

Abstract
Shikonin and its derivatives extracted from Lithospermeae plants’ red roots have current applications in food and pharmaceutical industries. Previous studies have cloned some genes related to shikonin biosynthesis. However, most genes related to shikonin biosynthesis remain unclear, because the lack of the genome/transcriptome of the Lithospermeae plants. Therefore, in order to provide a new understanding of shikonin biosynthesis, we obtained transcriptome data and unigenes expression profiles in three shikonin-producing Lithospermeae plants, i.e., Lithospermum erythrorhizon, Arnebia euchroma and Echium plantagineum. As a result, two unigenes (i.e., G10H and 12OPR) that are involved in “shikonin downstream biosynthesis” and “methyl jasmonate biosynthesis” were deemed to relate to shikonin biosynthesis in this study. Furthermore, we conducted a Lamiids phylogenetic model and identified orthologous unigenes under positive selection in above three Lithospermeae plants. The results indicated Boraginales was more relative to Solanales/Gentianales than to Lamiales.
Introduction
Shikonin and its derivatives, red naphthoquinones, are widely found in the epidermis of the roots of the Lithospermeae plants, such as genera Lithospermum, Arnebia and Echium 1–3. In particular, Lithospermum erythrorhizon, Arnebia euchroma and Echium plantagineum, which are known as “alkanets” or “gromwells”, were originally used as natural dyes and herbal medicines in both Europe and the Orient for many centuries1–3. Shikonin and its derivatives are currently used in the food industry, cosmetics production and modern pharmaceutical synthesis based on their various biological activities, such as antioxidant and antibacteria1–3. Recently, it was reported that shikonin and its derivatives could induce apoptosis of many type cancer cells and perform anticancer activities4, 5. To meet the increasing demand, numerous attempts to synthesize shikonin and its derivatives have been invented and reported, including attempts using chemosynthesis and two-stage culture biosynthesis1–3.
According to previous studies, the proposed biosynthesis of the shikonin and its derivatives stems from mevalonate biosynthesis (terpenoid backbone biosynthesis) and phenylpropanoid biosynthesis1, 6, 7. In the past 30 years, several genes that encoding enzymes directly involved in the shikonin biosynthesis, such as hydroxymethylglutaryl-CoA synthase gene (HMGS), hydroxymethylglutaryl-CoA reductase gene (HMGR), phenylalanine ammonia lyase (PAL), cinnamate 4-monooxygenase (C4H) and p-hydroxybenzoate geranyltransferase gene (PGT), have been cloned and characterized in L. erythrorhizon or A. euchroma 8–12. In addition, several genes indirectly related to shikonin biosynthesis have been discovered and cloned, such as L. erythrorhizon dark-inducible gene 1, 2, 3, 4, 5a/b/c (LeDI-1, 2, 3, 4, 5a/b/c) and L. erythrorhizon 1-aminocyclopropane-1-carboxylate synthase gene (LeACS)12–14. Moreover, several factors, such as light, methyl jasmonate, nitric oxide, ethylene and fungal elicitor are crucial regulators of shikonin biosynthesis12, 15–19. However, the complete pathway of the shikonin biosynthesis remains obscure, and most genes directly/indirectly related to shikonin biosynthesis remain unclear, particularly the enzymes/genes that participate in the shikonin downstream biosynthesis (i.e., the pathway from geranyl-hydroxy-benzoate to shikonin) and the biosynthesis of the regulatory factors (e.g., methyl jasmonate, nitric oxide, ethylene)1, 2.
At present, RNA sequencing (RNA-Seq) is an effective platform to acquire transcriptome information and gene sequences at a minimal cost compared with whole genome de novo sequencing. This technology provides new opportunities to explore and identify novel genes involved in natural product biosynthesis in plants. For instance, Zhang et al. constructed gene co-expression networks related to D-myo-inositol phosphates (IP) in maize by integrating transcriptome and metabolite data, and discovered three new genes related to IP metabolism20. Dugé et al. constructed an optimized transcriptomic resource for Catharanthus roseus by processing previous published transcriptome data, and provided new perspectives for the understanding of the monoterpenoid indole alkaloids biosynthesis (MIA) in C. roseus 21. In addition, RNA-Seq also provides new opportunities to perform multiple comparisons and phylogenetic analysis to discover genic molecular evolution in different species. For example, Yang et al. found that similar functional categories had undergone positive selection in high-altitude Phrynocephalus and Rana species by comparing transcriptome data from different low-/high-altitude poikilothermic species22. Wickett et al. assessed phylogenetic models to explore the origin and early diversification of land plants by using 92 plant transcriptome data plus 11 public plant genome data23.
Therefore, in order to provide a new understanding of shikonin biosynthesis, we performed transcriptome analysis for three Lithospermeae plants, i.e., L. erythrorhizon, A. euchroma and E. plantagineum. First, we detected shikonin and its derivatives in their green leaves/stems (GL, non-producing shikonin) and red roots (RR, producing shikonin) by high performance liquid chromatography (HPLC). Subsequently, we constructed their transcriptome libraries and compared unigenes expression profiles between GL and RR lines in the above three Lithospermeae plants. The results indicated that two unigenes (i.e., G10H and 12OPR) were related to shikonin biosynthesis in this present study. Furthermore, we conducted a Lamiids phylogenetic model based on above three Lithospermeae plants’ transcriptome data plus seven public plant genome data. The results showed that Boraginales might be resolved as a sister to Solanales/Gentianales rather than Lamiales. Moreover, we identified orthologous unigenes under positive selection (PSOs) in the above three Lithospermeae plants, and found that one PSO (i.e., 4CL) potentially influenced shikonin biosynthesis in this study.
Results and Discussion
Detection of shikonin and its derivatives in GL and RR lines by HPLC
Detection of shikonin and its derivatives in GL and RR lines from three Lithospermeae plants, i.e., L. erythrorhizon, A. euchroma and E. plantagineum, was performed using HPLC. The results shown that shikonin and its derivatives scarcely existed in all GL lines (Fig. 1a,b,c,e and g); expectedly, shikonin and its derivatives, e.g., shikonin, acetylshikonin, and isobutyrylshikonin, were detected in all RR lines as previously reported (Fig. 1a,b,d,f and h; Supplementary Table S1)24–26. Furthermore, we deduced that the peak* might be β-hydroxyisovalerylshikonin, which is consistent with the previous studies24–26. In summary, shikonin biosynthesis should exist only in the RR lines rather than GL lines in the three Lithospermeae plants assessed in this present study (Fig. 1 and Supplementary Table S1).
