

Abstract
The mycalesine butterfly Bicyclus anynana, the “Squinting bush brown,” is a model organism in the study of lepidopteran ecology, development, and evolution. Here, we present a draft genome sequence for B. anynana to serve as a genomics resource for current and future studies of this important model species. Seven libraries with insert sizes ranging from 350 bp to 20 kb were constructed using DNA from an inbred female and sequenced using both Illumina and PacBio technology; 128 Gb of raw Illumina data was filtered to 124 Gb and assembled to a final size of 475 Mb (∼×260 assembly coverage). Contigs were scaffolded using mate-pair, transcriptome, and PacBio data into 10 800 sequences with an N50 of 638 kb (longest scaffold 5 Mb). The genome is comprised of 26% repetitive elements and encodes a total of 22 642 predicted protein-coding genes. Recovery of a BUSCO set of core metazoan genes was almost complete (98%). Overall, these metrics compare well with other recently published lepidopteran genomes. We report a high-quality draft genome sequence for Bicyclus anynana. The genome assembly and annotated gene models are available at LepBase (http://ensembl.lepbase.org/index.html).
Keywords: bicyclus anynana; squinting bush brown; nymphalidae, nymphalid; satyrid; lepidopteran genome
Data Description
The squinting bush brown butterfly, Bicyclus anynana, is a member of the remarkably speciose nymphalid subtribe Mycalesina, which is distributed across the Old World tropics (Fig. 1). B. anynana is an important model organism for the study of lepidopteran ecology, development, speciation, behaviour, and evolution [1–6]. B. anynana are found primarily in woodland habitats across East Africa (from southern Sudan in the north to Swaziland in the south), and adults are typically observed flying close to the ground, where they feed on fallen fruit [1]. Strikingly, B. anynana exhibits seasonal polyphenism, a form of phenotypic plasticity whereby individuals that develop during the wet season differ in behaviour, appearance, and life history to those that develop during the dry season [7–9]. Wet season butterflies are smaller, have shorter lifespans, are more active, and show larger and more conspicuous eyespots on their wings in comparison to dry season individuals. The genetic basis of this plasticity and its impacts on various other life history and developmental characteristics are ongoing research questions to which the availability of a B. anynana reference genome will contribute [10–12].
Figure 1:

Wet-season morph of Bicyclus anynana (picture credit: William H. Piel and Antónia Monteiro).
Table 1:
Data counts and library information.
| Library type | Platform | Read length | Insert size (expected) | Number of reads (raw) | Number of reads (trimmed) | Number of bases (trimmed) | SRA run accessions |
|---|---|---|---|---|---|---|---|
| Short insert | Illumina HiSeq2500 | 125 bp paired-end | 350 bp | 271 808 057 pairs | 267 241 712 (98.3%) | 66 334 099 834 (97.6%) | ERR1102671-2, ERR1102675-6 |
| Short insert | Illumina HiSeq2500 | 125 bp paired-end | 550 bp | 241 050 065 pairs | 234 269 871 (97.2%) | 57 913 474 128 (96.1%) | ERR1102673-4, ERR1102677-8 |
