

Abstract
Ikaros is a hematopoietic cell-specific zinc finger DNA binding protein that plays an important role in lymphocyte development. Genetic disruption of Ikaros results in T-cell transformation. Ikaros null mice develop leukemia with 100% penetrance. It has been hypothesized that Ikaros controls gene expression through its association with chromatin remodeling complexes. The development of leukemia in Ikaros null mice suggests that Ikaros has the characteristics of a tumor suppressor gene. In this report, we show that the introduction of Ikaros into an established mouse Ikaros null T leukemia cell line leads to growth arrest at the G0/G1 stage of the cell cycle. This arrest is associated with up-regulation of the cell cycle-dependent kinase inhibitor p27kip1, the induction of expression of T-cell differentiation markers, and a global and specific increase in histone H3 acetylation status. These studies provide strong evidence that Ikaros possesses the properties of a bona fide tumor suppressor gene for the T-cell lineage and offer insight into the mechanism of Ikaros's tumor suppressive activity.
Cancer is a disease of uncontrolled cell growth. It is precipitated by genetic mutations that affect the regulation of cell cycle progression and genomic integrity. Tumor suppressor genes are a group of genes whose function is lost through mutation, leading to cancer (16). Studies of mouse model systems have identified the nuclear factor Ikaros as a novel candidate tumor suppressor involved in the regulation of T-cell proliferation (2, 56, 58). It is hypothesized that Ikaros functions through its ability to target chromatin remodeling complexes and their associated histone-modifying enzymes to specific genetic loci (24).
A decrease of Ikaros activity in mice, either by a complete knockout or expression of a dominant negative (DN) Ikaros isoform, results in T-cell leukemogenesis with 100% penetrance (56, 58). Data suggest that deregulation of T-cell receptor (TCR) signaling pathways may underlie T-cell transformation. Without exception, leukemic cells express a receptor linked to TCR signaling pathways (TCRαβ, TCRγδ, or pre-TCR), and this expression is essential to the leukemogenic process (57). When the Ikaros mutation was backcrossed onto a Rag 1−/− genetic background, which prevents somatic rearrangement of TCR genes, the resulting mice were completely protected from development of leukemia. Leukemogenesis again occurred with 100% penetrance when TCR expression was restored through expression of a TCR transgene in these mice.
In support of Ikaros's role as a regulator of TCR signaling pathways, prior to becoming transformed, Ikaros mutant T cells display augmented proliferative responses when stimulated via TCR complex engagement (2, 56, 58). This hyperresponsive phenotype is increasingly more dramatic as the levels of Ikaros activity are reduced (2), suggesting that Ikaros functions as a rheostat for TCR activation signals. Moreover, Ikaros mutant T cells require less TCR signal to drive them into cell cycle from the quiescent or G0 state and display a shortened G1 stage and premature entry into S phase (2). These data imply that Ikaros functions as a regulator of cell cycle progression and proliferation in T cells.
This function is likely due to Ikaros's role as a component of at least two chromatin remodeling complexes that are linked to histone acetylase (HAT) or histone deacetylase (HDAC) activity, thereby affecting chromatin structure and gene expression (24). It has been hypothesized that Ikaros's role in these complexes is to target them to specific genetic loci via its sequence-specific DNA binding ability. Significantly, it has been demonstrated that components of chromatin remodeling complexes are mutated in several forms of human cancer, including acute lymphocytic leukemia and acute myelogenous leukemia (41). The high degree of conservation of Ikaros in mice and humans (95% at the amino acid level) strongly suggests that Ikaros may also play a tumor suppressive role in human cells (31). In support of this, it has been shown that a high percentage of analyzed infant and childhood acute lymphocytic leukemias display defects in Ikaros gene expression that would result in a significant decrease in Ikaros activity (48, 49). Consequently, understanding the mechanism by which Ikaros mutations lead to T-cell leukemia in mice may contribute toward understanding how functionally similar events occur in humans.
Tumor suppressor genes are genes whose loss of function results in tumorigenesis. Moreover, reintroduction of their function into deficient cancer cells usually results in growth arrest (5, 14, 15, 21). In this report, we provide strong evidence that Ikaros fulfills this definition. Reintroduction of Ikaros activity into an Ikaros null T leukemia cell line arrests its uncontrolled proliferation. We show that growth arrest is accompanied by up-regulation of p27kip1 expression and initiation of a T-cell developmental program similar to that observed in developing T cells responding to TCR-mediated cues. Dramatically, these changes are accompanied by global changes in histone H3 acetylation status. These studies establish Ikaros as a tumor suppressor gene for the T-cell lineage and provide insight into the mechanism of Ikaros's tumor suppressive activity.
MATERIALS AND METHODS
Cell culture.
JE131 cells were derived from the thymus of an Ikaros null mouse that spontaneously developed T-cell leukemia. The thymus was aseptically removed and ground between two glass slides to release the thymocytes. Cells were plated, and within 2 weeks robustly proliferating cells were observed. JE131 cells were maintained in RPMI medium supplemented with 10% fetal bovine serum, 50 μM β-mercaptoethanol, and 500 U (each) of penicillin and streptomycin per ml (RPMI complete medium).
Retroviral constructs.
Sequences corresponding to Ikaros isoform Ik-1 were PCR amplified from cDNA generated from mouse T cells. Sequences corresponding to isoform Ik-7 were PCR amplified from DNA obtained from an Ik-7 transgenic mouse line (kindly provided by Katia Georgopoulos, Massachusetts General Hospital and Harvard Medical School). In both cases, sequences corresponding to the Flag epitope were introduced at the N terminus. FlagIk-1 and FlagIk-7 cDNA sequences were subcloned into murine stem cell virus internal ribosome entry site green fluorescent protein (MSCV-GFP-IRES) (Clontech) and MSCV-IRES-H-2Kk vectors (kind gift of Neil Clipstone, Feinberg School of Medicine). Sequencing was performed through the sequencing core facility at the University of Chicago to ensure fidelity of PCR amplification. cDNAs encoding p27kip1 were obtained from the American Tissue Culture Center (29). They were directly cloned into the MSCV-IRES-H-2Kk retroviral vector.
Retroviral transduction.
Retroviral constructs were transfected into the Phoenix ecotropic packaging cells by using Lipofectamine reagent (Invitrogen). Viral supernatants were collected at 48 and 72 h, passed through 0.22-μm-pore-size filters to remove virus-producing cells and stored at −80°C. Supernatants were used to infect JE131 cells by using 1 ml of viral supernatant per 2 × 106 cells. Cells were centrifuged in 24-well tissue culture dishes with viral supernatants supplemented with 8 μg of polybrene per ml at 500 × g for 120 min at 32°C. Supernatants were then removed and cells were maintained in RPMI complete medium for the duration of the experiment.
Cell sorting.