Figure 1.
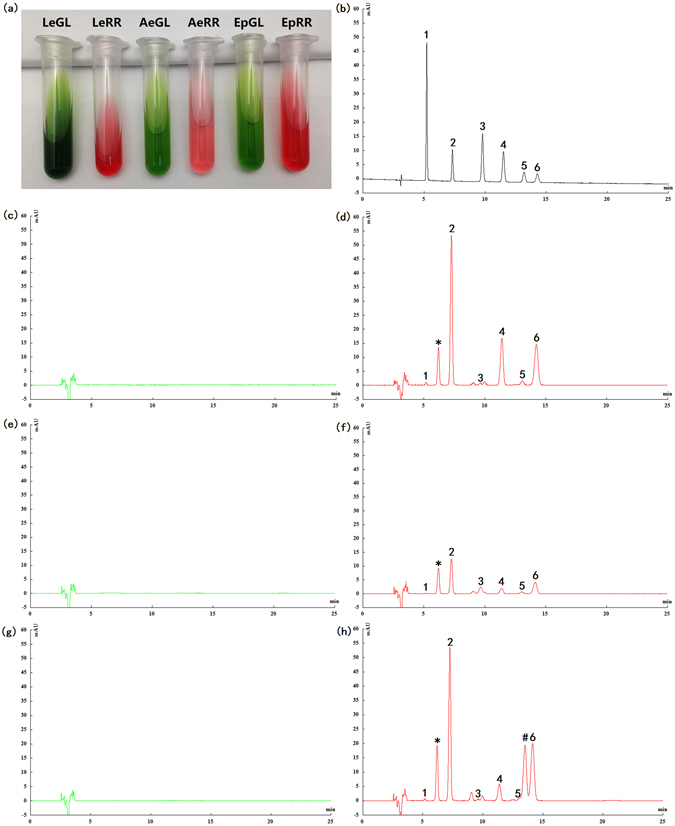
Detection of shikonin and its derivatives in the GL and RR lines of the three Lithospermeae plants (i.e., Lithospermum erythrorhizon, Arnebia euchroma and Echium plantagineum) by HPLC. (a) The ethanol extracts from the above three Lithospermeae plants, (b) standards, (c) LeGL, (d) LeRR, (e) AeGL, (f) AeRR, (g) EpGL, (h) EpRR; 1. Shikonin, 2. Acetylshikonin, 3. Deoxyshikonin, 4. Isobutylshikonin; 5. β, β’-Dimethylacrylshikonin, 6. α-Methyl-n-butyrylshikonin/Isovalerylshikonin (isomerized products can not be separated in this study). * β-Hydroxyisovalerylshikonin (inferential). #Unknown compound.
Transcriptome sequencing, assembly, annotation and KEGG classification
To obtain an overview of genes associated with shikonin biosynthesis, six cDNA libraries (i.e., LeGL, LeRR, AeGL, AeRR, EpGL and EpRR) were prepared from the GL and RR lines of the three Lithospermeae plants, and then were sequenced on an Illumina HiSeq™ 4000 platform. After raw data filtration and de novo assembly, 76455 unigenes with total length of 84.51 (Mb) and N50 length (median length of all non-redundant sequences) of 1856 (bp) were generated in the L. erythrorhizon libraries; and 89639 unigenes with total length of 87.19 (Mb) and N50 length of 1629 (bp) were generated in the A. euchroma libraries; and 54627 unigenes with total length of 55.59 (Mb) and N50 length of 1653 (bp) were generated in the E. plantagineum libraries (Table 1). Subsequently, the functional annotation of all assembled unigenes was performed using seven public databases, i.e., Non-redundant Protein Sequence (NR), Nucleotide Sequence (NT), SwissProt, InterPro, Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups of Proteins (COG) and Gene Ontology (GO). As a result, a total of 54128 unigenes (70.80%) in the L. erythrorhizon libraries, 61221 unigenes (68.30%) in the A. euchroma libraries and 41313 unigenes (75.62%) in the E. plantagineum libraries were annotated in at least one of the above databases (Table 1). Furthermore, we synthesized annotation information from five protein databases (i.e., NR, SwissProt, KEGG, InterPro, COG). The results indicated that a total of 17145 unigenes (22.42%) in the L. erythrorhizon libraries, 17237 unigenes (19.23%) in the A. euchroma libraries and 13333 unigenes (24.41%) in the E. plantagineum libraries were conjointly annotated by all five protein databases (Fig. 2). In addition, all assembled unigenes were performed coding sequence (CDS) prediction. As a result, a total of 53,675 (70.20%), 60728 (67.75%), 40441 (74.03%) CDS were generated in the above three Lithospermeae plants, respectively (Table 1). Subsequently, we classified all unigenes based on their function in KEGG database to better explore and understand unigenes potentially related to shikonin biosynthesis because KEGG is a highly integrated protein database designed to link genes to gene products (mostly proteins) in the metabolic pathways. As a result, a total of 38173 unigenes (49.93%) in the L. erythrorhizon, 43093 unigenes (48.07%) in the A. euchroma and 29514 unigenes (54.03%) in the E. plantagineum were assigned to 126 pathways, which were grouped into 20 sub categories and 7 categories (Supplementary Table S2).
Table 1.
Overview of transcriptome assembly, annotation and CDS prediction.
| Lithospermum erythrorhizon | Arnebia euchroma | Echium plantagineum | ||
|---|---|---|---|---|
| Assembly | Total Length (bp) | 84,508,068 | 87,189,611 | 55,592,752 |
| N50 Length (bp) | 1,856 | 1,629 | 1,653 | |
| GC (%) | 39.84 | 40.62 | 40.76 | |
| Total Unigenes | 76,455 | 89,639 | 54,627 | |
| Annotation | NR | 50,832 | 57,717 | 39,503 |
| NT | 41,021 | 45,267 | 32,705 | |
| SwissProt | 35,812 | 40,061 | 28,792 | |
| InterPro | 40,327 | 42,085 | 31,160 | |
| KEGG | 38,173 | 43,093 | 29,514 | |
| COG | 21,514 | 21,975 | 16,409 | |
| GO | 7,486 | 10,182 | 7,871 | |
| Overall | 54,128 | 61,221 | 41,313 | |
| CDS prediction | Blast CDS | 50,396 | 56,602 | 38,863 |
| ESTScan CDS | 3,279 | 4,126 | 1,578 | |
| Total CDS | 53,675 | 60,728 | 40,441 |
Figure 2.