| Mate pair | Illumina HiSeq2500 | 100 bp paired-end | 3 kb | 77 105 680 pairs | 31 848 200 (41.3%) | 5 758 856 502 (37.3%) | ERR1750945 |
| Mate pair | Illumina MiSeq | 100 bp paired-end | 3 kb | 5 641 764 pairs | 2 170 610 (38.5%) | 397 993 018 (35.3%) | ERR754051 |
| Mate pair | Illumina HiSeq2500 | 100 bp paired-end | 5 kb | 77 614 870 pairs | 45 676 725 (58.9%) | 8 203 769 131 (52.8%) | ERR1750946 |
| Mate pair | Illumina MiSeq | 100 bp paired-end | 5 kb | 7 939 601 pairs | 4 734 000 (59.6%) | 861 352 793 (54.2%) | ERR754052 |
| Long read | PacBio P6 | 0.80–50 kb | 10 kb | 1 388 796 | 1 199 064 (86.3%) | 4 086 394 966 | ERR1797559-74 |
Sampling and sequencing
Genomic DNA was extracted from a B. anynana female that had been inbred via 7 generations of brother-sister matings. The captive laboratory stock population from which these individuals originated was established in 1988 from 80 wild-caught individuals and has been maintained at large effective population sizes to minimise the loss of genetic diversity [1]. Two short-insert libraries with insert sizes of 350 and 550 bp were constructed using Illumina TruSeq Nano reagents and sequenced (125 base, paired-end) on an Illumina HiSeq2500 at Edinburgh Genomics (Edinburgh, UK). DNA from a sister to this focal animal was used to construct four long-insert (mate-pair) libraries with insert sizes of 3 and 5 kb (2 of each) at the Centre for Genomic Research, University of Liverpool (Liverpool, UK); libraries of both insert-sizes were then sequenced on an Illumina HiSeq2500 and an Illumina MiSeq at Edinburgh Genomics (Table 1). DNA from a female descendent of the same inbred line was used to construct 2 long read libraries with insert sizes of 10 and 20 kb, sequenced on the PacBio platform at the Genome Institute of Singapore at ∼×10 coverage using 16 P6 SMRT cells. All raw data have been deposited in the Short Read Archive under the accessions given in Table 1.
A total of 128.2 Gb of raw Illumina data was filtered for low-quality bases and adapter contamination using Skewer v. 0.2.2 [13], and both raw and trimmed reads were inspected using FastQC v. 0.11.4 [14]. Only 4 Gb of data (3.1%) was discarded, indicating the high quality of the raw data. Kmer frequency distributions were estimated using the “kmercountexact” program from the BBMap v. 36.02 package [15] and showed 2 major coverage peaks at ∼×105 and ∼×210 (Fig. 2). The first peak (×105) represents the proportion of the genome that is heterozygous and has an approximate span of 87.7 Mb (18.4% of the genome; calculated as one-half of the area under the ×105 curve, from ×50 to ×150). The expected proportion of heterozygous sites given 7 brother-sister (full-sib) matings is 0.75⁁7 = 13.3%, or 63.5 Mb. Thus, the greater than expected heterozygosity is likely to be due primarily to selection against highly inbred individuals during the course of the inbreeding regime [16].
Figure 2:
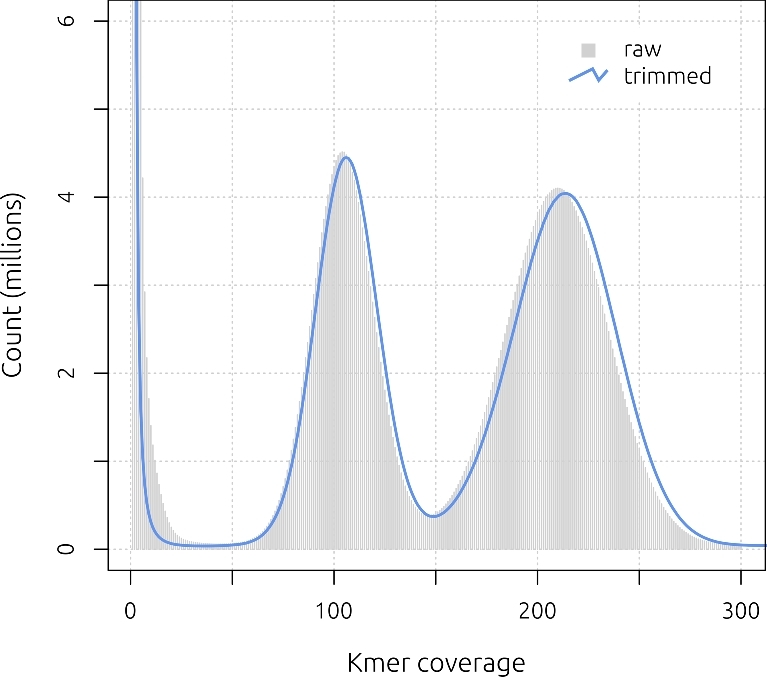
Kmer frequency distribution for B. anynana short-insert libraries (k = 31). The bimodality of the distribution, with peaks at approximately ×105 and ×210, is the result of heterozygosity in the sequence data.
