

Abstract
While a significant amount is known about the biochemical signaling pathways of the Rho family GTPase Cdc42, a better understanding of how these signaling networks are coordinated in cells is required. In particular, the predominant subcellular sites where GTP-bound Cdc42 binds to its effectors, such as p21-activated kinase 1 (PAK1) and N-WASP, a homolog of the Wiskott-Aldritch syndrome protein, are still undetermined. Recent fluorescence resonance energy transfer (FRET) imaging experiments using activity biosensors show inconsistencies between the site of local activity of PAK1 or N-WASP and the formation of specific membrane protrusion structures in the cell periphery. The data presented here demonstrate the localization of interactions by using multiphoton time-domain fluorescence lifetime imaging microscopy (FLIM). Our data here establish that activated Cdc42 interacts with PAK1 in a nucleotide-dependent manner in the cell periphery, leading to Thr-423 phosphorylation of PAK1, particularly along the lengths of cell protrusion structures. In contrast, the majority of GFP-N-WASP undergoing FRET with Cy3-Cdc42 is localized within a transferrin receptor- and Rab11-positive endosomal compartment in breast carcinoma cells. These data reveal for the first time distinct spatial association patterns between Cdc42 and its key effector proteins controlling cytoskeletal remodeling.
Cytoskeletal remodelling is a highly dynamic, tightly regulated process which drives the formation of cell protrusions necessary for cell movement. Members of the Rho family of GTPases regulate cytoskeletal dynamics by cycling between inactive GDP-bound and active GTP-bound states. Cdc42 activation results in a localized increase in actin polymerization and the formation of filopodia. Immediately downstream of Cdc42 are p21-activated kinase 1 (PAK1) as well as the Wiskott-Aldrich syndrome protein (WASP) family of proteins, both of which are activated through the binding of GTP Cdc42 via the Cdc42- and Rac-binding domain (CRIB or PBD). PAK1 has been reported to localize to regions of cytoskeletal assembly, and the activated form can induce cytoskeletal remodeling at the leading edge via downstream events such as microtubule growth and stathmin phosphorylation (11, 60). A close correlation has been found between the PAK1 activity state and the baseline invasiveness of human breast cancer cells as well as breast tumor grades (52). Paradoxically, the same work demonstrated that breast carcinoma cells expressing the GTPase binding-deficient H83L, H86L PAK1 mutant exhibited extensive membrane ruffling in the cell periphery. The lamellipodia formation and membrane ruffling observed in the presence of PAK1 were subsequently demonstrated by others to be independent of PAK catalytic activity (13). The relationship between the local level of PAK1 activity and the formation of specific subcellular structures is therefore unclear.
The WASP-family proteins and the Arp2/3 complex are important nucleators of new actin filaments in response to signals causing cell shape and motility changes. Recent in vitro biochemical studies have shown that a homolog of WASP, N-WASP, is locked in an inactive closed conformation by an intramolecular interaction between its GTPase-binding domain and the VCA domain (for “verprolin homology and central basic and acidic motifs”) (40). The binding of GTP-loaded Cdc42 and phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) to N-WASP synergistically enhances the activation of N-WASP-induced actin nucleation by the Arp2/3 complex in vitro (17), probably by releasing the GTPase-binding domain and basic region from the VCA domain. A similar paradox exists, however, for N-WASP, in terms of the spatial correlation between the activity state of this protein and the formation of specific cellular structures such as filopodia. In the N-WASP knockout fibroblasts, filopodia can still be induced at the cell periphery by microinjecting a mixture of a constitutively active Cdc42 (L61), dominant-negative Rac (to suppress lamellipodia) and C3-transferase (to inhibit Rho) (23, 46). These results indicate that proteins other than N-WASP can trigger F-actin assembly at the cell periphery. Physiological stimulation by epidermal growth factor (EGF) has been shown, using a fluorescence resonance energy transfer (FRET) biosensor (Raichu-Cdc42) coupled with multiphoton microscopy, to stimulate an increase of Cdc42 activity clearly at lamellipodia and membrane ruffles (21). A similar FRET biosensor (Stinger) for monitoring the N-WASP conformation or activity state in situ has been reported (58). EGF stimulation, however, does not significantly enhance the Stinger activity at peripheral membrane protrusion sites (58); instead, activated N-WASP was observed in both the nucleus and the cytoplasm of resting and EGF-stimulated cells.
In view of these recent findings, we have undertaken fluorescence lifetime imaging microscopy (FLIM)-based measurements (2, 16, 22, 34-36, 38, 39) to establish the spatial distribution of the activator-effector complexes of Cdc42 bound to its downstream effectors in response to cell signals in situ. The detection of FRET between a green fluorescent protein (GFP) donor and a Cy3 acceptor by FLIM requires a spatial separation between the fluorophores of no more than 9 nm (assuming an ability to resolve fluorescence lifetime changes of the order of 100 ps). FRET results in a shortening of the GFP (donor) fluorescence lifetime. Specifically, we monitored the occurrence of FRET to determine the subcellular site of the activated form of GFP-PAK1 [T(P)423] (44) bound to Cdc42, in comparison to the location of GFP-N-WASP complexed to the same upstream Rho GTPase in situ. Multiphoton FLIM-based measurements suggest that different Cdc42 effectors are triggered within different subcellular compartments in response to the same cytoskeletal remodeling stimulus and may mediate distinct functions in these subcompartments in breast cancer cells.
MATERIALS AND METHODS
Cells and antibodies.
All cells were maintained in Dulbecco minimal essential medium supplemented with 10% fetal calf serum. For FRET/FLIM analysis, cells were microinjected using an Eppendorf microinjection system. Postinjection, the cells were returned to the incubator and allowed to quiesce or express the protein of interest for 3 to 4 h. They were then fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100-phosphate-buffered saline (PBS), and then either viewed directly or stained with a Cy3-anti-HA immunoglobulin G (IgG) Fab fragment (and Cy5-labeled organelle marker antibodies where indicated). Organelle marker antibodies used were as follows: anti-transferrin receptor (Santa Cruz Biotechnology), mouse monoclonal anti-caveolin 2 (Transduction Laboratories), anti-TGN antibody (Transduction Laboratories), anti-N-WASP (a gift from T. Takenawa), and anti-Rab11 (a gift from S. Tooze). The anti-T(P)423 PAK1 polyclonal antibody was produced as follows. The phosphopeptide SKRST(P)MVGTPYC, derived from amino acid residues 419 to 429 in human PAK1 (43), was synthesized and coupled to keyhole limpet hemocyanin via a COOH-terminal linkage. The coupled peptide was injected into rabbits, and the resulting antisera were purified by protein A-Sepharose chromatography. The sera were then affinity purified, first by passage over a nonphosphorylated peptide column to remove antibodies that react with nonphosphorylated epitopes and then by passage over a phosphopeptide column. The sera were eluted in 1-ml fractions from this column with 0.1 M glycine (pH 2.5), immediately neutralized by the addition of 1 M Tris (pH 9.5), and extensively dialyzed against PBS.
Plasmids.
Full-length N-terminal GFP-N-WASP was generated by PCR and cloned into the XhoI and HindIII sites of pEGFPC1 (Clontech) by using sites introduced in the primers. The GFP-N-WASP H211D mutant was generated by QuikChange site-directed mutagenesis (Stratagene) using forward (5′ CCAAGCAATTTCCAA GACATTGGACATGTGGG 3′) and reverse (5′ CCCACATGTCCAAT GTCTTGGAAATTGCTTGG 3′) oligonucleotides, where bases shown in bold indicate the His211-to-Asp changes. Full-length wild-type (WT) Cdc42 and p16A were generated by PCR and cloned into the BamHI and EcoRI sites of pRK5 Myc vector with the Myc epitope tag at the N terminus. Full-length N-terminal Myc epitope-tagged PAK1 has been described previously (43). The NHE-1-HA construct was a gift from D. Barber. GFP-PAK-1 PIX-binding mutant constructs were generated using GFP-PAK1 as a template. QuikChange site-directed mutagenesis was performed as above, using forward (5′CCACCAGTGATTGCTCCAGGCGCAGAGCACACAAAATCTGTATAC3′) and reverse (5′GTATACAGATTTTGTGTGCTCTGCGCCTGGAGCAATCACTGGTGG 3′) oligonucleotides, where bases shown in bold indicate the amino acid changes.