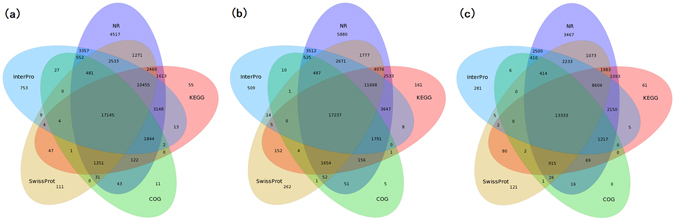
The Venn diagram of unigenes annotation from five public protein databases (The colors severally denote NR, SwissProt, KEGG, InterPro, COG databases). (a) Lithospermum erythrorhizon, (b) Arnebia euchroma, (c) Echium plantagineum.
Differentially expressed unigene (DEG) identification and KEGG enrichment
According to the general thresholds (i.e., |Log2(ratio)| ≥ 1 and FDR < 0.001), a total of 19447 unigenes were significantly differentially expressed in the L. erythrorhizon libraries, including 8144 up-expressed and 11303 down-expressed in the LeRR line; and a total of 15738 unigenes were significantly differentially expressed in the A. euchroma libraries, including 7599 up-expressed and 8139 down-expressed in the AeRR line, and a total of 15713 unigenes were significantly differentially expressed in the E. plantagineum libraries including 4538 up-expressed and 11175 down-expressed in the EpRR line (Fig. 3a). Subsequently, we classified all DEGs into KEGG classification using the same method and performed KEGG enrichment analysis for the pathways associated with each DEG. As a result, a total of 44 pathways in the L. erythrorhizon libraries, 45 pathways in the A. euchroma libraries and 22 pathways in the E. plantagineum libraries were significant enriched (Supplementary Table S2). Specifically, these enriched pathways mainly referred to “energy metabolism”, “carbohydrate metabolism”, “biosynthesis of other secondary metabolites” and “metabolism of terpenoids and polyketides” pathway sub-categories, particularly several pathways involved in “starch and sucrose metabolism”, “photosynthesis”, “phenylpropanoid biosynthesis” and “terpenoid backbone biosynthesis” (Supplementary Table S2). Therefore, KEGG enrichment revealed that photosynthesis metabolism and shikonin biosynthesis were the main distinctions between the RR and GL lines of the above three Lithospermeae plants. However, the DEGs generated according to the general thresholds were seemingly too numerous; thus, it was difficult to identify the major genes related to shikonin biosynthesis. Therefore, we defined highly significant differentially expressed unigenes (HDEGs) on the basis of the stringent thresholds (i.e., |Log2(ratio)| ≥ 1 and FDR < 0.001 and Max(FPKM) ≥ 100) in this present study. As a result, a total of 651 HDEGs including 287 up-expressed and 364 down-expressed were generated in the LeRR line of the L. erythrorhizon libraries; and a total of 717 HDEGs including 340 up-expressed and 377 down-expressed were generated in the AeRR line of the A. euchroma libraries; and a total of 688 HDEGs including 341 up-expressed and 347 down-expressed were generated in the EpRR line of the E. plantagineum libraries (Supplementary Table S3, Fig. 3b).
Figure 3.
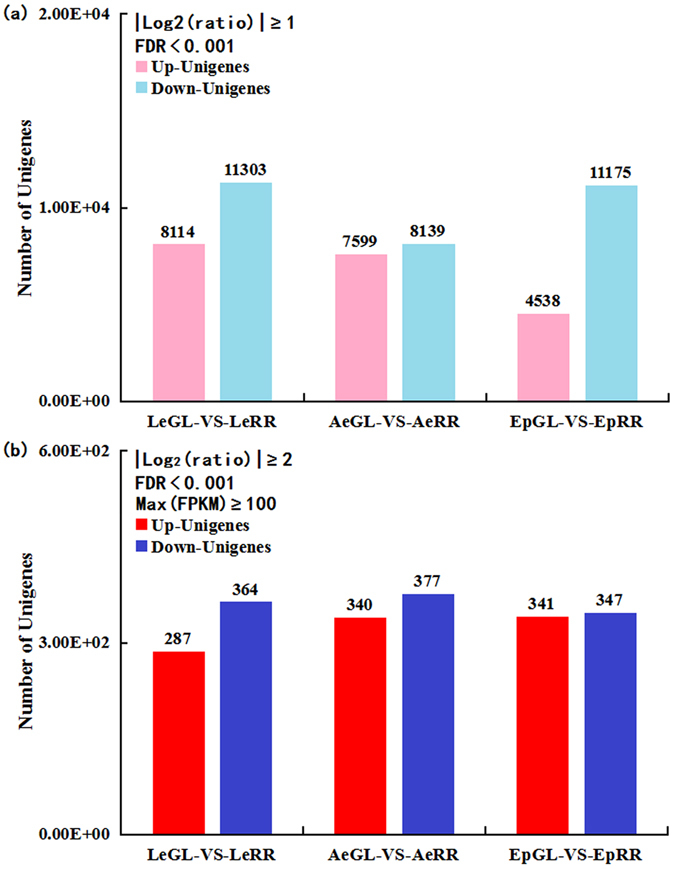
Unigene expression between GL and RR lines in Lithospermum erythrorhizon, Arnebia euchroma and Echium plantagineum. (a) Significantly differentially expressed unigenes (DEGs); (b) Highly significant differentially expressed unigenes (HDEGs).
Novel unigenes related to shikonin biosynthesis
According to the stringent thresholds, we found that a series of unigenes that were previously reported to be related to shikonin biosynthesis are up-expressed in all RR lines, such as HMGR, PAL, PGT, LeDI-1, LePR2 genes and so on (Supplementary Table S3). Moreover, we also identified two novel unigenes (i.e. G10H and 12OPR) related to shikonin biosynthesis in this present study (Supplementary Table S3).
