

Abstract
One hallmark of human immunodeficiency virus type 1 (HIV-1) infection is the dysregulation of cytokine gene expression in T cells. Transfection of T cells with human T-cell leukemia type 1 or 2 transactivator results in the induction of the T-cell-restricted cytokine interleukin-2 (IL-2) and its receptor (IL-2Rα). However, no T-cell-specific factor(s) has been directly linked with the regulation of IL-2 and IL-2Rα transcription by influencing the promoter activity. Thymocytes from SATB1 (special AT-rich sequence binding protein 1) knockout mice have been shown to ectopically express IL-2Rα, suggesting involvement of SATB1 in its negative regulation. Here we show that SATB1, a T-cell-specific global gene regulator, binds to the promoters of human IL-2 and IL-2Rα and recruits histone deacetylase 1 (HDAC1) in vivo. SATB1 also interacts with Tat in HIV-1-infected T cells. The functional interaction between HIV-1 Tat and SATB1 requires its PDZ-like domain, and the binding of the HDAC1 corepressor occurs through the same. Furthermore, Tat competitively displaces HDAC1 that is bound to SATB1, leading to increased acetylation of the promoters in vivo. Transduction with SATB1 interaction-deficient soluble Tat (Tat 40-72) and reporter assays using a transactivation-negative mutant (C22G) of Tat unequivocally demonstrated that the displacement of HDAC1 itself is sufficient for derepression of these promoters in vivo. These results suggest a novel mechanism by which HIV-1 Tat might overcome SATB1-mediated repression in T cells.
Interleukin-2 (IL-2) is the key growth factor for T lymphocytes that participates in the regulation of the duration and magnitude of the T-cell immune response (29). The IL-2 receptor (IL-2R) is present on the T-cell surface and is involved in the IL-2-mediated signal transduction for T-cell proliferation, differentiation, and functional activation (13, 29). The physiologically active, high-affinity form of IL-2R is comprised of an inducible 55-kDa protein encoded by the T-cell activation antigen or CD25 or IL-2Rα gene, a constitutive 70-kDa protein encoded by the IL-2Rβ gene, and a gamma chain that is common with other receptors, such as IL-7R (13). The expression of both IL-2 and IL-2Rα is tightly regulated and is induced by agents that mimic antigenic stimulation, including mitogens and lymphotropic retroviruses (13, 19, 29, 46). All transcription factors implicated in IL-2 (38) induction are ubiquitously expressed, and no T-lineage-specific factor that regulates its expression has yet been identified. IL-2Rα is ectopically expressed in SATB1 (special AT-rich sequence binding protein 1) knockout mice, suggesting involvement of SATB1 in its regulation (2). In regard to the mouse IL-2Rα locus, SATB1 recruits histone deacetylase 1 (HDAC1) from the nucleosome remodeling and histone deacetylase complex to its intronic binding site and mediates the deacetylation of histones over long distances within the locus (53). Since SATB1 is a T-lineage-specific global suppressor (2, 53), we explored its involvement in the regulation of human IL-2 and IL-2Rα expression. SATB1 participates in the maintenance of chromatin architecture in a cell-type-specific manner by organizing it into domains via periodic anchoring of base-unpairing regions (BURs) to the nuclear matrix (6). In thymocyte nuclei, SATB1 has a cage-like network distribution circumscribing heterochromatin and selectively tethers BURs onto its network resulting in coordinated regulation of distant genes (6). Studies of SATB1 knockout mice indicate that ∼2% of the genes, including cytokine receptor genes, were derepressed at appropriate stages of T-cell development in a spatiotemporal manner. The maturation of thymocytes was blocked mainly at the CD4+ CD8+ double-positive stage, indicating a key role for SATB1 in T-cell development and differentiation (2). SATB1 regulates large chromatin domains by acting as a “landing platform” for several chromatin remodeling enzymes in T cells (53). SATB1 may also regulate gene expression by directly influencing the promoter activity. SATB1 binds directly to multiple elements in the gp91(phox) promoter and thereby negatively regulates gp91(phox) expression (22).
Dysregulation of cytokine gene expression is a hallmark of human immunodeficiency virus type 1 (HIV-1) infection (5, 8). The retroviral transactivator protein (Tat) is implicated in much of the transcriptional dysregulation in host T cells (5, 7). Transfection of T cells with human T-cell lymphotropic virus type 1 or 2 (HTLV-1 or -2) transactivator (Tax) results in the induction of the IL-2 and IL-2Rα (19). Consistent with this, interaction of inducible nuclear proteins with discrete promoter elements has been implicated in the regulation of IL-2Rα transcription (28, 30). However, the identity of the host factor has remained unknown, especially since NF-κB-independent activation was observed (35). Here we show that SATB1, a T-cell-specific global gene regulator, binds to the human IL-2 and IL-2Rα promoters and recruits HDAC1 in vivo, causing the downregulation of the promoters. We found functional interaction between HIV-1 Tat and SATB1 both in vitro and in vivo, and we show that this interaction occurs via the PDZ-like domain of SATB1 and the HDAC1 corepressor also binds through the same. Our results suggest a novel mechanism by which HIV-1 Tat induces the expression of IL-2 and IL-2Rα via displacement of the SATB1-bound HDAC1 corepressor.
MATERIALS AND METHODS
EMSA.
Electrophoretic mobility shift assays (EMSA) were performed as described previously (27). The recombinant DNA-binding C-terminal half of SATB1 was expressed and purified as a glutathione S-transferase (GST) fusion and purified as described previously (10). Binding reactions were performed in a 10-μl total volume containing 10 mM HEPES (pH 7.9), 1 mM dithiothreitol, 50 mM KCl, 2.5 mM MgCl2, 10% glycerol, 0.5 μg of double-stranded poly(dI-dC), 10 μg of bovine serum albumin, and 1 to 5 μg of the recombinant protein. Samples were preincubated at room temperature for 5 min prior to the addition of the 32P-labeled probe. As probes we used 32P-labeled PCR-amplified products of IL-2 promoter and IL-2Rα promoter sequences. Some of the longer products were also digested with restriction enzymes, and the fragments were used as probes following gel purification. After 15 min of incubation at room temperature, the products of such binding reactions were then resolved by 6% native polyacrylamide gel electrophoresis (PAGE). The gels were dried under vacuum and exposed to X-ray film.
In vitro binding and displacement assays.
HEK 293 cells were transfected with FLAG-HDAC1 (provided by S. Schreiber), and lysate was incubated at 4°C for 1 h with 10 μl of M2-FLAG affinity gel (Sigma). Beads were washed three times in buffer containing 50 mM NaCl phosphate buffer. These beads were incubated with 5 and 10 μg of GST-Tat protein at 4°C for 8 h. Purified GST was used as a control. Bound proteins were eluted with sodium dodecyl sulfate (SDS)-loading buffer and analyzed by Western blotting with anti-SATB1 antibody or, in the case of in vitro-translated proteins, by exposure to X-ray film (Kodak).
Yeast two-hybrid analysis.
The DraI fragment of human SATB1 cDNA encoding near full-length SATB1, the PDZ domain, and the matrix attachment region (MAR)-binding domain plus homeodomain (MD+HD) domain of SATB1 were fused separately with the GAL4 activation domain (AD) in pGAD424 vector (Clontech) (16). Full-length Tat was cloned as a fusion with the GAL4 DNA-binding domain (DBD) in the pAS2.1 vector (Clontech). The AD and DBD fusion constructs were cotransformed in a pairwise fashion in yeast strain AH109 (Clontech) as described previously (16) and assayed for protein-protein interaction by using standard protocols, with HIS3 and LacZ as the proteins encoded by the reporter genes. The liquid β-galactosidase (β-Gal) assay was performed according to the manufacturer's protocol (Pierce), and the values represent an average of results from two independent experiments.
PBMC isolation, activation, and infection.
Human peripheral blood mononuclear cells (PBMCs) were isolated from blood samples of normal seronegative donors. The cells were activated with phytohemagglutinin (5 μg/ml) and kept for 36 to 48 h. The activated cells were infected with HIV-1 at a multiplicity of infection of 0.1 and were maintained in media containing human IL-2 at 20 U/ml (Roche Applied Bioscience, Mannheim, Germany). The infection was monitored by HIV-1 p24 antigen capture enzyme-linked immunosorbent assay of culture supernatants (Perkin-Elmer Life Science, Boston, Mass.).
Coimmunoprecipitation.
One hundred micrograms of protein in nuclear extract was diluted by adding 2 volumes of IP-150 buffer and precleared with rabbit immunoglobulin G (IgG; Sigma) and protein A/G plus beads (Pierce). Precleared extract was then incubated with either polyclonal anti-SATB1 (1:50), anti-Tat antibody (1:50; National Institutes of Health AIDS reagent program) (20), anti-HDAC1 (1:25; Santa Cruz), or anti-FLAG M2 monoclonal antibody (Sigma). Protein-antibody complexes were recovered with protein A/G beads and washed in phosphate-buffered saline (PBS) containing 0.5% NP-40. Soluble proteins were obtained with SDS loading buffer and identified by Western analysis with anti-HDAC1, anti-SATB1, and anti-Tat antibodies.
ChIP.
CEM-GFP cells were infected with the HIV-1 NL4.3 virus isolate (1) at a multiplicity of infection of 0.1 as described previously (32). We consistently observed that at least 70% of the infected cell population exhibited long terminal repeat (LTR)-driven green fluorescent protein expression. Uninfected and infected cells were cross-linked for 15 min at 37°C by adding formaldehyde (to a final concentration of 1%) directly to the culture medium, and chromatin immunoprecipitations (ChIP) were carried out as described previously (21). For immunoprecipitation of chromatin we used various antibodies as described above and also the anti-H3 acetylated lysine 9 (acetyl-H3K9) from Upstate Biotechnology (Lake Placid, N.Y.). One-fiftieth of the DNA from each pool was PCR amplified in 50-μl reaction mixtures containing 50 mM KCl, 10 mM Tris-HCl, 1.5 mM MgCl2, 0.1% Triton X-100, 1.0 U of Taq DNA polymerase (Promega), and a 1 μM concentration of each of the IL-2 promoter primer pairs or IL-2Rα promoter primers using 1 cycle of 95°C for 5 min and 30 cycles of 95°C for 1 min, 48°C for 1 min, and 72°C for 1 min. PCR products were resolved by native PAGE, stained with SYBR gold (molecular probes) and visualized under UV illumination. Quantitative PCRs were performed using the SYBR green IQ supermix (Bio-Rad) and the ICycler IQ real-time thermal cycler (Bio-Rad). The changes in threshold cycle (CT) values were calculated as follows: ΔCT = CT Target − CT Input.