Coimmunoprecipitation and Western analysis.
Immunoprecipitation and Western analysis were carried out as previously described (38).
Immunostaining and confocal microscopy.
Cells were permeabilized with 0.2% (vol/vol) Triton X-100-PBS following fixation in 4% (wt/vol) paraformaldehyde. Primary antibodies were diluted 1:200 to 1:500 in PBS containing 1% bovine serum albumin, except for the fluorophore-conjugated antibodies, which were used at 1:10 to 1:50. The Cy5-labeled conjugates were obtained from Jackson ImmunoResearch Laboratories. Slides used for FRET analysis were fixed again following antibody staining. Confocal images were acquired on a confocal laser-scanning microscope (model LSM 510; Carl Zeiss Inc.) equipped with both 40×/1.3Plan-Neofluar and 63×/1.4Plan-APOCHROMAT oil immersion objectives. Each image represents a two-dimensional projection of sections in the Z-series, taken across the depth of the cell at 0.2-μm intervals unless otherwise indicated. Colocalization coefficients (between N-WASP and organelle markers) were calculated using Zeiss colocalization coefficient function software, applying the following formula:
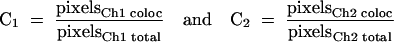 |
where all pixels above background are taken into account and the relative number of colocalizing pixels in channels (Ch1 and Ch2) are calculated compared to total number of pixels above threshold. Data are presented as a value between 0 and 1, where 0 indicates no colocalization and 1 indicates that all pixels colocalize.
Sucrose gradient fractionation.
Cells were cotransfected with GFP-N-WASP and Cdc42-HA and then homogenized for 30 min on ice in 10 mM Tris-HCl buffer containing 1 mM EDTA, 5% glycerol, 1% Triton X-100, 5mM β-mercaptoethanol, 0.15 M NaCl, 10 mM MgCl2, and protease inhibitors. Lysates were fractionated on a small-scale sucrose gradient (1.6 ml of 40% sucrose, 2 ml of 20% sucrose, 1.6 ml of 5% sucrose) and centrifuged in a Beckman SW55 rotor for 18 h at 35,000 rpm at 4°C. Fractions (410 μl) were collected and analyzed by immunoblotting.
Fluorescence lifetime measurements by time-correlated single-photon counting (TCSPC or time-domain FLIM) and analysis.
Time-domain FLIM was performed with a multiphoton microscope system, based on a modified Bio-Rad MRC 1024MP workstation, comprising a solid-state-pumped (10 W Millennia X, Nd:YVO4; Spectra-Physics), femtosecond Ti:Sapphire (Tsunami, Spectra-Physics) laser system, an afocal scan head, and an inverted microscope (TE200; Nikon) (1). Enhanced detection of the scattered component of the emitted (fluorescence) photons was afforded by the use of fast single-photon response (R7401-P; Hamamatsu) non-descanned detectors, developed in-house, situated in the reimaged objective pupil plane. Fluorescence lifetime imaging capability was provided by time-correlated single-photon counting electronics (SPC 700; Becker & Hickl). A 40× objective was used throughout (CFI60 Plan Fluor N.A. 1.3; Nikon), and data were collected at 500 ± 20 nm through a bandpass filter (35-50 40; Coherent Inc.). Laser power was adjusted to give average photon-counting rates of the order 104 to 105 photons s−1 (0.0001 to 0.001 photon count per excitation event) to avoid pulse pileup.
FLIM analysis can be enhanced by application of global analysis (i.e., assumption of globally invariant fluorescence lifetime components and calculation over all available data sets, or pixels) to data obtained using either time-domain-based (4, 5) or frequency domain-based (53-56) methods. The spatially invariant lifetimes are assumed to pertain to the donor molecular species in the presence or absence of FRET, with the relative amplitudes of the components being proportional to the molar fractions (hereafter referred to as the “populations”) of the two species.
Immunogold electron microscopy.
Cells were scraped carefully and fixed in 4% paraformaldehyde for 1 h before being processed for routine sectioning on a Leica ultracryotome for immunolabeling. Single and double labeling was carried out as described previously (45). In the double labeling, the first antibody used was a monoclonal anti-HA IgG recognized by 10-nm-diameter protein A-gold (PAG); this was followed, after an additional glutaraldehyde fixation step (1% monomeric glutaraldehyde for 5 min to cross-link the primary antibody), by a rabbit anti-GFP antiserum (a kind gift of D. Shima, ICRF) detected with 5-nm-diameter PAG. In some studies, the second antibody was omitted to control for cross-reactivity of the 5-nm PAG with the first antibody. After antibody labeling, sections were examined using a Jeol 1010 microscope.
RESULTS
Visualizing the Cdc42-bound PAK1 species by multiphoton FLIM.
MDA-MB-231 breast carcinoma cells expressing GFP-PAK1 and WT or dominant negative (N17) variants of Cdc42-HA were imaged by multiphoton FLIM, and the interaction between these two molecules was assessed by the degree of donor fluorescence lifetime shortening. In the uncomplexed state, GFP fluorescence generally undergoes a single exponential decay. When a significant proportion of GFP undergoes FRET, biexponential decay kinetics are observed. Lifetime maps (referred to hereafter as “lifetime”) shown for each example of FRET data indicate the lifetime of the GFP at each pixel as imaged (i.e., the raw data). For example, the global lifetime of GFP species not undergoing FRET in this case is 2.23 ns whereas the global lifetime of GFP undergoing FRET with the Cy3 acceptor is 0.74 ns. These data can subsequently be analyzed to determine the percentage of the population of GFP undergoing FRET at each image pixel (referred to hereafter as the “FRET population”). Figure 1 shows a reduction in the GFP lifetime of GFP-PAK1 in cells coexpressing the WT form of Cdc42. This reduction in lifetime, as well as the FRET population, was positioned strikingly at membrane protrusions (indicated by white arrows in Fig. 1). High-magnification imaging of membrane protrusions (bottom panels of Fig. 1) in cells overexpressing GFP-PAK1 and Cdc42-HA-Cy3 demonstrates a clear interaction between these two molecules along the lengths of cell protrusion structures. In contrast, when N17 Cdc42 was coexpressed with GFP-PAK1, no biexponential decay kinetics were observed and the resultant monoexponential lifetime indicates that there was no significant FRET population. GFP-PAK1 binding to Cdc42 is therefore dependent on the guanine nucleotide exchange of the Rho GTPase. No FRET was detected between GFP-PAK1 and another Cy3-labeled plasma membrane-targeted protein (NHE-1-HA-Cy3) (see Fig. S3 in the supplemental material).
FIG. 1.

Site of PAK1-Cdc42 interaction in cells. Multiphoton FLIM was undertaken to determine the extent of FRET between the GFP-PAK1 donor and anti-HA-Cy3 acceptor in cells coexpressing GFP-PAK1 and HA-tagged Cdc42 (WT and N17 variants). FRET results in a shortening of the GFP fluorescence lifetime (τ in nanoseconds). In the uncomplexed state, GFP fluorescence generally undergoes a single exponential decay. When a significant proportion of GFP undergoes FRET, biexponential decay kinetics are observed and can be analyzed to determine thepercentage of the population of GFP undergoing FRET at each image pixel (the global, i.e., spatially invariant lifetime of GFP undergoing FRET is τ1 = 0.73 ns, shortened from the normal GFP lifetime of τ2 = 2.23 ns). Where appropriate, the subcellular localization of maximal lifetime decrease and largest FRET population was indicated by a white arrow. For the N17 Cdc42-expressing cells, the normal, monoexponential decay GFP lifetime was observed.
