

Abstract
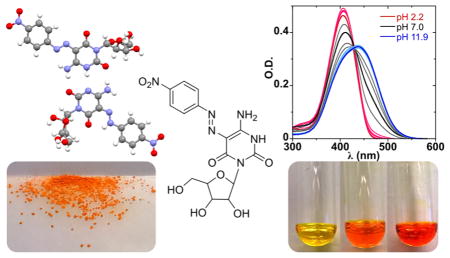
Nucleodyes, visibly colored chromophoric nucleoside analogues, are reported. Design criteria are outlined and the syntheses of cytidine and uridine azo dye analogues derived from 6-aminouracil are described. Structural analysis shows that the nucleodyes are sound structural analogues of their native nucleoside counterparts, and photophysical studies demonstrate that the nucleodyes are sensitive to microenvironmental changes. Quantum chemical calculations are presented as a valuable complementary tool for the design of strongly absorbing nucleodyes, which overlap with the emission of known fluorophores. Förster critical distance (R0) calculations determine that the nucleodyes make good FRET pairs with both 2-aminopurine (2AP) and pyrrolocytosine (PyC). Additionally, unique tautomerization features exhibited by 5-(4-nitrophenylazo)-6-oxocytidine (8) are visualized by an extraordinary crystal structure.
Graphical Abstract
INTRODUCTION
The ongoing challenge of studying nucleic acid and their cellular interactions, while minimally perturbing their constituents, has led to a growing interest in fluorescent nucleoside probes.1 The development of such emissive analogues has been triggered by the nonemissive nature of the natural nucleobases and has been accelerated by the increasing availability of highly sensitive fluorescence-based techniques.1b,2 Current efforts have concentrated on the development of smaller and responsive probes, displaying higher isomorphicity and sensitivity to polarity as well as favorable red-shifted absorption and emission bands.3,4 However appealing, efforts to develop fluorescent nucleoside probes are often hampered by synthetic challenges and the unpredictability of their spectroscopic properties, particularly features pertinent to their excited-state dynamics.5 Furthermore, the fluorescence of emissive nucleosides is frequently substantially quenched upon incorporation into oligonucleotides and even further upon duplex formation,1a,5a,6 rendering this development and use a tedious and empirical process.
The design and implementation of a visibly colored nucleoside probe, on the other hand, could be more straightforward since absorption properties are easier to predict a priori and can be inspired by known chromophores with high molar extinction coefficients. Although not as advantageous as having visible emission with little background interference, intense dyes can nevertheless become useful as sensitive solvatochromic probes and fluorescence quenchers for FRET-based constructs.7
Azo-based dyes are obvious candidate chromophores since they display high molar extinction coefficients and are established as dyes and indicators as well as photoswitches and fluorescence quenchers.8 Indeed, azo dyes have been incorporated into oligonucleotides and used mainly as photoswitches and quenchers.9 Their incorporation methodologies vary and include replacement of the natural nucleobases10 as well as conjugation through linkers to the sugar hydroxyl groups11 or to the nucleobase.12 While there are many examples of nucleoside azo dye derivatives, in only a selected few the nucleobase is electronically communicating to the azo-based chromophore.13
The fundamental design of nucleodyes outlined here requires electronic conjugation between the dye moiety and the heterocyclic nucleobase. In this fashion, one enhances the likelihood that the favorable and tunable photophysical features of azo dyes, including their solvatochromism and sensitivity to pH, become electronically coupled to the nucleobase’s environment. The key principle for the design and successful implementation of nucleodyes is to utilize visibly absorbing and responsive chromophores with high molar absorptivity while attempting to minimize structural perturbations of the native nucleobase. The established azo chromophore is a good candidate since one of the phenyl rings in the archetypal azobenzene core can be replaced by a native pyrimidine. Electronic communication and sensitivity to microenvironmental changes may be achieved by creating a conjugated donor–acceptor relationship with the polarizable nucleobase between a substituent on the phenyl ring and one of the heteroatoms of the nucleobase (Figure 1). To fulfill such a requirement in a uridine core, phenyl azo substitution at the 6 position should have an electron-donating substitution, thereby electronically communicating with the carbonyl at the 4 position (Figure 1a). Alternatively, phenyl azo substitution at the 5 position should have an electron-withdrawing substituent, thus interacting with N1 or with an electron-rich substituent at the 6 position (Figure 1b,c). In this fashion and assuming this does not impact the tautomeric preference, the uracil base-pairing face remains intact and capable of Watson–Crick base pairing.
