

Abstract
Histone N-terminal tails of nucleosomes are the sites of complex regulation of gene expression through post-translational modifications. Among these modifications, histone methylation had long been associated with permanent gene inactivation until the discovery of Lys-specific demethylase (LSD1), which is responsible for dynamic gene regulation. There are more than 30 members of the Lys demethylase (KDM) family, and with exception of LSD1 and LSD2, all other KDMs possess the Jumonji C (JmjC) domain exhibiting demethylase activity and require unique cofactors, for example, Fe(II) and a-ketoglutarate. These cofactors have been targeted when devising KDM inhibitors, which may yield therapeutic benefit. KDMs and their counterpart Lys methyltransferases (KMTs) regulate multiple biological processes, including oncogenesis and inflammation. KDMs’ functional interactions with retinoblastoma (Rb) and E2 factor (E2F) target promoters illustrate their regulatory role in cell cycle progression and oncogenesis. Recent findings also demonstrate the control of inflammation and immune functions by KDMs, such as KDM6B that regulates the pro-inflammatory gene expression and CD4+ T helper (Th) cell lineage determination. This review will highlight the mechanisms by which KDMs and KMTs regulate the target gene expression and how epigenetic mechanisms may be applied to our understanding of oral inflammation.
Keywords: epigenetics, histone lysine methylation, oncogenesis, oral inflammation
Introduction
DNA molecules wrap around histone octamers comprised of H2A, H2B, H3, and H4 to make up nucleosomes, which are the basic units of chromatin. Chromatin remodeling plays a critical role in permissivity of gene expression by allowing access to trans-regulators through dynamic processes (Voss and Hager, 2014). Chromatin remodeling can occur upon methylation of the 5-position of cytosine in guanine–cytosine (GC)-rich regions called CpG islands by DNA methyltransferases (DNMTs), resulting in transcriptional repression (Hung et al, 1999; Martinowich et al, 2003). In addition, chromatin remodeling may occur with post-translational modifications (PTMs) at the N-terminus region of histone tails by means of methylation, acetylation, or ubiquitination, as well as other modifications, for example, sumoylation and phosphorylation (Shiio and Eisenman, 2003; Desjarlais and Tummino, 2016).
Histone acetylation is a common histone modification and involves the transfer of an acetyl group from acetyl coenzyme A (acetyl-CoA) to lysine and arginine residues on histone proteins by histone acetyltransferases (HATs) (Roth et al, 2001). This modification results in transcriptional activation and can be reversed by histone deacetylases (HDACs). Histone methylation can also occur on the lysine and arginine residues through transfer of a methyl group from S-adenosyl-l-methionine (SAM) by histone methyltransferases (HMTs) and be reversed by histone demethylases (HDMs) (Morera et al, 2016). Histone acetylation and methylation may be influenced by phosphorylation of serine and threonine residues on histone tails. Histone kinases transfer a phosphate group from adenosine triphosphate (ATP) to serine and threonine side chain hydroxyl groups (Jeong et al, 2013). Phosphorylation of certain serine or threonine residues, such as serine 10 on histone 3 (H3S10) or threonine 6 on histone 3 (H3T6), can result in histone acetylation or histone demethylation, respectively (Lo et al, 2000; Metzger et al, 2010). Similar to histone acetylation and methylation, histone phosphorylation can be reversed by protein phosphatases. Histone proteins can also be ubiquitinated by ubiquitin ligases, resulting in transcriptional repression or activation upon ubiquitination of H2A or H2B, respectively (Wang et al, 2004; Lee et al, 2007). Histone ubiquitination is reversible through activity of deubiquitinating enzymes, thereby contributing to the dynamicity of epigenetic gene regulation. Histone sumoylation utilizes similar enzymes as ubiquitination to covalently bind small ubiquitin-like modifier (SUMO) proteins to lysine residues (Cimarosti et al, 2012). This modification results in transcriptional repression and can be reversed by sentrin/SUMO-specific proteases (SENPs) (Shiio and Eisenman, 2003; Cimarosti et al, 2012). Among the PTMs of histone tails for epigenetic regulation, histone acetylation and methylation are the most common and well studied (Koch et al, 2007). Histone acetylation by HATs generally leads to gene activation (Roth et al, 2001), and histone tail methylation may result in gene silencing or activation, depending on the location of the amino acid substrate for methylation and the valency, that is, mono-, di-, or tri-methylation (Black et al, 2012).
Histone methylation has long been thought to be involved with heterochromatin formation and permanent chromosome inactivation (Peters et al, 2002). For instance, di- and tri-methylation of histone 3 at Lys 9 (H3K9Me2/3) causes a heritable heterochromatic state by recruiting heterochromatin protein 1 (HP1), which facilitates gene silencing through DNA CpG methylation (Fuks et al, 2003; Audergon et al, 2015). However, the discovery of a Lys-specific demethylase 1 (LSD1 or KDM1A) specific for H3K4 and H3K9 mono- and di-methylation raised the possibility that histone methylation is a dynamic process in transcriptional regulation (Shi et al, 2004). Subsequent to this initial finding, numerous studies reported the presence of histone Lys demethylases (KDMs) that commonly contain Jumonji C (JmjC) domains responsible for catalyzing the demethylation reactions. To date, more than 30 KDM family members have been reported, among which the vast majority belong to JmjC-containing KDMs, except LSD1 and LSD2, which were the original two demethylases identified (Shi and Tsukada, 2013). In general, the gene regulatory roles of KDMs are positioned highly upstream in the signaling cascade, and KDMs possess multiple target genes involved in diverse biological processes. For instance, KDM4B and KDM6B regulate osteogenic differentiation capacities of human mesenchymal stem cells (MSCs) (Ye et al, 2012; Xu et al, 2013); KDM4A regulates metastatic colonization of oral squamous cell carcinomas (OSCCs) (Ding et al, 2013); and KDM6B determines senescence and inflammatory programs (Agger et al, 2009; Barradas et al, 2009; De Santa et al, 2009). Summarized in Table 1 are the Lys histone marks with the corresponding KDMs and Lys methyltransferases (KMTs), as well as examples of known biological effects.
Table 1.
