

Abstract
This is one of the few examples in which the diverse products have been synthesized just by changing the applied potential. The synthesis of sulfonyl derivatives of p-methylaminophenol were carried out by reaction of the electrogenerated p-methylquinoneimine with sulfinic acids. Various types of mono (MSP), bis (BSP) and tris (TSP) sulfonyl p-methyl aminophenols were obtained by changing the electrode potential, in one pot under green conditions. The mono sulfonyl-p-(methylamino)phenol derivatives (MSP) were assessed for their in vitro antibacterial activity against the gram positive (Staphylococcus aureus) and gram negative (Escherichia coli) strains. It was found that the tested compounds were more active against Staphylococcus aureus than Escherichia coli. We also found that the antimicrobial activity of MSP derivatives to vary in the order MSP4 (R = CH3) > MSP1 (R = p-tolyl) ≈ MSP2 (R = phenyl) > MSP3 (R = p-ClC6H4). Moreover, the observed homogeneous rate constants (k obs) of the reaction of p-methyl quinoneimine with sulfinic acids were estimated in various pH values, based on the EC and ECEC mechanisms, by comparing the simulated cyclic voltammograms with the experimental ones.
Introduction
The control of selectivity is one of the important challenges in organic syntheses1–6. To overcome to this problem, a number of organic and metal catalyst systems has been examined1–6. However, they have the disadvantages of safety problems and heavy metal pollution. Organic electrochemical synthesis provides a powerful strategy for the synthesis of organic compounds in both laboratory and industry scale7–13. In this method, the electrons are considered as clean reagents, so that this method can be considered as a green technique. Another important feature of this method, which is used in this work, is its selectivity towards synthesis of products. This unique feature arises from the fact that, the different active intermediates can be provided just by changing the electrode potential7–13.
p-Aminophenol is commercially significant as a versatile intermediate in the manufacture of chemical dye, drugs such as acetaminophen, photographic developer and anticorrosion agents14–17. In particular, it is known that some aminophenol derivatives have antiviral activity against flu A and simple herpes18. It is also known that, the aminophenols containing a sulphone group in addition to activity against flu A and simple herpes have excellent activity against the HIV infection19. On the other hand, it is found that, diphenylsulfone compounds exhibited antibacterial activity20. For example, 4,4-diaminodiphenylsulfone (dapsone) is a bacteriostatic drug that inhibits dihydrofolic acid synthesis by competition with para-aminobenzoic acid21. The oxidation of sulfides or sulfoxides using peracids or hydrogen peroxide, addition reactions to alkenes and alkynes, Friedel–Crafts-type sulfonylation of arenes in the presence of a Lewis or Brønsted acid catalyst, and alkylation of sulfinate salts, are four traditional methods for the synthesis of sulfones22. The excess oxidizing agent, high temperatures, harsh reaction conditions, low regioselectivity, the need for stoichiometric amounts of the catalyst, the generation of hazardous waste and formation of a mixture of isomers are the main disadvantages of these methods23–27.
We assume that the compounds containing both aminophenol and diphenylsulfone moieties, may show promising biological activities. So, the synthesis of these type of compounds by a simple method without the above discussed disadvantages using a green and controllable strategy is the main object of this paper.
Results and Discussion
Figure 1a shows the cyclic voltammogram (CV) of p-methylaminophenol (MAP) in aqueous phosphate buffer (c = 0.2 M, pH = 2.0). It includes a well-defined anodic (A1)/cathodic (C1) peak couple at E 1/2 = 0.44 V versus Ag/AgCl. These peaks (A1 and C1) are attributed to two-electron oxidation of MAP to p-methylquinoneimine (MQI) and vice versa, respectively. Under these conditions, I pC1/I pA1 is near to one, which can be regarded as a criterion for the consistency of electrogenerated MQI under the experimental conditions.
Figure 1.

Cyclic voltammograms of: (a) MAP (1.0 mM), (b) MAP (1.0 mM) + BS (1.0 mM) and (c) BS (1.0 mM) in aqueous phosphate buffer (c = 0.2 M, pH = 2.0). Working electrode: glassy carbon electrode. Scan rate 100 mV s−1. Temperature = 25 ± 1 °C.
Figure 1b shows the CV of MAP in the presence of benzensulfinic acid (BS) under the same conditions as Fig. 1a. Comparison of CVs 1a and 1b shows three important differences: (i) the I pC1/I pA1 is near to one in Fig. 1a, but is about 0.06 in Fig. 1b, (ii) contrary to Fig. 1b shows two couples of peaks (A1/C1 and A2/C2), (iii) E pA1 in Fig. 1b is less positive than that of Fig. 1a. In addition, our data show that two factors: (i) potential scan rate and (ii) BS concentration are effective in the peak current ratio (I pC1/I pA1) of MAP in the presence of BS, so that it decreases with increasing BS concentration and decreasing the potential scan rate.
All of these results are in agreement with the participation of electrogenerated MQI in the Michael addition reaction with BS 28–34. Fig. 1c is the cyclic voltammogram of BS which indicates that in the studied potential range, BS is not electroactive.