Figure 1.

Design strategy for three distinct uridine-based azo nucleodyes. Uracil replaces one of the phenyl rings in the archetypal azobenzene core, and a donor–acceptor relationship is introduced between a substituent on the phenyl ring and one of the heteroatoms of the nucleobase. R = ribofuranose or 2′-dexoyribofuranose.
In the current study, we exploit the versatility of 6-aminouracil to obtain both uridine and cytidine azo dye analogues. We prepared both 6-amino-5-(4-nitrophenylazo)-uridine (5) and 5-(4-nitrophenylazo)-6-oxocytidine (8) as new pyrimidine-based nucleodyes derived from 6-aminouracil. Their structural and photophysical features are characterized as well as their responsiveness to microenvironmental changes including polarity and pH. Our observations are supplemented by quantum chemical calculations of absorptive properties, which nicely predict these compounds to be highly absorbing visible dyes. We demonstrate that small nucleoside-based quenchers can be designed to electronically match emissive nucleosides and other known fluorescent probes.
RESULTS
Synthesis
Vorbrüggen glycosylation of 6-aminouracil (1) and N6-DMF 6-aminouracil (2) with β-D-ribofuranose 1-acetate 2,4,5-tribenzoate, followed by removal of all protecting groups in methanolic ammonia, gave 6-oxocytidine (7) and 6-aminouridine (4), respectively (Scheme 1). Their structures were confirmed by X-ray crystallography (Figure S1). The nucleosides were reacted with 4-nitrobenzenediazonium chloride under standard conditions to give the desired 6-amino-5-(4-nitrophenylazo)uridine (5) and 5-(4-nitrophenyla-zo)-6-oxocytidine (8), which were fully characterized by 1H and 13C NMR spectroscopy as well as by HRMS and X-ray crystallography (Figure 2).
Scheme 1. Synthesis of 6-Amino-5-(4-nitrophenylazo)uridine (5) and 5-(4-Nitrophenylazo)-6-oxocytidine (8)a.

aReagents and conditions: (a) dimethylformamide dimethyl acetal, DMF, 60 °C; (b) (i) N,O-bis(trimethylsilyl)acetamide, DCE, 50 °C; (ii) β-D-ribofuranose 1-acetate 2,4,5-tribenzoate, TMSOTf, DCE, 84 °C; (c) NH3/MeOH, 60 °C; (d) 4-nitrobenzenediazonium chloride, H2O. See the Experimental Section for procedures and analytical data.
Figure 2.

X-ray crystal structures: (a) 6-amino-5-(4-nitrophenylazo)uridine (5); (b) solid-state structure of 8 showing two tautomeric forms, 5-(4-nitrophenylazo)-6-oxocytidine (8a) and 5-(4-nitrophenylhydrazono)-6-oxocytidine (8b); (c) schematic depiction of the two tautomeric forms in the crystal packing illustrating the Watson–Crick-like base pairing; (d) overlap of the pyrimidine core of the two tautomers clearly visualizing the relative rotation of the ribose and the 4-nitrophenylazo substitutions. 8a in blue and 8b in orange. See the Supporting Information for side views (Figures S2 and S3).