Tat transduction.
Wild-type Tat containing the 86-amino-acid Tat protein of HIV-1 molecular clone NL4.3, Tat 1-48, Tat 40-72, and C22G mutant Tat were expressed as GST fusions in BL21 cells and purified using glutathione Sepharose (Amersham Biosciences) affinity chromatography. Tat was cleaved on the affinity column from the GST tag by using thrombin. GST was eluted by using glutathione-containing elution buffer. All recombinant proteins were dialyzed against PBS, sterilized by passing through a 0.22 μm-pore-size filter, and used immediately. Jurkat cells were transduced essentially as described previously (48), with few modifications. Briefly, cells were resuspended to a density of 5 × 105 per ml in fresh RPMI 1640 medium supplemented with 10% fetal calf serum, 100 μM chloroquine, and 200 ng of recombinant Tat protein/ml. Cells were incubated for 4 h at 37°C in a 5% CO2 atmosphere. Cells were then harvested by centrifugation and washed twice with PBS. Nuclear extracts were prepared using 0.4 M NaCl-containing extraction buffer as described previously (27), and total RNA was isolated using Trizol (Invitrogen). An aliquot of transduced cells was fixed by using 0.4% paraformaldehyde and stained by using either polyclonal anti-Tat antibody or rabbit IgG as a control. Cells were then incubated with anti-rabbit-fluorescein isothiocyanate (BD Biosciences) and acquired on a flow cytometer (FACSvantage; BD Biosciences). The efficiency of transduction was expressed as the percentage of cells stained with anti-Tat antibody.
Luciferase reporter assays.
Luciferase assays were performed using Luc Lite reagent (Perkin Elmer), and luciferase activity was measured using Top Count (Packard). The 555-bp IL-2Rα promoter region (9) spanning base pairs −445 to +110 was PCR amplified and cloned into pGL3 basic vector. The IL-2 promoter (45)-luciferase reporter construct was obtained from K. McGuire. For cotransfections we used p3XFLAG-CMV10-SATB1, pCDNA3.1-Tat (25), and pBJ5-HDAC1 at 1 to 2 μg/well either singly or in different combinations along with the reporter constructs at 1 μg/well. HEK 293 cells were seeded at 0.2 × 106 cells per well and transfected using Lipofectamine 2000 reagent (Invitrogen). Forty hours after transfection, the culture medium was removed; cells were washed with PBS and processed further. Cells were resuspended in 100 μl of reporter lysis buffer and kept at −70°C. After being freeze-thawed twice, the lysate was clarified by spinning at 8,000 × g for 15 min. For accurate quantitation of luciferase activity, an equal amount (50 μg) of the protein was assayed. Protein concentrations in the lysates were measured using Bradford reagent (Bio-Rad). Fold differences were calculated by normalizing the treatment values to the control value. The statistical significance of differences between the treatment groups was calculated using one-way analysis of variance (SigmaStat; SPSS Inc.), and the observed P values were always less than 0.001.
RESULTS
SATB1 directly binds to the upstream regulatory region of IL-2Rα.
We first investigated whether SATB1 binds directly to the enhancer elements within the upstream regulatory elements of human IL-2Rα by using EMSA. We generated a series of radiolabeled probes spanning the entire length of the 1.3-kb IL-2Rα promoter (9) (Fig. 1A). Of the seven probes used, only one probe (probe 4) containing the inverted direct repeat (IDR) (9, 19) in the IL-2Rα promoter was bound by SATB1 (Fig. 1B, lanes 24 and 25) but not by GST-poly(ADP-ribose) polymerase (PARP) (Fig. 1B, lanes 22 and 23) or GST alone (Fig. 1B, lanes 21 and 22). We next examined whether SATB1 is complexed with the IL-2Rα promoter in vivo by performing a ChIP assay using chromatin from CEM-GFP cells. We used chromatin from CEM-GFP, a stable CD4+ T-cell line containing a plasmid encoding the green fluorescent protein driven by the HIV-1 LTR promoter (17). This cell line has been used widely as a reporter cell line to monitor HIV infection. The cross-linked chromatin was immunoprecipitated with anti-SATB1, and the same set of primers was used for PCR amplification following reversal of cross-links. The PCR analysis was performed in different numbers of cycles ranging from 30 to 40. We observed that while most of the primer sets yielded amplified products at 35 and 40 cycles, only probe 4 primers amplified the chromatin at 30 cycles (Fig. 1C, lane 19), suggesting specific binding in vivo. We further confirmed the specificity of this observation by carrying amplification at 30 cycles and using chromatin immunoprecipitated by control rabbit IgG and anti-SATB1 (Fig. 1D, top and middle panels). We did not observe any amplification of this pool of chromatin; however, probe 4 primers amplified a specific product with anti-SATB1 immunoprecipitated chromatin (Fig. 1D, lane 4). To further narrow down the binding site and to verify the contribution of IDRs, we digested the probe 4 region with AluI and XmnI (Fig. 1E). The resulting 103-bp fragment (probe 8) corresponding to base pairs −323 to −226 of the IL-2Rα promoter containing both IDRs and the κB-like motif was radiolabeled and used for EMSA. SATB1 bound to this region in a dose-dependent manner (Fig. 1F, lanes 6 and 7). This region contains an 11-bp motif resembling the NF-κB consensus that is implicated in the inducible expression of IL-2Rα (28, 30). We therefore investigated whether this motif is bound by SATB1 by using competition assays. A 16-bp double-stranded oligonucleotide corresponding to base pairs −267 to −252 of the IL-2Rα upstream region bearing the κB-like motif effectively competed binding by SATB1 in a dose-dependent manner (Fig. 1F, lanes 8 to 10). However, SATB1 failed to bind directly to this duplex κB oligonucleotide (probe 9, Fig. 1F, lanes 12 to 14), suggesting that its binding site overlaps with the κB-like motif in the upstream region of IL-2Rα. The 3′ half of the duplex κB fragment overlaps with the first eight bases of the right half of IDR. It should be noted that SATB1 prefers to bind to DNA substrate harboring at least two binding sites that are separated by a 10- to 30-bp spacer (our unpublished observations). To further verify whether the IDR is essential for binding of SATB1 to the IL-2Rα promoter region, we performed EMSA analysis using smaller portions of the region corresponding to probe 4. The slightly shorter version of this probe (probe 10, spanning base pairs −445 to −167) was also bound by SATB1 (Fig. 1G, lanes 3 and 4). Two overlapping halves of this region (probe 11, spanning base pairs −445 to −243, and probe 12, spanning base pairs −295 to −167) were bound by SATB1 (Fig. 1G, lanes 8, 11, and 12). Interestingly, this overlapping region contained the IDR. We therefore prepared deletions in these probes wherein the IDR was eliminated (probe 13, spanning base pairs −445 to −294, and probe 14, spanning base pairs −240 to −167) and monitored SATB1 binding by using EMSA. SATB1 binding activity was abolished when IDR was eliminated from this region (Fig. 1G, lanes 15, 16, 19, and 20). Collectively, these results suggest that the IDR in the IL-2Rα promoter is a preferred target for SATB1.
FIG.1.

SATB1 binds to the human IL-2Rα promoter in vitro and in vivo. (A) Schematic representation of the IL-2Rα 1.3-kb promoter sequence spanning the base pair −1240 to +110 region. The positions of the probes used for EMSA and ChIP are as indicated. (B) Mapping of the binding site of SATB1 in the IL-2Rα promoter region. EMSA analysis was performed using bacterially expressed and purified GST-SATB1 and 32P-end-labeled probes. Probes 1 (−1240F to −937R), 2 (−957F to −725R), 3 (−746F to −473R), 4 (−493F to −167R), 5 (−187F to +110R), 6 (−957F to −473R), and 7 (−1240F to −725R) were amplified by PCR using specific primer sets. The probes were incubated with 1 μg (lanes 2, 5, 8, 11, 14, 17, and 24), 2 μg of GST-SATB1 (lanes 3, 6, 9, 12, 15, 18, and 25), 1 to 2 μg of GST alone (lanes 20 and 21), and 1 to 2 μg of GST-PARP (lanes 22 and 23). DNA-protein complexes were resolved on native polyacrylamide gels. Usage of probes was as indicated below the lanes. (C) SATB1 binds to the IL2Rα promoter in vivo. DNA purified from CEM-GFP chromatin immunoprecipitated with anti-SATB1 using seven sets of primers encompassing the entire promoter region of IL-2Rα were subjected to 30, 35, and 40 cycles of PCR amplification. Usage of probes was as indicated above the lanes. (D) SATB1 binds specifically to the base pair −493 to −167 region (probe 4) in the IL-2Rα promoter in vivo. The DNA in CEM-GFP cells was chromatin immunoprecipitated with rabbit IgG (R-IgG, top panel) and anti-SATB1 (middle panel) and was subjected to 30 cycles of PCR using a panel of primers as described above. The lower panel depicts the PCR-amplified DNA from total chromatin as a control. (E) Schematic representation of the IL-2Rα base pair −493 to −167 promoter sequence with positions of the probes used for EMSA. The relative positions of useful restriction sites are as indicated. The IDRs are underlined, and the position of the κB-like element is also indicated. (F) An SATB1 binding site encompasses the IDRs in IL-2Rα promoter. EMSA using bacterially expressed and purified SATB1 and the 32P-end-labeled 326-bp probe 4 (lanes 1 to 4) or 103-bp probe 8 generated by AluI and HinfI digestion of the same followed by polyacrylamide gel purification (base pair −328 to −226 fragment). GST-SATB1 was used at 0.5 (lane 2), 1 (lanes 3 and 6), and 2 μg (lanes 4 and 7 to 10) in the binding reactions. Incubation of the reaction mixture with 10-, 50-, and 100-fold concentrations of cold κB duplex oligonucleotide (oligo) as a competitor DNA resulted in loss of binding (lanes 8, 9, and 10, respectively). Similarly, increasing amounts of SATB1 (lane 12, 13, and 14) were incubated with the 32P-end-labeled duplex κB oligonucleotide (probe 9). (G) IDR is essential for binding of SATB1 to the IL-2Rα promoter region. EMSA analysis was performed using bacterially expressed and purified GST-SATB1 and 32P-end-labeled probes. Probe 10 (−445F to −167R), 11 (−445F to −243R), 12 (−295F to −167R), 13 (−445F to −294R), and 14 (−240F to −167R) were PCR amplified using specific primer sets. The probes were incubated with 1 μg of GST-SATB1 (lanes 3, 7, 11, 15, and 19), 2 μg of GST-SATB1 (lanes 4, 8, 12, 16, and 20), and 2 μg of GST alone (lanes 2, 6, 10, 14, and 18). DNA-protein complexes were resolved on native polyacrylamide gels and visualized by autoradiography. Usage of probes was as indicated at the top. Probes 13 and 14 are essentially the same as probes 10 and 11, respectively, except that they lack the IDR.