PAK activation occurs at the cell periphery after Cdc42 binding.
On binding to GTPases, PAK1 is known to autophosphorylate at several sites, including Thr423 within the activation loop of the kinase domain. Phosphorylation at Thr423 is strongly correlated with activation (44). A Thr-to-Glu (which mimics a phosphorylated Thr residue) substitution at this site renders the molecule constitutively active (27). Antibodies raised against the peptide sequence SKRST(P)MVGTPY from PAK1, which contains phospho-Thr423 [T(P)423], also cross-react with other PAK-related kinases such as Mst2 (44), making their use to specifically detect activated PAK1 in cells by immunofluorescence difficult. Donor FLIM, however, overcomes the nonspecific binding of phosphospecific antibodies by directly assessing the proportion of the specific protein of interest (GFP-PAK1) that is bound to the antibody (36). In the experiment whose results are shown in Fig. 2A, cells were coinjected with GFP-tagged WT PAK1 and WT, V12, or N17 Cdc42 constructs, and then stained with an antibody specific to the T(P)423 form of PAK1, directly labeled with Cy3 dye. Multiphoton FLIM images of GFP-PAK indicate that where the WT Cdc42 is coexpressed in cells, PAK1 activation occurs at the membrane, particularly at the tips of cell protrusion structures (indicated by white arrows in Fig. 2A). High-magnification images (bottom panels, Fig. 2A) demonstrate that PAK1 T423 phosphorylation takes place at the cell periphery and along the lengths of cell extensions, similar to those seen in Fig. 1. Expression of the N17 inactive Cdc42 did not significantly induce PAK T423 phosphorylation, and hence no FRET signal was detected in these samples. Cumulative efficiency data from these experiments (n = 7) were also compiled (Fig. 2B), where FRET efficiency = 1 − (τda/τd) (τda is the pixel-by-pixel fluorescence lifetime of the donor in the presence of acceptor, and τd is the average lifetime of the donor in the absence of acceptor). In these breast carcinoma cells, the extent of GFP- PAK activation was similar when coexpressed with either the WT or the V12 variant of Cdc42. Together with the GFP-PAK-Cdc42 binding data, these results indicate that active WT Cdc42 binds to and activates PAK1 while the inactive N17 variant does not.
FIG. 2.

PAK1 activation at cell protrusions is dependent on GTP-Cdc42. (A) MDA-MB-231 cells injected in the nuclei with GFP-PAK1 and Cdc42 constructs as specified. The cells were stained with PAK1 T423(P) phosphospecific antibody directly conjugated to Cy3. Multiphoton FLIMwas then undertaken to determine the localization of activated [T423(P)] PAK1. Where appropriate, the subcellular localization of maximal τ decrease and highest FRET population is indicated by a white arrow. (B) Cumulative FRET efficiency histogram compiled from all the data sets (n = 7) for each plasmid combination are shown in the bottom panel. FRET efficiency = 1 − τda/τdi, where τda is the pixel-by-pixel fluorescence lifetime of the donor in the presence of acceptor and τd is the average lifetime of the donor in the absence of acceptor (unstained controls).
PAK1-Cdc42 interaction is dependent on the PIX binding site in PAK.
PAK1 activation and recruitment to focal complexes and the cell periphery have previously been reported to be partially dependent on PAK1 binding to PAK-interacting exchange factor (αPIX) (29). To test the dependence of PAK1-Cdc42 complex formation on the recruitment of PAK1 to the membrane via PIX, we generated a GFP-tagged PIX-binding mutant of PAK1 (R193G/P194A). This mutant was expressed, and its ability to bind V12 Cdc42 was tested by multiphoton FLIM analysis. Figure 3A demonstrates that compared to WT PAK1, the PIX-binding deficient mutant no longer interacts with active Cdc42. Cumulative FRET efficiency histograms of WT PAK and Cdc42 variants versus the PAK PIX-binding mutant (Fig. 3B) demonstrate a significant shift in efficiency between WT PAK and mutant PAK (n = 6). Since there are currently no other known binding partners for PAK at this site, we conclude that the PAK1-Cdc42 interaction is dependent on the recruitment of PAK, via PIX, to focal complexes along cell protrusion structures. This is in agreement with a recent report which shows that the PAK-PIX association is required for PAK kinase activity in breast cancer cells (47).
FIG. 3.

The PAK1-Cdc42 interaction is dependent on the PIX-binding site in PAK. (A) MDA-MB-231 cells coinjected with WT or a PIX-binding-deficient mutant of GFP-PAK1 (R193G/P194A) and Cdc42-HA. The cells were stained with anti-HA-Cy3 and multiphoton FLIM was undertaken as described in the legend to Fig. 2A. (B) Cumulative FRET efficiency histogram compiled from all the data sets (n = 6) for each plasmid combination including data from Fig. 1 (Cdc42 variants). FRET efficiency = 1 − τda/τd, where τda is the pixel-by-pixel fluorescence lifetime of the donor in the presence of acceptor and τd is the average lifetime of the donor in the absence of acceptor (unstained controls).
GTP-dependent N-WASP-Cdc42 complex formation in situ.
In contrast to the above-described GFP PAK-Cdc42 complex localization, Fig. 4A shows an association of GFP-N-WASP with WT Cdc42 in punctate/vesicular cytoplasmic structures in MDA-MB-231 breast carcinoma cells (marked with white arrows). Our results indicate that both the localization of GFP-N-WASP and its association with Cdc42 in situ are dependent on the nucleotide state of Cdc42. The N17 inactive mutant of Cdc42, unlike its WT counterpart, was unable to associate with GFP-N-WASP to any significant extent. In the presence of a coexpressed N17 Cdc42, there was a significant accumulation of GFP-N-WASP in the nuclear region (marked with red arrows in Fig. 4A). This phenotype is similar to that observed for the GFP-N-WASP GTPase-binding-deficient mutant (H211D, the bovine equivalent of the H208D mutation in the rat [32]) in MDA-MB-231 cells. This mutant did not associate with coexpressed WT Cdc42 and was also predominantly retained in the nucleus. High-magnification analysis by FLIM (Fig. 4A, bottom panel) shows the GFP-N-WASP/Cdc42 HA-Cy3 interaction occurs specifically within endosomal vesicular structures but is not detectable at the cell periphery. In contrast, there was no significant lifetime reduction within the cell protrusion-targeted N-WASP subpopulation (indicated by red arrows). Cumulative analysis of FRET efficiency for each plasmid set (n = 6) is shown in Fig. 4B. Western analysis showed that coexpression of WT Cdc42 increased the amount of Cdc42 by four-fold over the endogenous protein level (see Fig. S1 in the supplemental material). The GFP-N-WASP/Cdc42 association was also confirmed by coimmunoprecipitation (Fig. 4C).
FIG. 4.