1H NMR spectroscopy and the crystal structure of 5 suggest a single tautomer in both solution and the solid state (Figure S24 and Figure 2a). In contrast, the 1H NMR spectrum of pure 8 in DMSO-d6 showed the presence of two species in a ~7:3 ratio, suggesting two tautomeric forms (Figure S30). This was further supported by the addition of acid or base, as the two species merged into one (Figures S32 and S33). Initial crystallization attempts in strong acidic conditions (ACN/EtOH/HCl or TFA/water) resulted in deamination to give crystals of 5-(4-nitrophenylhydrazono)-6-oxouridine (9), which was further characterized by HRMS (Scheme 2). Crystallization of 8 under mild acidic conditions (AcOH/EtOH) resulted in cocrystallization of the two tautomeric forms, 5-(4-nitro-phenylazo)-6-oxocytidine (8a) and 5-(4-nitrophenylhydrazo-no)-6-oxocytidine (8b) (Figure 2b), providing structural validation of the anticipated product, the substituted 6-amino-2,4(1H,3H)-pyrimidinedione analogue 8a, as well as the structure of its C-face analogue tautomer 8b as indicated by the 1H NMR spectra.
Scheme 2.

Crystallization of 5-(4-Nitrophenylazo)-6-oxocytidine (8) in Acidic Media Yields 5-(4-Nitrophenylhydrazono)-6-oxouridine (9)
Photophysical Evaluation
Ground-state absorption spectra of 5 and 8 in phosphate buffer (pH 7) show maxima at 397 nm (ε = 2.9 × 104 M−1 cm−1) and 411 (ε = 3.5 × 104 M−1 cm−1), respectively (Figures 3a and 3c). Compounds 5 and 8 exhibit favorable red-shifted absorption bands and higher extinction coefficients compared to 4-nitroazobenzene. The latter exhibits an absorption maximum at 338 nm (ε = 2.62 × 104 M−1 cm−1) in EtOH14 and 330 nm (ε = 2.56 × 104 M−1 cm−1) in cyclohexane.15 The spectral data measured for 5 and 8 in MeOH [397 nm (ε = 3.1 × 104 M−1 cm−1) and 402 nm (ε = 3.6 × 104 M−1 cm−1), respectively] and dioxane [405 nm (ε = 3.2 × 104 M−1 cm−1) and 402 nm (ε = 4.2 × 104 M−1 cm−1), respectively] clearly reflect substantial red-shifted wavelengths and higher extinction coefficients.
Figure 3.

(a) Absorption spectra as a function of pH for 6-amino-5-(4-nitrophenylazo)uridine 5 (5.9 × 10−6 M). See the Supporting Information for split figure (Figure S5). (b) Correlating absorption maxima to pH shows two transitions (pKa1 = 3.9 ± 0.2; pKa2 = 8.8 ± 0.1). (c) Absorption spectra as a function of pH for 5-(4-nitrophenylazo)-6-oxocytidine 8 (1.2 × 10−5 M). (d) Correlating absorption maxima to pH shows one major transition (pKa1 = 7.6 ± 0.1); a second one (pH < 2) may also be present. pKa values reflect the average over three independent measurements and are equal to the inflection point determined by Boltzmannic functions fitted to the curves in (b) and (d). See the Experimental Section for a detailed explanation.
To evaluate whether these nucleodyes are potentially capable of detecting microenvironmental changes through changes in absorbance wavelength and/or intensity, spectra in a wide range of pH and polarities were recorded.16 pH titration of 6-amino-5-(4-nitrophenylazo)uridine (5) (Figure 3a) reveals two deprotonation events (pKa1 = 3.9 ± 0.2 and pKa2 = 8.8 ± 0.1, Figure 3b). Acidic or basic changes to the neutral form result in a red shift of the absorption maximum. Acidic conditions (pH < 3.9) result in a sharper absorption band (λmax = 411 nm) with higher intensity and short tailing into the midvisible range, whereas basic conditions (pH > 8.8) exhibit a more predominant red shift (λmax = 434 nm) together with spectral widening and tailing into the midvisible range (Figure 3a). The pH titration absorption spectra of 5-(4-nitrophenylazo)-6-oxocytidine (8) appear to show a two-state transition with a well-defined isosbestic point (λ ≅ 429 nm) (Figure 3c). Fitting the wavelength maxima to pH suggests a single deprotonation event near neutral pH (pKa1 = 7.6 ± 0.1) (Figure 3d).17
To assess the impact of polarity, the absorption spectrum of 6-amino-5-(4-nitrophenylazo)uridine (5) was recorded in MeOH–dioxane mixtures (Figure S6a). As the polarity increases, 5 exhibits a slight hypsochromic shift along with a minor hypochromic effect (Figure 4a). A good linear correlation is observed for the hypochromic effect in the MeOH–dioxane mixtures (Figure 4b). We note that data collected in pure solvents (dioxane, dichloromethane, acetonitrile, MeOH, and water) do not provide a clearer correlation, likely due to solvent specific effects (Figures S6b and S7). The absorption spectra of 5-(4-nitrophenylazo)-6-oxocytidine (8) show no wavelength change in a range of MeOH–dioxane mixtures. However, pure water shows a notable 10 nm bathochromic shift (Figure S8 and Figure 4c). A hypochromic effect is observed, and the correlation with relative intensity (including the pure water sample) appears to be sigmoidal, suggesting a polarity-dependent transition (ET30 = 54.2 ± 0.1 kcal/mol) (Figure 4d).