Figure 3:

Taxon-annotated GC-coverage plots for (a) draft and (b) final B. anynana genome assemblies. Each contig/scaffold in the assembly is represented by a circle, coloured according to the best match to taxonomically annotated sequence databases (see legends) and distributed according to the proportion GC (x-axis) and read coverage (y-axis). The upper- and right-hand panels show the distribution of the total span (kb) of contigs/scaffolds for a given coverage (upper panel) or GC (right panel) bin. The heterozygosity in the sample is evident in the bimodal coverage distribution seen in (a). The cluster of orange-coloured contigs at a lower coverage and higher GC than the main cloud were likely derived from contaminant Enterococcus present in the sample. The final assembly (b) shows the effective collapse of heterozygous regions, the removal of contaminant sequences, and the scaffolding of contigs into long contiguous sequences. Note that only taxon annotations with a span > 1 Mb are shown in the legend for clarity.
Contaminant filtering and assembly
Short-insert libraries were screened for the presence of contaminant reads using Taxon-Annotated GC-Coverage (TAGC) plots, or “blobplots” [17]. An initial draft assembly was constructed using the CLC assembler (CLCBio, Copenhagen) and compared to the NCBI nucleotide database (nt) using Megablast v. 2.3.0+ [18], and against the UniRef90 protein database using Diamond v. 0.7.10 [19]. Read coverage for each contig was calculated by mapping both libraries to the CLC assembly using CLC mapper (CLCBio, Copenhagen), and blobplots were generated using Blobtools v. 0.9.19.4 [20] using the “bestsumorder” rule for taxonomic annotation of contigs (Fig. 3). Contigs that showed a substantially different coverage relative to that of the main cluster of contigs and/or good hits to sequences annotated as non-Arthropoda were classed as putative contaminants. A total of 237 394 pairs of reads (∼59 Mb) that were classed as either “mapped/mapped” or “mapped/unmapped” to a putative contaminant were subsequently discarded from further analysis.
Filtered libraries were reassembled using the heterozygous-aware assembler Platanus v. 1.2.4 [21], with default parameters. Contigs were further scaffolded with the mate pair libraries using SSPACE v. 3.0 [22] and with 35 747 assembled B. anynana transcripts [23] using a combination of L_RNA_scaffolder [24] and SCUBAT v. 2 [25]. A final round of scaffolding was performed with PacBio long reads (fastq files error-corrected using the RS_Preaassembler.2 protocol) using SSPACE-LongRead v. 1.1 [26]. Finally, gaps between scaffolds were filled using GapFiller v. 1.10 [27] and PBJelly v. 15.8.24 [28].
Our final assembly (v. 1.2) comprised 10 800 scaffolds spanning a total of 475.4 Mb, with a scaffold N50 of 638 kb (Table 2). The genome-wide proportion of G+C was 36.5%, while the number of undetermined bases (Ns) was 5.8 Mb (∼1.2% of the total span). We determined assembly completeness by mapping both genomic and transcriptomic reads from B. anynana (SRA whole genome sequencing accessions ERR1102671-8 and transcriptome accessions ERR1022636-7, ERR1022640-1, and ERR1022644-5, downloaded October 2016) to the genome using BWA mem v. 0.7.12 [29] and STAR v. 020201 [30], respectively. Over 99% of reads from the 2 short-insert libraries mapped to the assembly, suggesting that the vast majority of the genome represented by these data has been assembled. In addition, 94.9% of RNA-Seq reads mapped to the assembly, suggesting that the majority of transcribed genes are present. Gene-level completeness was assessed using CEGMA v. 2.5 [31] and BUSCO v. 2.0 [32]. The proportion of CEGMA genes “completely” recovered (n = 248) was 81%, increasing to 97% when partially recovered genes were included. The recovery of BUSCO genes specific to the metazoa (n = 978) was higher, at 98% for complete genes, increasing to 99% when partial genes were included. An almost complete set (99.2%) of BUSCO genes specific to the Arthropoda (n = 1066) was also recovered. In addition, CEGMA indicated a duplication rate of 1.1 while BUSCO estimated only ∼2% of genes were present in multiple copies. The high complete CEGMA/BUSCO scores suggestthat a good assembly has captured the majority of core metazoan/Arthropod genes in full length and that the fragmentation of genes across multiple scaffolds is low. In addition, the low duplication rates suggest that most genes are present in single copy, and thus that the genome does not include significant duplicated segments representing alternative haplotypes.