First, we found that a geraniol 10-hydroxylase unigene (G10H; P-450 monooxygenase) is up-regulated in all RR lines (Supplementary Tables S3 and S4, Fig. 4). In addition, Yamamoto et al. reported a geranyl-hydroquinone 3″-hydroxylase enzyme (G3″H) that participates in shikonin biosynthesis as a P-450 monooxygenase, but they failed to report its nucleotide and amino acid sequence27. Therefore, we deduced that the G10H in this present study might be the gene of G3″H, which participates in shikonin downstream biosynthesis, because the G10H and G3″H enzyme have a similar function and belong to the P-450 monooxygenase family27, 28. The reason why that G3″H was annotated as G10H in this present study might be the lack of the gene or protein information of the G3″H/G3″H in the above public databases.
Figure 4.
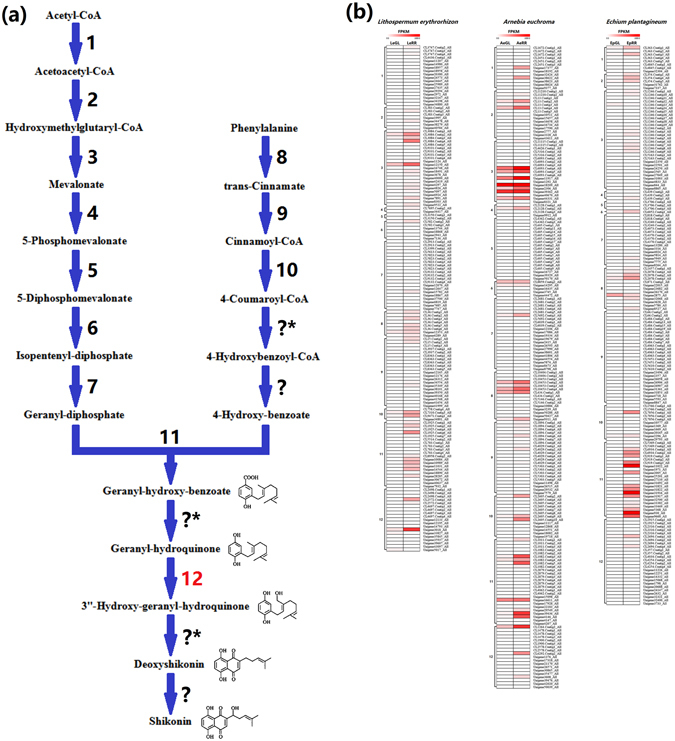
Unigene expression involved in shikonin biosynthesis in Lithospermum erythrorhizon, Arnebia euchroma and Echium plantagineum. (a) Shikonin biosynthesis; (b) Unigene expressions involved in shikonin biosynthesis? One step unknown?* Several steps unknown; 1. Acetoacetyl-coenzyme A thiolase gene (AACT), 2. Hydroxy-methylglutaryl CoA synthase gene (HMGS), 3. Hydroxy-methylglutaryl-CoA reductase gene (HMGR), 4. Mevalonate kinase gene (MK), 5. Phosphomevalonate kinase gene (PMK), 6. Mevalonate disphosphate decarboxylase gene (MVD), 7. Geranyl diphosphate synthase gene (GDS), 8. Phenylalanine ammonia-lyase gene (PAL), 9. 4-Coumarate:CoA ligase gene (4CL), 10. Cinnamic acid 4-hydroxylase gene (C4H), 11. p-Hydroxybenzoate geranyltransferase gene (PGT), 12. Geraniol 10-hydroxylase gene (G10H, predicted).
Second, we found that LeDI-5a and LeDI-5b, which were specifically highly expressed in all RR lines as previously reported12, were re-annotated as the 12-oxophytodienoate reductase gene (12OPR) (Supplementary Tables S3 and S5, Fig. 5). The 12OPR protein is a key enzyme in methyl jasmonate biosynthesis (Supplementary Table S5, Fig. 5)29, 30. In addition, previous studies reported that exogenous methyl jasmonate could improve shikonin biosynthesis15, 16. Thus, we speculated that up-expressed 12OPR unigenes (i.e., LeDI-5a/b gene) could promote MeJA biosynthesis in the above Lithospermeae plants, and endogenous MeJA accumulation could further promote shikonin biosynthesis.
Figure 5.
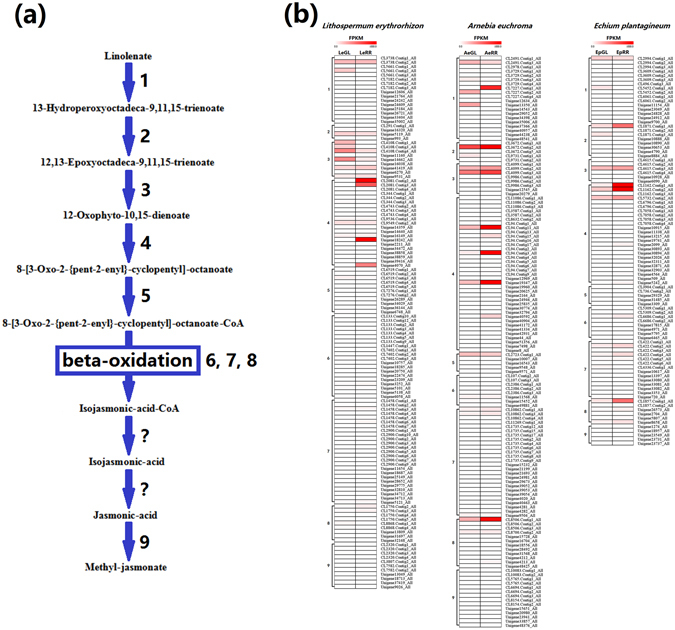
Unigene expression involved in methyl jasmonate biosynthesis in Lithospermum erythrorhizon, Arnebia euchroma and Echium plantagineum. (a) Methyl jasmonate biosynthesis, (b) Unigene expression involved in methyl jasmonate biosynthesis? One step unknown; 1. Lipoxygenase gene (LOX), 2. Allene oxide synthase gene (AOS), 3. Allene oxide cyclase gene (AOC), 4. 12-Oxophytodienoate reductase gene (12OPR), 5. CoA ligase gene (CL), 6. Acyl-CoA oxidase gene (ACX), 7. Fatty acid β-oxidation multifunctional protein MFP/AIM gene (MFP/AIM), 8. 3-ketoacyl-CoA thiolase gene (KAT), 9. Jasmonate O-methyltransferase gene (JMT).