Successfully retrovirally transduced cells were sorted by either flow cytometric cell sorting (Robert H. Lurie Comprehensive Cancer Center Flow Cytometry core facility) or the MiniMacs system (Miltenyi Biotec) for GFP and H-2Kk markers, respectively. When the MiniMacs system was used, cells were first trypansized to disperse clumps and this step was followed by centrifugation through Histopaque-1083 (Sigma) to remove dead cells. Cells were then incubated with H-2Kk antibodies coupled to magnetic beads (Miltenyi Biotec) and run over a magnetic column that retained the labeled H-2Kk-positive cells. Purity was assessed by using either the MACSelect control fluorescein isothiocyanate (FITC) antibody (Miltenyi Biotec) or anti H-2Kk FITC antibody (BD Biosciences). Purity was consistently greater than 90%.
RT-PCR.
mRNA was obtained from sorted retrovirally transduced cells 24 h postinfection with Trizol reagent (Invitrogen). cDNA was generated with a Superscript II kit (Invitrogen). The cDNA was then subjected to reverse transcription-PCR (RT-PCR) with the following primers (5′ to 3′): p27kip1, CCCGCCCGAGGAGGAAGATGTCAAAC and CCCTTTTGTTTTGCGAAGAAGAATCT; p21, CCGTGGACAGTGAGCAGTTG and TGGGCACTTCAGGGTTTTCT; cyclin D2, AGACCTTCATCGCTCTGTGC and TAGCAGATGACGAACACGCC; hypoxanthine phosphoribosyltransferase, GTTGGATACAGGCCAGACTTTGTTG and GAGGGTAGGCTGGCCTATAGGCT.
Protein preparation and Western blot analysis.
Cell extracts were prepared with whole cell lysis buffer (20 mM Tris [pH 7.5], 0.42 M NaCl, 0.1% bovine serum albumin [BSA], 1% NP-40, and 1 mM EDTA) supplemented with 4 μg of leupeptin per ml, 2 μg of aprotinin per ml, 1 μg of dithiothreitol per ml, and 5 μg of phenylmethylsulfonyl fluoride per ml. Cells were lysed on ice for 30 min and centrifuged to remove cellular debris, and supernatants were stored at −80°C. Protein concentration was determined with Bradford reagent (Bio-Rad). For immunoblotting, 10 μg of protein was separated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (SDS-10% PAGE) and transferred to polyvinylidene difluoride membrane. Membranes were blocked with Tris-buffered saline (TBS)-5% milk for 1 h and probed with antibody in TBS-5% milk for 1 h at room temperature, followed by incubation with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Enhanced chemiluminescence reagent was used to visualize proteins. For p27kip1 blots, the membrane was blocked in TBS-5% milk supplemented with 0.02% Tween-20 and 2% BSA. The p27kip1 antibody was diluted in 10% blocking buffer in TBS supplemented with 0.01% Tween-20.
Histones were prepared by using an acid extraction protocol as detailed on the Upstate Biotechnology web page. Briefly, cells were lysed by using whole-cell lysis buffer containing 0.2 M HCl. After debris was removed by centrifugation, acetone precipitation was performed on the supernatants. Pellets were resuspended in water. Protein concentration was determined by using the Bradford reagent (Bio-Rad). For immunoblotting, 10 μg of protein was separated by SDS-15% PAGE and transferred to polyvinylidene difluoride membrane. Membranes were blocked with TBS-3% milk for 1 h and probed with antibody in TBS-3% milk overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibody (Jackson Immunoresearch) for 1 h at room temperature. Enhanced chemiluminescence reagent was utilized to visualize proteins.
Densitometry was performed by using a Multi-Image light cabinet (Alpha Innotech Corporation).
Cell cycle analysis and Pyronin Y staining.
For cell cycle analysis, 106 successfully transduced cells were pelleted, washed in phosphate-buffered saline, and fixed in cold 70% ethanol overnight. Cells were stained with propidium iodide, followed by analysis on a FACSCalibur (BD Biosciences, Mountainview, Calif.) flow cytometer. The ModFit Cell Cycle analysis program was used to determine the percentages of cells at each stage of the cell cycle. Pyronin Y staining was performed as previously described (43). Briefly, sorted GFP+ and GFP− cells were fixed in 70% ethanol. Cells were incubated with 2 μg of Hoechst stain per ml and 4 μg of Pyronin Y per ml for 20 min prior to running on the flow cytometer (Beckman Coulter Elite ESP).
Antibodies.
All antibodies were purchased from eBioscience unless otherwise stated. The following antibodies were used for flow cytometric analysis: anti-CD4 (GK1.5), anti-CD8 (53-6.7), anti-TCRβ (H57-597), anti-CD5 (53-7.3), anti-CD69 (H1.2F3), MACSelect control FITC antibody (Miltenyi Biotec), and anti-H-2Kk (H100-27.R55 and AF3-12.1) (Miltenyi Biotec and BD Biosciences, respectively). Antibodies were used as allophycocyanin, FITC, or phycoerthrin conjugates. For Western blot analyses, anti-Ikaros monoclonal antibody (4E9) (kind gift of Katia Georgopoulos, Massachusetts General Hospital and Harvard Medical School) and anti-p27kip1 (Transduction Laboratories) as well as anti-H3, anti-H4, anti-acetyl H3, and anti-acetyl H4 (Upstate Biotechnologies) antibodies were used.
Cell staining and flow cytometry.
Cells were plated in microwell staining plates at a density of 5 × 105 to 1 × 106 cells/well. Fluorochrome-conjugated antibodies were added to cells and incubated on ice for 15 to 30 min. Cells were then washed and fixed in 75 μl of 2% paraformaldehyde, followed by the addition of an equal volume of phosphate-buffered saline-1% BSA. Fixed cells were analyzed 12 to 72 h later on a FACSCalibur (BD Biosciences) flow cytometer. Analyses were performed on Cellquest Pro software.
RESULTS
Retroviral transduction of Ikaros into an Ikaros null T-cell leukemia line.
An examination of the consequence of the reintroduction of Ikaros DNA-binding activity into an Ikaros null T-cell leukemia line required development of a system with which to introduce the Ikaros cDNA. A retroviral transduction system was chosen, taking advantage of the MSCV-IRES-GFP and the MSCV-IRES-H-2Kk retroviral vectors (39, 54). These were selected since the MSCV long terminal repeat promoter and enhancer elements drive high levels of expression in T cells (35, 51). The vectors contain IRES to allow translation of Ikaros and the GFP or H-2Kk, a truncated major histocompatibility complex class I protein, from one transcript. Expression of GFP or H-2Kk, as determined by flow cytometry, was used to identify successfully transduced cells. Retroviruses generated by using both of these constructs were utilized interchangeably in the following experiments with identical results.
cDNAs encoding two Ikaros isoforms, Ik-1 and Ik-7, were subcloned into the retroviral vectors to create MSCV-(Ik-1)-IRES-GFP (or H-2Kk) and MSCV-(Ik-7)-IRES-GFP (or H-2Kk). Ik-1 is the largest and most predominantly expressed Ikaros isoform in T cells. It has also been shown in ectopic expression and reporter assays to have the highest transcriptional activation and repression activity (25, 26, 30, 50). Ik-7 is an engineered (not naturally occurring) mutant Ikaros protein that lacks the N-terminal zinc finger DNA binding domain and, therefore, is unable to bind DNA (50). Ik-7 expression is used as a control to assign the effects of Ik-1 reintroduction on Ikaros's DNA binding ability. Recombinant retroviruses encoding Ik-1, Ik-7, or no Ikaros protein, as well as H-2Kk, were used to infect a thymocyte cell line (JE131) developed from an Ikaros null mouse with a spontaneously arising T-cell leukemia. At 24 h postinfection, protein extracts were prepared from JE131 cells that had been successfully transduced and, therefore, were expressing H-2Kk. Retrovirally expressed Ik-1 and Ik-7 were efficiently expressed at equivalent levels at this early time point as determined by Western blot analysis (Fig. 1). Importantly, levels of Ikaros approximate those observed in wild-type thymocytes (Fig. 1). Similar results were obtained with GFP retroviruses.