N-WASP associates with WT Cdc42 in an intracellular vesicular compartment. (A) Cells were microinjected with plasmids encoding GFP-N-WASP (WT or H211D non-GTPase binding mutant) and HA-tagged WT or N17 inactive mutant Cdc42 (plasmid ratio, 1:2), incubated for 4 h at 37°C, fixed, and stained with a Cy3-conjugated anti-HA IgG Fab fragment. Multiphoton donor FLIM was undertaken to determine the extent of FRET between GFP-N-WASP (donor) and anti-HA-Cy3 (acceptor). FRET population determination was possible only with the WT GFP-N-WASP/WT Cdc42 pair; biexponential decay kinetics were not observed with either the H211D non-GTPase binding GFP-N-WASP or the N17 inactive mutant of Cdc42 labeled with anti-HA Fab-Cy3. Where appropriate, the subcellular localization of maximal τ decrease and highest FRET population is indicated by a white arrow. Red arrows indicate the accumulation of nuclear GFP-N-WASP in the presence of a coexpressed or N17 Cdc42 or the GTPase-binding-deficient GFP-N-WASP variant. High-resolution imaging of GFP-N-WASP/WT Cdc42 demonstrates interaction in vesicles (bottom panels) but no change in the GFP lifetime of the cell protrusion-targeted subpopulation of N-WASP (as indicated by red arrows). (B) FRET efficiency histogram compiled from all the data sets (n = 6) for each N-WASP plasmid combination. FRET efficiency = 1 − τda/τd, where τda is the pixel-by-pixel fluorescence lifetime of the donor in the presence of acceptor and τd is the average lifetime of the donor in the absence of acceptor (unstained controls). (C) Coimmunoprecipitation of the N-WASP-Cdc42 complex. In panel I, cells were transfected with various constructs as indicated and lysed. Western blotting was carried out on transfected versus nontransfected cell extracts, and membranes were blotted for total Cdc42 to allow a comparison between transfected and endogenous Cdc42 levels. In panel II, cells were transfected with GFP-N-WASP and lysed. Lysates were immunoprecipitated using either a control IgG or anti-GFP antibody. A fraction (one-fifth) of unbound material (S) was run alongside the antibody-bound protein (P) and Western blotted for GFP and Cdc42 protein.
Previous studies, based mostly on yeast two-hybrid and biochemical analysis, suggest that the interaction between N-WASP and Cdc42 might be regulated (3,49) by the GTP-GDP state of Cdc42, as shown in Fig. 4A. To investigate more directly whether the GFP-N-WASP/WT Cdc42 association was GTP dependent, a myc-tagged Rho GTPase-activating protein domain (GAP) of RICH-1, which catalyzes GTP hydrolysis on Cdc42 and Rac1 but not on RhoA (41), was coinjected. In three independent experiments, less than half of the cells coinjected with the Rho-GAP construct survived and expressed the protein domain that was diffusely distributed throughout the cytoplasm, as seen by anti-myc monoclonal antibody immunostaining (data not shown). Multiphoton FLIM analysis of these surviving cells showed a reduction of FRET efficiency for the GFP-N-WASP/anti-HA Fab-Cy3 (WT Cdc42) pair in the presence of a coexpressed Rho-GAP domain (Fig. 5). Interestingly, in cells coexpressing Rho-GAP and exhibiting a partial reduction in FRET efficiency, some residual GFP-N-WASP/Cdc42 complex remained in vesicular structures (marked with red arrows in Fig. 4). Thus, we conclude that the interaction observed between WT Cdc42 and N-WASP is likely to be restricted to the GTP-bound form of Cdc42.
FIG. 5.

The Rho-GAP domain inhibits the association between GFP-N-WASP and Cdc42. Cells were injected with GFP-N-WASP and WT Cdc42-HA, with or without the Rho-GAP domain. Lifetime analysis and confocal imaging of the cells were carried out with different microscopes, and the images were aligned. Some between-cell (n = 5) heterogeneity in FRET efficiency for the GFP-N-WASP/anti-HA Fab-Cy3 (WT Cdc42) pair was observed in the absence of the RICH-1 Rho-GAP domain, but there was a clear partial reduction in FRET efficiency when the Rho-GAP domain was coexpressed, as demonstrated in the efficiency histogram (bottom panel). FRET efficiency = 1 − τda/τd, where τda is the pixel-by-pixel fluorescence lifetime of the donor in the presence of acceptor and τd is the average lifetime of the donor in the absence of acceptor (unstained controls in the upper panels).
The N-WASP-Cdc42 association occurs in a transferrin receptor-positive recycling endosomal compartment.
As shown in Fig. 2, PAK1 activation in the presence of GTP-loaded Cdc42 appears to occur at the membrane, maximally along the lengths of cell protrusion structures. We were interested to determine the precise localization of the N-WASP-Cdc42 complexes in the cell. Cells were injected with GFP-N-WASP and Cdc42-HA and immunostained using organelle-specific markers. FRET between GFP and anti-HA Fab-Cy3 was assessed using multiphoton FLIM as described above. Following this, the same cells were also imaged using a single-photon confocal microscope to visualize the organelle markers labeled with Cy5, in relation to the GFP intensity. Figure 6A shows both lifetime and confocal data indicating that the FRET population, i.e., Cdc42-interacting species of GFP-N-WASP, localized to a transferrin receptor-positive endosomal compartment (marked with red arrows).
FIG. 6.

Localization of the GFP-N-WASP/Cdc42 complex in cells. (A) MDA-MB-231 cells were microinjected with plasmids encoding GFP-N-WASP and a HA-tagged Cdc42 (plasmid ratio, 1:2), fixed, and dually stained with a Cy3-conjugated anti-HA IgG Fab fragment and an affinity-purified anti-transferrin receptor mouse IgG plus Cy5 secondary conjugate. The GFP intensity image, lifetime map, and distribution of FRET population in cells, as determined by multiphoton FLIM, are shown, along with the corresponding confocal images, which were obtained separately using a Zeiss LSM-510 microscope (G, GFP intensity; R, anti-transferrin receptor Cy5). Where appropriate, the subcellular localization of maximal τ decrease and largest FRET population and its colocalization with a transferrin receptor-positive compartment is indicated by a white arrow. Parallel samples were also dually stained using an anti-γ-tubulin (marker for MTOC) caveolin antibody or anti-TGN (marker for Golgi) antibody, and confocal images alone show that no significant colocalization occurs between GFP-N-WASP and these markers. (B) Confocal imagesof uninjected cells and cells overexpressing GFP-N-WASP on the same coverslip, costained with anti-N-WASP-Cy3 (red) and anti-transferrin receptor-Cy5 (blue). White arrows indicate some areas of colocalization between the N-WASp and transferrin receptor-positive compartments. (C) Biochemical identification of N-WASP-Cdc42 complex localization. Cells were cotransfected with GFP-N-WASP and WT Cdc42-HA, lysed, and subjected to sucrose gradient density fractionation as described in Materials and Methods. All 12 fractions were then run on sodium dodecyl sulfate-polyacrylamide gels, along with 1/10 of the remaining insoluble cell pellet, and Western blotted. The membranes were probed with various antibodies to identify the fractions containing GFP-N-WASP, Cdc42 (both expressed and endogenous Cdc42), transferrin receptor, Rab11, and the plasma membrane-associated Na/H exchanger NHE-1.
In HeLa cells, the V12 variant Cdc42 localizes to and recruits N-WASP to the Golgi complex (25). Anti-γ-tubulin (the marker for the microtubule-organizing center [MTOC]), anti-Golgi marker (trans-Golgi network [TGN]), and caveolin staining did not show any significant colocalization with the total population of GFP-N-WASP in any of the cells analyzed, although the presence of a small subpopulation of GFP-N-WASP which resides in these compartments cannot be ruled out (Fig. 6A). Colocalization coefficients for the different organelle markers (and GFP-N-WASP) were calculated to be 0.68 ± 0.16 for transferrin receptor, 0.21 ± 0.07 for γ-tubulin, 0.31 ± 0.10 for caveolin, and 0.19 ± 0.09 for TGN.
To ensure that our overexpression system was not causing mislocalization or aggregation of GFP-N-WASP compared to the endogenous protein, the localization of endogenous N-WASP was checked in MDA-MB-231 cells by using a specific antibody (a kind gift of T. Takenawa). Confocal images were acquired of uninjected cells and cells overexpressing GFP-N-WASP on the same coverslip costained with anti-N-WASP and anti-transferrin receptor antibodies. Figure 6B demonstrates that the anti-N-WASP antibody recognizes the GFP-N-WASP, which colocalizes significantly with the transferrin receptor staining in vesicular structures. Similarly, a significant portion of the endogenous N-WASP in these cells also localizes to the transferrin receptor-positive compartments. The structures seen for both GFP-N-WASP and endogenous N-WASP closely resemble those seen in Fig. 6A, where GFP-N-WASP and Cdc42-HA-Cy3 interact. In the absence of an ultrafast laser source in the 1,000- to 1,200-nm excitation range and using our existing multiphoton FLIM setup, it is unfortunately not possible to excite Cy5 emission from the labeled organelle markers. Nevertheless, we conclude that the localization of the GFP-N-WASP/Cdc42-HA-Cy3 FRET species to a transferrin receptor-positive vesicular compartment is not a consequence of protein overexpression.