SATB1 directly binds to the upstream regulatory region of IL-2.
Since a common transcription factor is implicated in the inducible expression of IL-2 and its receptor (44), we further investigated whether SATB1 is involved in the regulation of IL-2 by binding directly to the upstream regulatory elements of human IL-2. We analyzed the SATB1 binding sites in the IL-2 promoter using various restriction fragments of the promoter (45). The EMSA analysis with these probes suggested the presence of a binding site in each of the two halves of the promoter (Fig. 2A). The presence of multiple SATB1 binding sites in promoters is not unique since SATB1 is known to bind directly to multiple elements within the gp91(phox) promoter in vitro (22). We next examined whether SATB1 is complexed with the IL-2 promoter in vivo by performing a ChIP assay. The cross-linked chromatin from CEM-GFP cells was immunoprecipitated with anti-SATB1 and used for PCR amplification following reversal of cross-links. We used two-primer sets specific for the distal (P1) and proximal (P2) halves of the promoter and performed ChIP analysis using anti-SATB1. Interestingly, only the P1 primer set (Fig. 2B, lane 1, upper panel) and not P2 (Fig. 2B, lane 1, lower panel) yielded a specific product at 30 cycles of amplification. This binding is very specific since, although both the distal and proximal regions in the IL-2 promoter were bound by SATB1 in vitro, only the distal promoter was bound in vivo. Although amplification product using the P2 primer set was detected at cycles numbered 35 and above, it is possible that SATB1 is actually bound to only the distal promoter in vivo. While searching for canonical binding elements within the distal promoter, we came across a 37-nucleotide contiguous stretch of As, Ts, and Cs that agrees with the definition of the ATC context (10). The ATC context containing overlapping regions F4 (base pairs −477 to −345) and F5 (base pairs −626 to −441) were amplified by PCR using specific primers and labeled using 32P-dCTP. EMSA analysis using GST-SATB1 demonstrated binding of SATB1 to both these halves of the distal promoter (Fig. 2D, lanes 3, 4, 7, and 8). The overlapping region between these two probes corresponds to the 37-mer ATC region. To confirm whether SATB1 specifically recognizes the ATC context, we disrupted the continuity of ATC by changing A to G at two sites (Fig. 2E). EMSA analysis using the end-labeled wild-type and mutated 37-mer duplexes revealed specific binding of SATB1 to the wild-type sequence (Fig. 2D, lanes 11 and 12), whereas binding to the mutated 37-mer was completely abolished (Fig. 2D, lanes 15 and 16). Thus, the ATC context spanning nucleotides −477 to −441 from the translation start site of IL-2 harbors the preferred binding site for SATB1.
FIG. 2.

SATB1 binds to the human IL-2 promoter in vitro and in vivo. (A) Schematic representation of 0.62-kb IL-2 promoter sequence spanning base pair −626 to −2 region of translation start site. The positions of the probes used for EMSA and ChIP are as indicated. (B) SATB1 binds to the IL-2 promoter in vitro. Mapping of the binding site of SATB1 in the IL-2 promoter region (IL2P) was performed by EMSA analysis as described in Materials and Methods, using 2 μg of bacterially expressed and purified GST-SATB1 and 32P-labeled probes. An equal amount of GST was used as a control in binding reactions. The probes were generated using AflIII restriction digestion (F2 and F3) or PCR amplification using specific primer pairs (full length and P1). (C) In vivo binding of SATB1 to the distal IL-2 promoter. Anti-SATB1 immunoprecipitated chromatin from CEM-GFP cells was amplified with P1 (top) and P2 (bottom) primer sets for 30 (lane 1), 35 (lane 2), and 40 (lane 3) cycles. ChIP analysis was again performed using rabbit IgG (R-IgG, lane 4, upper panel) and anti-SATB1 (lane 5, upper panel) and subjected to 30 cycles of PCR amplification with the P1 primer set. Input (lanes 4 and 5, bottom panel) denotes amplification of the DNA in soluble chromatin prior to immunoprecipitation. (D) SATB1 binds to the 37-mer ATC context present in the distal promoter (P1 region) of the IL-2 promoter. The ATC context containing overlapping regions F4 (−477 to −345) and F5 (−626 to −441) were amplified by PCR using specific primers and labeled using [32P]dCTP. EMSA analysis was performed using 1 (lanes 3 and 7) or 2 (lanes 4 and 8) μg of GST-SATB1 or 2 μg of GST alone (lanes 2 and 5). Wild-type (Wt) and mutant (Mut) forward and reverse 37-mer oligonucleotides were annealed and end labeled with [32P]ATP. Wild-type annealed double-stranded 37-mer oligonucleotide (corresponding to nucleotides −477 to −441 of the IL-2 promoter) displays perfect ATC context whereas the mutant has two A-to-G changes that disrupt the ATC context. Binding reactions were performed as described for F4 and F5. (E) Nucleotide sequences of the wild-type and mutated 37-mer ATC context spanning the region from base pair −477 to base pair -441 from the translation start site of IL-2. The two mutated nucleotides are shown in boxes.
HIV-1 infection leads to upregulation of IL-2Rα and IL-2 in T cells.
The effect of HIV infection and Tat activity on upregulation of IL-2 and its receptor occurs in the context of activated T cells in synergy with T-cell activation signals (5, 8, 34, 49). We therefore used activated PBMCs for infection. CEM-GFP cells were also used as a representative CD4+ T-cell line. PBMCs and CEM-GFP cells were infected with the NL4.3 isolate of HIV-1 (1). Since SATB1 negatively regulates the expression of the mouse IL-2Rα (53), we tested whether the same is true in regard to the expression of the human IL-2Rα. The IL-2Rα is induced in Tat/Tax transfected cell lines (28, 30, 46). We first monitored the effect of activation and viral infection on the expression of IL-2Rα by Western blot analysis. We isolated PBMCs and treated them with phytohemagglutinin for 48 h. The resulting activated population was then infected with HIV-1 isolate NL4.3. The activation was sustained by adding IL-2 to the medium. We then analyzed the IL-2Rα levels by immunoblot analysis of infected cells at day 7 and compared them with those of uninfected activated cells (day 7) and control PBMCs. We observed that the level of IL-2Rα was increased in both day 7 activated uninfected and day 7 activated infected cells (Fig. 3A, upper panel, lanes 2 and 3, respectively). Interestingly, the level of IL-2Rα protein was upregulated by at least 2.5-fold in infected PBMCs compared to that in the uninfected activated PBMCs (Fig. 3A). We also monitored the expression of IL-2Rα by immunoblot analysis of control and NL4.3-infected CEM-GFP total cell-free lysates. We observed a sixfold induction of IL-2Rα expression in NL4.3-infected CEM-GFP cells (Fig. 3A, upper panel, lane 5) compared to that in uninfected CEM-GFP cells (Fig. 3A, upper panel, lane 4). Immunoblot analysis of actin was used as a loading control (Fig. 3A, lower panel).
FIG. 3.

Induction of IL-2Rα and IL-2 expression upon HIV infection. (A) Immunoblot analysis of total cell lysates from PBMCs (lane 1), activated PBMCs (lane 2), activated and HIV-1-infected PBMCs (lane 3), uninfected CEM-GFP cells (lane 4), and NL4.3-infected CEM-GFP cells (lane 5) using anti-IL-2Rα (upper panel) and anti-actin (lower panel). (B) RT-PCR analysis of PBMCs (lane 1), activated PBMCs (lane 2), activated and HIV-1-infected PBMCs (lane 3), uninfected CEM-GFP cells (lane 4), and NL4.3-infected CEM-GFP cells (lane 5) using primers specific for IL-2 (upper panel) and human GAPDH (hGAPDH, lower panel).
Since IL-2 is the principal cytokine in T cells that is regulated almost exclusively at the level of transcription and is not expressed in resting T cells (38), we monitored its expression in PBMCs and CEM-GFP cells infected with NL4.3. Reverse transcription-PCR (RT-PCR) analysis of mRNA isolated from control (Fig. 3B, upper panel, lane 1), activated (Fig. 3B, upper panel, lane 2), and activated infected (Fig. 3B, upper panel, lane 3) PBMCs indicated that IL-2 is upregulated in activated as well as infected PBMCs. RT-PCR analysis of mRNA isolated from control (Fig. 3B, upper panel, lane 4) and infected (Fig. 3B, upper panel, lane 5) CEM-GFP cells indicated that IL-2 is upregulated at least 10-fold upon HIV-1 infection, based on densitometric comparison of band intensities. The expression of IL-2 mRNA was normalized with respect to that of human GAPDH (Fig. 3B, lower panels). Thus, in uninfected cells IL-2 and IL-2Rα seem to be repressed. Since we observed that the upstream regulatory regions of both are directly bound by SATB1 in vitro and in vivo, these results collectively implicate SATB1 in the negative regulation of IL-2 and IL-2Rα in PBMCs and the CEM-GFP human T-cell line.
SATB1 interacts with the HDAC1 corepressor via its PDZ-like domain in vitro and in vivo.