FIG. 1.
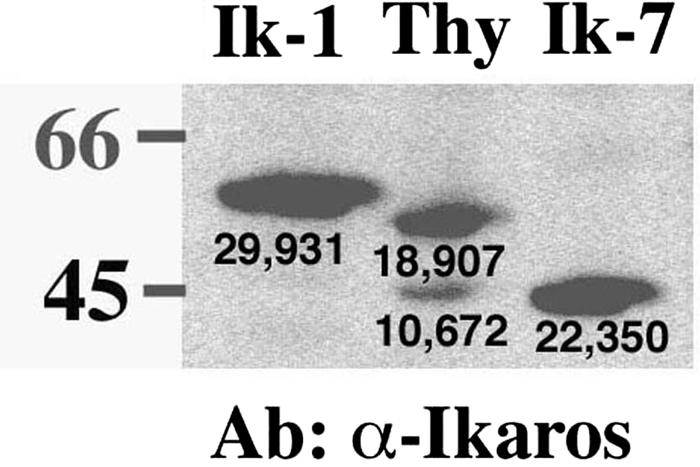
Retroviral expression levels of Ik-1 are comparable to expression levels of DNA-binding Ikaros isoforms in wild-type thymocytes. Whole-cell extracts of wild-type thymocytes and purified JE131 cells successfully transduced with the MSCV-(Ik-1)-IRES-H-2Kk (Ik-1) or the MSCV-(Ik-7)-IRES-H-2Kk (Ik-7) retrovirus were prepared. A total of 10 μg of protein was subjected to SDS-PAGE, and Western blotting was performed with an anti-Ikaros monoclonal antibody. The two Ikaros bands visible in the wild-type thymocytes correspond to the predominantly expressed Ikaros isoforms Ik-1 (top band) and Ik-2 and Ik-3, which comigrate (bottom band). The Ik-1 band in the retrovirally transduced cells is of slightly larger molecular weight due to the Flag epitope tag. Densitometry readings are shown under each band. This figure was generated by using Adobe Photoshop software. Thy, thymocytes; Ab, antibody.
Reintroduction of Ik-1 inhibits proliferation of an Ikaros null T-cell leukemia line.
The consequences of reintroducing Ik-1 activity into the Ikaros null T leukemia cell line JE131 were studied by infecting cells with the MSCV-(Ik-1)-IRES-GFP and MSCV-IRES-GFP (control) retroviruses. Six days after infection, successfully transduced cells were sorted from the cultures, and cell cycle analyses were performed. Ik-1-transduced cells displayed an increase in the percentage of cells in G0/G1 compared to cells transduced with control retrovirus (Fig. 2A). This effect was observed only in cells expressing Ik-1 since sorted GFP− cells from the Ik-1-transduced culture showed a similar percentage of cells at G0/G1 as their control retrovirus-transduced counterparts. This observation strongly suggests that the observed phenotype is dependent on the expression of Ik-1 within the cell and is not induced in a paracrine fashion on nontransduced cells by an Ik-1-induced secreted factor.
FIG. 2.

Reintroduction of Ikaros into the JE131 cells has a profound effect on their growth properties. (A) JE131 cells were infected with the MSCV-IRES-GFP or MSCV-(Ik-1)-IRES-GFP retrovirus. Successfully transduced cells were purified and stained with propidium iodide to measure DNA content as a measure of cell cycle status. Shown are percentages of cells that fall into the G0/G1, S, and G2/M phases of the cell cycle for successfully transduced (GFP+) and nontransduced (GFP−) cells from each culture. (B) JE131 cells were infected with the MSCV-IRES-H-2Kk (Ik−), MSCV-(Ik-7)-IRES-H-2Kk (Ik-7), or the MSCV-(Ik-1)-IRES-H-2Kk (Ik-1) retrovirus. Successfully transduced cells were purified and plated at 106 cells/well in a 24-well plate. Counts of viable cells were performed every 24 h. (C) H-2Kk expression as monitored every 24 h in the purified cell populations by flow cytometry. (D) After 8 days in culture, cells were resorted for H-2Kk expression, and whole-cell protein extracts were prepared from H-2Kk+ and H2-Kk− fractions. A total of 10 μg of protein was loaded onto an SDS-PAGE gel, and Western blotting was performed with an anti-Ikaros monoclonal antibody. This figure was generated by using CellQuest, Microsoft Excel, and Adobe Photoshop software. Ab, antibody.
Next, we tested whether this partial block in G0/G1 resulted in an overall reduction in the growth potential of the Ik-1-transduced cells compared to those transduced with either the Ik-7 or the control retrovirus. For these experiments, JE131 cells were infected with the H-2Kk-coexpressing retroviruses. Twenty-four hours after infection, successfully transduced cells were purified from the cultures and plated. Over the next 5 days, cell counts were performed, and expression of the H-2Kk marker was followed. These analyses demonstrate that Ik-1-transduced cells display slower growth kinetics compared to cells transduced with the Ik-7 or control retroviruses (Fig. 2B). In addition, in Ik-1-transduced cultures, a decrease in the percentage of cells expressing H-2Kk, as well as a decrease in levels of surface H-2Kk expression per cell, was observed over time. Therefore, Ik-1-transduced cells, defined as those cells expressing H-2Kk, were selected against in the culture (Fig. 2C). Moreover, cells that retain H-2Kk expression in Ik-1-transduced cultures show dramatically decreased levels of Ik-1 expression compared to levels of Ik-7 expression retained in Ik-7-transduced cultures (Fig. 2D). These data suggest that in order for Ik-1-transduced cells to proliferate in culture they cannot express high levels of Ik-1.
Ik-1 expression blocks cell cycle progression.
Ik-1-transduced cultures contain a unique population of cells with low forward and side scatter that appear 48 h after infection (Fig. 3A, R2). The majority of cells in this population express H-2Kk, suggesting that this phenotype is induced by expression of Ik-1 and does not occur in cells in the same culture that have not been successfully transduced (Fig. 3B). The small cell population was not observed in cells infected with the Ik-7 or control retrovirus, strongly suggesting that it is dependent on Ikaros's sequence-specific DNA binding domain (Fig. 3A).
FIG. 3.