To confirm biochemically that the N-WASP-Cdc42 interaction was occurring within a transferrin receptor-positive compartment, sucrose gradient cellular fractionation (Fig. 6C) was also performed on MDA-MB-231 cells transfected with GFP-N-WASP and Cdc42-HA. Figure 6C demonstrates that GFP-N-WASP and Cdc42 (predominantly fractions 3 and 4) cofractionate with the transferrin receptor and Rab11, markers of the recycling endosomal compartment. Blotting for the plasma membrane-associated NHE-1 Na/H transporter in parallel experiments demonstrates that the plasma membrane and recycling endosomal compartments are well separated in these samples. These data support the observation in Fig. 6A that the GFP-N-WASP/Cdc42 complex appears to colocalize with a transferrin receptor-positive compartment.
Colocalization of GFP-N-WASP and Cdc42 by immunoelectron microscopy.
The vesicular compartment where GFP-N-WASP and Cdc42 interact in cells was also investigated by electron microscopy of cryosections. Both proteins were found to be at plasma membrane protrusions. Coclusters of both HA-tagged Cdc42 (detected by an anti-HA MAb plus 10-nm PAG) and GFP-N-WASP (detected by a rabbit anti-GFP antiserum plus 5-nm PAG) can be found in the TGN (Fig. 7A) and subplasmalemmal endosomes (Fig. 7B). GFP-N-WASP labeling was also observed on clathrin-coated vesicles (Fig. 7C). In contrast, there was no detectable labeling of either protein in other membranous structures including the mitochondria and the nucleus. While we are unable to obtain a quantitative distribution of the coclustered anti-GFP-N-WASP/anti-HA-Cdc42 gold particles at each of these compartments, we conclude that some of the vesicular structures, where the interacting complexes were visualized in the FLIM and FRET experiments, can be precisely localized to the TGN and endosomes beneath the plasma membrane.
FIG. 7.

Colocalization of GFP-WASP and Cdc42 by immuno-EM. GFP-N-WASP and Cdc42 are enriched at plasma membrane protrusions (a and d), TGN (h and i), clathrin-coated vesicles (c), and peripheral endosomes (b, e, f, and g). The immunoelectron micrographs show the localization of HA-tagged Cdc42 (panels d, e, and f to i, 10-nm gold) and GFP-N-WASP (panels a to c, 10-nm gold; panels f to i, 5-nm gold) detected by primary anti-tag antibodies, as described in Materials and Methods, followed by a gold-conjugated secondary antibodies. Arrowheads point to sites of colocalization between GFP-N-WASP and Cdc42. Bars, 100 nm.
N-WASP-Arp2/3 association in the presence of WT Cdc42.
The Arp2/3 complex has been well characterized in the literature as being a downstream effector of active N-WASP, leading to local actin nucleation. We were interested to know if the Cdc42-bound N-WASP seen in endosomal compartments in these cells was also associated with an intact Arp2/3 complex. We tested the requirement of WT Cdc42 for the interaction between N-WASP and the Arp2/3 complex biochemically, by cotransfecting cells with GFP-N-WASP and myc-tagged p16A in the presence or absence of HA-tagged WT Cdc42. Figure 8 shows that GFP-N-WASP was coprecipitated with the expressed p16A only in the presence of coexpressed Cdc42. H211D GFP-N-WASP, which did not associate with the coexpressed Cdc42, also lacked the ability to coprecipitate the expressed p16A (data not shown). Endogenous Arp2 and p34 were also found in the N-WASP-p16A protein complex, indicating that the expressed p16A was functional and capable of being incorporated into the endogenous Arp2/3 actin-nucleating complex. We conclude that the N-WASP-Cdc42 complex localized within the recycling compartment is functional and linked physically to the Arp2/3 actin-nucleating machinery.
FIG. 8.

N-WASP/p16A colocalization in the presence of WT Cdc42. (A) Cells were cotransfected with GFP-N-WASP and myc-tagged p16A in the presence or absence of an HA-tagged WT Cdc42. GFP-N-WASP was immunoprecipitated from the detergent-soluble cell fraction by using an anti-GFP rabbit serum or a nonspecific IgG control. All the bound proteins on the protein G bead (bound) and one-fifth of the unbound proteins left in the cell extract supernatant after the first centrifugation postprecipitation (unbound) were loaded onto a 10 to 20% Tris-glycine PAGE gradient gel. After the transfer, the plots were probed with an anti-myc or anti-HA MAb. One of the membranes was stripped and reprobed with an anti-GFP MAb. Some of the membranes were also stripped and reprobed with an anti-p34 or anti-Arp2 rabbit serum IgG or nonspecific rabbit IgG. (B) MDA-MB-231 cells were microinjected with plasmids encoding GFP-N-WASP and a HA-tagged p16A (plasmid ratio, 1:2), fixed, and dually stained with a Cy3-conjugated anti-HA IgG Fab fragment and an affinity-purified anti-transferrin receptor mouse IgG-Cy5 secondary conjugate. The GFP intensity image, lifetime map, and distribution of FRET population in cells, as determined by multiphoton FLIM, are shown, along with the parallel confocal images which were obtained using a Zeiss LSM-510 microscope (G, GFP intensity; R, anti-transferrin receptor Cy5). The subcellular localization of maximal τ decrease, largest FRET population, and its colocalization with the transferrin receptor-positive compartment in a corresponding aligned confocal image of the cell is indicated by a white arrow.
Further clarification of the formation of functional complexes between GFP-N-WASP and Arp2/3 proteins was demonstrated using FRET. GFP-N-WASP and HA-p16A were overexpressed in MDA-MB-231 in the presence of WT Cdc42. The cells were fixed and stained with anti-HA-Cy3 and anti-transferrin receptor-Cy5; they were then imaged using multiphoton FLIM technology as above and confocal laser-scanning microscopy to acquire corresponding images of transferrin receptor staining. Figure 8B demonstrates an interaction between GFP-N-WASP and p16A-HA-Cy3 in vesicle structures, which partially colocalize with transferrin receptor staining. These structures are again reminiscent of those seen in Fig. 6A and B, suggesting that N-WASP interacts with Cdc42 in similar structures to those where N-WASP also interacts with the Arp2/3 complex proteins.
DISCUSSION
The application of multiphoton FLIM to accurately localize the GFP subpopulation that undergoes fast decay due to FRET in situ has enabled us to directly visualize the binding of active Cdc42 to two of its downstream effectors, namely, PAK1 and N-WASP, in breast carcinoma cells. Our findings demonstrate that the largest population of the Cdc42-bound and hence activated [T(P)423] species of PAK1 occurs along the lengths of cell protrusion structures, possibly at PIX-dependent focal contact sites, whereas the transferrin receptor-positive recycling endosomal compartment is an important cellular site for the GFP-N-WASP/Cdc42 complex.
Investigation of the spatiotemporal interactions of proteins in situ is crucial to our understanding of the fundamental dynamics of cellular processes. Here we have demonstrated the application of multiphoton FLIM, by TCSPC, to high-resolution FRET imaging. TCSPC is suited to multiphoton imaging where photon fluxes are low (typically ∼104 photons s−1) and when high temporal resolution (<100 ps) is imperative. Direct measurement of the excited-state kinetics enables robust determination of the interacting protein population, achieved by biexponential analysis and statistical interpretation of the result. This assumes the presence of distinct interacting and noninteracting populations, with statistically determined spatially invariant lifetimes denoted τ1 and τ2 (see the figure legends), respectively, within the resolved volume. In addition, multiphoton excitation enables the localization of protein interactions to femtoliter compartments of the cellular matrix without the use of a confocal aperture. The spatial resolution enhancement thereby afforded by multiphoton FLIM over wide-field FLIM enables a much more accurate visualization of protein interactions within individual cellular compartments, as shown by the clearly defined physical structures evident in the lifetime maps.