Furthermore, we verified the expression profiles of the G10H and 12OPR unigenes in above three Lithospermeae plants by using qRT-PCR analysis. The qRT-PCR results were consistent with the results from RNA-seq data (Fig. 6). This indicated that G10H and 12OPR unigenes should be up-expressed in all RR lines from above three Lithospermeae plants, and probably related to shikonin biosynthesis in this present study. Moreover, the functional validation of the G10H and 12OPR unigenes is now underway, according to the similar strategy as reported in our previous papers14, 31, 32.
Figure 6.
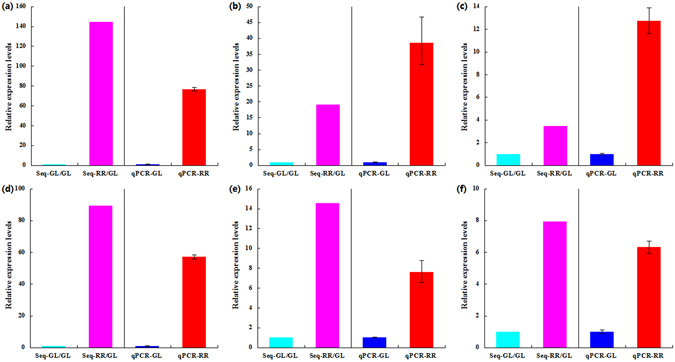
The qRT-PCR verification of the G10H and 12OPR unigenes’ expression profiles between GL and RR lines in Lithospermum erythrorhizon (Le), Arnebia euchroma (Ae) and Echium plantagineum (Ep). (a) G10H-LeCL2572.Contig1, (b) G10H-AeCL6292.Contig2, (c) G10H-EpCL2694.Contig1, (d) 12OPR-LeCL2081.Contig2, (e) 12OPR-AeCL94.Contig3, (f) 12OPR-EpCL1162.Contig2; Seq. data from RNA-seq, qPCR. data from qRT-PCR; The error bars in the qPCR results represent standard deviation of three biological replicates.
Boraginales’ evolutionary status and its unigenes under positive selection (PSOs)
A total of 973 orthologous unigenes were identified from the above three Lithospermeae plants’ transcriptomes and seven plants’ known genomes (i.e., Solanum lycopersicum, Coffea canephora, Salvia miltiorrhiza, Sesamum indicum, Erythranthe guttatus, Actinidia chinensis and Vitis vinifera) (Supplementary Table S6). Based on these probable orthologous unigenes, a phylogenetic tree of Lamiids was established (Fig. 7). As shown in Fig. 7, the above three Lithospermeae plants (i.e., L. erythrorhizon, A. euchroma and E. plantagineum) and S. lycopersicum probably exhibit a more close evolutionary relationship than other species. This suggested Boraginales might be closer to Solanales/Gentianales than to Lamiales in the evolutionary distance scale. In addition, Boraginales probably segregated from Solanales at about 76.1 (81.3–72.3) million years ago (Mya), which is approximately at the Campanian (from 83.6 ± 0.7 Mya to 72.1 ± 0.6 Mya) in the late Cretaceous33.
Figure 7.
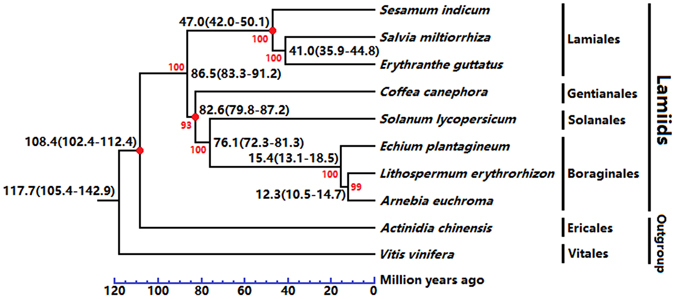
The phylogenetic model and divergence of Lamiids. (Black numbers) divergence times and their 95% confidence intervals, (Red numbers) maximum likelihood bootstrap support (MLBS), (Red nodes) divergence calibration points.
According to previous studies, Boraginales’ evolutionary status in Lamiids is still unclear. For instance, Refulio-Rodriguez et al. analysed the phylogeny of Lamiidae based on nine plastid regions and one mitochondrial region in 129 samples; they inferred that Boraginales might be resolved as a sister to Lamiales, but that result had only 65% maximum likelihood bootstrap support (MLBS)34. As another example, Maximilian et al. presented a phylogenetic analysis of Boraginales with four chloroplast locations, including 90 samples; they deduced that Boraginales might be a sister group relationship to Solanales/Gentianales, but that result had only tentative statistical support35. Although there is a lack of genome/transcriptome of taxa samples in this present study, our result supported that Boraginales should be resolved as a sister to Solanales/Gentianales rather than Lamiales, because our results exhibited high MLBS (Fig. 7), and our results were based on sufficient orthologous unigenes (973, Supplementary Table S6) rather than a few of plastid regions/locations.
Furthermore, according to the above phylogenetic model of Lamiids, we identified that 145 orthologous unigenes were under positive selection (PSOs) in the above three Lithospermeae plants (Supplementary Table S6). Comparing their expression level in GL and RR lines separately, two orthologous unigenes (i.e. leucine-rich repeat kinase/extensin unigene (LRE) and 4-coumarate:CoA ligase unigene (4CL)) exhibited highly specific expression in all RR lines (Supplementary Table S6). 4CL gene is directly involved in shikonin biosynthesis8. In the previous study, Yazaki et al. cloned two copies of 4CL gene from L. erythrorhizon (i.e., Le4CL-1 and Le4CL-2) and speculated that 4CL gene might not significantly influence shikonin biosynthesis8. However, through PSO analysis combined with gene expression comparison, we deduced that some 4CL unigenes (i.e., LeCL15, AeCL1894 and EpCL4063) probably influence shikonin biosynthesis, although their expression level is relatively limited (Supplementary Table S4, Fig. 4).