Controlled-potential coulometry at E app < E pA1 (E app = 0.40 V vs. Ag/AgCl) was used to determine the number of the electrons transferred (n) for the anodic oxidation of MAP in the presence of BS (Fig. 2I). The cyclic voltammetry during the progress of CPC indicates two significant changes: (i) decreasing I pA1 and (ii) increasing I pA2. The I pA1 reaches zero when the charge consumption was about 2e− (n = 1.95) per molecule of MAP.
Figure 2.

Cyclic voltammograms of MAP (0.25 mmol) in the presence of BS (0.5 mmol) during CPC at 0.40 (part I) and 0.55 (part II) V vs. Ag/AgCl. Part I: the consumed charge is (a) 0, (b) 16, (c) 32 and (d) 47 C. Part II: the consumed charge is (a) 0, (b) 40, (c) 70, (d) 90 and (e) 110 C. Other conditions are as same as Fig. 1.
The CPC was also performed at E app > E pA2 (E app = 0.55 V vs. Ag/AgCl) (Fig. 2II). In comparison with CPC at E app = 0.40, it shows two significant differences: first, the number of electrons is increased from two to four electrons (n = 4.5). Second, both anodic peaks have been removed. Another CPC was performed in the same solvent/electrolyte system containing MAP (0.25 mmol) in the presence of methanesulfinic acid (MS) (0.75 mmol) at E app = 0.55 V vs. Ag/AgCl) (Fig. 3). Contrary to Fig. 2, during the progress of CPC, a new anodic peak (A3) appeared at more positive potentials than the main wave. Under these conditions, all anodic and cathodic peaks disappear at the end of CPC after consumption of 151 C (n = 6.3) of electricity. The following evidences along with the spectroscopic data of the final products were used to propose the following mechanism for the electrochemical oxidation of MAP at different applied potentials in the presence of sulfone nucleophiles.
Figure 3.

Cyclic voltammograms of MAP (0.25 mmol) in the presence of MS (0.75 mmol) during CPC at 0.55 V vs. Ag/AgCl, after consumption of (a) 0, (b) 35, (c) 75, (d) 105, (e) 120, (f) 135 and (g) 151 C. Other conditions are as same as Fig. 1.
As indicated in Fig. 4, when the applied potential is 0.40 V vs. Ag/AgCl, the nucleophilic attack of the sulfone compounds on the electrogenerated MQI would result in INT1-4 which undergoes aromatization to afford the mono sulfonyl-p-(methylamino)phenol derivatives (MSP1–4) as the final products. MSP1–4 is structurally a p-methylaminophenol derivative, however, because of the presence of an electron-withdrawing sulfonyl group in its structure, its oxidation is more difficult than the oxidation of MAP (Fig. 5) and thus, its oxidation at the anode is avoided.
Figure 4.
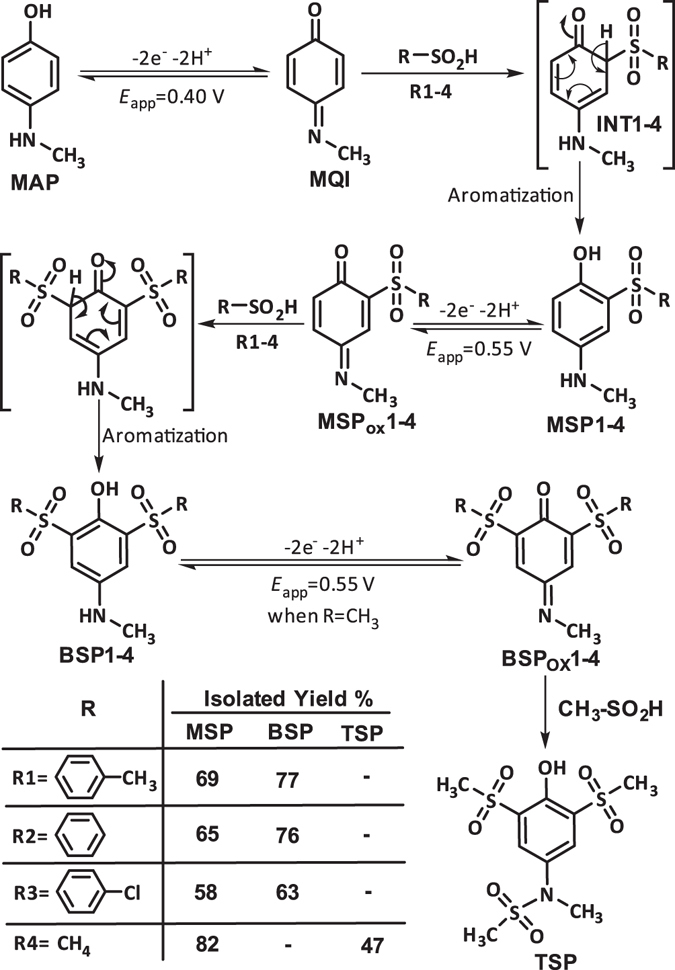
Electrochemical oxidation pathway of MPA in the presence of sulfone compounds.
Figure 5.