The Rho family of small GTPases are the main upstream activators of PAK1. Cdc42, Rac1/2/3, and TC10 all bind to the CRIB domain of PAK1 (20, 28, 33). Binding of active GTPase may act as more than just a switch to free the catalytic domain, by promoting the subsequent autophosphorylation events necessary for full kinase activity (7, 10, 61). Recruitment of PAK1 to cell protrusions is likely to be dependent on a number of factors. PAK1 binds to the adaptor protein Nck, which could potentially shuttle PAK1 between peripheral membrane structures and focal adhesions (24). However, a previous publication suggests that Nck itself is not directly necessary for recruitment of PAK1 to the membrane (6). Similarly, PAK1 binds via intermediary proteins to paxillin, which may also permit recruitment to active Cdc42 in localized cell membranes (50). However, as the current data demonstrate, the most likely candidate in our system is the exchange factor PIX. PIX has been shown to recruit PAK1 to focal complexes downstream of Cdc42 activation (29), and our data show that Cdc42-PAK interaction is dependent on this recruitment process. It is likely that PIX binding, PAK1 is available for GTPase interaction at juxtamembrane sites of action. An alternative explanation is that a high proportion of Cdc42 located in close proximity to its guanine nucleotide exchange factor, PIX, is in a favorable conformation (GTP bound) to bind to PAK1. Regardless of the mechanism, our demonstration of a substantial concentration of the activated, phosphorylated form of PAK1 in membrane protrusions lends credence to the notion that the primary function of the GTPase-bound of PAK1 is in cytoskeletal remodeling and subsequent cell protrusive events. These data, however, need to be carefully interpreted, taking into account the effect of neoplastic transformation. Some of the cell protrusion structures are indeed bulkier than the filopodia normally seen in fibroblasts. These features, along with the corresponding spatial distribution of various actin-remodeling proteins, are reminiscent of the more recently reported morphological features indicative of neoplastic cells containing, for instance, protrusive structures that are characteristic of PAK1 overactivity (15). The cell edge feature, which contains the Cdc42-bound activated PAK1 in breast carcinoma cells, is bulkier than filopodia and possibly represents a recently reported form of PAK1-dependent protrusions in neoplastic cells (15).
We postulate that in our cell system, the activated N-WASP is likely to be involved in active shuttling of recycling endosomes and the associated cargo proteins to and from the plasma membrane, thus facilitating the turnover of new protrusions. Both GFP-N-WASP and Cdc42 cofractionate with the transferrin receptor and Rab11, the latter being associated with the pericentriolar recycling compartment, post-Golgi vesicles, and the TGN (59). This is supported by our finding of a high degree of colocalization between the active N-WASP species and the transferrin receptor-positive compartment, as well as the recent demonstration that in yeast the WASP homolog Las17 powers the actin polymerization-dependent motility of endosomes via activation of the Arp2/3 complex (9). The Arp2/3 complex is also essential for Golgi polarization in wound edge NIH 3T3 cells (26). In N-WASP-deficient cells, the transit of hemagglutinin (as a protein marker) from the endoplasmic reticulum to the Golgi apparatus is reduced by 40%, demonstrating a specific role for N-WASP as a regulator of vesicle traffic between the endoplasmic reticulum and the Golgi complex (48). These findings are consistent with the notion that N-WASP may be involved in the regulation of peri-Golgi protein traffic (25) and possibly other endosomal transport processes (37). In support of the latter, depletion of endogenous N-WASP by mitochondrial sequestration leads to an impairment of endocytosis (19). Inhibition of endocytosis by the dominant inhibitory dynamin (K44A) mutant appears to significantly reduce the Cdc42-bound N-WASP species in cells (see Fig. S2 in the supplemental material), again supporting our notion that the preferred site of the N-WASP-Cdc42 interaction is in endosomes. Finally, our EM data precisely localize the coclusters of anti-GFP-WASP/anti-HA-Cdc42 gold particles to TGN as well as endosomes beneath the plasma membrane, and not to the nucleus or mitochondria. It is possible that the clathrin-coated vesicle-associated N-WASP is activated at these sites by Cdc42 and regulates actin dynamics coupled to clathrin-coated vesicle formation and its subsequent transport between the two subcellular structures.
It is possible that the ensemble fluorescence lifetime analysis may miss WASP-Cdc42 interactions that are rapidly turned over, particularly at the protruding zones of the leading edge of the cell. It is important to point out that as a result of the limitations imposed both by the temporal resolution of our current optical system in measuring small differences in ensemble average lifetime, calculated on a pixel-by-pixel basis, and by biological variability, these fluorescence lifetime assays will detect only a significant increase in FRET efficiency, above a certain threshold (of the order of 5% in our case). Lower-affinity interactions, where the mean separation between the donor and acceptor fluorophores is greater, and small FRET populations are both likely to be missed. This may also apply to interactions that are rapidly turned over, particularly at the protruding zones of the leading edge of the cell, to which actin is actively delivered (at speeds that exceed 5 μm/s) (62). This hypothesis is in keeping with the immunogold labeling of both HA-tagged Cdc42 and GFP-N-WASP at the membrane protrusions. Alternatively, recent immunofluorescence studies have identified several proteins (other than Cdc42) which colocalize with N-WASP in the actin bundles of microspikes or filopodia (18, 30). In fact, using purified proteins in the absence of Cdc42, a small number of components (WASP-coated beads, actin, Arp2/3 complex, and fascin) are found to be sufficient for the assembly of filopodium-like bundles (57). A good candidate for an alternative activator of N-WASP at the cell periphery in our system is WASP-interacting SH3 protein (WISH). Published data demonstrate that WISH can bind to N-WASP in microspikes and enhance Arp2/3 complex activation independently of Cdc42 (14).
The localization of the Cdc42-bound N-WASP species and hence its subcellular function are likely to be cell type dependent. For instance, in HeLa cells, the V12 variant Cdc42 is known to localize to, as well as recruit, N-WASP to the Golgi complex (25). In endothelial cells, the activated species of Cdc42 in response to fluid shear stress is localized to the MTOC (51). However, our data based on multi-photon FLIM or immuno-EM do not demonstrate a significant accumulation of the N-WASP-Cdc42 complex in proximity to the MTOC in a breast carcinoma cell model (Fig. 5 and data not shown). On the basis of our findings, we postulate that in breast cancer cells, the key role of the N-WASP-Cdc42 complex is to drive the actin polymerization-dependent movement of recycling endosomes carrying membrane receptors, such as various integrin receptors (35, 42), that are essential for directional cell motility. It will be interesting to determine whether other proteins of the WASP family, such as Scar/WAVE, associate directly or indirectly with the Rho GTPases (12) more readily in the cell periphery. Identification of the precise intracellular localization of these WASP protein-Rho GTPase complexes holds the key to a better understanding of the functional specialization among this important family of proteins in situ.
In breast malignancies, carcinoma cells are likely to receive autonomous stimulatory signals through both autocrine and paracrine mechanisms which could influence processes such as cell proliferation, polarization, and directional motility. The observed effect of the RICH Rho-GAP domain on N-WASP-Cdc42 complex formation suggests that by modulating the activities of Rho GTPase-activating proteins, one may be able to target the Rho-GTPase-dependent cell protrusion via downstream targets such as N-WASP in these cancer cells. Interestingly, aberrant expression levels of p190-B, a Rho-GAP that stimulates the intrinsic GTPase activities of Rho, Cdc42, and Rac, have been associated with a subset of mammary tumors that appear to be less well differentiated and potentially more aggressive (8). However, in our experiments where a cytosolic Cdc42 Rho-GAP was expressed, only a partial reduction in N-WASP-Cdc42 complex formation was obtained, with some residual FRET species detectable in vesicular structures within cell ruffles. In the therapeutic context, the engineering of a Cdc42 Rho-GAP with membrane-localizing property, such as that found in β2-chimerins (31), may greatly improve the pharmacological targeting of metastatic cancer cells.