Figure 4.

(a, b) Assessing the effect of solvent polarity on absorption of 5 in MeOH–dioxane mixtures: correlation of absorption energy maxima in kcal/mol (a) and absorption intensity (at the absorption maxima) (b) with ET(30) values illustrate that as polarity increases 5 exhibits a hypsochromic shift (R = 0.88) along with a hypochromic effect (R = 0.97). (c, d) Assessing the effect of solvent polarity on absorption of 8: correlation of (c) absorption wavelength maxima with ET(30) values for MeOH–dioxane solutions (circle) and water (square) and (d) absorption intensity (at the wavelength maxima) with ET(30) values suggests a polarity dependent transition.
Theoretical Calculations
The spectroscopically silent ribose moiety was replaced with a methyl group to facilitate a faster geometry optimization of the nucleobase in all calculations. AM1 geometry optimizations predict the structures of 5, 8a, and 8b to be overall planar. This planarity is in good agreement with the X-ray crystal structures (Figures S2 and S3). The electronic spectra of the AM1-optimized structures of compounds 5, 8a, and 8b were calculated using ZINDO/S by including the 15 lowest unoccupied and 15 highest occupied orbitals. The results of these calculations are summarized in Table 1. The lowest energy transition of the three compounds is predicted to have a high molar absorptivity (oscillator strength, f, calculated to be approximately 1 for the three compounds). Moreover, the positions of the lowest energy band of compounds 5 (λ = 386 nm) and 8a (λ = 382 nm) are, as can be expected from their structural similarity, predicted to be essentially the same. The alternative tautomeric form of 8, 8b, has a lowest energy band predicted to be blue-shifted approximately 20 nm compared to compound 5 and 8a.
Table 1.
Three Lowest Electronic Transitions of 5, 8a, and 8b and Their Oscillator Strengths (f)a Calculated Using the ZINDO/S Method in HyperChem on an AM1 Geometry Optimized Structure
| λb (f) | λb (f) | λb (f) | |
|---|---|---|---|
| 5 | 386 (1.004) | 303 (0.094) | 275 (0.270) |
| 8a | 382 (0.992) | 306 (0.010) | 302 (0.090) |
| 8b | 363 (1.062) | 303 (0.034) | 271 (0.071) |
Only considering oscillator strengths (f) > 0.01.
in nm.
To assess the potential of the 6-aminouracil nucleodyes as quenchers, we calculated their Förster critical distance (R0) for the commonly used 2-aminopurine (2AP) and pyrrolocytosine (PyC) as potential fluorescent donors (Table 2). With an emission maximum of 371 nm and quantum yield (QY) of 0.68, the nucleoside d-2AP shows good spectral overlap with the nucleodyes (Figure 5), with a calculated R0 of 43 Å. PydC displays moderate spectral overlap with the nucleodyes (Figure 5). With an emission maximum of 462 nm and QY of 0.05, the calculated R0 for FRET pairing 5 and 8 is 26 and 28 Å, respectively. 2AP and PyC exhibit substantial and moderate quenching, respectively, of the fluorescence upon incorporation into oligonucleotides, and further quenching of fluorescence is also observed upon duplex formation.5a,6,18 This results in decreased R0 values for the two FRET pairs inside oligonucleotide systems (see Table 2). Our R0 estimation shows that 5 and 8 are good FRET pair candidates with either 2AP or PyC also following incorporation into oligonucleotides.