To test whether the negative regulation by human SATB1 is manifested via recruitment of the HDAC1 corepressor, we explored the possibility of their direct interaction. We first performed coimmunoprecipitation analysis using Jurkat cell extracts and with antibodies against HDAC1, SATB1, and PARP. Analysis of the immunoprecipitate revealed that SATB1 was immunoprecipitated by anti-HDAC1 (Fig. 4A, lane 2) but not anti-PARP (Fig. 4A, lane 4). HDAC1 forms a complex with SATB1 in human T cells in vivo (Fig. 4A, upper and lower panels, lane 2), which is consistent with previous studies showing complex formation between mouse SATB1 and HDAC1 in vitro (53). We further delineated the region of SATB1 that interacts with HDAC1. The recombinant PDZ-like domain or MD+HD domain constituting the DNA-binding domain of SATB1 were separately incubated with cell extract from HEK 293 cells transfected with the construct expressing FLAG-HDAC1. Coimmunoprecipitation analysis using anti-FLAG antibody showed that HDAC1 is associated with the PDZ domain (Fig. 4B, lane 1) and full-length SATB1 (Fig. 4B, lane 2) but not with its DNA-binding domain (Fig. 4B, lane 3). Thus, human SATB1 may also act as a transcriptional repressor by recruiting HDAC1 corepressor to its specific genomic targets, including the IL-2 and IL-2Rα promoters. From these observations, it is apparent that for HIV-1 Tat to be able to upregulate the expression of genes such as IL-2 and IL-2Rα, the repression mediated by SATB1 should be alleviated.
FIG. 4.
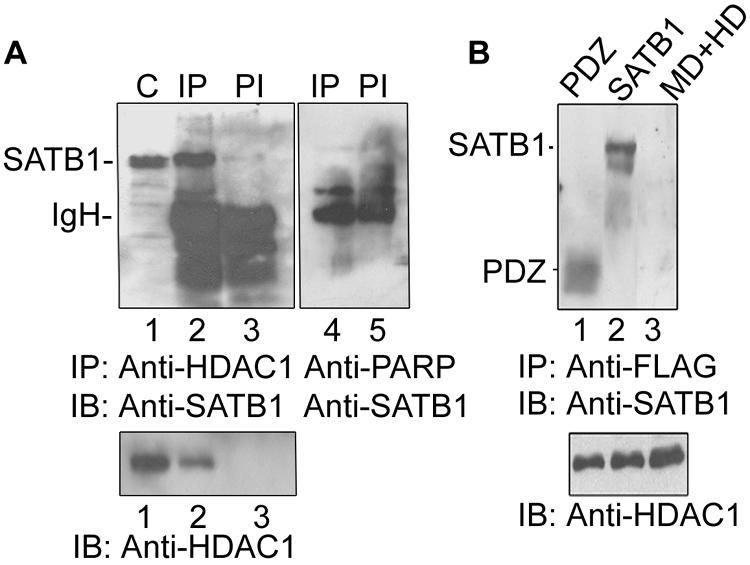
SATB1 directly interacts with the HDAC1 corepressor via its PDZ domain in vitro and in vivo. (A) In vivo coimmunoprecipitation analysis. SATB1 was immunoprecipitated from Jurkat cell extract using anti-HDAC1 antibody and detected by immunoblot analysis using anti-SATB1 (lane 2). C, control extract alone (lane 1); PI, coimmunoprecipitation using preimmune rabbit serum (lane 3). The lower panel represents an immunoblot (IB) of the same samples with anti-HDAC1, showing that HDAC1 is immunoprecipitated specifically by anti-SATB1 (lane 2) and not by preimmune serum (lane 3). Immunoprecipitation using anti-PARP (lane 4) serves as a negative control. (B) Interacting domain of SATB1 with FLAG-tagged HDAC1 was analyzed by a FLAG pulldown assay followed by immunoblotting using anti-SATB1 as described in Materials and Methods. Anti-FLAG pulled down the PDZ domain (lane 1) and full-length SATB1 (lane 2) but not the MD+HD domain (lane 3). The positions of molecular weight standards are indicated on the right. The lower panel represents an immunoblot with anti-HDAC1 as a loading control.
HIV-1 Tat associates with the PDZ-like domain of SATB1 in vitro and in vivo.
To investigate how Tat might overcome negative regulation of IL-2Rα by SATB1, we tested whether SATB1 specifically associates with HIV-1 Tat. Cell-free lysate from Jurkat (Fig. 5A, lanes 1 to 4 and 6 to 9) or HEK 293 (Fig. 5A, lanes 11 to 14) cells was passed over GST-Tat bound Sepharose matrix. The bound protein was eluted in a stepwise manner by using buffers containing 1 M NaCl or reduced glutathione. The eluted protein was analyzed by SDS-PAGE followed by immunoblotting using anti-SATB1 (Fig. 5A, lanes 1 to 5 and lanes 11 to 14, upper panel) or anti-GST (Fig. 5A, lanes 6 to 10) antibody. Their coelution (Fig. 5A, lanes 3 and 13, upper panel) suggested association between SATB1 with Tat. In HEK 293 cell-free lysates probed with anti-SATB1, we often observed two closely migrating bands (Fig. 5A, lane 11, upper panel). Control experiments using GST-bound glutathione Sepharose beads did not yield anti-SATB1 cross-reactive species (Fig. 5A, lane 5). Only GST was eluted from this column after elution with buffer containing glutathione (Fig. 5A, lane 10). We then tested the possibility of physical interaction between these two proteins by incubating in vitro-translated Tat with Jurkat cell-free lysate. By coimmunoprecipitation using polyclonal antibodies against each protein, we observed that SATB1 and Tat formed a complex in vitro (Fig. 5B, lanes 2 and 4). The interaction of these two proteins was further confirmed by coimmunoprecipitation using cell-free lysates from NL4.3-infected CEM-GFP cells. Anti-Tat antibody immunoprecipitated SATB1 in these lysates, indicating that SATB1 directly interacts with Tat in vivo (Fig. 5C, lane 2). Their in vivo functional interaction was further demonstrated by yeast two-hybrid assays. In a manner that is identical to coimmunoprecipitation of these with HDAC1 (Fig. 4B), we found that both the full-length SATB1 (Fig. 5D, panels 2 and 8) and the PDZ domain of SATB1 (Fig. 5D, panels 3 and 9) interacted with Tat. The MD and HD domains failed to interact with Tat (Fig. 5D, panels 4 and 10). The strength of this interaction was quantified by a β-Gal assay. The interaction of full-length SATB1 with Tat yielded a value of 41.2 U while that of PDZ domain and Tat yielded 26.7 U in contrast to the undetectable β-Gal activity of MD and HD, and Tat cotransformed yeast cells. Thus, these results suggest that functional interaction between HIV-1 Tat and SATB1 occurs via the PDZ-like domain. We then delineated the interaction domain of Tat with SATB1 by GST pulldown assay using truncations of Tat fused with GST. SATB1 was pulled down by Tat 20-72 (Fig. 5E, lane 1) and 1-45 (Fig. 5E, lane 2) but not by Tat 30-71 (Fig. 5E, lane 3) or Tat 40-72 (Fig. 5E, lane 4). Results of this experiment indicated that a region of 20 to 40 amino acids representing a part of transactivation domain of Tat is responsible for interaction with SATB1. Additionally, the transactivation-deficient mutant C22G is capable of binding to SATB1 (Fig. 5E, lane 5). Next we tested whether SATB1 and Tat associate with each other in vivo by indirect immunofluorescence using antibodies against each protein. When Tat was expressed in HEK 293 cells by transient transfection it was found to colocalize with endogenous SATB1 by indirect immunofluorescence analysis. Interestingly, when SATB1 was expressed exogenously along with Tat in the human breast cancer cell line SK-BR-3, which does not express SATB1 endogenously, a similar nuclear colocalization pattern was observed. In both cell types, the colocalization was evident in more than 90% of Tat-expressing cells (data not shown).
FIG. 5.

HIV-1 Tat physically interacts with SATB1 in vitro and in vivo. (A) SATB1 is eluted by GST-Tat affinity chromatography. Nuclear extracts from Jurkat and 293 cells were passed separately on GST-Tat and GST affinity columns. Bound proteins were eluted with phosphate buffer containing 1 M NaCl (lanes 3 and 13) or with reduced glutathione (lanes 9 and 14). Proteins from GST-bound Sepharose were eluted with reduced glutathione only (lanes 5 and 10). Column input (lanes 1, 6, and 11), flowthrough (lanes 2, 7, and 12), and wash using phosphate-buffered saline (lanes 4 and 8). (B) In vitro-translated 35S-labeled Tat and cold SATB1 were mixed and immunoprecipitated with normal rabbit serum (lane 1 and 3), anti-SATB1 (lane 2), and anti-Tat (lane 4). The immunoprecipitated proteins were detected by autoradiography (left panel) or immunoblot analysis using anti-SATB1 (right panel). NRS, normal rabbit serum; IP, immunoprecipitation, IB, immunoblot. (C) SATB1 immunoprecipitated using anti-Tat antibody in extract from NL4.3-infected CEM-GFP cells (lane 2) was detected by immunoblot analysis using anti-SATB1. Lane 3 (C, control) shows proteins in cell extract used for coimmunoprecipitation. The lower panel represents an immunoblot with anti-Tat. (D) Yeast two-hybrid analysis. Various constructs used for yeast two-hybrid analyses are schematically represented above the two plates depicting growth of cotransformants. Panels 1 and 2 and 7 and 8 represent the cotransformation of Gal4-AD:SATB1 and Gal4-AD:PDZ with Gal4-DBD:Tat, respectively. Panels 3 and 9 represent p53 and T antigen as positive controls, 4 and 10 represent p53 and lamin C as negative controls, 5 and 11 represent the Gal4-AD:MD+HD and Gal4-DBD:Tat cotransformation, and 6 and 12 represent the mock-transformed cells as controls. (E) Delineation of the interaction domain of Tat with SATB1. Nuclear extracts from Jurkat cells were passed separately on GST-fused Tat 20-72, 1-45, 30-72, and 40-72 truncations and GST-Tat C22G mutant affinity columns. Bound proteins were eluted from the GST-Tat affinity column with phosphate buffer containing 1 M NaCl (lanes 1 to 5, top panel) or with reduced glutathione (lanes 1 to 5, lower panel). Salt-eluted proteins were subjected to Western blotting using anti-SATB1 (upper panel), whereas proteins eluted with reduced glutathione were subjected to immunoblot analysis using anti-GST (lower panel).