Table 2:
Summary of B. anynana genome assembly and comparison to selected lepidopteran genomes.
| B. anynana | B. mori | D. plexippus | H. melpomene | M. cinxia | |
|---|---|---|---|---|---|
| Assembly version | 1.2 | ASM15162v1 | 3 | Hmel2 | MelCinx1.0 |
| Span | 475.4 Mb | 481.8 Mb | 248.6 Mb | 275.2 Mb | 389.9 Mb |
| Contigs | |||||
| Number | 23 699 | 88 673 | 10 682 | 3100 | 48 180 |
| N50a | 78.7 kb | 15.5 kb | 111.0 kb | 328.9 kb | 14.1 kb |
| NumN50b | 1543 | 8075 | 548 | 214 | 7366 |
| Scaffolds | |||||
| Number | 10 800 | 43 379 | 5397 | 795 | 8261 |
| N50 | 638.3 kb | 4008.4 kb | 715.6 kb | 2102.7 kb | 119.3 kb |
| NumN50 | 194 | 38 | 101 | 34 | 970 |
| N90 | 99.3 kb | 61.1 kb | 160.5 kb | 273.1 kb | 29.6 kb |
| NumN90 | 909 | 258 | 366 | 176 | 3396 |
| Shortest/longest | 201 b/5 Mb | 53 b/16.2 Mb | 300 b/6.2 Mb | 394 b/9.4 Mb | 1.5 kb/668 kb |
| G+C content | 36.5% | 37.7% | 31.6% | 32.8% | 32.6% |
| NNNs | |||||
| Span | 5.8 Mb (1.2%) | 50.1 Mb (10.4%) | 6.7 Mb (2.7%) | 986 kb (0.4%) | 28.9 Mb (7.4%) |
| N50 | 1.4 kb | 5.0 kb | 2.5 kb | 2.4 kb | 1.4 kb |
| CEGMAc (n = 248) | C: 81.1%; D: 1.1; F: 97.2% | C: 76.6%; F: 96.8% | C: 90.3%; F: 96% | C: 88.7%; F: 96.8% | NA |
| BUSCOc (n = 1066) | C: 98.3%; D: 1%; F: 99.2% | C: 97.5%; D: 0.5%; F: 98.4% | C: 97.4%; D: 8.6%; F: 98.5% | C: 98.8%; D: 0.7%; F: 99.3% | C: 85.7%; D: 0.2%; F: 91.8% |
aN50: the length of the contig/scaffold at which 50% of the genome span is accounted for, given a list of sequences sorted by length. bnumN50: the number of sequences required to reach the N50 sequence. cCEGMA/BUSCO notation: C, proportion (%) of genes completely recovered; D, duplication rate; F, proportion (%) of genes at least partially recovered (including complete genes); n, number of queries. Note that duplication rate (D) for CEGMA is given as the average number of (complete) genes recovered, whereas for BUSCO it is the proportion of complete genes recovered multiple times. BUSCO values are based on comparisons to the Arthropoda gene set.
Annotation
Prior to gene prediction, we masked the B. anynana assembly for repetitive elements to minimise the number of spurious open-reading frames due to low-complexity repeat regions or transposable elements. Repetitive motifs in the B. anynana assembly were modelled ab initio using RepeatModeler v. 1.0.5 (http://www.repeatmasker.org/RepeatModeler.html). Repeats occurring within genuine coding regions were excluded by querying the proteins from a previous B. anynana assembly (v. 0.1) versus the RepeatModeler database using BLAST, removing any sequences showing a match at the E-value ≤ 1e-10 threshold. The filtered RepeatModeler database was combined with known repeats from the Lepidoptera using RepBase v. 20.05 [33] and input to RepeatMasker v. 4.0.5 [34] to mask the assembly. Overall, approximately one-quarter of the assembly (122.6 Mb) was masked from gene prediction (Table 3).