Table 2.
Calculated R0a Values for FRET Pairs of 5 and 8 with d-2AP and PydC
Figure 5.
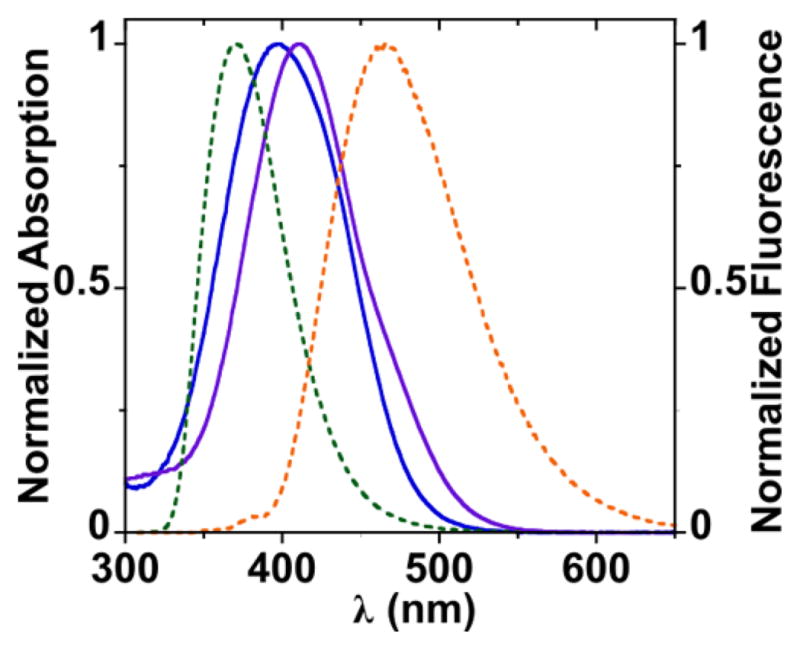
Normalized absorption (solid lines) and emission (dashed lines) spectra in water of 5 (blue), 8 (purple), d-2AP (green), and PydC (orange).
DISCUSSION
Synthetic Considerations and Strategy
The assembly of a uracil azo dye is influenced by the nature of the substituent on the phenylazo chromophore. Electron-donating phenylazo substitution at the 6 position (Figure 1a) could conceivably be constructed by diazotization of 6-aminouracil and subsequent reaction with electron-rich substituted benzene. Electron-withdrawing phenylazo substitution at the 5 position (Figure 1b,c) could be constructed differently by nucleophilic attack of the 5 position on a substituted benzenediazonium ion. Indeed, uracil reacts under basic conditions with benzenediazonium ion to give 5-phenylazouracil derivatives.19 However, 6-aminouracil does not react under diazotation conditions to give the corresponding uracil-6-diazonium salt, but rather yields the nitrosylation product at the highly nucleophilic 5 position.20 6-Aminouracil also readily reacts with benzenediazonium ions to afford 6-amino-5-phenylazouracil derivatives (Figure 1c).21
Although 5- and 6-phenylazouracil derivatives have been prepared, the corresponding nucleoside analogues have rarely been reported.13c,f–h Assembling a chromophore on a nucleoside with a native anomeric configuration is typically preferred over glycosylation of the modified heterocycle since the glycosylated nitrogen as well as the sugar configuration of the latter must then be confirmed by X-ray crystallography. Uridine has been reported to be unreactive toward benzenediazonium ion and does not give coupling or substitution products.19 However, the 5 position of 6-aminouridine is expected to be considerably more nucleophilic and should readily react to give 6-amino-5-phenylazouridine derivatives.