HIV-1 Tat competitively displaces SATB1-bound HDAC1 in vitro.
To evaluate the functional significance of the interaction of Tat with SATB1, we asked whether it would bind to SATB1 that had been complexed with HDAC1. We formed a complex between SATB1 and FLAG-HDAC1 in vitro, captured it with anti-FLAG affinity beads, and then incubated it with increasing amounts of purified GST-Tat. The protein bound to HDAC1 was collected using the FLAG beads and subjected to immunoblot analysis using anti-SATB1 (Fig. 6A, upper panel) or anti-HDAC1 (Fig. 6A, lower panel). Interestingly, GST-Tat displaced the HDAC1-bound SATB1 in a dose-dependent manner (Fig. 6A, lanes 2 and 3, upper panel), while GST alone at equivalent or higher concentrations did not (Fig. 6A, lanes 5 and 6, upper panel). To test whether the displacement of HDAC1 is really due to interaction of Tat with SATB1, we performed a displacement assay using the C22G mutant Tat, a point mutant of Tat that is transactivation defective (reviewed in reference 24), and truncations such as 20-72, 30-72, and 40-72. Strikingly, only the 20-72 truncated version of Tat, not the others, was able to displace HDAC1 from SATB1 (Fig. 6B, top panel, lane 4 versus lanes 8 and 10). This result is in perfect agreement with our protein interaction domain studies (Fig. 5E) suggesting that the displacement of HDAC1 is an outcome of SATB1-Tat interaction. Furthermore, the C22G mutant Tat displaces HDAC1, albeit with much lower efficiency than wild-type Tat (Fig. 6B, compare lanes 6 and 2). Despite the use of double the amount (8 μg) of C22G Tat compared to others, complete displacement is not observed, suggesting a weakened interaction.
FIG. 6.

Tat displaces SATB1-bound HDAC1 in vitro. (A) A competition assay was performed to analyze the binding of HDAC1 and Tat to SATB1 in vitro. The SATB1-HDAC1 complex was isolated using anti-FLAG affinity beads in the FLAG pulldown assay as described in Materials and Methods (lane 1). The complex was incubated with 2 and 4 μg of recombinant GST-Tat and pulled down using FLAG beads (lanes 2 and 3, respectively). Lane 4 represents the input lysate. As a control, the complex was incubated with 2 and 10 μg of purified GST and pulled down with FLAG beads (lanes 5 and 6). The lower panel represents an immunoblot analysis of these samples using anti-HDAC1. (B) A competition assay was performed to analyze the HDAC1-displacing ability of mutant and truncated Tat from the HDAC1-SATB1 complex in vitro. 293 cells were transiently transfected with FLAG-HDAC1, and the SATB1-HDAC1 complex was isolated using anti-FLAG affinity beads in the FLAG pulldown assay. The complex was incubated with 2 and 4 μg of recombinant wild-type GST-Tat (lanes 1 and 2), GST-Tat 20-72 (lanes 3 and 4), GST-Tat 30-72 (lanes 7 and 8), and GST-Tat 40-72 (lanes 9 and 10, respectively) and pulled down using FLAG beads. In the case of pulldown using the GST-Tat C22G mutant, 2 and 8 μg of protein were used (lanes 5 and 6, respectively). The middle panel represents the purified GST-Tat proteins stained with Coomassie brilliant blue. The lower panel represents an immunoblot analysis of these samples using anti-HDAC1 and serves as loading control.
Tat displaces SATB1-bound HDAC1 in HIV-1-infected T cells, resulting in increased acetylation of the promoters.
To ascertain whether the displacement occurs in vivo, we performed a coimmunoprecipitation assay with cell-free lysate from NL4.3-infected CEM-GFP cells. SATB1 was associated with HDAC1 in control uninfected CEM-GFP cells (Fig. 7A, lane 2). However, in NL4.3-infected cells, SATB1 was not immunoprecipitated by anti-HDAC1, indicating that HIV-1 Tat disrupts the SATB1-HDAC1 complex in vivo (Fig. 7A, lane 4). We then performed a ChIP assay to analyze whether SATB1 recruits HDAC1 to the IL-2 and IL-2Rα upstream regions in vivo. SATB1 was bound to these promoters in both uninfected (Fig. 7B and C, top panels, lanes 1) and NL4.3-infected (Fig. 7B and C, top panels, lanes 2) CEM-GFP cells. HDAC1 was recruited in vivo to the promoters in uninfected cells (Fig. 7B and C, middle panels, lanes 1). In contrast, HDAC1 was not recruited to the promoters in NL4.3-infected cells (Fig. 7B and C, middle panels, lanes 2). ChIP analysis using anti-Tat immunoprecipitated chromatin revealed that Tat is bound to the promoter only in infected cells (Fig. 7E and F, top panels, lanes 2) but not in uninfected cells (Fig. 7E and F, top panels, lanes 1), consistent with its expected expression pattern. We then monitored the acetylation status of IL-2 and IL-2Rα promoters by ChIP analysis using antibody against acetylated lysine 9 of histone H3 (H3-K9). We observed that the promoter chromatin was specifically acetylated in vivo in the infected cells (Fig. 7E and F, middle panels, lanes 2). In contrast, a minimal level of acetylation was observed in uninfected cells (Fig. 7E and F, middle panels, lanes 1). Interestingly, the proximal promoter region of IL-2 spanning base pairs −315 to + 1 was not occupied by SATB1 in both uninfected and infected CEM-GFP cells (Fig. 7D, top panel) while the distal IL-2 promoter spanning base pairs −575 to −292 is bound by SATB1 (Fig. 7D, middle panel). Strikingly, the same pattern of HDAC1 displacement at the IL-2 (Fig. 7H) and IL-2Rα (Fig. 7J) promoters was observed in NL4.3-infected primary T cells. Furthermore, ChIP analysis also revealed elevated levels of acetylation of the promoters (Fig. 7I and K, top panels) in infected PBMCs. To validate our findings further, we performed quantification of immunoprecipitated chromatin by real-time PCR analysis. Infection did not result in any significant change in SATB1 occupancy at both promoters (Fig. 7L and M, lanes 1 and 2). However, HDAC1 occupancy was decreased at least fourfold as evidenced by an increase in CT values by a factor of about 2 (Fig. 7L and M, lanes 3 and 4). A decrease in CT value of 1 U corresponds to a twofold increase in template copy number. As expected, Tat occupancy increased upon infection (Fig. 7L and M, lanes 6) and was undetectable in uninfected samples (Fig. 7L and M, lanes 5). Interestingly, when we used anti-histone H3 acetyl lysine 9 antibody to check the level of promoter acetylation, we observed a fourfold increase in the acetylation of IL-2Rα promoter upon infection (Fig. 7M, lanes 7 and 8), and about a sixfold increase in the acetylation of IL-2 promoter upon infection (Fig. 7L, lanes 7 and 8) as evidenced by the decrease in CT value. Collectively, these results indicated that Tat-mediated displacement of HDAC1 in the infected cells may be responsible for the observed derepression of IL-2 and IL-2Rα.
FIG. 7.

Tat displaces SATB1-bound HDAC1 in HIV-1 infected T cells, leading to increased acetylation of promoters. (A) Coimmunoprecipitation (Co-IP) analysis using extracts from uninfected (lanes 1 and 2) and NL4.3-infected (lanes 3 and 4) CEM-GFP cells. Immunoprecipitation was carried out using mouse IgG1 (lanes 1 and 3) and anti-HDAC1 antibody (lanes 2 and 4). As a control, a mixture of normal rabbit IgG and mouse monoclonal IgG1 was used (lane 3). IB, immunoblotting. (B and E) ChIP analysis of IL-2 promoter. PCR amplification of DNA isolated from immunoprecipitated chromatin was performed as described above by using the IL-2-P1 primer pair. The antibodies used for immunoprecipitation are as indicated. UI, uninfected; I, infected; C, control. (C and F) ChIP analysis of the IL-2Rα promoter. DNA isolated from immunoprecipitated chromatin from the uninfected and infected CEM cells using anti-SATB1, anti-HDAC1, anti-Tat, and anti-acetylated H3-K9 antibodies were PCR amplified. Input represents PCR amplification of the DNA in total cross-linked chromatin. (D) ChIP analysis of the IL-2 promoter was performed using anti-SATB1 immunoprecipitated chromatin and IL-2-P1 (middle panel) and IL-2-P2 (upper panel) primer pairs. (G) As a control, normal rabbit IgG (middle panel) and mouse monoclonal IgG1 (upper panel) were used for immunoprecipitation of cross-linked chromatin, and DNA in the immunoprecipitate was amplified using the IL-2-P1 primer pair. ChIP analysis of chromatin from uninfected and HIV-1-infected PBMCs was also performed using primers specific for IL-2 (H and I) and IL-2Rα (J and K) promoters. The antibodies used for immunoprecipitation were as indicated. Quantification of immunoprecipitated chromatin was performed by real-time PCR analysis. The changes in amount of immunoprecipitated chromatin (Δ Ct) were calculated as described in Materials and Methods. Values from three independent ChIP experiments were plotted for the IL-2 (L) and IL-2R (M) promoters. The antibodies used for ChIP are anti-SATB1 (1 and 2), anti-HDAC1 (3 and 4), anti-Tat (5 and 6), and anti-histone H3 acetyl lysine 9 (7 and 8). Solid and empty bars indicate infected and uninfected samples, respectively.
Soluble Tat upregulates IL-2 and IL-2R and increases the acetylation status of IL-2 and IL-2Rα promoters in Jurkat T cells.