Table 3:
Major types of repeat content for B. anynana.
| Repeat type | Span (Mb) | Proportion of genome |
|---|---|---|
| SINE | 10.8 | 2.3% |
| LINE | 15.3 | 3.2% |
| LTR elements | 1.1 | 0.2% |
| DNA elements | 0.8 | 0.2% |
| Small RNA | 10.8 | 2.3% |
| Unclassified | 86.2 | 18.1% |
| Total | 122.6 | 25.8% |
Gene finding was performed following a 2-pass approach [35]. Initial gene models were constructed with MAKER v. 2.31 [36] using HMMs derived from SNAP [37] and GeneMark-ES v. 4.3 [38] in conjunction with a recently published B. anynana transcriptome as evidence. MAKER gene models were then passed to AUGUSTUS v. 3.0.3 [39] for refinement, resulting in an initial set of 26 722 predicted protein-coding genes. A set of basic filters was applied to remove likely spurious gene models (Table 4), resulting in the deletion of 4080 gene models. Protein sequences from the filtered 22 642 genes were annotated using BLAST searches versus UniRef90 and the NCBI non-redundant protein database (nr), and domains/motifs were described using InterProScan5 [40]. Summary statistics for the 22 642 predicted gene models are given in Table 5.
Table 4:
Number of genes in potential error categories.
| Category | Description | Number of genes |
|---|---|---|
| (a) | Single-exon | 7112 |
| (b) | Small exon (<9bp) | 1866 |
| (c) | Small intron (≤40 bp) | 45 |
| (d) | Short (CDS < 120 bp) | 127 |
| (e) | No hit to nr | 6532 |
| (f) | Duplicate (≥98% identity over ≥98% query length) | 822 |
| Totala | 4080 |
aDefined as the non-redundant total of the intersection of each category (a) to (d) with category (e), plus the shorter of any duplicates identified in category (f).
Table 5:
Summary of B. anynana gene prediction.
| B. anynana | B. mori | D. plexippus | H. melpomene | M. cinxia | |
|---|---|---|---|---|---|
| Assembly version | 1.2 | ASM15162v1 | 3 | Hmel2 | MelCinx1.0 |
| Number of CDS | 22 642 | 19 618 | 15 130 | 13 178 | 16 668 |
| Mean length | 1.4 kb | 1.6 kb | 1.4 kb | 1.3 kb | 958 bp |
| Median length | 1.2 kb | 1.2 kb | 981 bp | 927 bp | 693 bp |
| Min/max | 84 bp/28.3 kb | 23 bp/60.3 kb | 9 bp/58.9 kb | 45 bp/46.4 kb | 6 bp/45.4 kb |
| Introns | |||||
| Mean number per gene | 4.4 | 9.9 | 5.7 | 5 | NAa |
| Length (mean/median) | 1.3/0.6 kb | 2.4/0.8 kb | 795/280 bp | 960/416 bp | NA |
| Exons | |||||
| Length (mean/median) | 208/126 bp | 283/161 bp | 206/149 bp | 284/157 bp | NA |
| Number of single-exon genes | 3571 | 1744 | 1461 | 3113 | NA |
| Transcript GC | 49.2% | 48.3% | 46.5% | 43% | 41.7% |
| Gene frequencyb (genes per Mb) | 47.7 | 32.1 | 60.9 | 55.5 | NA |
aGFF for M. cinxia not available. bDefined as the number of genes divided by the total genome span (Mb).