Supplementary Material
Acknowledgments
This study was supported by the United Kingdom Medical Research Council (in the form of a Clinician Scientist Grant awarded to T. Ng). We acknowledge financial support by the Research Councils UK Basic Technology Programme. L. M. Machesky is supported by an MRC Senior Research Fellowship. We also acknowledge financial support by Cancer Research UK (programme grant C133/A1812, B. Vojnovic). M. Peter is a recipient of a Human Frontier Science Program Long-Term Fellowship.
We are grateful to P. Aspenström for his kind gift of a RICH Rho-GAP domain construct. We thank J. Downward for contributing the HA-tagged Cdc42 constructs. We are grateful to P. Barber and R. Locke for technical expertise.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Ameer-Beg, S. M., P. R. Barber, R. J. Hodgkiss, R. J. Locke, R. G. Newman, G. M. Tozer, B. Vojnovic, and J. Wilson. 2002. Application of multiphoton steady-state and lifetime imaging to mapping of tumour vascular architecture in vivo. Proc. SPIE 4620:85-95. [Google Scholar]
- 2.Anilkumar, N., M. Parsons, R. Monk, T. Ng, and J. C. Adams. 2003. Interaction of fascin and protein kinase C{alpha}: a novel intersection in cell adhesion and motility. EMBO J. 22:5390-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aspenstrom, P., U. Lindberg, and A. Hall. 1996. Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Curr. Biol. 6:70-75. [DOI] [PubMed] [Google Scholar]
- 4.Beechem, J. M. 1989. A second generation global analysis program for the recovery of complex inhomogeneous fluorescence decay kinetics. Chem. Phys. Lipids 50:237-251. [DOI] [PubMed] [Google Scholar]
- 5.Beechem, J. M., and L. Brand. 1986. Global analysis of fluorescence decay: applications to some unusual experimental and theoretical studies. Photochem. Photobiol. 44:323-329. [DOI] [PubMed] [Google Scholar]
- 6.Brown, M. C., K. A. West, and C. E. Turner. 2002. Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol. Biol. Cell 13:1550-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchwald, G., E. Hostinova, M. G. Rudolph, A. Kraemer, A. Sickmann, H. E. Meyer, K. Scheffzek, and A. Wittinghofer. 2001. Conformational switch and role of phosphorylation in PAK activation. Mol. Cell. Biol. 21:5179-5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakravarty, G., D. Roy, M. Gonzales, J. Gay, A. Contreras, and J. M. Rosen. 2000. P190-B, a Rho-GTPase-activating protein, is differentially expressed in terminal end buds and breast cancer. Cell Growth Differ. 11:343-354. [PubMed] [Google Scholar]
- 9.Chang, F. S., C. J. Stefan, and K. J. Blumer. 2003. A WASp homolog powers actin polymerization-dependent motility of endosomes in vivo. Curr. Biol. 13:455-463. [DOI] [PubMed] [Google Scholar]
- 10.Chong, C., L. Tan, L. Lim, and E. Manser. 2001. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 276:17347-17353. [DOI] [PubMed] [Google Scholar]
- 11.Daub, H., K. Gevaert, J. Vandekerckhove, A. Sobel, and A. Hall. 2001. Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J. Biol. Chem. 276:1677-1680. [DOI] [PubMed] [Google Scholar]
- 12.Eden, S., R. Rohatgi, A. V. Podtelejnikov, M. Mann, and M. W. Kirschner. 2002. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418:790-793. [DOI] [PubMed] [Google Scholar]
- 13.Frost, J. A., A. Khokhlatchev, S. Stippec, M. A. White, and M. H. Cobb. 1998. Differential effects of PAK1-activating mutations reveal activity-dependent and -independent effects on cytoskeletal regulation. J. Biol. Chem. 273:28191-28198. [DOI] [PubMed] [Google Scholar]
- 14.Fukuoka, M., S. Suetsugu, H. Miki, K. Fukami, T. Endo, and T. Takenawa. 2001. A novel neural Wiskott-Aldrich syndrome protein (N-WASP) binding protein, WISH, induces Arp2/3 complex activation independent of Cdc42. J. Cell Biol. 152:471-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heckman, C. A., J. M. Urban, M. Cayer, Y. Li, N. Boudreau, J. Barnes, H. K. Plummer, III, C. Hall, R. Kozma, and L. Lim. 2004. Novel p21-activated kinase-dependent protrusions characteristically formed at the edge of transformed cells. Exp. Cell Res. 295:432-447. [DOI] [PubMed] [Google Scholar]
- 16.Herreros, J., T. Ng, and G. Schiavo. 2001. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol. Biol. Cell 12:2947-2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higgs, H. N., and T. D. Pollard. 2000. Activation by Cdc42 and PIP2 of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J. Cell Biol. 150:1311-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato, M., H. Miki, S. Kurita, T. Endo, H. Nakagawa, S. Miyamoto, and T. Takenawa. 2002. WICH, a novel verprolin homology domain-containing protein that functions cooperatively with N-WASP in actin-microspike formation. Biochem. Biophys. Res Commun. 291:41-47. [DOI] [PubMed] [Google Scholar]
- 19.Kessels, M. M., and B. Qualmann. 2002. Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J. 21:6083-6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knaus, U. G., Y. Wang, A. M. Reilly, D. Warnock, and J. H. Jackson. 1998. Structural requirements for PAK activation by Rac GTPases. J. Biol. Chem. 273:21512-21518. [DOI] [PubMed] [Google Scholar]
- 21.Kurokawa, K., R. E. Itoh, H. Yoshizaki, Y. O. Nakamura, and M. Matsuda. 2004. Coactivation of rac1 and cdc42 at lamellipodia and membrane ruffles induced by epidermal growth factor. Mol. Biol. Cell 15:1003-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Legg, J. W., C. A. Lewis, M. Parsons, T. Ng, and C. M. Isacke. 2002. A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat. Cell Biol. 27:399-407. [DOI] [PubMed] [Google Scholar]
- 23.Lommel, S., S. Benesch, K. Rottner, T. Franz, J. Wehland, and R. Kuhn. 2001. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2:850-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu, W., S. Katz, R. Gupta, and B. J. Mayer. 1997. Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr. Biol. 7:85-94. [DOI] [PubMed] [Google Scholar]
- 25.Luna, A., O. B. Matas, J. A. Martinez-Menarguez, E. Mato, J. M. Duran, J. Ballesta, M. Way, and G. Egea. 2002. Regulation of protein transport from the Golgi complex to the endoplasmic reticulum by CDC42 and N-WASP. Mol. Biol. Cell 13:866-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magdalena, J., T. H. Millard, S. Etienne-Manneville, S. Launay, H. K. Warwick, and L. M. Machesky. 2003. Involvement of the arp2/3 complex and scar2 in Golgi polarity in scratch wound models. Mol. Biol. Cell 14:670-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manser, E., H. Y. Huang, T. H. Loo, X. Q. Chen, J. M. Dong, T. Leung, and L. Lim. 1997. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 17:1129-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manser, E., T. Leung, H. Salihuddin, Z. S. Zhao, and L. Lim. 1994. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 367:40-46. [DOI] [PubMed] [Google Scholar]
- 29.Manser, E., T. H. Loo, C. G. Koh, Z. S. Zhao, X. Q. Chen, L. Tan, I. Tan, T. Leung, and L. Lim. 1998. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1:183-192. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Quiles, N., R. Rohatgi, I. M. Anton, M. Medina, S. P. Saville, H. Miki, H. Yamaguchi, T. Takenawa, J. H. Hartwig, R. S. Geha, and N. Ramesh. 2001. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat. Cell Biol. 3:484-491. [DOI] [PubMed] [Google Scholar]
