigen processing and presentation through the MHC class II pathway is critical for activating T helper cells. Angiotensin converting enzyme (ACE) is a carboxyl peptidase expressed by antigen presenting cells. By analysis of ACE null (knockout), wild type and ACE over-expressing (ACE10) mice and the antigen presenting cells derived from these mice, we found that ACE has a physiological role in the processing of peptides for MHC class II presentation. The efficiency of presenting MHC class II epitopes from ovalbumin and hen egg lysosome is markedly affected by cellular ACE levels. Mice over-expressing ACE in myeloid cells have a much more vigorous CD4+ T cell and antibody response when immunized with ovalbumin. ACE is present in the endosomal pathway where MHC class II peptide processing and loading occur. The efficiency of MHC class II antigen presentation can be altered by ACE over-expression or ACE pharmacological inhibition. Thus, ACE is a dynamic participant in processing MHC class II peptides. Manipulation of ACE expression by antigen-presenting cells may prove to be a novel strategy to alter the immune response.
Keywords: angiotensin converting enzyme, MHC class II, antigen presentation
INTRODUCTION
CD4+ T lymphocyte-dependent immune responses are critical to mediating autoimmune diseases and controlling pathogen infection. The MHC class II antigen processing pathway generates cell surface MHC class II peptide complexes essential for the CD4+ T cell responses (1). Unlike MHC class I molecules which are universally expressed in every nucleated cell in mammals, MHC class II molecules are predominantly expressed by professional antigen-presenting cells (APCs), such as dendritic cells and macrophages. The MHC class II-bound antigenic peptides are generated by proteolysis of self and non-self proteins in endosomes and lysosomes. MHC class II molecules must also access these compartments. Newly synthesized MHC class II molecules associate with invariant chain (Ii) in the endoplasmic reticulum (2). The N-terminal cytoplasmic domain of Ii targets the class II-Ii complex to the endocytic pathway. Ii is sequentially cleaved leaving a C-terminal portion of Ii, called class II-associated Ii peptide (CLIP), which protects the class II peptide binding groove from binding peptides (3). In the acidic environment of lysosomes, cathepsins and asparaginyl endopeptidase are activated and they cleave endocytosed exogenous proteins and endogenous proteins localized to this compartment, and generate peptides. In the lysosomal compartment, the class II-related molecule H2-M (HLA-DM in humans) interacts with class II-CLIP and facilitates the exchange of CLIP for peptides (4). MHC class II peptide complexes are then directed to the cell surface. MHC class II molecules can also recycle from the cell membrane and be reloaded with peptides in the endosome compartments (1). Understanding how peptide-MHC class II complexes are generated in antigen presenting cells provides insight into T cell function in normal and pathological states.
Angiotensin-converting enzyme (ACE) is a zinc-dependent carboxyl dipeptidase that belongs to the gluzincin family (6). The enzyme has an important role in the regulation of blood pressure through its cleavage of angiotensin I to the vasoconstrictor angiotensin II, and ACE inhibitors are widely used in treating hypertension and other cardiovascular disease. However, ACE has wide substrate specificity, including such diverse peptides as bradykinin, substance P and Ac-SDKP. ACE is a cell membrane–associated type I ectoenzyme, but functionally active ACE is also present in the endoplasmic reticulum where ACE participates in MHC class I antigen processing (7, 8). ACE is expressed by many different tissues (9), including APCs (10, 11). We observed the induction of ACE in APCs by either IFN-γ or L. monocytogenes, indicating a physiological advantage during immunologic challenge. Here, by studying mice and APCs with high, normal or no ACE expression, we show that under physiological conditions, ACE participates in MHC class II antigen presentation.
MATERIALS AND METHODS
Mice
ACE1 and ACE 10 mice have been described previously (12, 13). These mice have been backcrossed ten times to the C57BL/6J (H-2b) background. The WT mice were the littermates of either ACE1 or ACE 10 mice. OT-II mice were purchased from Jackson Laboratory. The mice used in the in vivo experiments were males aged between 8–10 weeks old. Mice were housed and bred in accordance with the guidelines of the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center and Zhejiang University.