Appearance of a small cell population in Ik-1-expressing JE131 cells. At 48 h after infection, JE131 cells infected with the MSCV-IRES-H-2Kk (Ik−), MSCV-(Ik-7)-IRES-H-2Kk (Ik-7), or MSCV-(Ik-1)-IRES-H-2Kk (Ik-1) retrovirus were stained with fluorochrome-conjugated anti-H-2Kk, and flow cytometric analysis was performed. (A) A unique population of cells with low forward and side scatter (R2) are observed in the Ik-1-transduced culture. (B) This effect is confined to successfully transduced cells, since the small cell population is almost uniformly H-2Kk+ and, therefore, Ik-1+. The larger cell population (R1) is already beginning to lose H-2Kk and, therefore, Ik-1 expression. This figure was generated by using CellQuest and Adobe Photoshop software. FSC, forward scatter; SSC, side scatter.
The decrease in growth potential and appearance of a small cell population in the Ik-1-transduced cells could be due to initiation of an apoptotic program, perturbation of cell cycle progression, or a combination of both. The small cell population is viable and not apoptotic since the cells fail to stain with annexin V, and they exclude propidium iodide as determined by flow cytometry (data not shown). In fact, there is no difference in the percentage of apoptotic cells in cells infected with MSCV-(Ik-1)-IRES-H-2Kk cells compared to those infected with the Ik-7 or control retrovirus (data not shown).
However, a difference in cell cycle phase distribution of the Ik-1-transduced cultures was observed. Seventy-two hours after infection, cell cycle analyses were performed with JE131 cells infected with the Ik-1 or the control retrovirus. Ik-1-transduced cells showed an increase in the percentage of cells in G0/G1 phase compared to cells transduced with the Ik-7 or the control retrovirus as well as nontransduced cells in the same culture. Interestingly, the small cell population (R2) contained cells exclusively in the G0/G1 phase of the cell cycle, whereas larger cells in the same culture do not show cell cycle distributions that are significantly different from their Ik-7- or control retrovirus-transduced counterparts (Fig. 4A). Six days postinfection, transduced cells were analyzed for cytoplasmic RNA content by staining with Pyronin Y, which can differentiate between G0 and G1 cell cycle status (43). In this experiment, the majority of Ik-1-transduced cells fell into the small cell population (80%) as determined by forward and side scatter profiles. Significantly, an equivalent percentage (89%) of cells did not stain with Pyronin Y, strongly suggesting that they have exited the cell cycle and are in a G0 state (Fig. 4B). Taken together, these data suggest that Ikaros may induce cell cycle arrest in G0 in the Ikaros null T-cell leukemia line JE131 without inducing apoptosis.
FIG. 4.

Small population of Ik-1-expressing cells are arrested at G0/G1. (A) Three days after infection of the JE131 cells with MSCV-IRES-H-2Kk (Ik−), MSCV-(Ik-7)-IRES-H-2Kk (Ik-7), or MSCV-(Ik-1)-IRES-H-2Kk (Ik-1) retrovirus, DNA content was analyzed by propidium iodide staining as a measure of cell cycle status. Cells that fall into the R1 gate show similar cell cycle profiles in all three transduced cultures. However, the unique small cell population (R2) observed in the Ik-1-transduced culture is blocked at the G0/G1 phase of the cell cycle. (B) At day 6 postinfection, successfully transduced cells were sorted from the cultures, and Pyronin Y staining was performed to measure cytoplasmic RNA content. Pyronin Y-negative cells are in G0, whereas Pyronin Y-positive cells are in G1, S, or G2/M phase. The majority of Ik-1-transduced cells fell into the small cell population and failed to stain with Pyronin Y. This figure was generated by using CellQuest and Adobe Photoshop software.
Ikaros induces expression of a critical cell cycle regulator.
The cell cycle arrest induced by Ik-1 expression suggests that Ikaros, similar to other tumor suppressor genes, may play a role in the regulation of cell cycle control factors. In order to investigate this possibility, JE131 cells successfully transduced with Ik-1 or the control retrovirus were purified from nontransduced cells 24 h after infection for analysis of the expression of known cell cycle regulators. Because of the G0/G1 block that was instituted upon Ik-1 expression, we focused our analysis on regulators of the phase transitions from G0 to G1 and G1 to S. By RT-PCR and/or Western blot analyses, no differences were observed in the expression levels of p15, p16, cyclin D, p21, Rb, or E2F between cells transduced with Ik-1 or the control retrovirus (Fig. 5 and data not shown). However, the cyclin-dependent kinase (CDK) inhibitor p27kip1 was up-regulated at both the RNA and protein levels in Ik-1-transduced cells (Fig. 5A and B). Infection of JE131 cells with a retrovirus expressing p27kip1 also dramatically slowed its growth. Significantly, the growth kinetics of p27kip1-transduced cells was very similar to those of Ik-1-transduced cells (Fig. 5C).
FIG. 5.

Ik-1-transduced JE131 cells display an increase in p27kip1 expression and can be slowed in their growth by retroviral transduction of p27kip1. JE131 cells with and without Ik-1 activity were analyzed for RNA and protein expression of p27kip1. (A) At 24 h after infection with the MSCV-IRES-GFP (Ik−) or MSCV-(Ik-1)-IRES-GFP (Ik+) retrovirus, successfully transduced JE131 cells were purified and cDNA was prepared. RT-PCR was performed by using a range of cDNA amounts. Shown here are the results of twofold dilutions (from 1:256 to 1:1024 for each sample). (B) At 24 h after infection with MSCV-(Ik-1)-IRES-GFP (Ik-1), MSCV-(Ik-7)-IRES-GFP (Ik-7), or MSCV-IRES-GFP (Ik−) retrovirus, successfully transduced JE131 cells were purified, and protein extracts were prepared. A total of 10 μg of protein was subjected to SDS-PAGE. Blots were probed with antibodies against p27kip1 or actin (as a loading control). Results from two representative experiments are shown. (C) JE131 cells were infected with the MSCV-IRES-H-2Kk (Ik−), MSCV-(Ik-7)-IRES-H-2Kk (Ik-7), MSCV-(Ik-1)-IRES-H-2Kk (Ik-1), or MSCV-(p27kip1)-IRES-H-2Kk (p27) retrovirus. Successfully transduced cells were purified and plated at 106 cells/well in a 24-well plate. Counts of viable cells were performed every 24 h. This figure was generated by using CellQuest, Microsoft Excel, and Adobe Photoshop software.
Restoration of Ik-1 expression leads to up-regulation of genes induced during T-cell development.
Cell cycle arrest is a hallmark of differentiation, and up-regulation of p27kip1 has been shown to parallel differentiation (7, 20, 40). In addition, reintroduction of the tumor suppressor genes WT1 and p53 to deficient cancer cell lines has been shown to induce differentiation in addition to cell cycle arrest (1, 9, 10, 27, 42, 44, 45). Taken together, this led us to hypothesize that the observed cell cycle arrest in Ik-1-transduced cells may precede or occur concomitantly with up-regulation of genes involved in T-cell differentiation.