- 31.Menna, P. L., G. Skilton, F. C. Leskow, D. F. Alonso, D. E. Gomez, and M. G. Kazanietz. 2003. Inhibition of aggressiveness of metastatic mouse mammary carcinoma cells by the beta2-chimaerin GAP domain. Cancer Res. 63:2284-2291. [PubMed] [Google Scholar]
- 32.Miki, H., T. Sasaki, Y. Takai, and T. Takenawa. 1998. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 391:93-96. [DOI] [PubMed] [Google Scholar]
- 33.Neudauer, C. L., G. Joberty, N. Tatsis, and I. G. Macara. 1998. Distinct cellular effects and interactions of the Rho-family GTPase TC10. Curr. Biol. 8:1151-1160. [DOI] [PubMed] [Google Scholar]
- 34.Ng, T., M. Parsons, W. E. Hughes, J. Monypenny, D. Zicha, A. Gautreau, M. Arpin, S. Gschmeissner, P. J. Verveer, P. I. Bastiaens, and P. J. Parker. 2001. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J. 20:2723-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng, T., D. Shima, A. Squire, P. I. H. Bastiaens, S. Gschmeissner, M. J. Humphries, and P. J. Parker. 1999. PKCa regulates b1 integrin-dependent motility, through association and control of integrin traffic. EMBO J. 18:3909-3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng, T., A. Squire, G. Hansra, F. Bornancin, C. Prevostel, A. Hanby, W. Harris, D. Barnes, S. Schmidt, H. Mellor, P. I. H. Bastiaens, and P. J. Parker. 1999. Imaging PKC alpha activation in cells. Science 283:2085-2089. [DOI] [PubMed] [Google Scholar]
- 37.Otsuki, M., T. Itoh, and T. Takenawa. 2003. Neural Wiskott-Aldrich syndrome protein is recruited to rafts and associates with endophilin A in response to epidermal growth factor. J. Biol. Chem. 278:6461-6469. [DOI] [PubMed] [Google Scholar]
- 38.Parsons, M., M. D. Keppler, A. Kline, A. Messent, M. J. Humphries, R. Gilchrist, I. R. Hart, C. Quittau-Prevostel, W. E. Hughes, P. J. Parker, and T. Ng. 2002. Site-directed perturbation of PKC-integrin interaction blocks carcinoma cell chemotaxis. Mol. Cell. Biol. 22:5897-5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parsons, M., and T. Ng. 2002. Intracellular coupling of adhesion receptors: molecular proximity measurements. Methods Cell Biol. 69:261-278. [DOI] [PubMed] [Google Scholar]
- 40.Prehoda, K. E., J. A. Scott, R. Dyche Mullins, and W. A. Lim. 2000. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science 290:801-806. [DOI] [PubMed] [Google Scholar]
- 41.Richnau, N., and P. Aspenstrom. 2001. Rich, a rho GTPase-activating protein domain-containing protein involved in signaling by Cdc42 and Rac1. J. Biol. Chem. 276:35060-35070. [DOI] [PubMed] [Google Scholar]
- 42.Roberts, M., S. Barry, A. Woods, P. van der Sluijs, and J. Norman. 2001. PDGF-regulated rab4-dependent recycling of alphavbeta3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr. Biol. 11:1392-1402. [DOI] [PubMed] [Google Scholar]
- 43.Sells, M. A., U. G. Knaus, S. Bagrodia, D. M. Ambrose, G. M. Bokoch, and J. Chernoff. 1997. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7:202-210. [DOI] [PubMed] [Google Scholar]
- 44.Sells, M. A., A. Pfaff, and J. Chernoff. 2000. Temporal and spatial distribution of activated Pak1 in fibroblasts. J. Cell Biol. 151:1449-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slot, J. W., and H. J. Geuze. 1985. A new method of preparing gold probes for multiple-labeling cytochemistry. Eur. J. Cell Biol. 38:87-93. [PubMed] [Google Scholar]
- 46.Snapper, S. B., F. Takeshima, I. Anton, C. H. Liu, S. M. Thomas, D. Nguyen, D. Dudley, H. Fraser, D. Purich, M. Lopez-Ilasaca, C. Klein, L. Davidson, R. Bronson, R. C. Mulligan, F. Southwick, R. Geha, M. B. Goldberg, F. S. Rosen, J. H. Hartwig, and F. W. Alt. 2001. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat. Cell Biol. 3:897-904. [DOI] [PubMed] [Google Scholar]
- 47.Stofega, M. R., L. C. Sanders, E. M. Gardiner, and G. M. Bokoch. 2004. Constitutive PAK activation in breast cancer cells as a result of mislocalization of PAK to focal adhesions. Mol. Biol. Cell 15:2965-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun, H., Martinez.M., Y. Wei, S. Snapper, and H. L. Yin. 2002. N-WASP deficiency reveals a role of N-WASP in the early secretory pathway. Mol. Biol. Cell 13(Suppl.):313a (abstract 1760). [Google Scholar]
- 49.Symons, M., J. M. Derry, B. Karlak, S. Jiang, V. Lemahieu, F. McCormick, U. Francke, and A. Abo. 1996. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 84:723-734. [DOI] [PubMed] [Google Scholar]
- 50.Turner, C. E., M. C. Brown, J. A. Perrotta, M. C. Riedy, S. N. Nikolopoulos, A. R. McDonald, S. Bagrodia, S. Thomas, and P. S. Leventhal. 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell Biol. 145:851-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tzima, E., W. B. Kiosses, M. A. del Pozo, and M. A. Schwartz. 2003. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J. Biol. Chem. 278:31020-31023. [DOI] [PubMed] [Google Scholar]
- 52.Vadlamudi, R. K., L. Adam, R. A. Wang, M. Mandal, D. Nguyen, A. Sahin, J. Chernoff, M. C. Hung, and R. Kumar. 2000. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J. Biol. Chem. 275:36238-36244. [DOI] [PubMed] [Google Scholar]
- 53.Verveer, P. J., and P. I. Bastiaens. 2003. Evaluation of global analysis algorithms for single frequency fluorescence lifetime imaging microscopy data. J. Microsc. 209:1-7. [DOI] [PubMed] [Google Scholar]
- 54.Verveer, P. J., A. Squire, and P. I. Bastiaens. 2000. Global analysis of fluorescence lifetime imaging microscopy data. Biophys. J. 78:2127-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verveer, P. J., A. Squire, and P. I. Bastiaens. 2001. Improved spatial discrimination of protein reaction states in cells by global analysis and deconvolution of fluorescence lifetime imaging microscopy data. J. Microsc. 202:451-456. [DOI] [PubMed] [Google Scholar]
- 56.Verveer, P. J., F. S. Wouters, A. R. Reynolds, and P. I. Bastiaens. 2000. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science 290:1567-1570. [DOI] [PubMed] [Google Scholar]
- 57.Vignjevic, D., D. Yarar, M. D. Welch, J. Peloquin, T. Svitkina, and G. G. Borisy. 2003. Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 160:951-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ward, M. E., J. Y. Wu, and Y. Rao. 2004. Visualization of spatially and temporally regulated N-WASP activity during cytoskeletal reorganization in living cells. Proc. Natl. Acad. Sci. USA 101:970-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilcke, M., L. Johannes, T. Galli, V. Mayau, B. Goud, and J. Salamero. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J. Cell Biol. 151:1207-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wittmann, T., G. M. Bokoch, and C. M. Waterman-Storer. 2003. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J. Cell Biol. 161:845-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zenke, F. T., C. C. King, B. P. Bohl, and G. M. Bokoch. 1999. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J. Biol. Chem. 274:32565-32573. [DOI] [PubMed] [Google Scholar]
- 62.Zicha, D., I. M. Dobbie, M. R. Holt, J. Monypenny, D. Y. Soong, C. Gray, and G. A. Dunn. 2003. Rapid actin transport during cell protrusion. Science 300:142-145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.