Cells and Cell Lines
Thioglycollate-elicited peritoneal exudate cells were collected via peritoneal lavage 4 days after a 2-ml injection of 3% thioglycollate broth intraperitoneally (i.p.) and were cultured in tissue culture-treated plates (1×106/ml) at 37 °C and 5% CO2 in 10% fetal calf serum RPMI 1640, 50 μM β-mercaptoethanol, 0.5 mM sodium pyruvate, 10 mM HEPES buffer, 50 units/ml penicillin, 50 μg/ml streptomycin, and 2 mM L-glutamine. For purification of thioglycollate-elicited peritoneal macrophages, peritoneal exudate cells were allowed to adhere for 2 hours, after which nonadherent cells were washed off to achieve a >95% purity of macrophages. For flow cytometry assay, peritoneal exudate cells were treated and cultured in Costar low adherence culture plates. Hen egg lysozyme-specific class II MHC-restricted hybridomas Hb1.9, H30.44, H46.13 and BO4 were generously donated by Dr. Lars Karlsson (R. W. Johnson Pharmaceutical Research Institute at San Diego). OT-II T cells were isolated from the spleen of OT-II transgenic mice by MACS with CD4+ T Cell Isolation kit (Miltenyi Biotec) using a negative selection strategy.
Antigens, Antibodies and Reagents
OVA323–339 peptide was synthesized by the Emory University Microchemical Core Facility. Ovalbumin was from Worthington Biochemical Corporation and hen egg lysozyme was from Sigma-Aldrich. Some reagents were purchased: mouse IFN-γ (PeproTech), and lisinopril (Sigma). The PE-conjugated anti-CD69, PE-conjugated anti-CD80 (16–10A1), APC-conjugated anti-CD3ε, APC-conjugated anti-CD86 (GL-1) and the ELISA kits for IL-2, IFN-γ and IL-17A were all purchased from eBioscience. Alexa Fluor 488-conjugated ovalbumin was from ThermoFisher. AF6–120.1 (anti-I-Ab),1G11 (anti-EEA1) and eBioH4A3 (anti-LAMP-1) were from eBioscience. A rabbit antiserum that recognizes mouse ACE was previously described (13). The FITC-conjugated mouse TCR Vβ Screening Panel was from BD Pharmingen. Flow cytometry stained samples were analyzed on CyAn ADP (Beckman Coulter) and data were analyzed with FlowJo software.
ACE Intracellular Staining
Macrophages derived from ACE10 mice were seeded into each chamber of 8-chamber culture slides (BD bioscience) in culture medium. Phagocytosis was performed by adding 1μm latex beads (Sigma,1:500 dilution) into the cells and co-incubating for 1 hour at 37 °C. After extensive washing with PBS, cells were fixed with 4% PFA/PBS for 10 min followed by blocking and permeabilization with 1% BSA and 0.1% Triton X-100 in PBS for 1 hour at room temperature (RT). Then, primary antibodies were used for staining at RT for 2 hours followed by treating the cells with secondary antibodies for another 2 hours at RT. After PBS washing, chamber walls were removed and coverslips were mounted with DAPI- mounting medium (Vector). Images were acquired using a laser scanning confocal microscope (Olympus FV10i). A secondary anti-rabbit antibody used to stain rabbit-anti-ACE IgG was Alexa fluor 488-conjugated (Abcam), while other secondary antibodies were all anti-mouse IgG conjugated with Alexa fluor 594 (Abcam).
In Vitro Antigen-Presenting
Macrophages were fed with either no antigen, 1 μg/ml OVA323–339, 100 μg/ml OVA or 5 mg/ml HEL for 2 hours according to the experiments. When using HEL as the antigen, 2 mM β-mercaptoethanol was added as well. In the experiments when OT-II T cells or T hybridoma cells were used to measure antigen presentation, cells were fixed with 1% paraformaldehyde, glycine/PBS washed, and then co-incubated with OT-II T or T hybridoma cells. OT-II T cells were stained with anti-CD69 after 4-hour co-incubation. Some co-incubation lasted for 18 hours and the supernatants were subject to ELISA examination of IL-2. Some cells were treated with 1 μM of the ACE inhibitor Lisinopril for 30 minutes before antigen uptake until fixation.