Summary of methylated Lys histone marks and corresponding KDMs/KMTs
| Histone Marks | KDMs | KMTs | Target Gene Effect | General Biological Effects |
|---|---|---|---|---|
| H3K4Me2 | KDM1A/LSD1 KDM1B/LSD2 |
SETD1A SET7 |
Activation | • Enriched over transcription binding regions of promotersa |
| H3K4Me3 | KDM2B/FBXL10/JHDM1B KDM5A/JARID1A KDM5B/JARID1B KDM5C/JARID1C KDM5D/JARID1D |
SETD1A SET7 |
Activation | • Enriched at the transcription start sites (TSS).b • Functions as the focal point for assembly of transcription preinitiation complex.c • Broad H3K4Me3 peak spans the enhancer regions of tumor suppressors.d |
| H3K9Me1 | KDM1A/LSD1 KDM3A/JMJD1/JHDM2A KDM7A/JHDM1D KDM7B/PHF8 |
SUVR5 SUV39H1 SUV39H2 |
Repression | • Establishes constitutive heterochromatin.e • Regulates rRNA synthesis.f |
| H3K9Me2 | KDM4A/JMJD2A/JHDM3A KDM4B/JMJD2B KDM4C/JMJD2C/JHDM3C KDM4D/JMJD2D KDM4E/JMJD2E |
G9A SETDB1 |
Repression | • Establishes constitutive heterochromatin.e • Linked with endotoxin-tolerant phenotype in immune cells.g • Mediates gene silencing at subtelomeric region.h • Regulates osteogenic differentiation.i |
| H3K27Me3 | KDM6A/UTX KDM6B/JMJD3 KDM6C/UTY KDM7A/JHDM1D |
EZH1 EZH2 |
Repression | • Linked with silencing of inflammatory mediators.j • Enriched over senescence gene promoters.k |
| H3K36Me2 | KDM2A/FBXL11/JHDM1A KDM2B/FBXL10/JHDM1B |
SET2 | Activation/Repression | • Determines rDNA promoter regulation.l |
| H3K36Me3 | KDM4A/JMJD2A/JHDM3A KDM4B/JMJD2B KDM4C/JMJD2C/JHDM3C KDM8/JMJD5 |
SET2 | Repression | • Associated with silencing subtelomeric transcripts.m • Regulates longevity through suppressing intragenic cryptic transcription.n |
| H3K56Me3 | KDM4E/JMJD2E | SUV39H | Repression | • Associated with pericentromeric heterochromatin.o |
| H4K20Me1/2 | KDM1A/LSD1 KDM7B/PHF8 |
SET8 SUV420H1 |
• Broadly distributed across genome.p • Involved in cell cycle regulation and DNA repair.p • Regulates chromatin structure during cell cycle progression.p |
|
| H4K20Me3 | KDM7C/PHF2 | SMYD5 SUV420H2 |
Repression | • Enriched at pericentric and subtelomeric heterochromatin.q |
| • Regulates the expression of TLR4-target inflammatory genes.r |
KDMs – basic mechanisms of enzyme functions
KDMs possess distinct functional domains and motifs that are critical to their enzyme activity, and are classified by the presence or absence of the JmjC domain. Lysine-specific demethylases (LSD) are those KDMs that lack the JmjC catalytic domain. LSD1, also known as KDM1A, instead possesses a C-terminal amine oxidase-like (AOL) domain that contains substrate binding and flavin-adenine dinucleotide (FAD)-binding domains (Chen et al, 2006a). The majority of KDMs, other than LSD1 and LSD2, possess the JmjC domain that confers demethylase activity and substrate specificity. This domain shares structural similarity to the cupin family of proteins and contains a cofactor coordinating region comprised of Fe(II) and α-ketoglutarate (α-KG) (Clissold and Ponting, 2001; Chen et al, 2006a,b). The JmjC domain also contains a zinc-finger region that brings the JmjC domain into close proximity with the C-terminal domain and plays an important role in structural stability and catalytic activity of the enzyme. The JmjC domain functions as a Fe(II) and α-KG-dependent oxygenase and confers the demethylase activity to the enzyme.
The PHD domain is commonly found in histone modifying enzymes due to its specificity to methylated lysines and also mediates substrate specificity for KDMs (Shi et al, 2006; Lan et al, 2007). Structural studies have revealed the functional interaction between PHD and JmjC domains for the recognition of methylated Lys residues, which may alter the enzyme activity. Horton et al (2010) showed that both PHD and JmjC domains of histone demethylase PHD Finger Protein 8 (PHF8) bind to H3K4Me3 and that such binding enhanced the rate of demethylation on its cognate substrate, H3K4Me2. In addition to these domains, KDMs contain chromodomains called ‘tudor’ motifs that consist of anti-parallel b-strands forming a tube-like structure, which is found in many proteins associated with chromatin (Maurer-Stroh et al, 2003; Sprangers et al, 2003). KDM2A contains a tandem repeat of two tudor motifs near the C-terminal region that bind to H3K4Me3 with specificity determined by side-chains from the tudor motifs (Huang et al, 2006). Hence, KDMs’ non-catalytic domains possess distinct functions that determine the substrate specificity and modulate the enzyme activity.
The first identified KDM containing the JmjC domain was FBXL11 (F-box and leucine-rich repeat protein 11), also named KDM2A, whose demethylase activity is conferred by the JmjC domain and is specific for H3K36Me2 (Tsukada et al, 2006). In addition to the JmjC domain, KDM2A also possesses other motifs, including the CxxC zinc-finger (ZF) domain, PHD domain, and three leucinerich repeats (Tsukada et al, 2006).
KDM2A directly binds to DNA via its ZF domain. An earlier study demonstrated that KDM2A is recruited to non-methylated CpG islands through the ZF domain and depletes the H3K36Me2 enrichment, causing a unique chromatin state at the CpG islands (Blackledge et al, 2010). KDM2A also binds unmethylated CpG islands of ribosomal DNA (rDNA) through its ZF domain during starvation, leading to reduced rDNA transcription by depletion of H3K36Me3 (Tanaka et al, 2014). Starvation would consequently cause reduced ribosome biogenesis and protein synthesis. Therefore, chromatin remodeling at the site of CpG islands by KDM2A is a dynamic process and responsive to environmental cues, rather than a permanent alteration.
The demethylase reaction mechanisms provide insights in design and discovery of potential inhibitors that modulate the biological functions of KDMs, which are involved in diverse human diseases. For instance, LSD1 and LSD2 can be targeted by tranylcypromine, a clinically validated inhibitor of monoamine oxidase (MAO), which structurally resembles LSDs and also requires FAD cofactor for the enzyme reaction (Binda et al, 2010). Likewise, the vast majority of inhibitors of JmjC-containing KDMs are the competitors of α-KG, for example, 5-carboxyl-8-hydroxyquinoline (8-HQ) compounds and GSK-J1, some of which are broad-spectrum inhibitors for all JmjC family KDMs (Hopkinson et al, 2013). Efforts in identifying non-competitive inhibitors of KDMs are also underway to enhance the drug specificity, such as amiodarone derivatives, which inhibit KDM5A activity by binding to the PHD domain (McAllister et al, 2015). Mechanism-based chemical inhibitors of KDMs are not favored as they target common KDM cofactors, such as FAD and a-KG, and thus lack target specificity. Nonetheless, KDMs represent promising therapeutic targets for a diverse array of human diseases as they regulate multiple biological processes, such as cell proliferation/differentiation, oncogenic transformation, and inflammation.
KDMs – master regulators of cell cycle progression and oncogenesis
The number of KDMs discovered so far is indicative of the diverse biological effects and target genes under regulation by the KDM family. A given KDM may elicit different biological effects based on the cell type, microenvironment, and the state of other interacting regulatory proteins. Biological effects of KDMs are further stratified by presence of non-histone target proteins that may be regulated through the demethylase activity. Importantly, individual KDMs are positioned highly upstream along the gene regulatory axis due to their mode of regulation involving chromatin state. Hence, KDMs do function as master regulators of multiple biological processes, among which regulation of retinoblastoma (Rb)-mediated cell cycle progression has been extensively studied.
LSD1, the first KDM identified possessing flavin-dependent mono-oxidase activity (Shi et al, 2004), regulates cell cycle progression by demethylating Lys 442 of myosin phosphatase target subunit 1 (MYPT1), a protein phosphatase that activates the growth inhibitory function of Rb by removing Ser 807/811 phosphorylation (Cho et al, 2011). Consequently, transcriptional activation of E2F target genes is induced by LSD1, resulting in cell cycle progression. For example, LSD1 is associated with Rb on the Epstein–Barr virus latency gene promoter to repress the promoter activity in a cell cycle-dependent manner (Chau et al, 2008). Evidence also indicates the presence of methylated Lys residues on Rb associated with LSD1, possibly modulating cell cycle progression. Hence, LSD1 provides an example in which non-histone proteins, such as MYPT1 and Rb, are the substrates for its demethylase activity and augment its primary biological effect, which is to suppress target gene expression through H3K4 demethylation.