The cell surface phenotype of the JE131 cell line (Thy1+ CD4− CD8− TCR− CD44− CD25+) places the cells at a late DN stage (DN3) of T-cell development (Fig. 6A and data not shown) (12, 13, 36). As thymocyte maturation proceeds from this stage, it is characterized by expression of the coreceptors CD4 and CD8, as well as a mature TCRαβ. In addition, down-regulation of CD25 (interleukin-2 receptor α) occurs. The result is progression to the CD4+ CD8+ TCR+ double positive (DP) stage of development. At the DP stage, thymocytes receive TCR signals that induce increased cell surface expression of the T-cell differentiation markers CD69, CD5, and TCR and lead to development of mature CD4 and CD8 T cells (17, 53, 55). In order to determine if reintroduction of Ikaros DNA binding activity into JE131 cells could result in induction of T-cell differentiation antigens, cell surface expression of these markers was evaluated after transduction of JE131 cells with MSCV-(Ik-1)-IRES-H-2Kk, MSCV-(Ik-7)-IRES-H-2Kk, or the MSCV-IRES-H-2Kk control retrovirus. Dramatically, from 2 to 3 days postinfection, H-2Kk+ cells transduced with the Ik-1, but not the Ik-7 or control retrovirus, displayed up-regulation of CD8 and, in most cases, CD4 to become either DP thymocytes or CD8 T cells (Fig. 6B). In addition, these cells exhibit up-regulation of TCRαβ, CD69, and CD5 and down-regulation of CD25 (Fig. 6C). Cell cycle arrest in the absence of Ik-1 activity is unable to induce this differentiation program as shown when the MSCV-(p27kip1)-IRES-GFP retrovirus was included in these experiments (Fig. 6B). Therefore, introduction of Ik-1 into an Ikaros null T-cell line recapitulates events that occur during T-cell development in the thymus.
FIG. 6.

Reintroduction of Ik-1 into JE131 cells induces a T-cell differentiation program. (A) Model depicting developmental progression of thymocytes. DN thymocytes can be broken down into four developmental stages based on cell surface expression of CD44 and CD25 as depicted here. (B and C) JE131 cells were infected with the MSCV-IRES-H-2Kk (Ik−), MSCV-(Ik-7)-IRES-H-2Kk (Ik-7), MSCV-(Ik-1)-IRES-H-2Kk (Ik-1), or the MSCV-(p27kip1)-IRES-GFP (p27) retrovirus. Two to three days postinfection, cells were stained with fluorochrome-conjugated anti-H-2Kk antibody to identify successfully transduced cells, together with antibodies against defined T-cell differentiation markers. This figure was generated by using CellQuest and Adobe Photoshop software.
Ikaros expression leads to widespread changes in histone acetylation patterns.
The panoply of effects initiated by the reintroduction of Ik-1 into the JE131 cell line suggests that Ikaros activity is responsible for initiating a complex program of gene expression resulting in cell cycle exit and T-cell differentiation. These effects could be the result of the association of Ikaros with the NuRD and/or Swi/Snf chromatin remodeling complexes (24). Chromatin remodeling complexes are linked to the activities of histone-modifying enzymes, such as HATs and HDACs. The acetylation status of histones affects chromatin structure and DNA accessibility, which, in turn, plays a central role in gene expression (47). It is hypothesized that Ikaros regulates gene expression in large part by targeting chromatin remodeling complexes and their associated histone-modifying enzymes. Therefore, we investigated whether reintroduction of Ikaros activity into the JE131 cell line could alter the levels of histone H3 and/or H4 acetylation.
The JE131 cell line was infected with the MSCV-(Ik-1)-IRES-H-2Kk or the MSCV-IRES-H-2Kk control retrovirus. Seventy-two hours later, histones were prepared from successfully transduced cells purified from the cultures, and Western blot analyses were performed. Through these analyses, it was shown that the reintroduction of Ikaros activity significantly increases the global level of H3 acetylation (Fig. 7). This effect was unique for histone H3 since no global change in H4 acetylation levels was observed (Fig. 7).
FIG. 7.

Expression of Ik-1 in JE131 cells induces widespread changes in histone acetylation. JE131 cells were infected with the MSCV-IRES-H-2Kk (Ik−), MSCV-(Ik-1)-IRES-H-2Kk (Ik-1), or the MSCV-(p27kip1)-IRES-GFP (p27) retrovirus. At 3 days postinfection, successfully transduced cells were purified. Histones were prepared by using an acid extraction procedure. A total of 10 μg of protein extract was subjected to SDS-PAGE. The blots in panels A and B are representative of six and two independent experiments, respectively. This figure was generated by using Adobe Photoshop software.
In order to determine if the widespread increase in H3 acetylation levels in Ik-1-transduced cells could be the consequence of cell cycle exit, JE131 cells were infected with the MSCV-(p27)-IRES-H-2Kk retrovirus or the MSCV-IRES-H-2Kk control retrovirus. Seventy-two hours later, histones were prepared from successfully transduced cells purified from the cultures, and Western blot analyses were performed. As shown in Fig. 7, cell cycle exit in the absence of Ikaros (as indicated by the p27-transduced cells) does not result in increased H3 acetylation levels, strongly suggesting that this phenotype is Ikaros dependent.
DISCUSSION
T-cell leukemia arises with 100% penetrance in mice genetically engineered to lack expression of the Ikaros protein, suggesting that Ikaros may function as a tumor suppressor for the T-cell lineage (56). In this report, we show that restoration of Ikaros to an Ikaros null T-cell leukemia line can arrest cell growth, a hallmark of a tumor suppressor gene. This phenotype provides evidence that the lack of Ikaros per se rather than downstream mutational events is responsible for the development of leukemia. Significantly, we have identified the p27kip1 gene as a potential gene target for Ikaros regulation, which may explain the mechanism of its effect on the cell cycle. Since p27kip1 mRNA and protein levels increase upon transduction with the Ik-1 retrovirus, it is our hypothesis that Ikaros activity is necessary to maintain high levels of expression of p27kip1 in quiescent T cells.
CDK inhibitors are key regulators of cell cycle progression. They negatively regulate CDKs, preventing cell cycle progression until appropriate signals are received. p27kip1 regulates the functions of cyclin D- and E-dependent kinases, whose functions are essential for G1 phase progression. It is known that p27kip1 is important in the regulation of T-cell proliferation. p27kip1 is expressed at high levels in quiescent, or G0, T cells (11, 33). Down-regulation of p27kip1 is required for cell cycle progression from quiescence through G1 phase after TCR complex engagement (33, 38, 52). These data suggest that p27kip1 may act to hold T cells in a quiescent state. Therefore, it would be expected that decreases in p27kip1 expression would affect the cell cycle progression of an activated T cell. Significantly, it has been shown that Ikaros mutant T cells display abnormal cell cycle progression (2). They are able to be activated and move into cell cycle from G0 with less TCR signal than wild-type T cells, suggesting a lowered threshold of activation for the G0 to G1 phase transition. It has also been shown that rapamycin, a pharmacological inhibitor of mammalian target of rapamycin, which blocks proliferation of activated wild-type T cells, has no effect on the ability of Ikaros mutant T cells to proliferate, even at high concentrations (2). Rapamycin blocks proliferation by preventing degradation of p27kip1. Rapamycin-sensitive T-cell lines lose sensitivity when antisense p27kip1 is introduced, indicating that basal levels of p27kip1 are a limiting factor in determining the rapamycin sensitivity of T cells (23). We hypothesize that p27kip1 levels are sufficiently low in Ikaros mutant T cells such that less p27kip1 degradation is required for cell cycle progression. In support of a direct role, the p27kip1 promoter region contains seven potential Ikaros DNA binding sites.