To address the question of whether Tat alone could induce the expression of IL-2 and its receptor, and also to rule out any other effects of exposure to the virions, we performed a transduction experiment. The uptake, internalization, and nuclear translocation of Tat can be used as a tool for protein delivery (39, 42). We prepared recombinant wild-type Tat and its truncations as well as mutations as described above (Fig. 8A) and incubated Jurkat cells in media containing these. Immunoblot analysis using nuclear extracts of Jurkat cells proved that the proteins had actually entered into the nuclei of treated cells (Fig. 8B). Flow cytometric analysis of transduced cells stained using anti-Tat antibody revealed that about 85% of the cells were positive for Tat (Fig. 8C). Incubation of cells with Tat resulted in upregulation of IL-2 and IL-2R as evidenced by an increase in mRNA levels shown by RT-PCR analysis (Fig. 8D, lane 3), and RNA from cells incubated with GST alone served as a control for this RT-PCR experiment (Fig. 8D, lane 1). However, when cells were incubated with the 40-72 truncation of Tat that is deficient in interaction with SATB1 (Fig. 5E), such an increase in the levels of IL-2 or IL-2R transcript levels was not observed (Fig. 8D, lane 2). Interestingly, under these conditions we found a reciprocal relationship between the HDAC1 occupancy and acetylation level of H3-K9 (Fig. 8E). We observed an increase in the level of acetylation of H3-K9 specifically in the presence of Tat (Fig. 8E, lanes 2 and 8) and its SATB1-interacting derivatives (lanes 6 and 12), including the C22G Tat mutant (lanes 5 and 11). Interestingly, Tat protein deficient in interaction with SATB1 (Tat 40-72) is unable to displace HDAC1 and induce acetylation at the promoters (Fig. 8E, lanes 4 and 10), whereas transactivation-defective Tat C22G is capable of displacing HDAC1 and inducing promoter acetylation, albeit to a lesser extent than wild-type Tat. These results indicate that Tat alone is responsible for induction of IL-2 and its receptor and that the interaction of Tat with SATB1 is necessary for this upregulation.
FIG. 8.

Transduction of soluble Tat upregulates IL-2 and IL-2R and increases the acetylation status of IL-2 and IL-2Rα promoters in Jurkat T cells. (A) SDS-15% PAGE analysis of purified recombinant Tat proteins. Wild-type Tat (lane 5), Tat 1-48 (lane 3), Tat 40-72 (lane 4), and C22G mutant Tat (lane 7) were expressed as GST fusions in BL21 cells and purified as described in Materials and Methods. Tat (lane 6) and GST (lane 2) were obtained by thrombin cleavage. B. Recombinant GST-Tat (lane 1) and Tat (lane 2) proteins were transduced separately into Jurkat cells, and nuclear lysates were prepared as described in Materials and Methods. The nuclear lysates were resolved by electrophoresis in 15% SDS-polyacrylamide gels followed by Western blot analysis using anti-Tat antibody. (C) Flow cytometric (FACS) analysis of transduced cells. Cells incubated with 100 ng of recombinant Tat for 4 h were immunostained using rabbit IgG (upper histogram) or anti-Tat (lower histogram) and acquired on a flow cytometer as described in Materials and Methods. (D) RT-PCR analysis of mRNA from transduced cells. Total mRNA was isolated from Jurkat cells incubated with GST (lane 1), GST-Tat 40-72 (lane 2), and GAT-Tat (lane 3). RT-PCR analysis was performed using IL-2 (upper panel), IL-2R (middle panel), and GAPDH (lower panel) cDNA primers. (E) HDAC1 occupancy and promoter acetylation are inversely correlated. To monitor the acetylation status of IL-2 and IL-2R promoters independent of HIV-1 virion exposure, we transduced the wild-type Tat (lanes 2 and 8), GST (lanes 3 and 9), Tat-40-72 (lanes 4 and 10), Tat-C22G (lanes 5 and 11), and Tat-1-48 (lanes 6 and 12) proteins into Jurkat cells as described in Materials and Methods. Chromatin was then prepared and subjected to ChIP analysis using anti-SATB1, anti-HDAC1, and anti-H3 acetylated lysine 9 antibodies. As negative and positive controls we used rabbit IgG (R-IgG, lanes 1 and 7) and input chromatin (bottom panels), respectively.
Tat-mediated displacement of HDAC1 causes derepression of IL-2 and IL-2Rα in vivo.
For direct assessment of the regulatory activity of SATB1 and Tat at the IL-2 and IL-2Rα promoters, we performed luciferase reporter assays. The 632-bp promoter region of IL-2 (Fig. 9A) and the 555-bp promoter region of IL-2Rα (Fig. 9B) were used in conjunction with the promoterless luciferase reporter vector pGL3basic. We transiently expressed SATB1 in 293 cells and cotransfected them with constructs expressing HDAC1 and Tat. SATB1 expression (Fig. 9A and B, lanes 2) downregulated these promoters individually and synergistically with HDAC1 (Fig. 9A and B, lanes 5). However, when cells were cotransfected with Tat, the repression mediated by SATB1 was alleviated (Fig. 9A and B, lanes 7) and was comparable to that of Tat alone (Fig. 9A and B, lanes 4). This may be attributed to the displacement of endogenous HDAC1 that is complexed with exogenously introduced SATB1. HDAC1 alone was not able to repress the transactivation by Tat to a significant extent (Fig. 9A and B, lanes 6). To unequivocally demonstrate the role of Tat in displacement of HDAC1 and thereby the derepression of the IL-2 and IL-2Rα, we used the transactivation-deficient mutant Tat C22G. We performed reporter assays using wild-type Tat and the above-mentioned mutants of Tat. We found that although C22G Tat is defective in transactivation, it is able to derepress both IL-2 and IL-2Rα in a dose-dependent manner (Fig. 9A and B, lanes 5 and 6). In fact, with 2 μg of C22G Tat, the derepression occurs such that the activity is almost back to the level of SATB1 alone (Fig. 9A and B, lanes 6). This dose-dependent effect further supports our finding that Tat and HDAC1 compete for binding to SATB1 in vivo. Wild-type Tat derepresses IL-2 and IL-2Rα by about fourfold (Fig. 9A and B, lanes 7). Collectively, these results suggest that the T-lineage-specific protein SATB1 is the common factor involved in the regulated expression of IL-2 and IL-2Rα and that Tat causes derepression and induction of IL-2 and IL-2Rα in the presence of SATB1.
FIG. 9.

Tat-mediated derepression of IL-2 and IL-2Rα transcription in 293 cells exogenously expressing SATB1, HDAC1, and Tat. The effect of Tat mutants in the derepression of IL-2 (A) and IL-2Rα (B) is shown. A luciferase (Luc) reporter assay was performed upon cotransfections of 1 to 2 μg of various expression constructs along with IL-2-luciferase (A) or IL-2Rα-luciferase (B) reporter constructs. Cells were harvested 40 h after transfection, and luciferase activity was measured using 50 μg of protein from each cell lysate. Luciferase activity is expressed as increase or decrease compared to the control (lane 1), which was set to baseline. Lane 1, reporter construct alone; lane 2, SATB1; lane 3, SATB1 + HDAC1; lane 4, Tat C22G; lane 5, SATB1 + HDAC1 + C22G (1 μg); lane 6, SATB1 + HDAC1 + C22G (2 μg); lane 7, wild-type Tat (WT-Tat); lane 8, SATB1 + HDAC1 + Tat. Data from triplicates are plotted, and relative luciferase units are represented as increase or decrease in activity with respect to the reporter alone. The statistical significance of differences between the treatment groups was calculated using one-way analysis of variance, and the observed P values were always less than 0.001. (C) Model depicting HDAC1 displacement following Tat transfection. In the absence of Tat, SATB1 represses transcription of the reporter by recruiting the HDAC1 corepressor. When Tat is introduced, it binds to the PDZ-like domain of SATB1 and displaces HDAC1, thereby stimulating transcription. For details, see the text.
DISCUSSION
HIV-I- and HTLV-1-infected CD4+ T cells are activated and proliferate during the initial phase of infection that is accompanied by dysregulation of multiple genes (7, 18). The retroviral infection is limited by the quiescence of most circulating T cells that are therefore nonsupportive of viral replication. Formation of integrated provirus is obligatory for viral replication and subsequent development of AIDS (13, 31, 47, 51, 54). The barrier for integration observed in host peripheral T cells can be overcome by T-cell activation (47). Recent studies have shown that expression of the earliest HIV gene products, Nef and Tat, can increase T-cell activity (34, 41, 50). Nef acts by lowering the activation threshold in T cells (41), while Tat acts by deregulating the expression of several host genes, including proto-oncogenes and genes encoding metabolic enzymes, cytokines, and cytokine receptors (5, 7, 12, 18, 40). Recent studies by Wu and Marsh solved the puzzle of how these proteins would function in the absence of viral replication. They reported that nef and tat genes are selectively transcribed in infected cells before integration (52). Thus, the early events of HIV infection induce the IL-2 and IL-2R protein levels that are required for rapid proliferation of active T-cells which subsequently become the most preferable target cells for HIV infection. In the later episodes of HIV infection, infected T cells undergo apoptosis, and this may be responsible for the sudden decrease in T-cell numbers and the resulting drop in immunity of the infected patient. This is one of the host-pathogen interactions in which virus initially takes advantage of the host machineries for its efficient infection and rapid proliferation of host T cells. However, the molecular mechanisms of how this is accomplished, and especially the contribution of host factors, are not completely understood.
Tat activates host and viral transcription by recruiting host transcription factors and coactivators (7). Contradictory observations describe the regulation of IL-2 and its receptor by Tat (19, 46, 36, 37). However, in studies where the expression of IL-2 and its receptor have been shown to be downregulated by Tat, stable expression of Tat was employed (36, 37). Lymphocytes from HIV-1 infected patients are known to express increased amounts of IL-2 and IL-2Rα (18, 43). We therefore argued that the molecular mechanism of this phenomenon could not be elucidated using only Tat-transfected T cells. Here, we have used HIV-1 infected T cells and evaluated the role of SATB1, a T-lineage-specific transcription factor (2), in the regulation of IL-2 and IL-2Rα expression. Studies utilizing SATB1 null mice have established SATB1 as a new type of gene regulator responsible for a novel cagelike nuclear architecture, providing sites for tissue-specific organization of DNA sequences and regulating region-specific histone modification (6). SATB1 is a predominant architectural protein in T cells (2) that functions as a global suppressor by targeting chromatin remodeling to regulate genes over long distances (53). We therefore reasoned that Tat would have to overcome the transcriptional suppression mediated by SATB1 in a manner that would allow activation of genes required for viral replication and T-cell activation.