It appears that Rb plays a central role in the epigenetic regulation of cell proliferation through its interactions with multiple KDMs. In addition to LSD1, the active form of Rb recruits KDM5B, a JmjC-containing demethylase specific for H3K4Me3/2/1, to the promoter regions of E2F-responsive genes, including Cdc25c, Cdc2, Mcm3, Mcm5, resulting in gene silencing (Nijwening et al, 2011). Using an immortalized mouse cell line with the temperature-sensitive SA40 large T antigen (LTA), the authors showed rampant occurrence of senescence-like growth arrest at a non-permissive temperature, at which LTA was inactivated. However, KDM5B knockdown abrogated this senescence program, allowing for continued proliferation at the non-permissive temperature. Enrichment of H3K4Me3 on E2F-target gene promoters was markedly reduced during senescence, presumably due to demethylase activity of KDM5B. Likewise, Beshiri et al (2012) showed that KDM5A is highly enriched on E2F-target promoters and that H3K4 methylation was lost during growth arrest induced by terminal differentiation in embryonic stem cells. During quiescence or senescence, Rb-mediated cell cycle arrest is facilitated by the assembly of a multi-subunit complex of DP1, Rb-like 2 (RBL2/p130), E2F4, and MuvB (RBBP4, LIN9, LIN37, LIN52, and LIN54), called the DREAM complex (Osterloh et al, 2007). Cell cycle gene suppression by the DREAM complex is mediated in part by recruitment of KDM5A to the promoter regions of the DREAM target genes, causing H3K4 demethylation (Beshiri et al, 2012). These findings demonstrate the role of the dynamic functional interaction between KDMs and Rb in the epigenetic regulation of E2F target gene expression (Figure 1).
Figure 1.
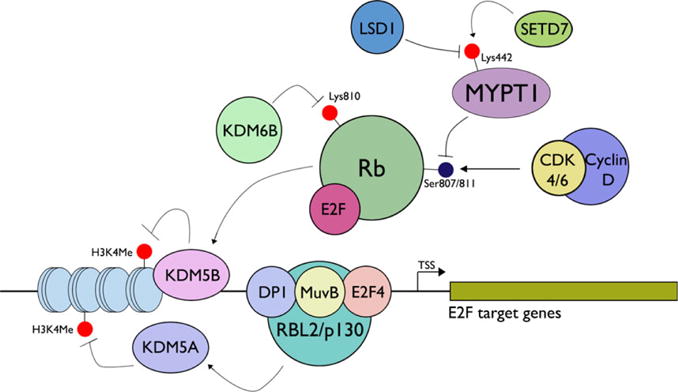
Functional interaction of KDMs with Rb-mediated control of E2F target genes. SETD7 methylates Lys442 of MYPT1, a protein phosphatase targeting Rb at Ser807/811. Demethylation of MYPT1 by LSD1 downregulates MYPT1, causing increased Rb phosphorylation, thereby inactivating Rb (Cho et al, 2011). The active form of Rb also recruits KDM5B to E2F-responsive promoters to demethylate H3K4Me2/3, resulting in gene silencing. During senescence or quiescence, Rb-mediated cell cycle arrest is achieved through the assembly of a multi-subunit complex of DP1, Rb-like 2 (RBL2/p130), E2F4, and MuvB, called the DREAM complex (Osterloh et al, 2007). The DREAM complex recruits KDM5A to the promoter regions of DREAM target genes, causing H3K4 demethylation and gene silencing (Beshiri et al, 2012).
One of the distinguishing features of senescence-associated cell division arrest is induction of the INK4A-ARF locus, resulting in an elevation of p16INK4A, a G1/S-specific cyclin-dependent kinase (CDK) inhibitor targeting CDK4 and CDK6 (Loughran et al, 1996). As a result, CDK4/6-cyclinD complex cannot phosphorylate Rb, allowing Rb (or Rb-like proteins, e.g., p130) to remain complexed with E2F, thereby silencing E2F target genes. The importance of p16INK4A for senescence has been validated in replicative senescence (RS), oncogene-induced senescence (OIS), and stress-induced premature senescence (SIPS) (Collins and Sedivy, 2003). Molecular mechanisms governing the activation of the INK4A-ARF loci during senescence involve epigenetic regulation by Bmi-1, a polycomb-group (PcG) protein component of the polycomb repressive complex (PRC) 1, a multi-subunit epigenetic complex with H2A ubiquitinase activity. To initiate target gene silencing, PRC1 functions in tandem with PRC2, possessing methyltransferase activity on H3K27 through the enhancer of zeste homologue 2 (EZH2). PRC1 subunit RING finger protein 1 (RING1B) is the catalytic unit displaying E3 ubiquitin ligase activity (Li et al, 2006). As p16INK4A elicits, its senescence-inducing effect through modulating Rb phosphorylation, altered H3K27 methylation at INK4A-ARF loci also regulates the Rb-mediated growth suppression.
Regulation of INK4A-ARF loci by PcG proteins suggested the role of KDM6A or KDM6B in regulation of senescence because these KDMs share the same histone substrate (H3K27Me3) as EZH2. This possibility was tested in OIS induced by BRAF oncogene in human diploid fibroblasts (HDFs); KDM6B was induced in cells undergoing OIS, while no changes occurred with KDM6A (Agger et al, 2009). Subsequent functional experiments revealed that loss of H3K27Me3 on the INK4A promoter by KDM6B led to upregulated p16INK4A expression. A similar senescence-inducing role of KDM6B was also demonstrated in HDFs through overexpression of H-Ras oncogene (Barradas et al, 2009). Interestingly, a recent study showed that Rb is a direct substrate for KDM6B demethylase activity during OIS (Zhao et al, 2015). This study showed that demethylation of Rb at Lys 810 by KDM6B reduced Rb phosphorylation at Ser 807/811 by CDK4/6, thereby activating the Rb-mediated suppression of E2F target genes. Therefore, KDM6B utilizes its demethylase activity both on H3K27Me3 and Rb, as a non-histone substrate, to impose its effects on senescence, and joins several other KDMs, including LSD1, KDM2A, KDM5A, and KDM5B, that regulate Rb-mediated growth suppression.
Gene regulation through H3K27 methylation also extends to SIPS, which can be induced by exposure to ionizing radiation. A senescent phenotype is induced in normal human keratinocytes (NHK) exposed to ionizing radiation, concomitant with elevated KDM6B levels and a loss of global H3K27Me3 levels, as well as reduction in PcG factors, such as Bmi-1 and EZH2 (Dong et al, 2011). Overexpression of Bmi-1 resulted in suppressed KDM6B and p16INK4A levels in the irradiated NHK, and these cells bypassed SIPS. Such radioprotective effects of Bmi-1 in NHK were not only due to suppression of INK4A but also increased expression of oxidase genes, for example, Lpo, p22-phox, p47-phox, and Gp1, which neutralize reactive oxygen species. Consequently, the cells expressing Bmi-1 demonstrate enhanced DNA repair capacities and increased survival upon irradiation.
PcG-mediated gene silencing is also linked to carcinogenesis. Using a transgenic mouse model, an earlier study found that aberrant Bmi-1 overexpression was sufficient for T-cell lymphoma development (Haupt et al, 1993). Aberrant overexpression of Bmi-1 and EZH2 has been reported in human cancers, including oral squamous cell carcinomas (OSCCs), and is known to promote the self-renewal capacities of cancer stem cells (Kang et al, 2007; Song et al, 2010). In addition, by suppressing the INK4A-ARF loci, Bmi-1 overexpression evades the senescence program and leads to cellular immortalization of human oral keratinocytes (Kim et al, 2010). A single heterozygous mutation in H3 genes at Lys 27 to methionine (K27M) is associated with the formation of an aggressive brain tumor known as diffuse intrinsic pediatric glioma (Chan et al, 2013). The glioma cancer cells harboring such a mutation demonstrated global reduction of H3K27Me3 expression but increased local enrichment of H3K27Me3 and EZH2 at the gene promoters specific to tumor suppression, including INK4A. A subsequent study confirmed the oncogenic potential of the H3.3K27M mutation in normal neuronal precursor cells (NPC), and the transformed NPC demonstrated elevated genes involved in self-renewal and proliferation, including Lin28B and PLAG1 (Funato et al, 2014). As such, the aberration of epigenetic alterations by PcG proteins is strongly implicated in carcinogenesis through suppression of the senescence growth checkpoints and inducing stem characteristics.