Glycosylation of 6-aminouracil takes place at the N3 to give the widely explored 6-oxocytidine.24 It has been reported, however, that glycosylation of N6 DMF-protected 6-amino-uracil has resulted in alkylation at the N1 position to give 6-aminouridine.25 Interestingly, assembling an electron-withdrawing phenylazo substitution at the 5 position of cytidine as well as 6-oxocytidine coincides nicely with our nucleodye design concept (Scheme 1 and Figure S9). Cytidine has also been reported to be unreactive toward benzenediazonium ion,19 and we therefore examined this reaction on 6-oxocytidine.
Considering the unreactivity of uridine and cytidine toward benzenediazonium ion19 and the preference to modify a nucleoside with a determined sugar configuration, the nucleophilicity of the 5 position and the versatility to obtain both uridine and cytidine derivatives makes 6-aminouracil a favorable precursor for assembling pyrimidine-based nucleodyes. To the best of our knowledge, this approach has been explored only once for the reported synthesis of 6-amino-5-(4-chlorophenylazo)uridine.13h
Synthesis
Protected nucleosides 3 and 6 were prepared from 6-aminouracil by modification of the reported procedures. The glycosylation of 1 has been reported as a two-step procedure involving prior activation in hexamethydisilazane (HMDS) followed by glycosylation with protected ribofuranose and trimethylsilyl triflate (TMSOTf) as catalyst for 24 h.24 The preparation of 3 has been reported starting from the activated silylated intermediate N6-(N,N-dimethylformimidamide)-O,O′-bis(trimethylsilyl)-6-aminouracil.25 The glycosylation was carried out under conditions similar to those reported for 1, and 94% yield of desired product 3 was reported as well as <5% of 6; however, full procedures were not disclosed for the activated starting material.
For the glycosylation of 1 and 2 we applied a two-step one-pot procedure. First, the nucleobase was activated with N,O-bis(trimethylsilyl)acetamide (BSA) in anhydrous 1,2-dichloro-ethane and then reacted with β-D-ribofuranose 1-acetate 2,4,5-tribenzoate in the presence of TMSOTf. Standard purification yielded 3 and 6 in moderate yields (54% and 70%, respectively). The glycosylation reaction of 2 gave also 6 as well as an N1 and N3 doubly glycosylated pyrimidine byproduct.
The DMF protection of the exocyclic amine of 6-aminouracil 1 was achieved using dimethylformamide dimethyl acetal overnight at 60 °C. However, 2 was found to be difficult to purify from unreacted starting material and byproducts and was thus used without purification. Attempts to drive the reaction to completion resulted in over-reaction byproducts and lower yields as determined by crude 1H NMR spectra of 2. It can be obtained in decent purity (with delectable amounts of starting material), albeit with lower yield, by washing the crude product with MeOH.
Structure Elucidation
The tautomerism of 6-amino-5-phenylazouracil derivatives has been a topic of interest and the focus of a recent report.21,26 The crystal structure and 1H NMR spectra of 5 indicate one tautomeric form corresponding to 6-amino-5-(4-nitrophenylazo)uridine. In contrast, two tautomeric forms were observed for 8 in both the solid state and solution. The chemical shifts observed in the 1H NMR spectra for the tautomeric hydrazone proton (=N–NH–) and amide proton (NH–C=O) correspond to the two tautomeric forms seen the crystal structure (Figure 2b and Figure S30).26a,27 One tautomer, 5-(4-nitrophenylazo)-6-oxocytidine (8a, Figure 2c), assumes a tautomeric form similar to that of 5 in which the azo group is hydrogen bonded to the neighboring exocyclic amine. In the other tautomer, 5-(4-nitrophenylhydrazono)-6-oxocytidine (8b, Figure 2c), the sugar, and the 4-nitrophenylhydrazone group are both rotated roughly 180° (183° ± 1° and 178° ± 1°, respectively), and the latter is hydrogen bonded to the neighboring exocyclic carbonyl (Figure 2c). The distinct solid-state conformation of the two tautomeric forms 8a and 8b is highlighted in Figure 2d. They are hydrogen bonded to one another in a Watson–Crick-like pairing (Figure 2b,c). The ~180° rotation of the 4-nitrophenyl hydrazone and the ribofuranose generates a pseudo symmetrical arrangement, which apparently enables crystal packing (Figure 2b). To the best of our knowledge, this is the first nucleoside to exhibit cocrystallization of two tautomeric forms that hydrogen bond to each other in a Watson–Crick base-pairing manner, and the only report of such example to include both tautomeric and conformational changes. The crystal structures of isocytosine28 and alloguanine29 show similar tautomeric Watson–Crick base pairing.