Comparison to other lepidopteran genomes
To ascertain the relative quality of the B. anynana v. 1.2 assembly, we compared our results to 9 other published lepidopteran genomes available on LepBase (http://lepbase.org/) [41]: Bombyx mori ASM15162 v. 1 [42], Danaus plexippus v. 3 [43], Heliconius melpomene Hmel2 [44,45], Lerema accius v. 1.1 [46], Melitaea cinxia MelCinx1.0 [47], Papilio glaucus v. 1.1 [48], Papilio polytes Ppol 1.0 [49], Papilio xuthus Pap_xu_1.0 [49], and Plutella xylostella DBM_FJ_v1.1 [50]. The B. anynana v. 1.2 assembly was of high quality compared to other published genomes, with the majority of the genome represented in a relatively small number of scaffolds despite being only marginally smaller than the largest lepidopteran genome, B. mori (Fig. 4a). Interestingly, B. anynana v. 1.2 encodes the highest number of proteins of the 10 species compared (Fig. 4b). Despite measures to eliminate potentially spurious ORFs caused by annotation error or by duplication, B. anynana encodes ∼3250 more genes than the diamondback moth P. xylostella, and ∼10 400 more than the swallowtail P. polytes. It is tempting to attribute the apparently high number of genes to the developmental plasticity and alternative seasonal forms with divergent morphologies and life histories in B. anynana. However, it remains to be determined whether the number of genes predicted in B. anynana is a function of its larger genome size or unusual life history characteristics, or if further curation of the v. 1.2 gene models will reduce the number of inferred genes.
Figure 4:

Assembly and gene prediction comparison among 10 lepidopteran genomes. (a) Cumulative assembly curves showing the relationship between the number of scaffolds (x-axis) and the cumulative span of each assembly (y-axis), coloured by species. Higher-quality assemblies are represented by an almost-vertical line (e.g., H. melpomene Hmel2 assembly in black), indicating that a relatively small number of scaffolds is required to reach the final genome span; conversely, a long tail indicates that the assembly includes a large number of smaller scaffolds. The curve for B. anynana (brown and bold) suggests a good assembly for this species, with the majority of the assembly comprised of relatively few scaffolds. (b)B. anynana v. 1.2 encodes the greatest number of genes of the 10 genomes and is particularly different from B. mori, which is of equivalent length. Species names/colours are as follows: “bicyclus” (brown), B. anynana; “bombyx” (blue), B. mori; “danaus” (light green), D. plexippus; “heliconius” (black), H. melpomene; “lerema” (dark green), L. accius; “melitaea” (orange), M. cinxia; “glaucus” (red), P. glaucus; “polytes” (pink), P. polytes; “xuthus” (violet), P. xuthus; “plutella” (grey), P. xylostella.
Concluding remarks
We present a high-coverage, high-quality draft assembly and annotation of the mycalesine butterfly B. anynana. The assembly will be a core resource for ongoing analyses of population genomics, discovery of cis-regulatory elements of wing patterning and other genes, functional genetics and functional ecology of complex gene families, and the evolution of novel and plastic lifecycle strategies in lepidopterans and other arthropods.
Abbreviations
BUSCO: Benchmarking Universal Single-Copy Orthologs; CEGMA: Core Eukaryotic Genes Mapping Approach; CDS: coding sequence; ORF: open reading frame.
Supplementary Material
Acknowledgments
We thank Edinburgh Genomics and the Genome Institute of Singapore for genome sequencing, initial QC, and data delivery. We also thank 2 reviewers for helpful comments on a previous version of this manuscript. Funding for the Bicyclus anynana genome project was provided by the ERC Advanced Grant number 250325 (EMARES) to P.M.B. and by the South East Asian Biodiversity Genomics Center (NUS grants R-154-000-648-646 and R-154-000-648-733) to A.M. Funding for LepBase was provided by BBSRC grant number BB/K020161.
Availability of supporting data
All raw sequence data have been deposited in the Short Read Archive (SRA) and are available for download using the accession numbers provided in Table 1. The B. anynana v. 1.2 assembly, as well as final predicted gene models and protein annotations, are publicly available for viewing and download via LepBase [41], an Ensembl [51] genome database for the Lepidoptera (http://ensembl.lepbase.org/index.html). Data supporting the manuscript, including annotations as well as BUSCO and CEGMA results, are also available via the GigaScience database, GigaDB [52]. A previous B. anynana assembly (nBa.0.1) is also available on LepBase.