To the contrary, KDM6B, which counteracts the PcG-mediated gene silencing, may elicit tumor suppressive effects. For instance, KDM6B expression is lost during malignant transformation of pancreatic ductal adenocarcinoma, and KDM6B knockdown led to enhanced tumorigenic ability of cells (Yamamoto et al, 2014). One mechanism of tumor suppression underlies induction of senescence (Lin et al, 2012), while another study illustrated a chromatin-independent mechanism that involves KDM6B interaction with p53 (Ene et al, 2012). KDM6B overexpression led to growth arrest in INK4-null glioblastoma stem cells by p21WAF1 induction through a p53-dependent mechanism. Co-immunoprecipitation revealed that KDM6B binds p53 as a substrate for demethylation at Lys 372, resulting in p53 nuclear localization. Interestingly, KDM6B may utilize its same biological functions to elicit tumorigenic effects under different pathological settings. KDM6B is strongly induced by the human papilloma virus (HPV) 16 E7 oncoprotein in human foreskin keratinocytes and then leads to elevation of p16INK4A expression through demethylation of H3K27Me3 at the INK4A promoter region (McLaughlin-Drubin et al, 2011). In HPV+ cervical cancer cells, elevated p16INK4A levels were found to be required for cancer cell survival in order to neutralize the hyperactive CDK4 and CDK6, which would lead to cell death under conditions of Rb inactivation by E7 (McLaughlin-Drubin et al, 2013). Consequently, KDM6B inhibition by means of siRNA or a small molecule inhibitor, such as GSK-J4, led to cell death of HPV+ cancer cells.
The above studies illustrate the mechanistic involvement of KDMs in diverse cellular processes, primarily through epigenetic regulation of target gene expression by histone Lys demethylation, and secondarily through modulation of non-histone target proteins. In particular, multiple KDMs regulate cell cycle progression through functional interactions with Rb and/or E2F. These interactions elicit broad phenotypic effects in cellular senescence and carcinogenesis. Cellular senescence is also closely linked with inflammation in that senescence triggers secretion of a discrete pattern of inflammatory cytokines, such as interleukin-6 (IL-6), IL-8, and GRO-α, collectively named senescence-associated secretory phenotype (SASP) originally observed in culture models exposed to genotoxic stress (Coppe et al, 2008). A recent study showed that KDM6B overexpression induced senescence and SASP in glioma cell lines through its demethylase activity (Perrigue et al, 2015). Likewise, senescence-associated secretion of MCP-1 in human mesenchymal stem cells (MSCs) was suppressed by Bmi-1 through the loss of H2AK119 ubiquitination (Jin et al, 2015). Hence, there is accumulating evidence that cellular inflammatory responses are regulated epigenetically by KDMs.
Emerging roles of KDMs in regulation of inflammation and immune functions
The first evidence supporting epigenetic regulation of inflammation was made in cultured human monocyte-derived dendritic cells (DCs) exposed to bacterial endotoxin lipopolysaccharides (LPS) (Saccani and Natoli, 2002). In this study, exposure of DCs to LPS led to rapid or delayed induction of various inflammatory cytokines, IL-8, MIP-1α, MDC, and ELC during the 72 hrs post-treatment. Among them, several genes encoding for the cytokines with delayed induction had transiently lost H3K9 methylation at the promoter regions during the 72-h period of exposure to LPS, while induction of other epigenetic marks, such as H3K4 methylation and acetylated H3 and H4, remained stable. Furthermore, altered H3K9 methylation at these promoter sites upon LPS treatment coincided with RNA polymerase II recruitment. A subsequent study showed enhanced binding of G9a, a Lys-specific methyltransferase, at the promoter region of tumor necrosis factor alpha (TNF-α) in human monocytes with an endotoxin tolerant phenotype, resulting in H3K9 methylation (El Gazzar et al, 2008). As endotoxin tolerance is linked with a loss of inducibility of cytokine expression after prior exposure to LPS, the above study indicated the mechanistic role of KTMs in such a phenomenon. In addition, G9a recruited at promoter sites forms a complex with HP1 and Dnmt3a/b, and suppresses target gene expression through hypermethylation at CpG islands. The functional significance of G9a enrichment for cytokine gene silencing was shown by knockdown of G9a and consequent reactivation of TNF-α expression in endotoxin tolerant cells. These studies suggest that epigenetic modifications triggered by H3K9 methylation played the primary role in silencing cytokine gene expression during endotoxin tolerance. The tolerant phenotype is associated with increased expression of RelB, a repressive subunit of nuclear factor-κB (NF-κB), as observed in blood mononuclear cells of sepsis patients, resulting in the suppression of inflammatory cytokines (Yoza et al, 2006). It was then found that RelB interacts with and recruits G9a in the tolerant phenotype, thereby causing target gene suppression via H3K9 dimethylation (Chen et al, 2009). These studies demonstrate that target gene regulation by histone Lys methylation is a dynamic process that modulates inflammatory responses through interactions with known inflammatory mediators.
To identify the KDMs that mediate inflammatory responses, De Santa et al (2009) reported screening of KDMs induced by acute LPS treatment in the Raw264.7 mouse macrophage cell line. Within 2 hrs of LPS exposure, KDM6B was strongly induced while KDM6A, a related KDM with the same substrate specificity (H3K27Me3), remained unchanged. KDM6B induction was dependent upon NF-κB, as overexpression of IκBα or deletion of IKKγ in cells abolished the inducing effect of LPS. The first intronic region of the Kdm6b gene was found to contain the conserved κB sites with a confirmed increase in p65 binding after LPS exposure. Chromatin immunoprecipitation (ChIP) assay combined with high throughput sequencing (ChIP-Seq) revealed enrichment of KDM6B near the target promoters at the genomic level (De Santa et al, 2009). ChIP assay allows for survey of DNA-protein interactions. Hence, sequencing of all DNA elements that co-precipitate with specific proteins, for example, KDM6B, would indicate potential genomic target sequences. Upon activation of macrophages with LPS and IFN-γ, KDM6B was preferentially recruited to the transcription start site (TSS) regions of immune regulatory genes and inflammatory mediators, and those genes involved in antimicrobial defense. Also, the vast majority (73%) of the genes with KDM6B enrichment correlated with the recruitment of RNA polymerase II (RNA Pol II) in the activated macrophages, indicative of a gene activator role of KDM6B in the inflammatory queue. As KDM6B demethylates H3K27Me3 and opposes PcG-mediated gene silencing, inflammatory signals would be diminished by PRC2 subunits possessing KMT activity, such as EZH2. In fact, Peng et al (2015) reported suppression of Th1-type chemokines, CXCL9 and CXCL10, in colon cancer tissues exhibiting high expression of EZH2, SUZ12, and EED, supporting the role of PcG proteins in the silencing of inflammatory genes. Apparently, the interplay between PcG and KDM6B determines the cellular responses to inflammatory and senescing signals, as well as other epigenetic modifiers, including G9a and Dnmt3a/b (Figure 2). A recent study by Kruidenier et al (2012) demonstrated the first small molecule inhibition of KDM6B by GSK-J4, which binds to the enzyme’s active site and prevents incorporation of α-KG, a required cofactor for KDM6B demethylase activity. Cell-based assay revealed reduced expression of LPS-inducible genes, such as TNF-α and inflammatory cytokines, and recruitment of RNA Pol II to the TNF-α TSS in macrophages exposed to GSK-J4 or related compounds. Besides KDM6B, a subsequent study found the involvement of PHF2, a KDM specific for H4K20Me3, in the activation of TLR4-target inflammatory genes, TNF and CXCL10, upon exposure to LPS (Stender et al, 2012). It is not known whether KDM6B and PHF2 have redundant or complementary effects on epigenetic inflammatory signaling. Also, there may be other KDMs that control the expression of inflammatory mediators through modifying different histone marks. However, KDMs in general may be therapeutic targets of novel anti-inflammatory approaches, and further research will be necessary to elucidate the therapeutic benefits of these compounds.
Figure 2.