In Vivo Immunization and Immune Response Assays
100 μg OVA was dissolved in 50 μl PBS and emulsified with an equal volume of CFA (0.05%, MP Biomedicals); 100 μl was injected s.c. into the dorsal flank of mouse. Nine days later, the spleens and draining lymph nodes (inguinal) were collected and the cells were restimulated with OVA323–339 for 3 days. The supernatants were collected for IFN-γ and IL-17A ELISA. In some experiments when Alum was used as adjuvant, mice were immunized i.p. with a mixture of 50 μg OVA and 75 μl (3 mg) of Imject Alum (Pierce Biochemicals). For measuring antibody production, 14 days after the immunization, 50 μg OVA alone was injected i.p. as the boost. Seven days after the boost, plasma was prepared from the recipients and their anti-OVA antibody titers were evaluated by ELISA.
Statistics
All P values were calculated using an unpaired 2-tailed Student’s t test with equal or unequal variance as appropriate. All figures show standard error of the mean (SEM).
RESULTS
Effects of ACE on MHC Class II Antigen Presentation
To test whether ACE functions in the processing and presentation of MHC class II peptides, we first examined the effects of ACE on the presentation of OVA323–339, the epitope of ovalbumin (OVA) presented by I-Ab. We used thioglycollate-induced peritoneal macrophages as the APCs since they are activated inflammatory macrophages and express relatively abundant MHC class II molecules. Macrophages derived from ACE knock-out (KO), wild type (WT) and ACE10, a mouse line with ACE over-expressed in macrophage lineage cells, were compared due to their expression of null, normal and increased level of ACE, respectively (12, 13) (Supplementary figure 1). After a 2-hour incubation with OVA protein, macrophages were fixed with paraformaldehyde and the ability to present OVA323–339 by MHC class II was measured by surface CD69 upregulation (Supplementary figure 2) and IL-2 secretion from OT-II T cells. For both CD69 expression and secretion of IL-2, there was a consistent pattern (Figure 1A, B). The ACE null cells gave the lowest values. Increasing ACE concentrations (WT vs. ACE10) resulted in increasing activation of the OT-II cells. In fact, there was an over 3-fold difference in both CD69 and IL-2 expression between ACE null and ACE over-expressing cells (p<0.001). If the background levels of expression are subtracted, the difference between the response to ACE null cells vs WT and ACE10 cells was about 3-fold and 8-fold (p<0.001). To investigate if the difference is due to ACE changing the fundamental ability of macrophages to present antigens to T cells, we repeated the experiment by feeding the ACE KO, WT and ACE10 macrophages with the mature MHC class II peptide OVA323–339. No difference was found between the 3 groups of macrophages (Figure 1A, B). These data suggest that the basic ability of APCs to present MHC class II peptides is not affected by ACE levels. These data imply that ACE participates in the processing of MHC class II antigens.
FIGURE 1. The effects of ACE on ovalbumin MHC class II antigen presentation in vitro.
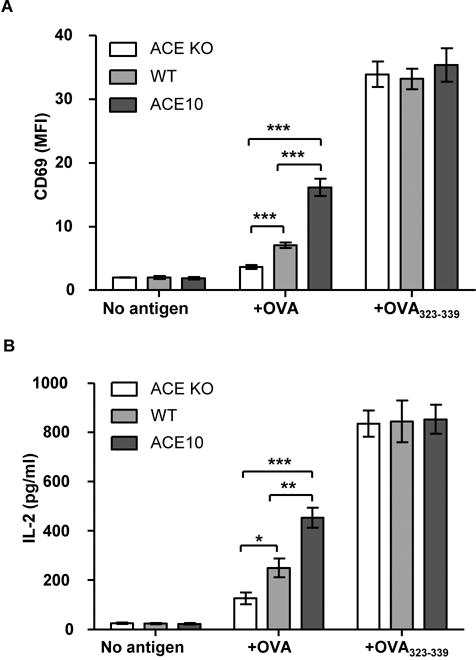
The efficiency of ACE KO, WT and ACE10 macrophages in presenting OVA329–339, when fed with no antigen, OVA protein and OVA329–339 peptide was evaluated by upregulation of CD69 (A) and secretion of IL-2 (B) by OT-II T cells. n=6. *P<0.05, **P<0.01, ***P<0.001. This figure, and all figures, show the standard error of the mean (SEM).