Materials and Methods
Plants materials
Seeds of L. erythrorhizon, A. euchroma and E. plantagineum were germinated according to the methods, as previously reported14, 26. The germinated seeds were then transferred into square plastic pots (50 * 30 * 30 cm) with peat growing medium (Pindstrup, Denmark) and were cultured in a greenhouse at 23 ± 1 °C under a 16 h/day photoperiod for approximately 60 to 90 days. Subsequently, GL (green leaves/stems) and RR (red roots) samples from the above three Lithospermeae plants were collected, separately (Supplementary Table S7). All samples were immediately transferred into liquid nitrogen and stored in a ‒80 °C freezer. To minimize inter-individual differences, three biological replications of each sample were mixed together.
HPLC analysis and the extraction of shikonin and its derivatives
Shikonin and its derivatives were extracted from all samples according to the following method. Each sample was ground in a grinding bowl with liquid nitrogen, and the ground powder was transferred into alcohol (99.7%, analytical reagent) at 25 ± 2 °C on a shaker at 120 rpm/min for 6 hours(samples:alcohol (w/v) = 0.1 g:2 mL)26. Subsequently, the sample was centrifuged at 10,000 g for 10 minutes. The supernatant was subject to HPLC analysis using an Agilent 1200 system (Agilent Technologies, USA). Separation was achieved using a Thermo Gold C18 column (4.6 × 250 mm, 5 μm, Thermo Fisher Scientific, USA). The mobile phase consisted of solvent A (HPLC water with 0.1% trifluoroacetic acid) and solvent B (acetonitrile) (A:B (v/v) = 30:70). The column oven temperature was at 40 °C; the flow rate was 1.0 mL/min; the injection volume was 10 μL; and the detection wavelength was at 515 nm, as previously reported24–26. Peaks were identified by comparing their retention times with the standard chromatogram of six shikonin and its derivatives (Supplementary Table S1), which were purchased from Nanjing PuYi Biological Technology CO., LTD (Nanjing, China).
RNA extraction and sequencing
Total RNA was extracted using the TRIzol® reagent (Invitrogen, Carlsbad, USA). The quality and quantity of the extracted total RNAs were detected and assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA) and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) (Supplementary Table S8). Subsequently, cDNA libraries from each sample were prepared individually according to the following method: poly(A) enrichment, RNA fragmentation, cDNA synthesis, linker ligation, length selection, PCR purification and PCR amplification. Finally, the cDNA libraries were sequenced on an Illumina HiSeq 4000TM platform, and the sequencing raw data were deposited in the NCBI’s Short Read Archive (SRA) under the following accession numbers: SRX1980116, SRX1980117, SRX2026182, SRX2026183, SRX2026190 and SRX2026191.
Transcriptome assembly, annotation and CDS prediction
After raw data were filtered by removing adapter sequences, reads containing ploy-N, and low-quality sequences (Q < 20), the filtered clean reads were used to perform transcriptome de novo assembly using the Trinity program combining three independent software modules: Inchworm, Chrysalis, and Butterfly36–38. In addition, short contigs (<200 bp) were filtered automatically36–38. And then the CD-HIT-EST program was used for clustering assembled contigs to unigenes with an identity threshold of 95%39. Subsequently, the assembled unigenes were annotated through BLAST analysis against the seven public databases: NR (e-value of 1E−5); NT (e-value of 1E−10), SwissProt (e-value of 1E−5), InterPro (e-value of 1E−5), KEGG (e-value of 1E−5), COG (e-value of 1E−5) and GO (e-value of 1E−5)40–44. Furthermore, the unigenes’ translated amino acid sequences that could be mapped to functional annotation databases (in a priority order of NR, SwissProt, KEGG, COG) were defined as BLASTed coding sequences (CDSs); on the other hand, the unigenes that could not be aligned to any functional annotation databases were predicted as scanned CDSs by ESTScan software45.
DEG identification
The expression level of each unigenes was calculated using RNA-Seq quantification analysis as the fragments per kb per million mapped fragments (FPKM) method46, 47. A rigorous algorithm was then used to identify differentially expressed unigenes (DEGs) based on the method of Audic and Claverie48. In addition, false discovery rate (FDR) was used to confirm the p-value in multiple tests49, 50. |Log2(ratio)| ≥ 1 and FDR < 0.001 were used as general thresholds to define significant differences in gene expression48, and |Log2(ratio)| ≥ 1 and FDR < 0.001 and Max(FPKM) ≥ 100 were used as stringent thresholds to define highly significant differences in gene expression in this present study.
KEGG classification and enrichment
According to unigenes’ function in KEGG database, we classified all unigenes and DEGs, and then performed KEGG enrichment analysis for the pathways associated with each DEG using phyper modules from R software40. In addition, we calculate FDR for each p-value in KEGG enrichment analysis49. Finally, p-value < 0.05 was used as a threshold to define significant enrichment, becasue we found that the general threshold (FDR < 0.05) might be too stringent and not suitable in this present study.
qRT-PCR verification
Total RNAs were extracted using TRIzol® reagent (Invitrogen, Carlsbad, USA) and then treated with ReverTra Ace® qPCR RT Kit (TOYOBO, Osaka, Japan) to reverse transcribe into cDNA. qRT-PCR was conducted by SYBR Green Realtime PCR Master Mix™ (TOYOBO, Osaka, Japan) on a BIO-RAD CFX Connect™ Optics Module system (BIO-RAD, USA). Three G10H copies, namely LeCL2572.Contig1, AeCL6292.Contig2 and EpCL2694.Contig1 (Supplementary Table S4), were selected for representing G10H unigenes in this study; and three 12OPR copies, namely LeCL2081.Contig2, AeCL94.Contig3 and EpCL1162.Contig2 (Supplementary Table S5), were selected for representing 12OPR unigenes in this study. The glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) was used as internal reference as previously reported14, 32. All primers were listed in Supplementary Table S8. Reactions were performed in three replicates. Three biological replicates were used to quantify relative gene expression levels, according to the 2−ΔΔCt method51 (Supplementary Table S9).