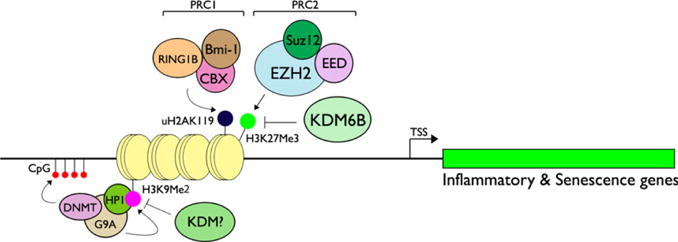
KDMs and KMTs regulating inflammatory and senescence genes through epigenetic modifications. PRC2 subunit EZH2, a KMT specific for H3K27, triggers K27Me3, which is recognized by CBX subunit of PRC1, which also contains Bmi-1 and RING1B. These protein subunits of PRC1 demonstrate E3 ubiquitin ligase activity for H2AK119, causing monoubiquitination, which then inhibits Pol II elongation (Li et al, 2006; Zhou et al, 2008). During inflammatory signaling or senescence, KDM6B is induced and demethylates H3K27Me3 and suppresses PcG-mediated gene silencing. Inflammatory cytokines are also regulated through G9a, a KMT specific for H3K9Me2 that recruits HP1 and Dnmt3a/b, resulting in hypermethylation of CpG islands (Han et al, 2016). During inflammation, demethylation of H3K9Me2 has been found strongly associated with RNA Pol II recruitment to the pro-inflammatory cytokines (Saccani and Natoli, 2002), although the corresponding KDM is yet to be identified.
A recent study demonstrated that KDM6B and KDM6A play pivotal roles in CD4+ T-cell development through epigenetic regulation of S1pr1 and Klf2 expression, both of which are necessary for Th cell maturation and egress from the thymus (Manna et al, 2015). KDMs also modulate immune functions by determining CD4+ Th cell lineage differentiation, thereby vastly affecting immune responses in multiple biological systems. Naïve CD4+ T cells from the thymus can differentiate into several different Th cell lineages, including Th1, Th2, Th3, Th17, natural killer T, and regulator T helper (Treg) (reviewed in Kennedy and Celis, 2008), and would facilitate distinct immune responses based on the cytokine and gene expression profiles, as well as its interaction with other immune cells. For instance, CD4+ naïve T cells differentiate into the Th1 lineage through transcription regulator T-bet expression and produce IFN-γ and TNF-α to provoke pro-inflammatory responses (Zhu et al, 2010). Conversely, the IL-4 and IL-2 cytokines drive the differentiation of CD4+ naïve T cells to the Th2 lineage by the action of GATA3 (Zheng and Flavell, 1997). Th2 cells with discrete cytokine profiles, for example, IL-4, IL-5, and IL-13, play pivotal roles in humoral immunity through B cells against certain pathogens and allergic reactions. The Th17 lineage is induced by TGF-β and IL-6 through transcription factors RORct and pSTAT3, while the immune-suppressive Treg cell lineage is induced by TGF-β through Foxp3 (Kimura and Kishimoto, 2011). Th17 cells play critical roles in pro-inflammatory reactions in autoimmue disorders, such as rheumatoid arthritis and systemic lupus erythematosis (Singh et al, 2014). As such, perturbation of CD4+ Th cell lineage differentiation could impair the immune system with diverse health consequences.
KDM6B was found to play critical roles in lineage determination for CD4+ Th cell differentiation. Li et al (2014) utilized CD4-specific deletion of the Kdm6b gene and showed that CD4+ naïve T cells preferentially undertook the Th2 and Th17 lineages while Th1 and Treg pathways were suppressed. This finding is in keeping with the prior discussion that KDM6B in general regulates pro-inflammatory cytokine gene expression in response to LPS challenge (De Santa et al, 2009). Kdm6b ablation in CD4+ Th cells led to congruent alterations in T-cell lineage factors; T-bet expression was reduced while GATA3 was induced, shifting the lineage to Th2. Also, KDM6B appears to interact with key transcription factors for Th lineage determinants. KDM6B interacts with Smad2/3 and is recruited to the Nodal target gene promoters to activate gene expression through H3K27Me3 demethylation (Dahle et al, 2010). KDM6B interaction with Smad3 was also reported by Li et al (2014) in the context of H3K4 methylation at the Foxp3 promoter, which drives the Treg lineage. These authors found that KDM6B also interacts with Ash2L, a KMT specific for H3K4 methylation sites that promotes gene activation, and recruits the KMT complex to the Smad3 target promoter, like Foxp3. Kdm6b ablation would therefore disrupt this Smad-Ash2l KMT complex formation at Foxp3, thereby suppressing Treg lineage differentiation. These findings demonstrate the critical role of KDM6B in CD4+ Th lineage determination through its histone demethylase activity and direct interactions with key transcription modulators.
KDMs – role in craniofacial and mineralized tissue formation in the oral cavity
In addition to regulating systemic developmental and diseases processes, KDMs have also been found to regulate biological processes in the oral cavity. In particular, KDMs regulate dental mesenchymal stem cell (dMSC) and bone marrow mesenchymal stem cell (BMMSC) proliferation and differentiation, which play an instrumental role in tooth and alveolar bone formation and craniofacial development. For instance, KDM2A has been found to promote ameloblast and odontoblast differentiation and suppress adipogenic and chrondogenic differentiation of stem cells from the apical papilla (SCAP), indicating its importance in tooth development (Dong et al, 2013; Yi et al, 2016). Zheng et al demonstrated that KDM5B and KDM6B are upregulated during tooth development (Zheng et al, 2014). KDM6B has been shown to transcriptionally activate bone morphogenetic protein 2 expression and promote odontogenic differentiation of dMSCs and mineralized tissue formation (Xu et al, 2013).
KDMs play an important role in alveolar bone formation and craniofacial development. Yang et al showed that KDM6A transcriptionally activates runt-related transcription factor 2 (Runx2) and Osterix, important transcription factors for osteoblast differentiation, thereby promoting mineralized tissue formation (Yang et al, 2015). KDM4B and KDM6B have also been shown to enhance osteogenic differentiation of BMMSCs by demethylating H3K9me3 to activate DLX gene expression and demethylating H3K27me3 to activate HOX expression, respectively (Ye et al, 2012). PHF8 regulates jaw development and neuronal survival through Msh homeobox 1 (MSX1), transcription factor regulating craniofacial and neuronal development, and its analog MSXB (Qi et al, 2010). Unlike these KDMs, which promote osteoblast and mineralized tissue formation, KDM2B has been shown to suppress the osteogenic differentiation of MSCs by demethylating H3K4 and H3K36 residues on key target gene promoters. Dysregulation KDM2B and its failure to demethylate these sites has been implicated in the development of oculo-facio-cardio-dental (OFCD) syndrome, an inherited disease that results in severe craniofacial and cardiac abnormalities (Fan et al, 2009).
The application of dMSCs for tissue regeneration has been an area of great interest due to ease of access and their immunomodulatory properties (Pierdomenico et al, 2005). KDM6B has been shown to play an important role in this phenomenon by transcriptionally activating insulin-like growth factor-binding protein 5 (IGFBP5), which is highly expressed in MSCs undergoing osteogenic differentiation (Liu et al, 2015). Liu et al demonstrated that IGFBP5 promotes the anti-inflammatory and immunomodulatory effect of dMSCs by downregulating NF-κB signaling, and that IGFBP5 expression is downregulated in periodontal tissues collected from patients with periodontitis. Thus, KDM6B is critical for the anti-inflammatory effect of IGFBP5 in periodontal tissues.