To further investigate whether ACE expression affects the basic machinery of MHC class II antigen presentation, we evaluated the surface expression of MHC class II and co-stimulatory molecules CD80 and CD86 on ACE KO and ACE10 macrophages (Figure 2A and Supplementary figure 3). The expression of these molecules does not differ when ACE expression changes. Further, the soluble antigen studied above was taken up by APC pinocytosis before going through antigen processing and presentation by MHC class II molecules, we studied whether protein uptake was affected by null or high levels of ACE. To investigate this, we incubated macrophages with Alexa Fluor 488-conjugated OVA for 1 hour. Cells were then studied by flow cytometry. No difference was observed between ACE KO, WT and ACE10 macrophages in taking up soluble antigen (Figure 2B).
FIGURE 2. The effects of ACE on macrophage surface molecules and pinocytosis.

(A) Expression of MHC class II molecular I-Ab and co-stimulatory factors CD80 and CD86 on the surface of macrophages from ACE KO, WT and ACE10 mice. N=5. (B) Pinocytosis of fluorescein conjugated OVA by ACE KO, WT and ACE10 macrophages was measured by flow cytometric analysis of mean fluorescence intensity (MFI). n=5. *P<0.05, **P<0.01, ***P<0.001.
To further understand the effects of ACE on MHC class II antigen presentation, we tested another model antigen, hen egg lysozyme (HEL), by feeding this protein to macrophages. The presentation of four MHC class II epitopes of HEL, i.e. HEL20–35, HEL30–53, HEL46–64 and HEL74–88, by I-Ab was evaluated using hybridomas Hb1.9, H30.44, H46.13 and BO4 (14) (Figure 3). Because three of these epitopes contain cysteine residues (5), we added β-mercaptoethanol to the coincubation of macrophages and HEL to increase the efficiency of HEL presentation. Again, ACE affected the antigen presentation. With HEL, 3 of the 4 epitopes tested showed a direct or inverse relationship between APC expression of ACE and the efficiency of surface class II peptide expression. Specifically, higher ACE expression increased the presentation of HEL30–53, and decreased the presentation of epitopes HEL20–35 and HEL74–88. For HEL46–64, there was no difference in presentation between ACE KO, WT and ACE10 cells. For each of the three epitopes affected by ACE, there was a consistent pattern that epitope presentation by null cells and ACE 10 cells over expressing ACE showed the opposite effect. Thus, these data again indicate that ACE takes part in the processing of MHC class II peptides. Moreover, ACE has different effects on different peptide epitopes, as would be expected given substrate preferences of the enzyme.
FIGURE 3. The effects of ACE on HEL MHC class II antigen presentation in vitro.

The efficiency of ACE KO, WT and ACE10 macrophages in presenting HEL-associated epitopes by I-Ab was measured by the secretion of IL-2 from their corresponding hybridomas. n=6. *P<0.05, **P<0.01, ***P<0.001.
ACE Promotes OVA Presentation to CD4+ T Cells In Vivo
The previous in vitro studies suggest that ACE over-expression influences, and can even augment, the presentation of certain antigens to CD4+ T cells in vivo. Next, we used an immunization strategy to investigate this hypothesis. Given that ACE KO mice have hypotension and other physiologic defects (12, 15), we used WT and ACE10 mice to study the immune response to immunization. OVA protein was emulsified in complete Freund’s adjuvant (CFA) and injected s.c. into cohorts of WT and ACE10 mice. After 9 days, spleens and draining lymph nodes were collected and lymphocytes were restimulated with OVA323–339 peptide. The expression of IFN-γ and IL-17A by CD4+ T cells, which represents the activation of Th1 and Th17 cells respectively, was measured by ELISA (Figure 4A, B and Supplementary figure 4A). The spleens and lymph nodes derived from ACE10 mice consistently produced a greater CD4+ T cell response compared to their counterparts derived from WT mice. For example, splenocytes from ACE10 mice produced 5.52-fold IFN-γ and 5.48-fold IL-17 as compared to WT cells (p<0.001). Data from cells from lymph nodes was similar, though more variable.