Deregulation of critical checkpoints in cell cycle progression likely contributes to the 100% penetrance of leukemogenesis observed in Ikaros null mice. Emphasizing the central importance of p27kip1 to T-cell growth control is the observation that decreasing levels of p27kip1 have been shown to contribute to the mechanism of leukemogenesis induced by the oncogene Bcr/Abl (22). Yet, p27kip1-deficient mice do not develop leukemia. Therefore, deregulated expression of p27kip1 cannot be solely responsible for the T-cell leukemogenesis that occurs in Ikaros null mice. However, we have uncovered a dramatic phenotype involving alteration of widespread histone modification levels that occurs upon reintroduction of Ikaros and that may be responsible for globally altering chromatin structure, a function that may also contribute to Ikaros's role as a tumor suppressor.
Regulated progression through the cell cycle requires changes in the programs of gene expression at critical checkpoints. It is clear that chromatin remodeling complexes and their associated histone-modifying enzymes, such as HATs and HDACs, are essential in coordinating these changes. In particular, the acetylation status of lysine residues in the amino termini of two of the core histones, H3 and H4, has been shown to play a fundamental role in transcriptional regulation (46). Sequence-specific DNA binding factors, such as c-Myc and E2F, have been shown to play important roles in this process via recruitment of complexes containing HAT activity to target genes (19, 28, 32, 37). However, to our knowledge, no mammalian DNA binding factor has yet been defined whose expression alters widespread histone acetylation levels. In this report, we demonstrate that the restoration of Ikaros function to an Ikaros null T-cell line results in a global increase in levels of histone H3 acetylation, while having no effect on levels of histone H4 acetylation. Interestingly, this suggests a role for Ikaros in targeting HAT complexes, although it does not distinguish between a direct and an indirect role.
Acetylation of histones has historically been associated with increases in gene expression, whereas Ikaros has been more often thought of as a transcriptional repressor. This is due to its association with the NuRD complex and the corepressors Sin3 and CtBP, its colocalization with silenced genes in regions of pericentric heterochromatin, and its ability, as a Gal4 fusion protein, to repress transcription from a subset of viral promoters in reporter plasmids in transient transfection assays (4, 6, 24-26). However, Ikaros has also been shown to activate transcription by using reporter plasmids in transient transfection assays and has been linked to the activation of cellular genes, such as, most recently, the CD8α gene (18, 50). In this report, we demonstrate that reintroduction of Ikaros leads to up-regulation of p27kip1 as well as genes associated with T-cell differentiation. Taken together, these observations support a role for Ikaros as a transcriptional activator in vivo and suggest that Ikaros's role as activator may be facilitated through its effect on histone acetylation levels.
Reintroduction of Ikaros into the Ikaros null T-cell line has a two-pronged effect. First, it leads to cell cycle arrest in the absence of apoptosis. Secondly, it leads to the initiation of a genetic program that results in up-regulation of T-cell differentiation markers. The pattern of gene expression induced by Ikaros expression is similar to that induced as cells move from the DN to the DP stage in the thymus. This is a powerful observation since it suggests that Ikaros activity alone is sufficient to induce expression of a gene subset correlated with a specific T-cell differentiation event and that this program can be initiated in a transformed cell line. In this way, reintroduction of Ikaros is similar to the effect of the reintroduction of the tumor suppressors WT1 and p53 into WT1- and p53-deficient cell lines, respectively. Reintroduction of WT1 into WT1-deficient leukemia lines leads to up-regulation of myeloid differentiation markers (9). Moreover, p53 has been shown to play a role in the differentiation process of a number of different tissues including pancreatic cells, muscle cells, keratinocytes, neurons, thyroid cells (1, 27, 45), and various hematopoietic cells (3, 8, 10, 42, 44).
In previous studies, it was shown that the absence of Ikaros results in up-regulation of CD4 and CD8 coreceptor molecules on the surface of Ikaros null × Rag1−/− thymocytes (57). This result may at first seem contradictory to data presented in this study, which demonstrate that restoration of Ik-1 to an Ikaros null T-cell line results in this same phenotype. It is likely that the differences in Ikaros function in these two studies reflect the fact that (i) JE131 cells are at a different developmental stage (pre-TCR+) than Rag1−/− thymocytes (pre-TCR−) and (ii) JE131 cells are proliferating robustly at the time of Ikaros reintroduction whereas Rag1−/− cells are in G0. We hypothesize that Ikaros is required for the repression of developmental genes (i.e., CD4 and CD8) prior to pre-TCR signaling. Its absence, consequently, would lead to inappropriate expression of these genes as seen in the Ikaros null × Rag1−/− thymuses as previously reported (57). Therefore, in nonproliferating early progenitor cells, Ikaros may function as a repressor for certain developmental genes. However, once a program of proliferation is initiated by pre-TCR signals, we hypothesize that Ikaros changes from a repressor to an activator for a certain subset of T-cell developmental genes. One example of this phenomenon is the basic helix-loop-helix transcription factor RBP-Jκ (recombination binding protein), which functions as a repressor in the absence of Notch signal but is converted to an activator in the presence of Notch signal (34). In the case of Ikaros, pre-TCR signals may change Ikaros from a repressor to an activator. Its role may be to lock in expression of CD4 and CD8 (perhaps by targeting chromatin remodeling complexes and histone-modifying enzymes that fix chromatin structure) as well as to initiate expression of other markers that are induced after pre-TCR signals (CD5, CD69, and TCR), as observed when Ikaros is reintroduced into the proliferating JE131 cells.
Several points support the physiological relevance of our observations based on the ex vivo system. First, the levels of retrovirally expressed Ikaros protein are similar to those expressed in primary thymocytes (Fig. 1). Secondly, the lineage-specific differentiation induced by Ikaros in the JE131 cell line correlates with markers up-regulated during T-cell differentiation in vivo (Fig. 6). Thirdly, although relatively few transcriptional target genes of Ikaros have been identified, one of these, CD8α, is induced by Ikaros in our assays (Fig. 6).
Although it has long been known that Ikaros deficiency leads to T-cell leukemogenesis in mouse model systems, prior to this report no mechanism for this role has been identified. In this report, we have provided compelling evidence that Ikaros is a tumor suppressor gene for the T-cell lineage. We have shown that reintroduction of Ikaros activity, following in the footsteps of known tumor suppressors, induces cell cycle arrest and differentiation in a robustly proliferating deficient leukemia cell line. We suggest that the mechanism of cell cycle arrest is Ikaros regulation of the p27kip1 gene, although whether Ikaros's role is direct or indirect has yet to be determined.
Acknowledgments
We acknowledge Mary Paniagua from the Robert H. Lurie Comprehensive Cancer Center for help with cell cycle analyses and Pyronin Y staining. We thank N. Clipstone, K. Rundell, L. Laimins, P. Stein, and G. Kansas for critical reading of the manuscript.