Orthologous genes identification, phylogenetic analysis and selection identification
Single-copy orthologous unigenes were identified by using InParanoid52 and MultiParanoid53 from the transcriptome data of above three Lithospermeae plants plus the public genome data of seven plants (i.e., S. lycopersicum, C. canephora, S. miltiorrhiza, S. indicum, E. guttatus, A. chinensis and V. vinifera; A. chinensis and V. vinifera are used as an outgroup)54–60. Subsequently, these orthologous unigenes were aligned and trimmed using PRANK and GBlocks programs61, 62. The tandem 4D-sites of the orthologous unigenes were then generated, and were used to support phylogenetic analysis and divergence time estimation. Then, the Phyml program was used to build a phylogenetic tree based on the maximum-likelihood (ML) method63. The divergence times were estimated and adjusted using MCMCtree and BEAST programs64, 65 based on the timing calibration points as previously reported54–60. A improved branch-site model that containing the null model (sites evolve under neutral or purifying selection) and the alternative model (sites evolve under positive selection on the foreground branch) was used to identify orthologous unigenes under positive selection22, 66, 67. The likelihood ratio test was used to distinguish the alternative model of each orthologous unigenes22, 66, 67. Finally, p-value < 0.05 was used as a threshold to define significant positive selection in this present study, becasue the general threshold (FDR < 0.05) might be too strict.
Electronic supplementary material
Acknowledgements
This work was financially supported by the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R27) and the National Natural Science Foundation of China (NSFC; 31670298 and 31470384).
Author Contributions
C.T., Y.P. and Y.Y. contributed to the experiment management. F.W., Y.G., Z.B. and C.T. performed the experiments. C.T., J.F., G.L. and J.Q. participated in the data analyses. F.W. and C.T. wrote the manuscript. All authors carefully checked and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Feng-Yao Wu and Cheng-Yi Tang contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04750-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Cheng-Yi Tang, Email: tangchengyi_nju@163.com.
Yan-Jun Pang, Email: pangyj@nju.edu.cn.
Yong-Hua Yang, Email: yangyh@nju.edu.cn.
References
- 1.Papageorgiou VP, Assimopoulou AN, Couladouros EA, Hepworth D, Nicolaou KC. Chemistry and biology of alkannin, shikonin, and related naphthazarin natural products. Cheminform. 1999;30:270–301. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 2.Malik S, Bhushan S, Sharma M, Ahuja PS. Biotechnological approaches to the production of shikonins: a critical review with recent updates. Crit. Rev. Biotechnol. 2016;36:327–340. doi: 10.3109/07388551.2014.961003. [DOI] [PubMed] [Google Scholar]
- 3.Tappeiner J, et al. Quantitative determination of alkannins and shikonins in endemic mediterranean alkanna species. Biomed. Chromatogr. 2014;28:923–933. doi: 10.1002/bmc.3096. [DOI] [PubMed] [Google Scholar]
- 4.Gao D, Hiromura M, Yasui H, Sakurai H. Direct reaction between shikonin and thiols induces apoptosis in HL60 cells. Biol. Pharm. Bull. 2002;25:827–832. doi: 10.1248/bpb.25.827. [DOI] [PubMed] [Google Scholar]
- 5.Mao X, Yu CR, Li WH, Li WX. Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 2008;18:879–888. doi: 10.1038/cr.2008.86. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto T, Yazaki K, Tabata M. Biosynthesis of shikonin derivatives from l-phenylalanine via deoxyshikonin in Lithospermum cell cultures and cell-free extracts. Phytochemistry. 1995;38:83–88. doi: 10.1016/0031-9422(94)00621-Y. [DOI] [Google Scholar]
- 7.Brigham LA, Michaels PJ, Flores HE. Cell-specific production and antimicrobial activity of naphthoquinones in roots of Lithospermum erythrorhizon. Plant Physiol. 1999;119:417–428. doi: 10.1104/pp.119.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yazaki K, Ogawa A, Tabata M. Isolation and characterization of two cDNAs encoding 4-coumarate:CoA ligase in Lithospermum cell cultures. Plant Cell Physiol. 1995;36:1319–1329. [PubMed] [Google Scholar]
- 9.Yazaki K, Kataoka M, Honda G, Severin K, Heide L. cDNA cloning and gene expression of phenylalanine ammonia-lyase in Lithospermum erythrorhizon. Biosci. Biotechnol. Biochem. 1998;61:1995–2003. doi: 10.1271/bbb.61.1995. [DOI] [PubMed] [Google Scholar]
- 10.Yazaki K, Kunihisa M, Fujisaki T, Sato F. Geranyl diphosphate: 4-hydroxybenzoate geranyltransferase from Lithospermum erythrorhizon: cloning and characterization of a ket enzyme in shikonin biosynthesis. J. Biol. Chem. 2002;277:6240–6246. doi: 10.1074/jbc.M106387200. [DOI] [PubMed] [Google Scholar]
- 11.Singh RS, et al. Expression of 3-hydroxy-3-methylglutaryl-CoA reductase, p-hydroxybenzoate-m-geranyltransferase, and genes of phenylpropanoid pathway exhibits positive correlation with shikonins content in Arnebia. BMC Mol. Biol. 2010;11:88. doi: 10.1186/1471-2199-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yazaki K, Matsuoka H, Ujihara T, Sato F. Shikonin biosynthesis in Lithospermum erythrorhizon: light-induced negative regulation of secondary metabolism. Plant Biotechnology. 1999;16:335–342. doi: 10.5511/plantbiotechnology.16.335. [DOI] [Google Scholar]
- 13.Yazaki K, Matsuoka H, Shimomura K, Bechthold A, Sato F. A novel dark-inducible protein, LeDI-2, and its involvement in root-specific secondary metabolism in Lithospermum erythrorhizon. Plant Physiol. 2001;125:1831–1841. doi: 10.1104/pp.125.4.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang R, et al. Transgenic analysis reveals LeACS-1, as a positive regulator of ethylene-induced shikonin biosynthesis in Lithospermum erythrorhizon hairy roots. Plant Mol. Biol. 2016;90:345–358. doi: 10.1007/s11103-015-0421-z. [DOI] [PubMed] [Google Scholar]
- 15.Yazaki K, Takeda K, Tabata M. Effects of methyl jasmonate on shikonin and dihydroechinofuran production in Lithospermum cell cultures. Plant & Cell Physiology. 1997;38:776–782. doi: 10.1093/oxfordjournals.pcp.a029235. [DOI] [Google Scholar]
- 16.Hao H, et al. Effects of exogenous methyl jasmonate on the biosynthesis of shikonin derivatives in callus tissues of Arnebia euchroma. Appl. Biochem. Biotechnol. 2014;173:2198–2210. doi: 10.1007/s12010-014-1025-9. [DOI] [PubMed] [Google Scholar]
- 17.Wu SJ, et al. Nitric oxide regulates shikonin formation in suspension-cultured Onosma paniculatum cells. Plant Cell Physiol. 2009;50:118–128. doi: 10.1093/pcp/pcn178. [DOI] [PubMed] [Google Scholar]