FIGURE 4. ACE over-expression enhances OVA presentation to CD4+ T cells in vivo.

WT and ACE10 mice were immunized with OVA-CFA. The spleens and draining lymph nodes (LN) were harvested 9 days later. OVA329–339-specific CD4+ T cell activation was evaluated by production of the cytokines IFN-γ (A) and IL-17A (B). n=6–7. WT and ACE 10 mice were immunized with OVA-CFA (C) or OVA-Alum (D) followed by a boost of OVA alone 14 days later. Seven days after the boost, the titers of anti-OVA IgG subtypes were evaluated by ELISA. n=5. *P<0.05. **P<0.01, ***P<0.001.
Antibody production by B cells requires help from CD4+ T cells. Thus, we also measured the production of anti-OVA antibodies. For all IgG subtypes evaluated, the ACE10 mice produced more anti-OVA antibodies compared to WT mice after CFA-OVA immunization and OVA enhancement (Figure 4C). In fact, for the most common IgG subtype, IgG1, ACE 10 mice averaged more than a 20-fold higher antibody titer (p<0.001). The adjuvant Alum exerts its effects through a different mechanism than CFA (16). To exclude the possibility that the over-production of anti-OVA antibody by ACE10 mice is adjuvant-dependent, we followed the same protocol but used an Alum-OVA mixture for the initial immunization (Figure 4D). Again, the ACE10 mice produced more anti-OVA antibodies. Here, each class of IgG was significantly different between ACE10 and WT. To ensure this change, we also measured the total IgG titer in the plasma of unimmunized WT and ACE10 mice, and no baseline difference was noticed (Supplemental figure 4B). Thus, consistent with the in vitro antigen presentation studies, ACE over-expression can affect several of the CD4+ T cell responses to MHC class II-presented antigen in vivo.
ACE is Present in the MHC Class II Pathway and Its Catalytic Activity is Required for Antigen Processing
The discovery that ACE can participate in MHC class II antigen presentation prompted the question of where ACE encounters MHC class II antigens. ACE is typically exported to the cell membrane where it is bound by its C-terminal hydrophobic sequence. Thus, it is conceivable that the cell membrane is the place where ACE trims peptides presented by MHC class II molecules. It is also possible that in the formation of the endosome/phagosome, vacuolar ACE is internalized with its catalytic domains exposed to the lumen of the endosome/phagosome. To investigate the distribution of ACE in MHC class II compartments inside cells, we treated macrophages with 1 μm latex beads for 1 hour to induce phagocytosis. Cells were permeabilized and co-stained for ACE and the MHC class II molecule I-Ab. Upon phagocytosis, ACE accumulated around latex beads in some intracellular organelles or vacuoles where MHC class II molecules localize, which indicates that ACE is present in MHC class II compartments. (Figure 5A). With the same protocol, we also co-stained for ACE and phagosomal markers. As shown in Figures 5B and 5C, after phagocytosis, ACE exhibited partial co-localization with the early endosome marker EEA1 and late endosome/lysosome marker LAMP1 around latex beads. Therefore, ACE is present in the MHC class II antigen presentation pathway where it may be available to modify intracellular peptides, including peptides for the formation of mature MHC class II.
FIGURE 5. ACE is present in MHC class II pathway and its catalytic activity is required for antigen processing.