Cyclic voltammograms of: MAP (1.0 mM), MSP1 (0.7 mM), BSP1 (0.5 mM), MSP4 (0.5 mM) and TSP (0.25 mM). Solvent for MAP: aqueous phosphate buffer, c = 0.2 M, pH = 2.0. Solvent for MSP1, BSP1, MSP4 and TSP: aqueous phosphate buffer (c = 0.2 M, pH = 2.0)/ethanol (80/20, v/v). Working electrode: glassy carbon electrode. Scan rate 100 mV s−1. Temperature = 25 ± 1 °C.
With increasing the applied potential to 0.55 V vs. Ag/AgCl, the oxidation of MSP1–4 becomes possible and MSP ox 1-4 is formed. The addition of sulfone nucleophile to MSP ox 1-4 followed by aromatization, converts MSP ox 1-4 into bis sulfonyl-p-(methylamino)phenol derivatives (BSP1-4). Since the oxidation of BSP1-4 is more difficult than the oxidation of MSP1–4 and MAP (Fig. 5), its oxidation was stopped due to the presence of the two electron-withdrawing sulfonyl groups as well as the insolubility of BSP1-4 in the electrolysis medium. A remarkable finding in this study is related to the role of methanesulfinic acid (MS) as a nucleophile. Despite several attempts to synthesize BSP4, no bis sulfonyl derivative has been isolated and unexpectedly a tris sulfonyl compound, TSP, which is a new sulphonamide molecule was obtained via the oxidation of BSP4 and attack of MS to electrogenerated BSP ox 4. The oxidation of BSP4 at 0.55 V vs. Ag/AgCl, can be related to the lower electron-withdrawing ability of the methyl-sulfony group compared with aryl-sulfony groups that causes the oxidation potentials of bis and tris sulfonyl compounds do not have any significant differences. In addition, higher nucleophilicity and lower steric effect of MS compared with aryl-sulfone nucleophiles and more solubility of the products containing MS, can also be effective in the observed behavior. According to Fig. 4, the anodic peaks A1, A2 and A3 pertain to the oxidation of MAP, MSP and BSP to the MQI, MSP ox and BSP ox, respectively.
MQI is a bis-Michael acceptor (ortho and meta of the phenolic group) and can be attacked by the sulfone nucleophiles from two sites to yield two isomers (for example, 4-(methylamino)-m-(methylsulfonyl)phenol and 4-(methylamino)-o-(methylsulfonyl) phenoltypes). However, the comparison of simulated 1H NMR35 for the possible compounds and experimental 1H NMR of the final product (See SI), confirms synthesis of ortho derivative. The protonation of the amino group in MAP at pH = 2.0, makes the ortho position in MQI more reactive than meta position for the addition reaction.
The compounds MSP1–4 were tested for the antibacterial activity against the Gram positive (Staphylococcus aureus) (Fig. 6) and Gram negative (Escherichia coli) strains. The results indicated that Staphylococcus aureus was more sensitive to MSP1–4 than Escherichia coli (See SI). The existence of an exterior membrane along with a collection of resisting pumps against drugs in the negative gram bacteria, makes a very effective barrier from this group of bacteria against antibacterial growth and operation. We also found that the antimicrobial activity of MSP derivatives to vary in the order MSP 4 (R=CH3) > MSP 1 (R=p-tolyl) > MSP 2 (R=phenyl) > MSP 3 (R=p-ClC6H4).
Figure 6.

Inhibition zone diameters (mm) obtained of Staphylococcus aureus in disc diffusion test for MSP1–4 (5 mg) and solvent.
The observed homogeneous rate constants (k obs) of the reaction of MQI with the sulfinic nucleophiles was studied based on ECEC (Fig. 7, parts I–IV) and EC (Fig. 7, parts V–VIII) mechanisms, by computer simulation of the experimental cyclic voltammograms. Our data shows that, k obs is strongly dependent to pH so that, it increases with decreasing pH value. The protonation of the nitrogen atom in MQI, makes it more reactive toward the addition reaction and is responsible for increasing k obs. In addition, the results displays that, k obs is also dependent directly to the electron donating ability of the nucleophile, so that, it varies in the order methanesulfinic acid > p-toluenesulfinic acid > banzenesulfinic acid > p-chlorosulfinic acid (Fig. 8).
Figure 7.

Experimental (a) and simulated (b) cyclic voltammograms of MAP (1 mM) in the presence of (I and V) p-toluenesulfinic acid (0.5 mM), (II and VI) benzensulfinic acid (0.5 mM), (III and VII) p-chlorosulfinic acid (0.5 mM) and (IV and VIII) methanesulfinic acid (0.5 mM) at glassy carbon electrode. In I–IV; solvent, aqueous HClO4 (0.1 M) and scan rate: 80 mV/s. In V–VIII, solvent, aqueous phosphate buffer (pH = 6.0, c = 0.2 M) and scan rate: 10 mV/s. Temperature = 25 ± 1 °C.
Figure 8.