Competing interests
The authors declare that they have no competing interests.
Author contributions
P.M.B. and M.B. designed the study; A.M. and B.R.W. collected samples and produced the inbred line; A.E.V.H., I.J.S., and H.C. extracted DNA samples; R.W.N., B.E., and M.B. worked on the genome assembly and annotation; V.O., B.J.Z., C.W., and M.S. contributed transcriptome data; A.M., H.C., and M.L.A. contributed PacBio data; S.K. and R.J.C. uploaded the assembly to LepBase. R.W.N., V.O., A.M., P.M.B., and M.B. wrote the manuscript. All authors read and approved the final version of the manuscript.
References
- 1. Brakefield PM, Beldade P, Zwaan BJ. The African butterfly Bicyclus anynana: a model for evolutionary genetics and evolutionary developmental biology. Cold Spring Harb Protoc. 2009; doi:10.1101/pdb.emo122–2. [DOI] [PubMed] [Google Scholar]
- 2. Brakefield PM. Radiations of mycalesine butterflies and opening up their exploration of morphospace. Am Nat 2010;176(suppl 1):S77–87. [DOI] [PubMed] [Google Scholar]
- 3. Prudic KL, Jeon C, Cao H et al. . Developmental plasticity in sexual roles of butterfly species drives mutual sexual ornamentation. Science 2011;331:73–5. [DOI] [PubMed] [Google Scholar]
- 4. Westerman EL, Hodgins-Davis A, Dinwiddie A et al. . Biased learning affects mate choice in a butterfly. Proc Natl Acad Sci U S A 2012;109:10948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Monteiro A. Origin, development, and evolution of butterfly eyespots. Annu Rev Entomol 2015;60:253–71. [DOI] [PubMed] [Google Scholar]
- 6. Aduse-Poku K, Brakefield PM, Wahlberg N et al. . Expanded molecular phylogeny of the genus Bicyclus (Lepidoptera: Nymphalidae) shows the importance of increased sampling for detecting semi-cryptic species and highlights potentials for future studies. System Biodivers 2017;15:115–30. [Google Scholar]
- 7. Brakefield PM, Reitsma N. Phenotypic plasticity, seasonal climate and the population biology of Bicyclus butterflies (Satyridae) in Malawi. Ecol Entomol 1991;16:291–303. [Google Scholar]
- 8. Brakefield PM, Gates J, Keys D et al. . Development, plasticity and evolution of butterfly eyespot patterns. Nature 1996;384:236–42. [DOI] [PubMed] [Google Scholar]
- 9. Monteiro A, Tong X, Bear A et al. . Differential expression of ecdysone receptor leads to variation in phenotypic plasticity across serial homologs. PLoS Genet 2015;11:e1005529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beldade P, Mateus ARA, Keller RA. Evolution and molecular mechanisms of adaptive developmental plasticity. Mol Ecol 2011;20:1347–63. [DOI] [PubMed] [Google Scholar]
- 11. Oostra V, Brakefield PM, Hiltemann Y et al. . On the fate of seasonally plastic traits in a rainforest butterfly under relaxed selection. Ecol Evol 2014;4:2654–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dion E, Monteiro A, Yew JY. Phenotypic plasticity in sex pheromone production in Bicyclus anynana butterflies. Sci Rep 2016;6:39002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang H, Lei R, Ding S-W et al. . Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 2014;15:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andrews S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (1 May 2015, date last accessed).
- 15. Bushnell B. BBMap short read aligner, and other bioinformatic tools. https://sourceforge.net/projects/bbmap/ (4 June 2015, date last accessed).
- 16. Saccheri IJ, Brakefield PM, Nichols RA. Severe inbreeding depression and rapid fitness rebound in the butterfly Bicyclus anynana (Satyridae). Evolution 1996;50:2000–13. [DOI] [PubMed] [Google Scholar]
- 17. Kumar S, Jones M, Koutsovoulos G et al. . Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet 2013;4:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Altschul SF, Madden TL, Schaffer AA et al. . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12:59–60. [DOI] [PubMed] [Google Scholar]
- 20. Laetsch DR. Blobtools: application for the visualisation of draft genome assemblies and general QC. https://github.com/DRL/blobtools (15 July 2015, date last accessed).