The activation dependence and cell type specificity of IL-2 expression are mediated through a dynamic assembly of diverse transcription factors, including NF-AT, NF-κB, AP-1, and Oct-1 at its promoter (38). Likewise, several studies have demonstrated the presence of multiple protein binding sites within the IL-2Rα upstream regulatory sequences in the region of base pairs −476 to −225 (28, 30). We have identified, for the first time, SATB1 binding sites in the promoters of IL-2 and its receptor. Furthermore, we narrowed down these binding sites to the IDR in the IL-2Rα promoter and a 37-mer ATC sequence in the IL-2 promoter. SATB1 may also bind to additional regions within these promoters; however, the presence of these two elements is essential since deletions of these elements abolish binding to longer overlapping regions. Studies using chromatin from thymocytes of wild-type and SATB1 null mice demonstrated the presence of a strong intronic SATB1 binding site within the IL-2Rα locus and a weak binding site within the promoter (53). In the present study, we have narrowed down the binding site for SATB1 in the human IL-2Rα promoter and demonstrate that it consists of the IDRs flanking the κB-like motif. Whether a similar binding site preference is exhibited by SATB1 in the mouse IL-2Rα promoter remains to be investigated. In most of the earlier studies, contribution of specific transcription factors has been evaluated by using their minimal binding sites in the upstream regulatory regions of IL-2 and IL-2Rα (9, 19, 28, 30, 38, 46). We used regions of more than 500 bp to analyze the role of SATB1, ensuring that the promoters may be simultaneously occupied by other transcription factors whose contribution was considered as the baseline. Our data demonstrate that SATB1 binds to the upstream regulatory elements of IL-2 and IL-2Rα and negatively regulates their expression. We monitored IL-2 levels in infected PBMCs by RT-PCR analyses and observed that IL-2 expression peaks around day 4 postinfection and then gradually decreases with time (our unpublished observation). However, monitoring the expression of IL-2 in PBMCs infected in the presence of IL-2 is more complicated due to the fact that exogenously added IL-2 exerts diverse effects on virus production (3) and the impaired IL-2 production during HIV disease may be attributed to a feedback mechanism wherein IL-2 induces CD8+ HIV suppressors (26). However, Tat was previously shown to derepress the IL-2 promoter (12), and the derepression is very clearly observed in the case of infected CEM cells.
SATB1 contains an MD and an HD towards the carboxy-terminal half, which confer the ability to recognize the core-unwinding element within a BUR element (10, 33). SATB1 harbors a novel dimerization domain in its N-terminal region, which is homologous to PDZ domains (16). Recently SATB2, a homolog of SATB1, was identified; it also possesses the PDZ-like domain (our unpublished observations). However, SATB2 is specifically expressed in brain, testis, kidney, pre-B cells, and B cells but not in T cells (11); therefore, it may not play a role in HIV pathogenesis at least during the early stages of infection. PDZ domains are protein interaction modules that are implicated in signal transduction (14). Both HDAC1 and Tat bind SATB1 via this protein interaction domain. SATB1 interacts with the transactivation domain of Tat, a domain that is known to associate with many of Tat's interaction partners (reviewed in reference 24). Thus, the competition within these factors would eventually determine the outcome of such interactions. Through direct physical interaction, Tat competitively displaces SATB1-bound HDAC1 in vivo. Strikingly, the transcriptional upregulation of IL-2 and IL-2Rα does not require the transactivation function of Tat. In the case of Tat-mediated regulation of LTR, the transcriptional activation is associated with remodeling of nucleosome 1 by acetylation. ChIP studies by He and Margolis suggested that histone acetyltransferases or other factors recruited to LTR by Tat may interact with HDAC1, YY1, or both and may regulate their functions or interaction (23). Furthermore, treatment with trichostatin A or Tat transfection decreased HDAC1 occupancy at the LTR, presumably via displacement. However, further study is required to define the mechanisms by which trichostatin A and Tat decrease HDAC1 occupancy at the LTR.
Histone acetylation is typically associated with transcriptional activation (reviewed in reference 4). Consequently, we reasoned that the loss of HDAC1 from the IL-2 and IL-2Rα promoters might likely lead to an increase in acetylation at the regulatory regions. We observed that the promoter chromatin was specifically acetylated in vivo in the infected cells, suggesting remodeling of the promoter to favor transcription. In the absence of Tat, SATB1 represses transcription of IL-2 and its receptor by recruiting the HDAC1 corepressor, which could be in the form of a larger repressor complex, and thereby promotes deacetylation of the promoter and represses transcription. When Tat is introduced either via HIV-1 infection or by transfection or transduction, it binds to the PDZ-like domain of SATB1 and competitively displaces HDAC1. Tat may further recruit the CBP/p300 histone acetyltransferase to promote acetylation of the promoter, thereby stimulating transcription (model depicted in Fig. 9C). Interestingly, transduction experiments using soluble Tat and its derivatives clearly demonstrated an inverse correlation between HDAC1 occupancy and acetylation of promoter chromatin and resulted in induction of IL-2 and its receptor, corroborating our hypothesis that competitive displacement of SATB1-bound HDAC1 by Tat is the cause for upregulation of IL-2 and its receptor. Our results further suggest that the induction of transcription may occur by abolition of the repressor function in addition to the induction of host factors upon viral infection. We propose that this mechanism may be employed by HIV-1 to overcome the SATB1-mediated repression of genes essential for its propagation in T cells.
Acknowledgments
We are grateful to K. Muniyappa for critical reading of the manuscript, G. C. Mishra for support and encouragement, T. Kohwi-Shigematsu for the gift of SATB1 cDNA and antibody, K. N. Ganesh for oligonucleotide synthesis, S. Schreiber for the FLAG-HDAC1 construct, Kuan-Teh Jeang for the GST-20-72, 1-45, 30-72, and 40-72 Tat constructs, and K. McGuire for the IL-2-Luc reporter construct. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: the molecular clone NL4.3, anti-Tat antibody from B. Cullen, CEM-GFP reporter cell line from J. Corbeil, and GST-fused Tat C22G mutant from Andrew Rice. We thank N. Sonawane for technical assistance. We thank the reviewers for their valuable comments.
P. P. Kumar and P. K. Purbey are supported by fellowships from the Council of Scientific and Industrial Research, New Delhi, India. D. S. Ravi is supported by a fellowship from the University Grants Commission, New Delhi, India. This work is partly supported by grants from the Department of Biotechnology, New Delhi, India, to S.G. S. Galande is an International Senior Research Fellow of the Wellcome Trust.
REFERENCES
- 1.Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and M. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez, J. D., D. H. Yasui, H. Niida, T. Joh, D. Y. Loh, and T. Kohwi-Shigematsu. 2000. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 14:521-535. [PMC free article] [PubMed] [Google Scholar]
- 3.Bahr, G. M., E. C. Darcissac, and Y. Mouton. 2003. Discordant effects of interleukin-2 on viral and immune parameters in human immunodeficiency virus-1-infected monocyte-derived mature dendritic cells. Clin. Exp. Immunol. 132:289-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger, S. L. 2002. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12:142-148. [DOI] [PubMed] [Google Scholar]
- 5.Buonaguro, L., G. Barillari, H. K. Chang, C. A. Bohan, V. Kao, R. Morgan, R. C. Gallo, and B. Ensoli. 1992. Effects of the human immunodeficiency virus type 1 Tat protein on the expression of inflammatory cytokines. J. Virol. 66:7159-7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai, S., H. J. Han, and T. Kohwi-Shigematsu. 2003. Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat. Genet. 34:42-51. [DOI] [PubMed] [Google Scholar]
- 7.Chang, H. K., R. C. Gallo, and B. Ensoli. 1995. Regulation of cellular gene expression and function by the human immunodeficiency virus type 1 Tat protein. J. Biomed. Sci. 2:189-202. [DOI] [PubMed] [Google Scholar]
- 8.Clerici, M., and G. M. Shearer. 1993. A Th1 to Th2 switch is a critical step in the etiology of HIV infection. Immunol. Today 14:107-111. [DOI] [PubMed] [Google Scholar]
- 9.Cross, S. L., M. B. Feinberg, J. B. Wolf, N. J. Holbrook, F. Wong-Staal, and W. J. Leonard. 1987. Regulation of the human interleukin-2 receptor alpha chain promoter: activation of a non-functional promoter by the transactivator gene of HTLV-I. Cell 49:47-56. [DOI] [PubMed] [Google Scholar]
- 10.Dickinson, L. A., C. D. Dickinson, and T. Kohwi-Shigematsu. 1997. The nuclear matrix attachment region (MAR)-binding protein SATB1 contains a homeodomain that promotes specific recognition of the core unwinding element of a MAR. J. Biol. Chem. 272:11463-11470. [DOI] [PubMed] [Google Scholar]
- 11.Dobreva, G., J. Dambacher, and R. Grosschedl. 2003. SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev. 15:3048-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehret, A., M. Li-Weber, R. Frank, and P. H. Krammer. 2001. The effect of HIV-1 regulatory proteins on cellular genes: derepression of the IL-2 promoter by Tat. Eur. J. Immunol. 31:1790-1799. [DOI] [PubMed] [Google Scholar]
- 13.Englund, G., T. S. Theodore, E. O. Freed, A. Engelman, and M. A. Martin. 1995. Integration is required for productive infection of monocyte-derived macrophages by human immunodeficiency virus type 1. J. Virol. 69:3216-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fanning, A. S., and J. M. Anderson. 1996. Protein-protein interactions: PDZ domain networks. Curr. Biol. 6:1385-1388. [DOI] [PubMed] [Google Scholar]