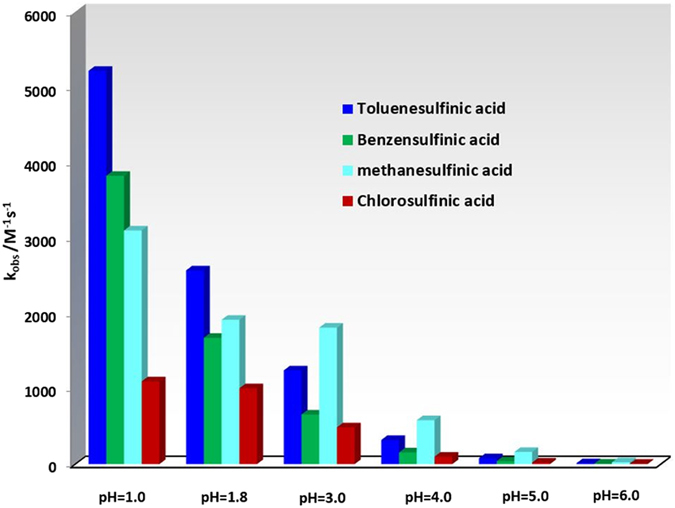
Observed homogeneous rate constants (k obs) of the reaction of MQI with the sulfone nucleophiles at different pH values.
Conclusions
In summary, the electrochemical synthesis of the title compounds has two advantages over conventional methods. Firstly, in this method, a variety of products could be formed just by changing the applied potential. In this work, the synthesis of some mono and bis (or tris) sulfonyl compounds were carried out by electrochemical oxidation of MAP in the presence of arylsulfinic acids only with changing the applied potential from 0.40 to 0.55 V vs. Ag/AgCl. Secondly, this method proceeds in a single step, in ambient conditions, without dealing with strong acids/base, organic solvent and catalysis, under green and mild conditions with high atom economy.
Materials and Methods
Apparatus and Reagents
Cyclic voltammetry, controlled-potential coulometry and preparative electrolysis were performed using a Zahner pp201 potentiostat/galvanostat. Macro-scale electrolysis and controlled-potential coulometry were carried out with a three electrode system, using a Behpajooh C 2056 potentiostat equipped with a digital coulometer. The working electrode used in the voltammetry experiments was a glassy carbon disc (1.8 mm2 area) and platinum wire was used as counter electrode. The working electrode used in controlled-potential coulometry and synthesis was an assembly of three ordinary soft carbon plates (20 mm length, 10 mm width and 40 mm height), and large stainless steel cylinder (25 cm2 area) constituted the counter electrode. The working electrode potentials were measured versus Ag/AgCl (all electrodes from AZAR electrode). The electrochemical synthesis was performed under controlled-potential condition in a simple cell equipped with a magnetic stirrer. More details are described in our previous paper36.
p-Methylaminophenol (MAP), p-toluenesulfinic acid, benzensulfinic acid, p-chlorosulfinic acid, methanesulfinic acid, phosphoric acid and ethanol were obtained from commercial sources. These chemicals were used without further purification. The glassy carbon electrode was polished using alumina slurry (from Iran Alumina Co.).
Electroorganic Synthesis of MSP1–4
An aqueous phosphate buffer solution (c = 0.2 M, pH = 2.0) (ca. 100 mL), containing MAP (0.25 mmol) and sulfone nucleophiles (p-toluenesulfinic acid, benzensulfinic acid, p-chlorosulfinic acid, methanesulfinic acid), (0.25 mmol), was electrolyzed at 0.40 V vs. Ag/AgCl, at 25 °C. The electrolysis was terminated when the decay of the current became more than 95%. At the end of electrolysis, the cell was placed in a refrigerator overnight. The precipitated solid was collected by filtration, washed copiously with distilled water. The products (MSP1–4) were characterized by their physical and spectroscopic data.
Electroorganic Synthesis of BSP1-3
The synthesis of BSP1–3 derivatives were carried out under the same conditions as described for MAP1-4 in a solution containing MAP (0.25 mmol) and sulfone nucleophiles (p-toluenesulfinic acid, benzensulfinic acid, p-chlorosulfinic acid), (0.5 mmol), at 0.55 V. At the end of electrolysis, the cell was placed in a refrigerator overnight. The precipitated solid was collected by filtration, washed copiously with distilled water.
Electroorganic Synthesis of TSP
The synthesis of TSP was carried out under the same conditions as described for MAP1-4 in a solution containing MAP (0.25 mmol) and methanesulfinic acid (0.75 mmol), at 0.55 V vs. Ag/AgCl, at 25 °C. At the end of electrolysis, the cell was placed in a refrigerator overnight. The precipitated solid was collected by filtration, washed copiously with distilled water.
4-(Methylamino)-2-tosylphenol (MSP1)
Isolated yield 69%; mp: 210–212 °C (dec.). 1H NMR (400 MHz, DMSO-d
6): δ 2.38 (s, 3H, methyl), 2.66 (s, 3H, methyl), 6.68 (m, 2H, aromatic), 7.07 (d, J = 2.4 Hz, 1H, aromatic), 7.37 (d, J = 8 Hz, 2H, aromatic), 7.76 (d, J = 8 Hz, 2H, aromatic), 9.53 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d
6): δ 21.5 (C-1), 30.8 (C-10), 110.3 (C-8), 118.8 (C-5), 119.9 (C-8), 126.8 (C-7), 128.2 (C-4), 129.7 (C-3), 139.3 (C-11), 143.3 (C-6,C-9), 143.8 (C-12); FT-IR (KBr): 3487, 3416 medium, O-H), 3091 (weak C-H), 1615 (medium N-H), 1492 (medium C=C), 1316, 1289 (weak S=O), 1141, 1087 (strong, S=O), 960, 818, 710, 656, 533 cm−1; MS (EI, 70 eV): m/z (relative intensity): 277 (M, 38), 65 (100), 93 (90), 78 (74), 51 (29).