This work was supported by the Blowitz-Ridgeway Foundation, American Cancer Society Illinois Division (grant BR-01), and the American Cancer Society (grant RSG GMC-105938). K.K. was supported by the Chicago Baseball Cancer Charities.
REFERENCES
- 1.Almog, N., and V. Rotter. 1997. Involvement of p53 in cell differentiation and development. Biochim. Biophys. Acta 1333:F1-F27. [DOI] [PubMed] [Google Scholar]
- 2.Avitahl, N., S. Winandy, C. Friedrich, B. Jones, Y. Ge, and K. Georgopoulos. 1999. Ikaros sets thresholds for T cell activation and regulates chromosome propagation. Immunity 10:333-343. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee, D., H. J. Lenz, B. Schnieders, D. J. Manno, J. F. Ju, C. P. Spears, D. Hochhauser, K. Danenberg, P. Danenberg, and J. R. Bertino. 1995. Transfection of wild-type but not mutant p53 induces early monocytic differentiation in HL60 cells and increases their sensitivity to stress. Cell Growth Differ. 6:1405-1413. [PubMed] [Google Scholar]
- 4.Brown, K. E., S. S. Guest, S. T. Smale, K. Hahm, M. Merkenschlager, and A. G. Fisher. 1997. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 91:845-854. [DOI] [PubMed] [Google Scholar]
- 5.Chen, P. L., Y. M. Chen, R. Bookstein, and W. H. Lee. 1990. Genetic mechanisms of tumor suppression by the human p53 gene. Science 250:1576-1580. [DOI] [PubMed] [Google Scholar]
- 6.Cobb, B. S., S. Morales-Alcelay, G. Kleiger, K. E. Brown, A. G. Fisher, and S. T. Smale. 2000. Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev. 14:2146-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deschenes, C., A. Vezina, J. F. Beaulieu, and N. Rivard. 2001. Role of p27(Kip1) in human intestinal cell differentiation. Gastroenterology 120:423-438. [DOI] [PubMed] [Google Scholar]
- 8.Ehinger, M., E. Nilsson, A. M. Persson, I. Olsson, and U. Gullberg. 1995. Involvement of the tumor suppressor gene p53 in tumor necrosis factor-induced differentiation of the leukemic cell line K562. Cell Growth Differ. 6:9-17. [PubMed] [Google Scholar]
- 9.Ellisen, L. W., N. Carlesso, T. Cheng, D. T. Scadden, and D. A. Haber. 2001. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 20:1897-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinstein, E., G. Cimino, R. P. Gale, G. Alimena, R. Berthier, K. Kishi, J. Goldman, A. Zaccaria, A. Berrebi, and E. Canaani. 1991. p53 in chronic myelogenous leukemia in acute phase. Proc. Natl. Acad. Sci. USA 88:6293-6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firpo, E. J., A. Koff, M. J. Solomon, and J. M. Roberts. 1994. Inactivation of a Cdk2 inhibitor during interleukin 2-induced proliferation of human T lymphocytes. Mol. Cell. Biol. 14:4889-4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Godfrey, D. I., J. Kennedy, T. Suda, and A. Zlotnik. 1993. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8− triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J. Immunol. 150:4244-4252. [PubMed] [Google Scholar]
- 13.Godfrey, D. I., and A. Zlotnik. 1993. Control points in early T-cell development. Immunol. Today 14:547-553. [DOI] [PubMed] [Google Scholar]
- 14.Groden, J., G. Joslyn, W. Samowitz, D. Jones, N. Bhattacharyya, L. Spirio, A. Thliveris, M. Robertson, S. Egan, M. Meuth, et al. 1995. Response of colon cancer cell lines to the introduction of APC, a colon-specific tumor suppressor gene. Cancer Res. 55:1531-1539. [PubMed] [Google Scholar]
- 15.Haber, D. A., S. Park, S. Maheswaran, C. Englert, G. G. Re, D. J. Hazen-Martin, D. A. Sens, and A. J. Garvin. 1993. WT1-mediated growth suppression of Wilms tumor cells expressing a WT1 splicing variant. Science 262:2057-2059. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan, D., and R. A. Weinberg. 2000. The hallmarks of cancer. Cell 100:57-70. [DOI] [PubMed] [Google Scholar]
- 17.Hare, K. J., R. W. Wilkinson, E. J. Jenkinson, and G. Anderson. 1998. Identification of a developmentally regulated phase of postselection expansion driven by thymic epithelium. J. Immunol. 160:3666-3672. [PubMed] [Google Scholar]
- 18.Harker, N., T. Naito, M. Cortes, A. Hostert, S. Hirschberg, M. Tolaini, K. Roderick, K. Georgopoulos, and D. Kioussis. 2002. The CD8α gene locus is regulated by the Ikaros family of proteins. Mol. Cell 10:1403-1415. [DOI] [PubMed] [Google Scholar]
- 19.Hassan, A. H., K. E. Neely, M. Vignali, J. C. Reese, and J. L. Workman. 2001. Promoter targeting of chromatin-modifying complexes. Front. Biosci. 6:D1054-D1064. [DOI] [PubMed] [Google Scholar]
- 20.Hauser, P. J., D. Agrawal, M. Flanagan, and W. J. Pledger. 1997. The role of p27kip1 in the in vitro differentiation of murine keratinocytes. Cell Growth Differ. 8:203-211. [PubMed] [Google Scholar]
- 21.Huang, H. J., J. K. Yee, J. Y. Shew, P. L. Chen, R. Bookstein, T. Friedmann, E. Y. Lee, and W. H. Lee. 1988. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 242:1563-1566. [DOI] [PubMed] [Google Scholar]
- 22.Jonuleit, T., H. van der Kuip, C. Miething, H. Michels, M. Hallek, J. Duyster, and W. E. Aulitzky. 2000. Bcr-Abl kinase down-regulates cyclin-dependent kinase inhibitor p27 in human and murine cell lines. Blood 96:1933-1939. [PubMed] [Google Scholar]
- 23.Kawamata, S., H. Sakaida, T. Hori, M. Maeda, and T. Uchiyama. 1998. The upregulation of p27Kip1 by rapamycin results in G1 arrest in exponentially growing T-cell lines. Blood 2:561-569. [PubMed] [Google Scholar]
- 24.Kim, J., S. Sif, B. Jones, A. Jackson, J. Koipally, E. Heller, S. Winandy, A. Viel, A. Sawyer, T. Ikeda, R. Kingston, and K. Georgopoulos. 1999. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 10:345-355. [DOI] [PubMed] [Google Scholar]
- 25.Koipally, J., and K. Georgopoulos. 2000. Ikaros interactions with CtBP reveal a repression mechanism that is independent of histone deacetylase activity. J. Biol. Chem. 275:19594-19602. [DOI] [PubMed] [Google Scholar]
- 26.Koipally, J., A. Renold, J. Kim, and K. Georgopoulos. 1999. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J. 18:3090-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang, D., S. J. Miknyoczki, L. Huang, and B. A. Ruggeri. 1998. Stable reintroduction of wild-type P53 (MTmp53ts) causes the induction of apoptosis and neuroendocrine-like differentiation in human ductal pancreatic carcinoma cells. Oncogene 16:1593-1602. [DOI] [PubMed] [Google Scholar]