- 15.Fung, M. R., and W. C. Greene. 1990. The human interleukin-2 receptor: insights into subunit structure and growth signal transduction. Semin. Immunol. 2:119-128. [PubMed] [Google Scholar]
- 16.Galande, S., L. A. Dickinson, I. S. Mian, M. Sikorska, and T. Kohwi-Shigematsu. 2001. SATB1 cleavage by caspase 6 disrupts PDZ domain-mediated dimerization, causing detachment from chromatin early in T-cell apoptosis. Mol. Cell. Biol. 21:5591-5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gervaix, A., D. West, L. M. Leoni, D. D. Richman, F. Wong-Staal, and J. Corbeil. 1997. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. USA 94:4653-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graziosi, C., K. R. Gantt, M. Vaccarezza, J. F. Demarest, M. Daucher, M. S. Saag, G. M. Shaw, T. C. Quinn, O. J. Cohen, C. C. Welbon, G. Pantaleo, and A. S. Fauci. 1996. Kinetics of cytokine expression during primary human immunodeficiency virus type 1 infection. Proc. Natl. Acad. Sci. USA 93:4386-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greene, W. C., W. J. Leonard, Y. Wano, P. B. Svetlik, N. J. Peffer, J. G. Sodroski, C. A. Rosen, W. C. Goh, and W. A. Haseltine. 1986. Trans-activator gene of HTLV-II induces IL-2 receptor and IL-2 cellular gene expression. Science 232:877-880. [DOI] [PubMed] [Google Scholar]
- 20.Hauber, J., A. Perkins, E. P. Heimer, and B. R. Cullen. 1987. Trans-activation of human immunodeficiency virus gene expression is mediated by nuclear events. Proc. Natl. Acad. Sci. USA 84:6364-6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauser, C., B. Schuettengruber, S. Bartl, G. Lagger, and C. Seiser. 2002. Activation of the mouse histone deacetylase 1 gene by cooperative histone phosphorylation and acetylation. Mol. Cell. Biol. 22:7820-7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawkins, S. M., T. Kohwi-Shigematsu, and D. G. Skalnik. 2002. The matrix attachment region-binding protein SATB1 interacts with multiple elements within the gp91phox promoter and is down regulated during myeloid differentiation. J. Biol. Chem. 276:44472-44480. [DOI] [PubMed] [Google Scholar]
- 23.He, G., and D. M. Margolis. 2002. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 22:2965-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeang, K. T., H. Xiao, and E. A. Rich. 1999. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 274:28837-28840. [DOI] [PubMed] [Google Scholar]
- 25.Joseph, M. A., S. J. Ladha, M. Mojamdar, and D. Mitra. 2003. Human immunodeficiency virus-1 nef protein interacts with Tat and enhances HIV-1 gene expression. FEBS Lett. 548:37-48. [DOI] [PubMed] [Google Scholar]
- 26.Kinter, A. L., S. M. Bende, E. C. Hardy, R. Jackson, and A. S. Fauci. 1995. Interleukin 2 induces CD8+ T cell-mediated suppression of human immunodeficiency virus replication in CD4+ T cells and this effect overrides its ability to stimulate virus expression. Proc. Natl. Acad. Sci. USA 92:10985-10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohwi-Shigematsu, T., I. de Belle, L. A. Dickinson, S. Galande, and Y. Kohwi. 1998. Identification of base-unpairing region (BUR)-binding proteins and characterization of their in vivo binding sequences. Methods Cell Biol. 53:323-354. [DOI] [PubMed] [Google Scholar]
- 28.Leung, K., and G. J. Nabel. 1988. HTLV-1 transactivator induces interleukin-2 receptor expression through an NF-kappa B-like factor. Nature 333:776-778. [DOI] [PubMed] [Google Scholar]
- 29.Lin, J. X., and W. J. Leonard. 1997. Signaling from the IL-2 receptor to the nucleus. Cytokine Growth Factor Rev. 8:313-332. [DOI] [PubMed] [Google Scholar]
- 30.Lowenthal, J. W., E. Bohnlein, D. W. Ballard, and W. C. Greene. 1988. Regulation of interleukin 2 receptor alpha subunit (TAC or CD25 antigen) gene expression: binding of inducible nuclear proteins to discrete promoter sequences correlates with transcriptional activation. Proc. Natl. Acad. Sci. USA 85:4468-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellors, J. W., C. R. Rinaldo, Jr., P. Gupta, R. M. White, J. A. Todd, and L. A. Kingsley. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272:1124-1125. [DOI] [PubMed] [Google Scholar]
- 32.Mitra, D., M. Steiner, D. H. Lynch, L. Staino-coico, and J. Laurence. 1996. HIV-1 upregulates Fas ligand expression in CD4+ T cells in vitro and in vivo: association with Fas-mediated apoptosis and modulation by aurintricarboxylic acid. Immunology 87:512-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagomi, K., Y. Kohwi, L. A. Dickinson, and T. Kohwi-Shigematsu. 1994. A novel DNA-binding motif in the nuclear matrix attachment DNA-binding protein SATB1. Mol. Cell. Biol. 14:1852-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ott, M., S. Emiliani, C. Van Lint, G. Herbein, J. Lovett, N. Chirmule, T. McCloskey, S. Pahwa, and E. Verdin. 1997. Immune hyperactivation of HIV-1-infected T cells mediated by Tat and the CD28 pathway. Science 275:1481-1485. [DOI] [PubMed] [Google Scholar]
- 35.Phares, W., B. R. Franza, Jr., and W. Herr. 1992. The kappa B enhancer motifs in human immunodeficiency virus type 1 and simian virus 40 recognize different binding activities in human Jurkat and H9 T cells: evidence for NF-kappa B-independent activation of the kappa B motif. J. Virol. 66:7490-7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puri, R. K., P. Leland, and B. B. Aggarwal. 1995. Constitutive expression of human immunodeficiency virus type gene inhibits interleukin 2 and interleukin 2 receptor expression on human CD4+ T lymphoid (H9) cell line. AIDS Res. Hum. Retrovir. 11:31-40. [DOI] [PubMed] [Google Scholar]
- 37.Purvis, S. F., D. L. Georges, T. M. Williams, and M. M. Lederman. 1992. Suppression of interleukin-2 and interleukin-2 receptor expression in Jurkat cells stably expressing the human immunodeficiency virus protein. Cell. Immunol. 144:32-42. [DOI] [PubMed] [Google Scholar]
- 38.Rothenberg, E. V., and S. B. Ward. 1996. A dynamic assembly of diverse transcription factors integrates activation and cell-type information for interleukin 2 gene regulation. Proc. Natl. Acad. Sci. USA 93:9358-9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rusnati, M., D. Coltrini, P. Oreste, G. Zoppetti, A. Albini, D. Noonan, F. d'Adda di Fagagna, M. Giacca, and M. Presta. 1997. Interaction of HIV-1 Tat protein with heparin. Role of the backbone structure, sulfation, and size. J. Biol. Chem. 272:11313-11320. [DOI] [PubMed] [Google Scholar]
- 40.Scala, G., M. R. Ruocco, C. Ambrosino, M. Mallardo, V. Giordano, F. Baldassarre, E. Dragonetti, I. Quinto, and S. Venuta. 1994. The expression of the interleukin 6 gene is induced by the human immunodeficiency virus 1 TAT protein. J. Exp. Med. 179:961-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scharager, J. A., and J. W. Marsh. 1999. HIV-1 Nef increases T cell activation in a stimulus-dependent manner. Proc. Natl. Acad. Sci. USA 96:8167-8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwarze, S. R., A. Ho, A. Vocero-Akbani, and S. F. Dowdy. 1999. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569-1572. [DOI] [PubMed] [Google Scholar]
- 43.Scott-Algara, D., F. Vuillier, M. Marasescu, J. de Saint Martin, and G. Dighiero. 1991. Serum levels of IL-2, IL-1 alpha, TNF-alpha, and soluble receptor of IL-2 in HIV-1-infected patients. AIDS Res. Hum. Retrovir. 7:381-386. [DOI] [PubMed] [Google Scholar]
- 44.Shibuya, H., M. Yoneyama, and T. Taniguchi. 1989. Involvement of a common transcription factor in the regulated expression of IL-2 and IL-2 receptor genes. Int. Immunol. 1:43-49. [DOI] [PubMed] [Google Scholar]
- 45.Siebenlist, U., D. B. Durand, P. Bressler, N. J. Holbrook, C. A. Norris, M. Kamoun, J. A. Kant, and G. R. Crabtree. 1986. Promoter region of interleukin-2 gene undergoes chromatin structure changes and confers inducibility on chloramphenicol acetyltransferase gene during activation of T cells. Mol. Cell. Biol. 6:3042-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siekevitz, M., M. B. Feinberg, N. Holbrook, F. Wong-Staal, and W. C. Greene. 1987. Activation of interleukin 2 and interleukin 2 receptor (Tac) promoter expression by the trans-activator (tat) gene product of human T-cell leukaemia virus, type I. Proc. Natl. Acad. Sci. USA 84:5389-5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson, M., T. L. Stanwick, M. P. Dempsey, and C. A. Lamonica. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 9:1551-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyagi, M., M. Rusnati, M. Presta, and M. Giacca. 2001. Internalization of HIV-1 Tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 276:3254-3261. [DOI] [PubMed] [Google Scholar]
- 49.Vacca, A., M. Farina, M. Maroder, E. Alesse, I. Screpanti, L. Frati, and A. Gulino. 1994. Human immunodeficiency virus type-1 tat enhances interleukin-2 promoter activity through synergism with phorbol ester and calcium-mediated activation of the NF-AT cis-regulatory motif. Biochem. Biophys. Res. Commun. 205:467-474. [DOI] [PubMed] [Google Scholar]
- 50.Wang, J. K., E. Kiyokawa, E. Verdin, and D. Trono. 2000. The Nef protein of HIV-1 associates with rafts and primes T cells for activation. Proc. Natl. Acad. Sci. USA 97:394-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiskerchen, M., and M. A. Muesing. 1995. Identification and characterization of a temperature-sensitive mutant of human immunodeficiency virus type 1 by alanine scanning mutagenesis of the integrase gene. J. Virol. 69:597-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu, Y., and J. W. Marsh. 2001. Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science 293:1503-1506. [DOI] [PubMed] [Google Scholar]
- 53.Yasui, D., M. Miyano, S. Cai, P. Varga-Weisz, and T. Kohwi-Shigematsu. 2002. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 10:641-645. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, Z., T. Schuler, M. Zupancic, S. Wietgrefe, K. A. Staskus, K. A. Reimann, T. A. Reinhart, M. Rogan, W. Cavert, C. J. Miller, et al. 1999. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286:1353-1357. [DOI] [PubMed] [Google Scholar]