4-(Methylamino)-2-(phenylsulfonyl)phenol (MSP2): Isolated yield 65%; mp = 201-203 °C (dec.). 1H NMR (400 MHz, DMSO-d
6): δ 2.66 (s, 3H, methyl), 6.74 (m, 2H, aromatic), 7.08 (d, J = 2.4 Hz, 1H, aromatic), 7.58 (t, J = 7.6 Hz, 2H, aromatic), 7.66 (t, J = 7.4 1H, aromatic), 7.89 (d, J = 7.6 Hz, 2H, aromatic), 5.79 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d
6): δ 30.8 (C-9), 110.3 (C-8), 118.9 (C-11), 120.1 (C-10), 126.5 (C-6), 128.1 (C-4), 129.2 (C-3), 133.4 (C-1), 142.1 (C-8), 143.3 (C-4), 146.5 (C-5); FT-IR (KBr): 3494 (medium, O-H), 3098 (weak C-H), 1615 (medium N-H), 1489 (medium C=C), 1315, 1289 (weak S=O), 1144, 1087 (strong, S=O), 954, 872, 733.41, 596, 510 cm−1; MS (EI, 70 eV): m/z (relative intensity): 263 (M, 100), 93 (54), 78 (36), 51 (29), 122 (16).
2-((4-Chlorophenyl)sulfonyl)-4-(methylamino)phenol (MSP3): Isolated yield 58%; mp = 139-141 °C (dec.). 1H NMR (400 MHz, DMSO-d
6): δ 2.77 (s, 3H, methyl), 6.87 (m, 2H, aromatic), 7.19 (d, J = 2.4 Hz, 1H, aromatic), 7.76 (t, J = 8.8 Hz, 2H, aromatic), 8.00 (t, J = 8.4 1H, aromatic), 7.88 (d, J = 7.6 Hz, 2H, aromatic); 13C NMR (100 MHz, DMSO-d
6): δ 30.4 (C-9), 109.7 (C-7), 118.4 (C-10), 120.0 (C-11), 126.5 (C-6), 128.9 (C-3), 129.7 (C-2), 137.9 (C-1), 140.4 (C-4), 142.7 (C-8), 146.2 (C-5); FT-IR (KBr): 3487 (medium, O-H), 3087 (weak C-H), 1613 (medium N-H), 1474 (medium C=C), 1321 (strong S=O), 1152, 1089 (strong, S=O), 933, 822, 754, 596, 483 cm−1; MS (EI, 70 eV): m/z (relative intensity): 297.3 (M,100), 93 (90), 159 (36), 41 (29), 229 (5).
4-(Methylamino)-2-(methylsulfonyl)phenol (MSP4): Isolated yield 82%; mp = 187–189 °C (dec.). 1H NMR (400 MHz, DMSO-d
6): δ 2.71 (s, 3H, methyl), 3.28 (s, 3H, methyl), 6.83 (m, 1H, aromatic), 6.94 (d, J = 8.8 Hz, 1H, aromatic), 6.97 (d, J = 2.8 Hz, 2H, aromatic; 13C NMR (100 MHz, DMSO-d
6): δ 30.3 (C-6), 42. 3 (C-1), 109.3 (C-4), 118.2 (C-7), 118.8 (C-8), 126.5 (C-3), 142.8 (C-2), 145.8 (C-5); FT-IR (KBr): 3428, 3256 (medium, O-H), 3006 (weak C-H), 1610 (medium N-H), 1489 (medium C=C), 1302 (strong S=O), 1133, 1083 (strong, S=O), 960, 819, 749, 568, 538 cm−1; MS (EI, 70 eV): m/z (relative intensity): 201 (M, 43), 122 (100), 92 (52), 80 (45), 108 (23).
4-(Methylamino)-2,6-ditosylphenol (BSP1): Isolated yield: 77%; mp = 185–187 °C (dec.). 1H NMR (400 MHz, CDCl3): δ 2.36 (s, 6H, methyl), 2.75 (s, 3H, methyl) 7.19 (s, 2H, aromatic), 7.23 (d, J = 8 Hz, 4H, aromatic), 7.74 (d, J = 8.4 Hz, 4H, aromatic), 9.06 (s, 1H, OH); 13C NMR (100 MHz, CDCl3): δ 21.7 (C-1), 31.0 (C-10), 117.8 (C-8), 127.9 (C-4), 128.6 (C-7), 129.8 (C-3), 137.5 (C-5), 142.7 (C-6), 144.6 (C-9), 145.0 (C-2); FT-IR (KBr): 3550, 3470 (medium, O-H), 2924 (weak C-H), 1617 (medium C=C), 1503 (medium C=C), 1317, 1288 (weak S=O), 1138 (strong S=O), 1083, 807, 656, 571, 526 cm−1; MS (EI, 70 eV): m/z (relative intensity): 431 (M., 100), 182 (61), 91 (38), 139 (28), 65 (25), 211(14).