- 21. Kajitani R, Toshimoto K, Noguchi H et al. . Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 2014;24:1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boetzer M, Henkel CV, Jansen HJ et al. . Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011;27:578–9. [DOI] [PubMed] [Google Scholar]
- 23. Oostra V, Saastamoinen M, Zwaan BJ et al. . Extensive phenotypic plasticity in a seasonal butterfly limits potential for evolutionary responses to environmental change. bioRxiv 2017; doi: https://doi.org/10.1101/126177 (23 June 2017, date last accessed). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue W, Li J-T, Zhu Y-P et al. . L_RNA_scaffolder: scaffolding genomes with transcripts. BMC Genomics 2013;14:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koutsovoulos G. SCUBAT2. https://github.com/GDKO/SCUBAT2 (25 July 2015, date last accessed).
- 26. Boetzer M, Pirovano W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 2014;15:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol 2012;13:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. English AC, Richards S, Han Y et al. . Mind the Gap: upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS One 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobin A, Davis CA, Schlesinger F et al. . STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genornes. Bioinformatics 2007;23:1061–7. [DOI] [PubMed] [Google Scholar]
- 32. Simão FA, Waterhouse RM, Ioannidis P et al. . BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- 33. Jurka J, Kapitonov VV, Pavlicek A et al. . Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005;110:462–7. [DOI] [PubMed] [Google Scholar]
- 34. Smit A, Hubley R, Green P. RepeatMasker. http://www.repeatmasker.org (8 August 2015, date last accessed).
- 35. Koutsovoulos G. CGP-Pipeline. https://gist.github.com/GDKO/ (8 August 2015, date last accessed).
- 36. Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 2011;12:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korf I. Gene finding in novel genomes. BMC Bioinformatics 2004;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ter-Hovhannisyan V, Lomsadze A, Chernoff YO et al. . Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 2008;18:1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stanke M, Diekhans M, Baertsch R et al. . Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008;24:637–44. [DOI] [PubMed] [Google Scholar]
- 40. Jones P, Binns D, Chang H-Y et al. . InterProScan 5: genome-scale protein function classification. Bioinformatics 2014;30:1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Challis RJ, Kumar S, Dasmahapatra KKK et al. . Lepbase: the Lepidopteran genome database. bioRxiv 2016; doi: http://dx.doi.org/10.1101/056994 (23 June 2017, date last accessed). [Google Scholar]
- 42. Duan J, Li R, Cheng D et al. . SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res 2010;38:D453–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhan S, Merlin C, Boore JL et al. . The monarch butterfly genome yields insights into long-distance migration. Cell 2011;147:1171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heliconius Genome Consortium Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 2012;487:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davey JW, Chouteau M, Barker SL et al. . Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 million years of butterfly evolution. G3 2016;6:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cong Q, Borek D, Otwinowski Z et al. . Skipper genome sheds light on unique phenotypic traits and phylogeny. BMC Genomics 2015;16:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahola V, Lehtonen R, Somervuo P et al. . The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nat Commun 2014;5:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cong Q, Borek D, Otwinowski Z et al. . Tiger swallowtail genome reveals mechanisms for speciation and caterpillar chemical defense. Cell Rep 2015;10:910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nishikawa H, Iijima T, Kajitani R et al. . A genetic mechanism for female-limited Batesian mimicry in Papilio butterfly. Nat Genet 2015;47:405–9. [DOI] [PubMed] [Google Scholar]
- 50. You M, Yue Z, He W et al. . A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet 2013;45:220–5. [DOI] [PubMed] [Google Scholar]
- 51. Yates A, Akanni W, Amode MR et al. . Ensembl 2016. Nucleic Acids Res 2016;44:D710–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nowell RW, Elsworth B, Oostra V et al. . Supporting data for “A high-coverage draft genome of the mycalesine butterfly Bicyclus anynana.” GigaScience Database. 2017; http://dx.doi.org/10.5524/100280 (23 June 2017, date last accessed). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.