KDMs and other epigenetic modulators of oral inflammation
Chronic inflammation by means of periodontal or root canal infection allows abscess formation, causing gross tissue destruction and tooth loss. Earlier studies indicated the possible role of Th1 pro-inflammatory cytokines, including IL-1β and TNF-α, in the pathogenesis of human apical periodontitis linked to chronic inflammation (Matsuo et al, 1994; Formigli et al, 1995). Analysis of human periapical granulomas revealed significant bone resorbing activity, which could be neutralized by anti-IL-1β and anti-TNF-β antibodies (Wang and Stashenko, 1993). In rats, homologous forms to these mediators, IL-1α and TNF-α, are strongly induced during the early stages of apical periodontitis in a murine model of pulp exposure and were primarily secreted by macrophages and fibroblasts (Tani-Ishii et al, 1995). The functional significance of other types of Th1 and Th2 cytokines was assessed in the pulp exposure model with gene knockouts. These studies revealed that IFN-γ, IL-12, and IL-18 had little effect on apical periodontitis, while IL-6 and IL-10 showed protective effects against bone destruction in these pulp exposure models (Sasaki et al, 2000, 2004; Balto et al, 2001). The pro-inflammatory cytokine IL-17, produced principally by CD4+ Th17 cells and neutrophils, is elevated in inflammatory bone lesions, including periodontitis and periapical lesions (Vernal et al, 2005). Ablation of the IL-17 signaling by IL-17RA knockout or IL-17 neutralizing antibody showed protective effects against apical periodontitis in the pulp exposure model, confirming its role in mediating inflammation-induced bone destruction (AlShwaimi et al, 2013). In another study, CD4+ Treg cells expressing the master regulator Foxp3 were shown to accumulate in the periapical lesions, in parallel to an increase in the bony defect, suggesting a possible role of Treg cells in the balance of periapical inflammation (AlShwaimi et al, 2009). Thus, the progression of infection-related bone lesions in the oral cavity involve a complex interplay among pro-inflammatory and immunomodulatory cytokines, as well as lineage-specific differentiation of CD4+ Th subsets.
Mechanistic studies involving KMT/KDMs in oral inflammation are very limited, while studies pertaining to other epigenetic modifications, such as histone acetylation and DNA methylation, demonstrate their regulatory roles in the disease process. Xuan et al (2016) showed indirect evidence of KDM6B involvement in periodontal inflammation through regulation of M2 macrophage polarization, although the detailed mechanisms by which KDM6B regulates periodontal tissue responses to infection remains unknown. Downregulated expression of Lys methyltransferase EZH2 and concordant induction of KDM6B were noted in infected dental pulp, resulting in a loss of differentiation capacities of dental pulp cells exposed to inflammatory mediators, including LPS and TNF-α (Hui et al, 2014). Acetylation of histones is generally associated with active gene expression. All Lys sites, for example, K5, K8, K12, and K16, of H4 are acetylated in euchromatin, whereas Lys acetylation is strongly suppressed on H4 in heterochromatin (O’Neil and Turner, 1995). While H3/H4 acetylation is generally linked with gene transcription, recent studies demonstrate site-specific roles of histone acetylation or deacetylation for gene activity (Shahbazian and Grunstein, 2007). Hence, like KMT/KDMs, histone acetylation/deacetylation is considered a dynamic process to modulate gene activity. In periodontitis models, H3 acetylation was found to be induced in gingival epithelial cells through upregulation of histone acetyltransferase p300/CBP, although identification of specific gene targets mediating the periodontal inflammatory response requires further investigation (Martins et al, 2016). The role of DNA methylation at CpG-rich promoter regions has been more extensively studied in oral inflammatory conditions than that of histone modifications. Several studies showed reduced methylation of CpG islands on the promoter regions of inflammatory genes, such as TLR2, TLR4, and IL-6, in oral epithelial tissue with periodontitis, which presumably result from diminished expression of DNMT1 and DNMT3a (Ishida et al, 2012; De Camargo et al, 2013; Martins et al, 2016). Notably, DNA CpG hypermethylation is mechanistically connected with KMT activity, such as G9a, and H3K9 methylation and recruitment of HP1 and DNMT lead to CpG island hypermethylation and gene silencing (Fuks et al, 2003; Audergon et al, 2015). Likewise, EZH2, the KMT subunit of PRC2, directly modulates the DNA methylation state of the target promoter through protein interactions with DNMTs (Viré et al, 2006). These findings demonstrate epigenetic cross talk between histone and DNA modifications to tighten the control of target gene expression through epigenetic modulators. Further studies will be required to elucidate the details of epigenetic mechanisms underlying inflammatory lesions throughout the oral cavity and to determine whether they are similar or unique to such processes elsewhere in the body. In doing so, it may be possible to develop novel therapeutics in the management of inflammatory conditions in the oral cavity.
Footnotes
Conflict of interest
None to disclose.
Author contributions
MKK, NHP, CYW provided concept of review. MKK, SM, and CYW wrote the manuscript. SM proof read the manuscript. CYW designed the review and revised the content of this manuscript.
References
- Agger K, Cloos PA, Rudkjaer L, et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23:1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlShwaimi E, Purcell P, Kawai T, et al. Regulatory T cells in mouse periapical lesions. J Endod. 2009;35:1229–1233. doi: 10.1016/j.joen.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlShwaimi E, Berggreen E, Furusho H, et al. IL-17 receptor A signaling is protective in infection-stimulated periapical bone destruction. J Immunol. 2013;191:1785–1791. doi: 10.4049/jimmunol.1202194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult N, Van Beneden A, Decottignies A. Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1a. Nat Struct Mol Biol. 2012;19:948–956. doi: 10.1038/nsmb.2364. [DOI] [PubMed] [Google Scholar]
- Audergon PN, Catania S, Kagansky A, et al. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science. 2015;348:132–135. doi: 10.1126/science.1260638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balto K, Sasaki H, Stashenko P. Interleukin-6 deficiency increases inflammatory bone destruction. Infect Immun. 2001;69:744–750. doi: 10.1128/IAI.69.2.744-750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barradas M, Anderton E, Acosta JC, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–1182. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshiri ML, Holmes KB, Richter WF, et al. Coordinated repression of cell cycle genes by KDM5A and E2F4 during differentiation. Proc Natl Acad Sci USA. 2012;109:18499–18504. doi: 10.1073/pnas.1216724109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda C, Valente S, Romanenghi M, et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc. 2010;132:6827–6833. doi: 10.1021/ja101557k. [DOI] [PubMed] [Google Scholar]
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell. 2010;38:179–190. doi: 10.1016/j.molcel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Gene Dev. 2013;27:985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau CM, Deng Z, Kang H, Lieberman PM. Cell cycle association of the retinoblastoma protein Rb and the histone demethylase LSD1 with the Epstein-Barr virus latency promoter Cp. J Virol. 2008;82:3428–3437. doi: 10.1128/JVI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang Y, Wang F, et al. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc Natl Acad Sci USA. 2006a;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zang J, Whetstine J, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006b;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Chen X, El Gazzar M, Yoza BK, McCall C. The NF-jB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem. 2009;284:27857–27865. doi: 10.1074/jbc.M109.000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HS, Suzuki T, Dohmae N, et al. Demethylation of Rb regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011;71:655–660. doi: 10.1158/0008-5472.CAN-10-2446. [DOI] [PubMed] [Google Scholar]