4-(Methylamino)-2,6-bis(phenylsulfonyl)phenol (BSP2): Isolated yield 76%; mp = 200–202 °C (dec.). 1H NMR (400 MHz, CDCl3): δ 3.22 (s, 3H, methyl), 7.33 (s, 2H, aromatic), 7.51 (t, J = 7.8 Hz, 4H, aromatic), 7.62 (t, J = 7.4 Hz, 2H, aromatic), 7.95 (d, J = 8 Hz, 4H, aromatic), 1.03 (s, 1H, OH); 13C NMR (100 MHz, CDCl3): δ 31.0 (C-9), 118.0 (C-7), 127.8 (C-3), 128.3 (C-4), 129.2 (C-2), 133.9 (C-1), 140.5 (C-6), 142.8 (C-5), 144.7 (C-8); FT-IR (KBr): 3427 (medium, N-H), (3344, medium, OH) 3066 (weak C-H) 1619 (weak N-H), 1501 (medium C=C), 1311, 1293 (strong S=O), 1144, 1082 (strong, S=O), 811, 726, 609, 577, 542 cm−1; MS (EI, 70 eV): m/z (relative intensity): 403 (M., 100), 77 (87), 168 (67), 51 (50), 125 (32), 262(6).
2,6-Bis((4-chlorophenyl)sulfonyl)-4-(methylamino)phenol (BSP3): Isolated yield 54%; mp = 166–168 °C (dec.). 1H NMR (400 MHz, CDCl3): δ 2.89 (s, 3H, methyl), 7.41 (s, 2H, aromatic), 7.52 (d, J = 8.8 Hz, 4H, aromatic), 7.90 (d, J = 8.4 Hz, 4H, aromatic); 13C NMR (100 MHz, CDCl3): δ 31.1 (C-9), 118.2 (C-7), 128.2 (C-6), 129.3 (C-3), 129.6 (C-2), 138.8 (C-1), 140.8 (C-4), 142.7 (C-5), 144.6 (C-8); FT-IR (KBr): 3410 (medium, N-H), 3307 (medium, OH), 3091 (weak C-H), 1619 (weak N-H), 1573 (medium C=C), 1318, 1299 (strong S=O), 1144, 1091 (strong, S=O), 1022, 806, 752, 618, 581 cm−1; MS (EI, 70 eV): m/z (relative intensity): 471 (M., 91), 111 (100), 75 (75), 159 (52), 131(42), 296(8).
N
-(4-Hydroxy-3,5-bis(methylsulfonyl)phenyl)-
N
-methylmethanesulfonamide: Isolated yield 47%; mp = 213–215 °C (dec.). 1H NMR 400 MHz, (DMSO-d
6): δ 3.03 (s, 3H, methyl), 3.27 (s, 3H, methyl), 3.38 (s, 6H, methyl), 8.02 (s, 2H, aromatic); 13C NMR 100 MHz,(DMSO-d
6): δ 35.1 (C-6), 37.6 (C-7), 42.8 (C-1), 130.8 (C-3), 132.3 (C-4), 132.7 (C-5), 152.8 (C-2); FT-IR (KBr): 3442 (medium, OH), 3026 (weak C-H), 1573 (medium C=C), 1314, 1341(strong S=O), 1149, 1126 (strong, S=O), 996, 812, 526, 497 cm−1; MS (EI, 70 eV): m/z (relative intensity): 357 (M., 25), 278 (100), 79 (95), 199 (75), 120(52), 90(45).
Antibacterial Susceptibility Assay: The samples were tested to determine the antibacterial susceptibility by Kirby-bauer disk diffusion method. Escherichia coli (E. coli) ATCC 35218 (gram negative) and Staphylococcus aureus (S.aureus) ATCC 6538 (gram positive) were used as test organisms. First, the colony of the bacterias of this study were cultured in the sterile Nutrient Broth and after 18–24 hours incubation at 37 °C microbial suspension was prepared from each bacteria balanced with turbusion 0.5 McFarland standard to 1.5 × 108 cfu.ml−1 with distillated water and was cultured on Muller Hinton agar (MHA) (Merck, KGaA) culture condition in the form of lawn culture. To solve the present samples, dimethyl sulfoxide 10% in distillated water was used. Then 5 mg from each sample was poured on sterile blank discs, and after setting discs in aseptic conditions on cultured Muller Hinton, bacterias were incubated 24 hours at 37 °C in the presence of studied derivatives. Afterwards, the diameter of the inhabitation zone surrounding of the discs were measured by special ruler. Also, to study the lack of influence of the solution containing 10% dimethyl sulfoxide on the bacterias of the study, 20 microlitre of the solution was poured on the discs and set on cultured Muller Hinton agar and incubated.