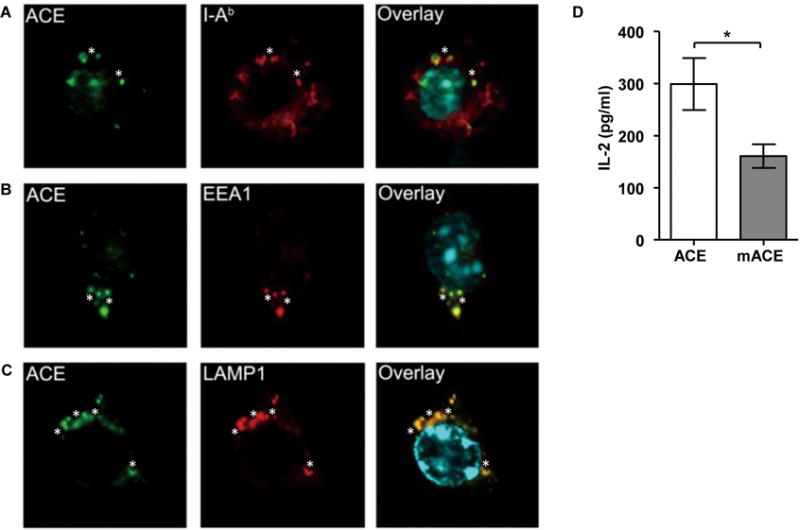
One hour after macrophage phagocytosis was induced with latex beads, ACE co-localized in the compartments of MHC class II (A), early endosome (B) and late endosome/lysosome (C). Stars indicate latex beads which were pinpointed under bright field. (A-C) Representative pictures of 3 independent experiments are shown. (D) WT macrophages were transfected with constructs encoding either wild-type ACE (ACE) or catalytic domain mutated ACE (mACE). Their presentation efficiency of OVA323–339 following OVA administration was assessed by IL-2 secretion of OT-II T cells. n=6. *P<0.05.
To further investigate the relationship between ACE expression and MHC class II antigen presentation, we used nucleofection to transfect WT bone marrow-derived macrophages with an expression construct for catalytically active ACE or with a mutated ACE cDNA (mACE) encoding a protein in which the two catalytic domains of ACE were rendered inactive by point mutations (8). Twenty-four hours after transfection, live attached cells were primed with IFN-γ for 12 hr to upregulate the MHC class II machinery. Cells were then fed with OVA protein, and I-Ab presentation of OVA323–339 was measured by OT-II T cells. Transfection of the catalytically active ACE construct resulted in 2-fold more I-Ab-OVA323–339 presentation than that of cells expressing mACE (Figure 5D). These data strongly indicate that ACE catalytic activity participates in MHC class II antigen processing of at least some epitopes.
The Inhibition of ACE Activity Alters MHC Class II Antigen Presentation
ACE inhibitors have been widely used in treating cardiovascular diseases. To understand whether pharmacological ACE inhibition would affect MHC class II antigen presentation, we treated WT and ACE10 peritoneal macrophages with the ACE inhibitor lisinopril for 2 hours before co-incubating them with OVA. Inhibiting ACE activity completely abrogated the enhanced ability of ACE10 cells in presenting OVA323–339, as measured by the upregulation of CD69 and secretion of IL-2 by OT-II cells (Figure 6A). Indeed, the conversion of ACE 10 cells from cells that over-present OVA323–339 to cells equivalent to WT after short term inhibitor treatment implicates ACE catalytic activity as responsible for the ACE 10 phenotype.
FIGURE 6. MHC II antigen presentation is altered by ACE inhibition.

(A) Macrophages purified from WT and ACE10 mice were pre-treated with or without lisinopril for 2 hours and then co-incubated with OVA. OVA323–339 presentation was assessed through surface CD69 upregulation (top) or IL-2 secretion (bottom) by OT-II T cells. (B) WT and ACE10 mice were treated with lisinopril for 7 days followed by s.c. immunization of OVA-CFA. After another 9 days of lisinopril treatment, the spleens and the draining lymph nodes were harvested and the OVA323–339-specific T cell responses were evaluated for IFN-γ (top) and IL-17 (bottom) production. n=6–7. *P<0.05. **P<0.01, ***P<0.001, NS: Not significant.