- Cimarosti H, Ashikaga E, Jaafari N, et al. Enhanced SUMOylation and SENP-1 protein levels following oxygen and glucose deprivation in neurons. J Cereb Blood Flow Metab. 2012;32:17–22. doi: 10.1038/jcbfm.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold PM, Ponting CP. JmjC: cupin metalloenzyme-like domains in jumonji, hairless and phospholipase A2beta. Trends Biochem Sci. 2001;26:7–9. doi: 10.1016/s0968-0004(00)01700-x. [DOI] [PubMed] [Google Scholar]
- Collins CJ, Sedivy JM. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell. 2003;2:145–150. doi: 10.1046/j.1474-9728.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahle O, Kumar A, Kuehn MR. Nodal signaling recruits the histone demethylase Jmjd3 to counteract polycomb-mediated repression at target genes. Sci Signal. 2010;3:ra48. doi: 10.1126/scisignal.2000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camargo Pereira G, Guimaraes GN, Planello AC, et al. Porphyromonas gingivalis LPS stimulation downregulates DNMT1, DNMT3a, and Jmjd3 gene expression levels in human HaCaT keratinocytes. Clin Oral Investig. 2013;17:1279–1285. doi: 10.1007/s00784-012-0816-z. [DOI] [PubMed] [Google Scholar]
- De Santa F, Narang V, Yap ZH, et al. Jmjd3 contributes to the control of gene expression in LSP-activated macrophages. EMBO J. 2009;28:3341–3352. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P, Chen QM, Hong C, Wang CY. Histone methyl-transferases are demethylases: regulators in balancing osteogenic and adipogenic differentiation of mesenchymal stem cells. Int J Oral Sci. 2015;7:197–204. doi: 10.1038/ijos.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjarlais R, Tummino PJ. The role of histone-modifying enzymes and their complexes in regulation of chromatin biology. Biochemistry. 2016;55:1584–1599. doi: 10.1021/acs.biochem.5b01210. [DOI] [PubMed] [Google Scholar]
- Ding X, Pan H, Li J, et al. Epigenetic activation of AP1 promotes squamous cell carcinom metastasis. Sci Signal. 2013;6:ra28.1–13. S0–15. doi: 10.1126/scisignal.2003884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Q, Oh JE, Chen W, et al. Radioprotective effects of Bmi-1 involve epigenetic silencing of oxidase genes and enhanced DNA repair in normal human keratinocytes. J Invest Dermatol. 2011;131:1216–1225. doi: 10.1038/jid.2011.11. [DOI] [PubMed] [Google Scholar]
- Dong R, Yao R, Du J, Wang S, Fan Z. Depletion of histone demethylase KDM2A enhanced the adipogenic and chrondogenic differentiation potentials of stem cells from apical papilla. Exp Cell Res. 2013;319:2874–2882. doi: 10.1016/j.yexcr.2013.07.008. [DOI] [PubMed] [Google Scholar]
- El Gazzar M, Yoza B, Chen X, Hu J, Hawkins G, McCall CE. C9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J Biol Chem. 2008;283:32198–32208. doi: 10.1074/jbc.M803446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene CI, Edwards L, Riddick G, et al. Histone demethylase Jumonji D3 (Jmjd3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS One. 2012;7:e51407. doi: 10.1371/journal.pone.0051407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Yamaza T, Lee JS, et al. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol. 2009;11:1002–1009. doi: 10.1038/ncb1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formigli L, Orlandini SZ, Tonelli P, Gianelli M, Martini M, Brandi M. Osteolytic processes in human radicular cysts: morphological and biochemical results. J Oral Pathol Med. 1995;24:216–220. doi: 10.1111/j.1600-0714.1995.tb01170.x. [DOI] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nuc Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Li W, Yang J, et al. Epigenetic response to environmental stress: assembly of BRG1-G9a/GLP-DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta. 2016;1863:1772–1781. doi: 10.1016/j.bbamcr.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Bath ML, Harris AW, Adams JM. bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene. 1993;8:3161–3164. [PubMed] [Google Scholar]
- Hopkinson RJ, Tumber A, Yapp C, et al. 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation. Chemical Science. 2013;4:3110–3117. doi: 10.1039/C3SC51122G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol. 2010;17:38–43. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of Jmjd2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Hui T, A P, Zhao Y, et al. ZH2, a potential regulator of dental pulp inflammation and regeneration. J Endod. 2014;40:1132–1138. doi: 10.1016/j.joen.2014.01.031. [DOI] [PubMed] [Google Scholar]
- Hung MS, Karthikeyan N, Huang B, Koo HC, Kiger J, Shen CJ. Drosophila proteins related to vertebrate DNA (5-cytosine) methyltransferases. Proc Natl Acad Sci USA. 1999;96:11940–11945. doi: 10.1073/pnas.96.21.11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Kobayashi T, Ito S, et al. Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J Periodontol. 2012;83:917–925. doi: 10.1902/jop.2011.110356. [DOI] [PubMed] [Google Scholar]
- Jack AP, Bussemer S, Hahn M, et al. H3K56me3 is a novel, conserved heterochromatic mark that largely but not completely overlaps with H3K9me3 in both regulation and localization. PLoS ONE. 2013;8:e51765. doi: 10.1371/journal.pone.0051765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong MW, Kang TH, Kim W, Choi YH, Kim KT. Mitogen-activated protein kinase phosphatase 2 regulates histone H3 phosphorylation via interaction with vaccinia-related kinase 1. Mol Biol Cell. 2013;24:373–384. doi: 10.1091/mbc.E12-06-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JH, Lee HJ, Heo J, et al. Senescence associated MCP-1 secretion is dependent on a decline in BMI1 in human mesenchymal stromal cells. Antioxid Redox Signal. 2015;24:471–485. doi: 10.1089/ars.2015.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen S, Schotta G, Sørensen CS. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–2806. doi: 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MK, Kim RH, Kim SJ, et al. Elevated Bmi-1 expression is associated with dysplastic cell transformation during oral carcinogenesis and is required for cancer cell replication and survival. Br J Cancer. 2007;96:126–133. doi: 10.1038/sj.bjc.6603529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–144. doi: 10.1111/j.1600-065X.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- Kim RH, Kang MK, Shin KH, et al. Bmi-1 cooperates with human papillomavirus type 16E6 to immortalize normal human oral keratinocytes. Exp Cell Res. 2010;313:462–472. doi: 10.1016/j.yexcr.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Kimura A, Kishimoto T. Th17 cells in inflammation. Int Immunopharmacol. 2011;11:319–322. doi: 10.1016/j.intimp.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Koch CM, Andrews RM, Flicek P, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L, Chung CW, Cheng Z, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Collins RE, De Cegli R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, et al. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Li Z, Cao R, Wang M, Myers MP, Zhang Y, Xu RM. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem. 2006;281:20643–20649. doi: 10.1074/jbc.M602461200. [DOI] [PubMed] [Google Scholar]
- Li Q, Zou J, Wang M, et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat Commun. 2014;5:5780. doi: 10.1038/ncomms6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, Cheng YC, Yang HC, et al. Loss of the candidate tumor suppressor BTG3 triggers acute cellular senescence via the ERK-JMJD3-p16(INK4A) signaling axis. Oncogene. 2012;31:3287–3297. doi: 10.1038/onc.2011.491. [DOI] [PubMed] [Google Scholar]
- Liu D, Wang Y, Jia Z, et al. Demethylation of IGFBP5 by histone demethylase KDM6B promotes mesenchymal stem cell-mediated periodontal tissue regeneration by enhancing osteogenic differentiation and anti-inflammation potentials. Stem Cells. 2015;33:2523–2536. doi: 10.1002/stem.2018. [DOI] [PubMed] [Google Scholar]
- Lloret-Llinares M, Perez-Lluch S, Rossell D, et al. dKDM5/LID regulates H3K4me3 dynamics at the transcription-start site (TSS) of actively transcribed developmental genes. Nucleic Acids Res. 2012;40:9493–9505. doi: 10.1093/nar/gks773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Trievel RC, Rojas JR, et al. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- Loughran O, Malliri A, Owens D, et al. Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: relationship of dysfunction to immortality and neoplasia. Oncogene. 1996;13:561–568. [PubMed] [Google Scholar]
- Manna S, Kim JK, Bauge C, et al. Histone H3 Lysine 27 demethylases Jmjd3 and Utx are required for T-cell differentiation. Nat Commun. 2015;6:8152. doi: 10.1038/ncomms9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Martins MD, Jiao Y, Larsson L, et al. Epigenetic modifications of histones in periodontal disease. J Dent Res. 2016;95:215–222. doi: 10.1177/0022034515611876. [DOI] [PubMed] [Google Scholar]