Digital Simulation
Digital simulation was performed using the DigiElch SB simulation software version 2.037. The simulation was carried out assuming semi-infinite one-dimensional diffusion and planar electrode geometry. The experimental parameters entered for digital simulation consisted of the following: (i) the transfer coefficient (α) was assumed to be 0.5. (ii) The formal potentials were obtained experimentally as the midpoint potential between the anodic and cathodic peaks (E mid). (iii) Analytical concentration of MAP, E start, E switch, E end and temperature = 25 °C. All these parameters were kept constant throughout the fitting of the digitally simulated voltammogram to the experimental data. Electrochemical oxidation of MAP was examined by digital simulation in the pH range 1–6. The simulation was performed based on the proposed EC and ECEC mechanisms (for simplifying, the proton transfers processes are not included). In these mechanisms, E is electrochemical oxidation of MAP to MAP OX, and C is the addition reaction that happens after the electrooxidation process. In these mechanisms, the peak current ratio (I pC/I pA1) is a criterion for the chemical reaction rate between MAP OX and sulfinic ion nucleophiles.
Electronic supplementary material
Acknowledgements
We acknowledge the Bu-Ali Sina University Research Council and Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) for their support of this work.
Author Contributions
D.N. conceived and designed the study. M.R., S.K. and S.M. did the experiments. D.N. wrote the manuscript. D.N. directed the research.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04581-0
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Afewerki S, Córdova A. Combinations of aminocatalysts and metal catalysts: A powerful cooperative approach in selective organic synthesis. Chem. Rev. 2016;116:13512–13570. doi: 10.1021/acs.chemrev.6b00226. [DOI] [PubMed] [Google Scholar]
- 2.Just-Baringo X, Clark J, Gutmann MJ, Procte DJ. Selective synthesis of cyclooctanoids by radical cyclization of seven-membered lactones: neutron diffraction study of the stereoselective deuteration of a chiral organosamarium intermediate. Angew. Chem. Int. Ed. 2016;55:12499–12502. doi: 10.1002/anie.201606792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landry ML, Hu DX, McKenna GM, Burns NZ. Catalytic enantioselective dihalogenation and the selective synthesis of (−)-deschloromytilipin A and (−)-danicalipin A. J. Am. Chem. Soc. 2016;138:5150–5158. doi: 10.1021/jacs.6b01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim T, et al. J. Am. Chem. Soc. 2016;138:8368–8371. doi: 10.1021/jacs.6b04545. [DOI] [PubMed] [Google Scholar]
- 5.Funken N, Mühlhaus F, Gansäuer A. General, highly selective synthesis of 1,3- and 1,4-difunctionalized building blocks by regiodivergent epoxide opening. Angew. Chem. 2016;128:12209–12013. doi: 10.1002/ange.201606064. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Moselage M, González MJ, Ackermann L. Selective synthesis of indoles by cobalt(III)-catalyzed C–H/N–O functionalization with nitrones. ACS Catal. 2016;6:2705–2709. doi: 10.1021/acscatal.5b02937. [DOI] [Google Scholar]
- 7.Lund, H. & Hammerich, O. Organic Electrochemistry. (Marcel Dekker, 1991).
- 8.Osa, T. New Challenges in Organic Electrochemistry. (Gordon & Breach Science Publishers, 1998).
- 9.Brittonm W. E. & Fry, A. J. Topics in Organic Electrochemistry. (Springer Science, 1986).
- 10.Fry, A. J. Synthetic Organic Electrochemistry. (John Wiley & Sons, 1989).
- 11.Fuchigami, T., Atobe, M. & Inagi, S. Fundamentals and Applications of Organic Electrochemistry. (Wiley, 2015).
- 12.Landau, U., Yeager, E. & Kortan, D. Electrochemistry in Industry: New Directions. (Plenum Press, 1982).
- 13.Yoshida J, Kataoka K, Horcajada R, Nagaki A. Modern strategies in electroorganic synthesis. Chem. Rev. 2008;108:2265–2299. doi: 10.1021/cr0680843. [DOI] [PubMed] [Google Scholar]
- 14.Min KI, et al. p-Aminophenol synthesis in an organic/aqueous system using Pt supported on mesoporous carbons. Appl. Catal. A: Gen. 2008;337:97–104. doi: 10.1016/j.apcata.2007.12.004. [DOI] [Google Scholar]
- 15.Wang S, He B, Wang Y, Zhao X. MgAPO-5-supported Pt–Pb-based novel catalyst for the hydrogenation of nitrobenzene to p-aminophenol. Catal. Commun. 2012;24:109–113. doi: 10.1016/j.catcom.2012.03.024. [DOI] [Google Scholar]
- 16.Sun W, Jiang Q, Wang Y, Jiao K. Electrochemical behaviors of metol on hydrophilic ionic liquid 1-ethyl-3-methylimidazolium tetrafluoroborate-modified electrode. Sens. Actuators B. 2009;136:419–424. doi: 10.1016/j.snb.2008.10.003. [DOI] [Google Scholar]
- 17.Löbbert, G. Photography in Ullmann’s Encyclopedia of Industrial Chemistry. (Wiley-VCH, 2005).