To evaluate the effects of an ACE inhibitor in vivo, we treated WT and ACE10 mice with lisinopril for one week with a dose (6 mg/kg/day) which was sufficient to lower blood pressure by about 28 mmHg. This dose achieved complete inhibition of ACE and is higher than the typical dose in humans of about 0.5 mg/kg/day which decreases blood pressure by about 5 mmHg in normotensive subjects. The mice were then immunized s.c. with OVA-CFA. We continued ACE inhibition until 9 days after immunization. On harvesting, CD4+ T cells in the spleen and draining lymph nodes were evaluated in response to OVA323–339 challenge (Figure 6B). ACE inhibition significantly decreased CD4+ T cell responses to OVA antigen in ACE10 mice and there was no difference between lisinopril-treated WT and lisinopril-treated ACE 10 mice. Of note, ACE inhibition also suppressed the presentation efficiency of WT cells. To investigate the possibility that the difference between the WT mice in the presence and the absence of ACE activity may be caused by T cell receptor (TCR) repertoire change, we assessed the spectrum of CD4+ TCR β-chain variable region (Vβ) in splenocytes isolated from WT and ACE KO mice. No difference was found in the use of CD4+ TCR Vβ in presence or absence of ACE (Supplemental Figure 5). Thus, both in vitro and in vivo studies consistently indicate that ACE affects the peripheral activation of CD4+ T cells by APCs.
DISCUSSION
In this report, we have presented several different experimental approaches which consistently show that the modulation of ACE expression or ACE catalytic activity results in changes in the ability of cells to present MHC class II peptides. A variety of epitopes were evaluated; ACE increases the presentation of some epitopes and decreases the presentation of others. The dramatic effect on antigen processing of the ACE inhibitor lisinopril provides strong evidence that it is ACE catalysis that mediates this processing. These studies culminated with in vivo data showing enhanced generation of antigen-specific CD4+ T cells cytokines and antibodies in mice over-expressing ACE. To our knowledge, these studies are the first to show an enzymatic effect of ACE on MHC class II actions.
Our earlier studies showed that ACE expression is upregulated when myeloid cells mature, monocytes differentiate to macrophages, and dendritic cells are stimulated with IFN-γ or Listeria monocytogenes (8). These observations indicate a physiological advantage of ACE expression in antigen presenting cells during immunologic challenge. In these cells, it is well established that peptides presented on MHC class II molecules are products of the continual degradation of extracellular proteins by cathepsins in the endosome/lysosome pathway. The predominant action of ACE is to cleave C-terminal dipeptides from oligopeptide substrates. ACE has wide substrate specificity. Some studies have shown that ACE can also remove C-terminal tripeptides (from substance P) and cleave N-terminal tripeptides (from luteinizing hormone–releasing hormone). However, the in vivo importance of these unusual ACE catalytic activities is not known. What is clear is that ACE can cleave peptides ranging from 3 amino acids (hippuryl-histidyl-leucine) to over 40 amino acids (amyloid-β peptide) (6). It is important in the future to understand to what extend ACE contributes to the diversity and quality of the MHC class II peptide repertoire. However, the fact that MHC class II molecules have an open peptide-carrying grove with variable peptide overhangs makes this question somewhat more difficult to evaluate. It will be important in future studies to understand how the effects of ACE on antigen presenting cells translates into an effect on the pathology of autoimmune disease and the response to infectious pathogens. It has long been recognized that serum ACE levels are elevated in some patients with sarcoidosis and that ACE expression is up regulated by macrophages and giant cells within granuloma (17.18)
Our data show that pharmacological inhibition of ACE alters immune responses in vivo. While this observation is striking, one should be careful extending these data to human use of an ACE inhibitor. As indicated in the Results, the dose we used in mice was much higher than the normal dose for human. As a result, the blood pressure of normotensive mice will drop by about 28 mmHg. Thus, this dose achieves complete inhibition of ACE, something not generally seen in humans. Also, whether ACE inhibitor treatment could be used to alleviate immune overactivation, such as that in the setting of autoimmune diseases, requires further evaluation.