- 28.Marmorstein, R. 2001. Protein modules that manipulate histone tails for chromatin regulation. Nat. Rev. Mol. Cell Biol. 2:422-432. [DOI] [PubMed] [Google Scholar]
- 29.Matsuoka, S., M. C. Edwards, C. Bai, S. Parker, P. Zhang, A. Baldini, J. W. Harper, and S. J. Elledge. 1995. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 9:650-662. [DOI] [PubMed] [Google Scholar]
- 30.Molnar, A., and K. Georgopoulos. 1994. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol. Cell. Biol. 14:8292-8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molnar, A., P. Wu, D. A. Largespada, A. Vortkamp, S. Scherer, N. G. Copeland, N. A. Jenkins, G. Bruns, and K. Georgopoulos. 1996. The Ikaros gene encodes a family of lymphocyte-restricted zinc finger DNA binding proteins, highly conserved in human and mouse. J. Immunol. 156:585-592. [PubMed] [Google Scholar]
- 32.Narlikar, G. J., H. Y. Fan, and R. E. Kingston. 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108:475-487. [DOI] [PubMed] [Google Scholar]
- 33.Nourse, J., E. Firpo, W. M. Flanagan, S. Coats, K. Polyak, M. H. Lee, J. Massague, G. R. Crabtree, and J. M. Roberts. 1994. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 372:570-573. [DOI] [PubMed] [Google Scholar]
- 34.Osborne, B., and L. Miele. 1999. Notch and the immune system. Immunity 11:653-663. [DOI] [PubMed] [Google Scholar]
- 35.Ouyang, W., S. H. Ranganath, K. Weindel, D. Bhattacharya, T. L. Murphy, W. C. Sha, and K. M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 9:745-755. [DOI] [PubMed] [Google Scholar]
- 36.Pearse, M., L. Wu, M. Egerton, A. Wilson, K. Shortman, and R. Scollay. 1989. A murine early thymocyte developmental sequence is marked by transient expression of the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 86:1614-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson, C. L., and J. L. Workman. 2000. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr. Opin. Genet. Dev. 10:187-192. [DOI] [PubMed] [Google Scholar]
- 38.Polyak, K., J. Y. Kato, M. J. Solomon, C. J. Sherr, J. Massague, J. M. Roberts, and A. Koff. 1994. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8:9-22. [DOI] [PubMed] [Google Scholar]
- 39.Porter, C. M., and N. A. Clipstone. 2002. Sustained NFAT signaling promotes a Th1-like pattern of gene expression in primary murine CD4+ T cells. J. Immunol. 168:4936-4945. [DOI] [PubMed] [Google Scholar]
- 40.Quaroni, A., J. Q. Tian, P. Seth, and C. Ap Rhys. 2000. p27(Kip1) is an inducer of intestinal epithelial cell differentiation. Am. J. Physiol. Cell Physiol. 279:C1045-C1057. [DOI] [PubMed] [Google Scholar]
- 41.Redner, R. L., J. Wang, and J. M. Liu. 1999. Chromatin remodeling and leukemia: new therapeutic paradigms. Blood 94:417-428. [PubMed] [Google Scholar]
- 42.Rizzo, M. G., A. Zepparoni, B. Cristofanelli, R. Scardigli, M. Crescenzi, G. Blandino, S. Giuliacci, S. Ferrari, S. Soddu, and A. Sacchi. 1998. Wt-p53 action in human leukaemia cell lines corresponding to different stages of differentiation. Br. J. Cancer 77:1429-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmid, I., S. W. Cole, Y. D. Korin, J. A. Zack, and J. V. Giorgi. 2000. Detection of cell cycle subcompartments by flow cytometric estimation of DNA-RNA content in combination with dual-color immunofluorescence. Cytometry 39:108-116. [DOI] [PubMed] [Google Scholar]
- 44.Shaulsky, G., N. Goldfinger, A. Peled, and V. Rotter. 1991. Involvement of wild-type p53 in pre-B-cell differentiation in vitro. Proc. Natl. Acad. Sci. USA 88:8982-8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soddu, S., G. Blandino, R. Scardigli, S. Coen, A. Marchetti, M. G. Rizzo, G. Bossi, L. Cimino, M. Crescenzi, and A. Sacchi. 1996. Interference with p53 protein inhibits hematopoietic and muscle differentiation. J. Cell Biol. 134:193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 403:41-45. [DOI] [PubMed] [Google Scholar]
- 47.Struhl, K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12:599-606. [DOI] [PubMed] [Google Scholar]
- 48.Sun, L., P. A. Goodman, C. M. Wood, M. L. Crotty, M. Sensel, H. Sather, C. Navara, J. Nachman, P. G. Steinherz, P. S. Gaynon, N. Seibel, A. Vassilev, B. D. Juran, G. H. Reaman, and F. M. Uckun. 1999. Expression of aberrantly spliced oncogenic Ikaros isoforms in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 17:3753-3766. [DOI] [PubMed] [Google Scholar]
- 49.Sun, L., N. Heerema, L. Crotty, X. Wu, C. Navara, A. Vassilev, M. Sensel, G. H. Reaman, and F. M. Uckun. 1999. Expression of dominant-negative and mutant isoforms of the antileukemic transcription factor Ikaros in infant acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 96:680-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun, L., A. Liu, and K. Georgopoulos. 1996. Zinc finger-mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. EMBO J. 15:5358-5369. [PMC free article] [PubMed] [Google Scholar]
- 51.Szabo, S. J., S. T. Kim, G. L. Costa, X. Zhang, C. G. Fathman, and L. H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100:655-669. [DOI] [PubMed] [Google Scholar]
- 52.Toyoshima, H., and T. Hunter. 1994. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78:67-74. [DOI] [PubMed] [Google Scholar]
- 53.Vanhecke, D., B. Verhasselt, M. De Smedt, G. Leclercq, J. Plum, and B. Vandekerckhove. 1997. Human thymocytes become lineage committed at an early postselection CD69+ stage, before the onset of functional maturation. J. Immunol. 159:5973-5983. [PubMed] [Google Scholar]
- 54.Van Parijs, L., Y. Refaeli, A. K. Abbas, and D. Baltimore. 1999. Autoimmunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity 11:763-770. [DOI] [PubMed] [Google Scholar]
- 55.von Boehmer, H. 1994. Positive selection of lymphocytes. Cell 76:219-228. [DOI] [PubMed] [Google Scholar]
- 56.Wang, J. H., A. Nichogiannopoulou, L. Wu, L. Sun, A. H. Sharpe, M. Bigby, and K. Georgopoulos. 1996. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity 5:537-549. [DOI] [PubMed] [Google Scholar]
- 57.Winandy, S., L. Wu, J. H. Wang, and K. Georgopoulos. 1999. Pre-T cell receptor (TCR) and TCR-controlled checkpoints in T cell differentiation are set by Ikaros. J. Exp Med. 190:1039-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winandy, S., P. Wu, and K. Georgopoulos. 1995. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell 83:289-299. [DOI] [PubMed] [Google Scholar]