- 18.Shadyro O, et al. Synthesis and study of antiviral and anti-radical properties of aminophenol derivatives. Bioorg. Med. Chem. Lett. 2008;18:2420–2423. doi: 10.1016/j.bmcl.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 19.Bazyl OK, et al. Electronic structure and spectroscopic properties of anti-HIV active aminophenols. Opt. Spectrosc. 2012;112:223–232. doi: 10.1134/S0030400X12010031. [DOI] [Google Scholar]
- 20.Kuo CJ, Liang PH. Characterization and inhibition of the main protease of severe acute respiratory syndrome coronavirus. Chem. Bio. Eng. Rev. 2015;2:118–132. [Google Scholar]
- 21.Broussalis E, Trinka E, Kraus J, McCoy M. & Killer, M. Treatment strategies for vasculitis that affects the nervous system. Drug Discov. Today. 2013;18:818–835. doi: 10.1016/j.drudis.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Liu NW, Liang S, Manolikakes G. Recent advances in the synthesis of sulfones. Synthesis. 2016;48:1939–1973. doi: 10.1055/s-0035-1560444. [DOI] [Google Scholar]
- 23.Posner GH, et al. Antimalarial sulfide, sulfone, and sulfonamide trioxanes. Bioorg. Med. Chem. 2000;8:1361–1370. doi: 10.1016/S0968-0896(00)00079-1. [DOI] [PubMed] [Google Scholar]
- 24.Pritzius AB, Breit B. Asymmetric rhodium-catalyzed addition of thiols to allenes: synthesis of branched allylic thioethers and sulfones. Angew. Chem. Int. Ed. 2015;54:3121–3125. doi: 10.1002/anie.201411402. [DOI] [PubMed] [Google Scholar]
- 25.Tozkoparan B, Küpeli E, Yeşilada E, Ertan M. Preparation of 5-aryl-3-alkylthio-l,2,4-triazoles and corresponding sulfones with antiinflammatory-analgesic activity. Bioorg. Med. Chem. 2007;15:1808–1814. doi: 10.1016/j.bmc.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 26.Catarinella M, Grüner T, Strittmatter T, Marx A, Mayer TU. BTB-1: A Small molecule inhibitor of the mitotic motor protein Kif18A. Angew. Chem. Int. Ed. 2009;48:9072–9076. doi: 10.1002/anie.200904510. [DOI] [PubMed] [Google Scholar]
- 27.Sipe, H. J., Clary, D. W., Jr. & White, S. B. An improved synthesis of aryl sulfones. Synthesis 283–284 (1984).
- 28.Bard, A. J. & Faulkner, L. R. Electrochemical Methods. 2 edn, p. 496 (Wiley, 2001).
- 29.Khazalpour S, Nematollahi D. Electrochemical and chemical synthesis of different types of sulfonamide derivatives of N,N-dimethyl-1,4-benzenediamine using 4-nitroso-N,N-dimethylaniline. Green Chem. 2015;17:3508–3514. doi: 10.1039/C5GC00438A. [DOI] [Google Scholar]
- 30.Beiginejad H, Nematollahi D. Electrochemical synthesis of sulfonamide derivatives based on the oxidation of 2,5-diethoxy-4-morpholinoaniline in the presence of arylsulfinic acids. J. Org. Chem. 2014;79:6326–6329. doi: 10.1021/jo500812d. [DOI] [PubMed] [Google Scholar]
- 31.Nematollahi D, Baniardalan M, Khazalpour S, Pajohi-Alamoti MR. Product diversity by changing the electrode potential. Synthesis, kinetic evaluation and antibacterial activity of arylsulfonyl-4,4’-biphenol and bis-arylsulfonyl-4,4’-biphenol derivatives. Electrochim. Acta. 2016;191:98–105. doi: 10.1016/j.electacta.2015.12.230. [DOI] [Google Scholar]
- 32.Nematollahi D, Nikpour F, Salahifar E. Electrochemical synthesis of 1-N-phenyl-4-(sulfonyl)benzene-1,2-diamine derivatives. A mild and regioselective protocol. New J. Chem. 2016;40:5442–5447. doi: 10.1039/C5NJ02748A. [DOI] [Google Scholar]
- 33.Sharafi-Kolkeshvandi M, Nematollahi D, Nikpour F, Bayat M, Soltani E. Electrochemical behavior of 2-aminodiphenylamine and efficient factors on the site-selectivity of sulfonylation reaction: Experimental and theoretical studies. Electrochim. Acta. 2016;222:845–855. doi: 10.1016/j.electacta.2016.11.046. [DOI] [Google Scholar]
- 34.Jamshidi M, Nematollahi D, Bayat M, Salahifar E. Unsymmetrical diaryl sulfones through electrochemical oxidation of fast violet b in the presence of aryl sulfinic acids. J. Electrochem. Soc. 2016;163:G211–G218. doi: 10.1149/2.1431614jes. [DOI] [Google Scholar]
- 35.ChemBioDraw Ultra 12.0, Cambridgesoft, Cambridge, MA, USA.
- 36.Nematollahi D, Dehdashtian S, Niazi A. Electrochemical oxidation of some dihydroxybenzene derivatives in the presence of indole. J. Electroanal. Chem. 2008;616:79–86. doi: 10.1016/j.jelechem.2008.01.012. [DOI] [Google Scholar]
- 37.Rudolph, M. Digital simulations on unequally spaced grids: Part 1. Critical remarks on using the point method by discretisation on a transformed grid. J. Electroanal. Chem. 529, 97–108, http://www.digielch.de (2002).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.