- 18.Touno K, Jin T, Ohashi Y, Shimomura K. Ethylene induced shikonin biosynthesis in shoot culture of Lithospermum erythrorhizon. Plant Physiol. Biochem. 2005;43:101–105. doi: 10.1016/j.plaphy.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Dong JK, Chang HN. Increased shikonin production in Lithospermum erythrorhizon, suspension cultures with in situ, extraction and fungal cell treatment (elicitor) Biotechnology Letters. 1990;12:443–446. doi: 10.1007/BF01024401. [DOI] [Google Scholar]
- 20.Zhang S, et al. Analysis of weighted co-regulatory networks in maize provides insights into new genes and regulatory mechanisms related to inositol phosphate metabolism. BMC Genomics. 2016;17:129. doi: 10.1186/s12864-016-2476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dugé dBT, et al. Characterization of a second secologanin synthase isoform producing both secologanin and secoxyloganin allows enhanced de novo assembly of a Catharanthus roseus transcriptome. BMC Genomics. 2015;16:619. doi: 10.1186/s12864-015-1678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang YZ, et al. Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude Lizard living in the Qinghai-Tibet Plateau. BMC Evol. Biol. 2015;15:101. doi: 10.1186/s12862-015-0371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wickett NJ, et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA. 2014;111:4859–4868. doi: 10.1073/pnas.1323926111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albreht A, Vovk I, Simonovska B, Srbinoska M. Identification of shikonin and its ester derivatives from the roots of Echium italicum, L. J. Chromatogr. A. 2009;1216:3156–3162. doi: 10.1016/j.chroma.2009.01.098. [DOI] [PubMed] [Google Scholar]
- 25.Yusai I, Kenichi O, Takeshi Y, Yoko K. Tigloylshikonin, a new minor shikonin derivative, from the roots and the commercial root extract of Lithospermum erythrorhizon. Chem. Pharm. Bull. 2011;59:117–119. doi: 10.1248/cpb.59.117. [DOI] [PubMed] [Google Scholar]
- 26.Zhu X, et al. Identification and localization of bioactive naphthoquinones in the roots and rhizosphere of paterson’s curse (Echium plantagineum), a noxious invader. J. Exp. Bot. 2016;67:3777–3788. doi: 10.1093/jxb/erw182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto H, Inoue K, Li SM, Heide L. Geranylhydroquinone 3″-hydroxylase, a cytochrome P-450 monooxygenase from Lithospermum erythrorhizon, cell suspension cultures. Planta. 2000;210:312–317. doi: 10.1007/PL00008139. [DOI] [PubMed] [Google Scholar]
- 28.Miettinen K, et al. The seco-iridoid pathway from Catharanthus roseus. Nat Commun. 2014;5:3606. doi: 10.1038/ncomms4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaller F, Biesgen C, Müssig C, Altmann T, Weiler EW. 12-Oxophytodienoate reductase 3 (OPR3) is the isoenzyme involved in jasmonate biosynthesis. Planta. 2000;210:979–984. doi: 10.1007/s004250050706. [DOI] [PubMed] [Google Scholar]
- 30.Wasternack C. Jasmonates: an update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann Bot. 2007;100:681–697. doi: 10.1093/aob/mcm079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao H, et al. Overexpression of LeMYB1 enhances shikonin formation by up-regulating key shikonin biosynthesis-related genes in Lithospermum erythrorhizon hairy roots. Biologia Plantarum. 2015;59:429–435. doi: 10.1007/s10535-015-0512-3. [DOI] [Google Scholar]
- 32.Fang R, et al. Transgenic studies reveal the positive role of LeEIL-1 in regulating shikonin biosynthesis in Lithospermum erythrorhizon hairy roots. BMC Plant Biology. 2016;16:121. doi: 10.1186/s12870-016-0812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gradstein FM, Ogg JG, Hilgen FJ. A geologic time scale. Newsletters on Stratigraphy. 2012;45:171–188. doi: 10.1127/0078-0421/2012/0020. [DOI] [Google Scholar]
- 34.Refulio-Rodriguez NF, Olmstead RG. Phylogeny of Lamiidae. Am. J. Bot. 2014;101:287–299. doi: 10.3732/ajb.1300394. [DOI] [PubMed] [Google Scholar]
- 35.Maximilian W, et al. From capsules to nutlets - phylogenetic relationships in the Boraginales. Cladistics. 2014;30:508–518. doi: 10.1111/cla.12061. [DOI] [PubMed] [Google Scholar]
- 36.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pertea G, et al. TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 38.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanehisa M, et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quevillon E, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatusov RL, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 44.Ye J, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999;99:138–148. [PubMed] [Google Scholar]
- 46.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 47.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to Multiple Testing. Royal Statistical Society. 1995;57:289–300. [Google Scholar]
- 50.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Remm M, Storm CE. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 53.Alexeyenko A, Tamas I, Liu G, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs shared by multiple proteomes. Bioinformatics. 2006;22:9–15. doi: 10.1093/bioinformatics/btl213. [DOI] [PubMed] [Google Scholar]
- 54.Consortium TTG. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denoeud F, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science. 2014;345:1181–1184. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- 56.Xu H, et al. Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant. 2016;9:949–952. doi: 10.1016/j.molp.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L, et al. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 2014;15:1–13. doi: 10.1186/gb-2014-15-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hellsten U, et al. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl. Acad. Sci. USA. 2013;110:19478–19482. doi: 10.1073/pnas.1319032110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang S, et al. Draft genome of the kiwifruit Actinidia chinensis. Nat. Commun. 2013;4:2640. doi: 10.1038/ncomms3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaillon O, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 61.Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 63.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of Phyml 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 64.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 65.Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 67.Yang Z, Wong W, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.