- Matsuo T, Ebisu S, Nakanishi T, Yonemura K, Harada Y, Okada H. Interleukin-1 alpha and interleukin-1 beta in periapical exudates of infected root canals: correlations with the clinical findings of the involved teeth. J Endod. 1994;20:432–435. doi: 10.1016/s0099-2399(06)80032-4. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The Tudor domain “royal family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- McAllister TE, England KS, Hopkinson RJ, Brennan PE, Kawamura A, Schofield CJ. Recent progress in histone demethylase inhibitors. J Med Chem. 2015;59:1308–1329. doi: 10.1021/acs.jmedchem.5b01758. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci USA. 2011;108:2130–2135. doi: 10.1073/pnas.1009933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Park D, Munger K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc Natl Acad Sci USA. 2013;110:16175–16180. doi: 10.1073/pnas.1310432110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Imhof A, Patel D, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464:792–796. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- Morera L, Lübbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijwening JH, Geutjes EJ, Bernards R, Beijersbergen RL. The histone demethylase Jarid1b (Kdm5b) is a novel component of the Rb pathway and associates with E2f-target genes in MEFs during senescence. PLoS One. 2011;6:e25235. doi: 10.1371/journal.pone.0025235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’;Neil LP, Turner BM. Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J. 1995;14:3946–3957. doi: 10.1002/j.1460-2075.1995.tb00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh L, von Eyss B, Schmit F, et al. The human syn-Muv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J. 2007;26:144–157. doi: 10.1038/sj.emboj.7601478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrigue PM, Silva ME, Warden CD, et al. The histone demethylase jumonji coordinates cellular senescence including secretion of neural stem cell-attracting cytokines. Mol Cancer Res. 2015;13:636–650. doi: 10.1158/1541-7786.MCR-13-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, Mermoud JE, O’Carroll D, et al. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet. 2002;30:77–80. doi: 10.1038/ng789. [DOI] [PubMed] [Google Scholar]
- Pierdomenico L, Bonsi L, Calvitti M, et al. Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation. 2005;80:836–842. doi: 10.1097/01.tp.0000173794.72151.88. [DOI] [PubMed] [Google Scholar]
- Qi HH, Sarkissian M, Hu GQ, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Ann Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002;16:2219–2224. doi: 10.1101/gad.232502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Hou L, Belani A, et al. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J Immunol. 2000;165:3626–3630. doi: 10.4049/jimmunol.165.7.3626. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Balto K, Kawashima N, et al. Gamma interferon (IFN-gamma) and IFN-gamma-inducing cytokines interleukin-12 (IL-12) and IL-18 do not augment infection-stimulated bone resorption in vivo. Clin Diagn Lab Immunol. 2004;11:106–110. doi: 10.1128/CDLI.11.1.106-110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Dang W, Donahue G, et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015;29:1362–1376. doi: 10.1101/gad.263707.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Shi YG, Tsukada Y. The discovery of histone demethylases. Cold Spring Harb Perspect Biol. 2013;5:pii, a017947. doi: 10.1101/cshperspect.a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA. 2003;100:13225–13330. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinchi Y, Hieda M, Nishioka Y, et al. SUV420H2 suppresses breast cancer cell invasion through down regulation of the SH2 domain-containing focal adhesion protein tensin-3. Exp Cell Res. 2015;334:90–99. doi: 10.1016/j.yexcr.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Singh RP, Hasan S, Sharma S, et al. Th17 cells in inflammation and autoimmunity. Autoimmun Rev. 2014;13:1174–1181. doi: 10.1016/j.autrev.2014.08.019. [DOI] [PubMed] [Google Scholar]
- Song J, Chang I, Chen Z, Kang M, Wang CY. Characterization of side populations in HNSCC: highly invasive, chemoresistant and abnormal Wnt signaling. PLoS One. 2010;5:e11456. doi: 10.1371/journal.pone.0011456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Groves MR, Sinning I, Sattler M. High-resolution X-ray and NMR structures of the SMN Tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues. J Mol Biol. 2003;327:507–520. doi: 10.1016/s0022-2836(03)00148-7. [DOI] [PubMed] [Google Scholar]
- Stender JD, Pascual G, Liu W, et al. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone K4K20. Mol Cell. 2012;48:28–38. doi: 10.1016/j.molcel.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Kato H, Suzuki Y, et al. Histone H3K36 trimethylation is essential for multiple silencing mechanisms in fission yeast. Nucleic Acids Res. 2016;44:4147–4162. doi: 10.1093/nar/gkw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Umata T, Okamoto K, Obuse C, Tsuneoka M. CxxC-ZF domain is needed for KDM2A to demethylate histone in rDNA promoter in response to starvation. Cell Struc Funct. 2014;39:79–92. doi: 10.1247/csf.13022. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Yano H, Ogasawara S, et al. Mild glucose starvation induces KDM2A-mediated H3K36me2 demethylation through AMPK to reduce rRNA transcription and cell proliferation. Mol Cell Biol. 2015;35:4170–4184. doi: 10.1128/MCB.00579-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani-Ishii N, Wang CY, Stashenko P. Immunolocalization of bone-resorptive cytokines in rat pulp and periapical lesions following surgical pulp exposure. Oral Microbiol Immunol. 1995;10:213–219. doi: 10.1111/j.1399-302x.1995.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Vernal RN, Dutzan A, Chaparro J, Puente M, Antonieta Valenzuela M, Gamonal J. Levels of interleukin-17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol. 2005;32:383–389. doi: 10.1111/j.1600-051X.2005.00684.x. [DOI] [PubMed] [Google Scholar]
- Viré E, Brenner C, Deplus R, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet. 2014;15:69–81. doi: 10.1038/nrg3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Stashenko P. Characterization of bone-resorbing activity in human periapical lesions. J Endod. 1993;19:107–111. doi: 10.1016/S0099-2399(06)80503-0. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li X, Hu H. H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics. 2014;103:222–228. doi: 10.1016/j.ygeno.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Xu J, Yu B, Hong C, Wang CY. KDM6B epigenetically regulates odontogenic differentiation of dental mesenchymal stem cells. Int J Oral Sci. 2013;5:200–205. doi: 10.1038/ijos.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan D, Han Q, Tu Q, et al. Epigenetic modulation in periodontitis: interaction of adiponectin and Jmjd3-IRF4 Axis in macrophages. J Cell Physiol. 2016;231:1090–1096. doi: 10.1002/jcp.25201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Tateishi K, Kudo Y, et al. Loss of histone demethylase KDM6B enhances aggressiveness of pancreatic cancer through downregulation of C/EBPα. Carcinogenesis. 2014;35:2404–2414. doi: 10.1093/carcin/bgu136. [DOI] [PubMed] [Google Scholar]
- Yang D, Okamura H, Teramachi J, Haneji T. Histone demethylase Utx regulates differentiation and mineralization in osteoblasts. J Cell Biochem. 2015;116:2628–2636. doi: 10.1002/jcb.25210. [DOI] [PubMed] [Google Scholar]
- Ye L, Fan Z, Yu B, et al. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11:50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Q, Cao Y, Liu OS, et al. Spatial and temporal expression of histone demethylase, Kdm2a, during murine molar development. Biotech Histochem. 2016;91:138–144. doi: 10.3109/10520295.2015.1106586. [DOI] [PubMed] [Google Scholar]
- Yoza BK, Hu JY, Cousart SL, Forrest LM, McCall CE. Induction of RelB participates in endotoxin tolerance. J Immunol. 2006;177:4080–4085. doi: 10.4049/jimmunol.177.6.4080. [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhang Y, Gao Y, et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ. 2015;22:1630–1640. doi: 10.1038/cdd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- Zheng LW, Zhang BP, Xu RS, Xu X, Ye L, Zhou XD. Bivalent histone modifications during tooth development. Int J Oral Sci. 2014;6:205–211. doi: 10.1038/ijos.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Zhu P, Wang J, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29:69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]