In summary, our data show that the over-expression of ACE in APCs is associated with changes in the processing and presentation of some MHC class II antigens. This is due to the peptidase activity of ACE. Depending on the peptide epitope, ACE expression can have a positive or negative contribution to the generation of individual class II epitopes. In ACE10 mice, over-expression of ACE by monocytes and macrophages is associated with enhanced resistance to the growth of melanoma (13). Whether manipulation of peptidase expression by APCs can be used as a form of immune enhancement for tumors or chronic infections remains to be investigated.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China 31270950 (to XZS), NIH grant R21 AI114965 and PO1 HL129941 (to KEB), National Basic Research Program of China (973 Program) 2015CB553706 (to JF) and China Scholarship Council funding for study abroad (to TZ). The authors thank Ellen Bernstein and Brian Taylor for their assistance in animal husbandry and administration. We also thank You Li for his assistance in confocal microscopy.
Footnotes
DISCLOSURE/CONFLICTS OF INTEREST: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Alfonso C, Han JO, Williams GS, et al. The impact of H2-DM on humoral immune responses. J Immunol. 2001;167(11):6348–6355. doi: 10.4049/jimmunol.167.11.6348. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein KE, Ong FS, Blackwell WL, et al. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev. 2013;65(1):1–46. doi: 10.1124/pr.112.006809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busch R, Rinderknecht CH, Roh S, et al. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunological reviews. 2005;207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 4.Cole J, Ertoy D, Lin H, et al. Lack of angiotensin II-facilitated erythropoiesis causes anemia in angiotensin-converting enzyme-deficient mice. J Clin Invest. 2000;106(11):1391–1398. doi: 10.1172/JCI10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cresswell P. Invariant chain structure and MHC class II function. Cell. 1996;84(4):505–507. doi: 10.1016/s0092-8674(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 6.Danilov SM, Sadovnikova E, Scharenborg N, et al. Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Experimental hematology. 2003;31(12):1301–1309. doi: 10.1016/j.exphem.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 7.Esther CR, Jr, Howard TE, Marino EM, et al. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Laboratory investigation; a journal of technical methods and pathology. 1996;74(5):953–965. [PubMed] [Google Scholar]
- 8.Harmer D, Gilbert M, Borman R, et al. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002;532(1–2):107–110. doi: 10.1016/s0014-5793(02)03640-2. [DOI] [PubMed] [Google Scholar]
- 9.Kool M, Soullie T, van Nimwegen M, et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205(4):869–882. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neefjes J, Jongsma ML, Paul P, et al. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 11.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. 2015;15(4):203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saijonmaa O, Nyman T, Fyhrquist F. Atorvastatin inhibits angiotensin-converting enzyme induction in differentiating human macrophages. Am J Physiol Heart Circ Physiol. 2007;292(4):H1917–1921. doi: 10.1152/ajpheart.00920.2006. [DOI] [PubMed] [Google Scholar]
- 13.Shen XZ, Billet S, Lin C, et al. The carboxypeptidase ACE shapes the MHC class I peptide repertoire. Nat Immunol. 2011;12(11):1078–1085. doi: 10.1038/ni.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen XZ, Li P, Weiss D, et al. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol. 2007;170(6):2122–2134. doi: 10.2353/ajpath.2007.061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen XZ, Lukacher AE, Billet S, et al. Expression of angiotensin-converting enzyme changes major histocompatibility complex class I peptide presentation by modifying C termini of peptide precursors. J Biol Chem. 2008;283(15):9957–9965. doi: 10.1074/jbc.M709574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh R, Cresswell P. Defective cross-presentation of viral antigens in GILT-free mice. Science. 2010;328(5984):1394–1398. doi: 10.1126/science.1189176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shorr AF, Torrington KG, Parker JM. Serum angiotensin converting enzyme does not correlate with radiographic stage at initial diagnosis of sarcoidosis. Respir Med. 1997;91(7):399–401. doi: 10.1016/s0954-6111(97)90252-3. [DOI] [PubMed] [Google Scholar]
- 18.Kahkouee S, Samadi K, Alai A, et al. Serum ACE Level in Sarcoidosis Patients with Typical and Atypical HRCT Manifestation. Pol J Radiol. 2016;81:458–461. doi: 10.12659/PJR.897708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.