Crystal structures of 5 and the C-face tautomer 8b display an anti orientation at the glycosidic linkage similar to the preference seen with the native nucleobases uridine and cytidine. This is of significance since the pyrimidine core of these modified nucleosides is substituted at the neighboring 6 position. Indeed, 9 as well as the substituted 6-amino-2,4(1H,3H)-pyrimidinedione analogue tautomer 8a exhibit a syn orientation at the glycosidic linkage.
Overlaying the structures of uridine and 5 shows minimal root-mean-square deviation (rmsd) of the pyrimidine core (0.0416 Å, Figure S10a) but a notable impact on the sugar pucker (rmsd 0.33 Å, Figure S10b). The relatively close dihedral angle χ (−152.96° and −119.7° for uridine and 5, respectively) accounts for an overall moderate deviation of both the sugar and pyrimidine core (0.538 Å, Figure S10c). Overlaying the structures of cytidine and 8b shows minor rmsd of the sugar pucker (0.0743 Å, Figure S11b) and minimal deviation of the pyrimidine core (0.0486 Å, Figure S11a). The difference in the dihedral angle χ (−162.43° and −106.6° for cytidine and 8b, respectively) accounts for the deviation observed for the overlay of both the sugar and pyrimidine core (0.492 Å, Figure S11c). The divergence observed in the ribofuranose conformation as well as in the dihedral angle are likely a result of crystal-packing forces.30 The exclusive crystal structure of 8 demonstrates the influence of crystal packing and hydrogen bonding on the dihedral angle and molecular conformation of the ribofuranose. Importantly, the uridine and cytidine analogues 5 and 8b exhibit an anti orientation at the glycosidic linkage as well as a reasonable overlay of the native nucleoside core (ribose and nucleobase). These structural features are a good indication that these nucleodyes are likely to be viable structural surrogates for their native counterparts.
Photophysical Features
Nucleodyes 5 and 8 exhibit photophysical properties similar to those reported for the closely related chromophores 6-amino-5-(4-nitrophenylazo) uracil (unsubstituted) and 1,3-dimethyl-6-amino-5-(4-nitrophenylazo)uracil (disubstituted).21,26a The favorable spectral properties of nucleodyes 5 and 8 compared to 4-nitroazobenzene can be attributed to a stronger donor–acceptor interaction between the 6-aminouracil heterocycle and the nitro group as articulated above. The differences in the extinction coefficients between 5 and 8 and the notable changes with polarity compared to 4-nitroazobenzene confirm that the dye moiety is electronically communicating with the nucleobase and sensitive to its environment.
The pH titration absorption spectra of 5 suggests a two-state transition between the acidic and the neutral form reflected by a blue shift along with decreased intensity and widening of the Gaussian, generating two isosbestic points (λ ≅ 380, λ ≅ 447 nm). However, the spectra of the basic pH do not appear to share an isosbestic point with the neutral form (Figure 3a). Considering that each form may represent a contribution of absorption bands of several possible tautomers, classical spectral pH transitions with defined isosbestic points are not expected. Correlation of the wavelength maximum to pH clearly depicts two deprotonation events (Figure 3b). The first transition, from the acidic to neutral form (pKa1 = 3.9 ± 0.2), observed also by the isosbestic points, likely corresponds to the deprotonation of the protonated azo group. The second transition, from the neutral to basic form (pKa2 = 8.8 ± 0.1), likely corresponds to deprotonation of the N3 of the U base-pairing face. In contrast to the dramatic effect of pH, absorption spectra of 5 in pure solvents as well as in MeOH–dioxane mixtures exhibit a rather minimal wavelength and intensity changes (396–406 nm; ~10%, respectively).
The pH titration absorption spectra of 8 appear to reflect a two-state transition with a well-defined isosbestic point (λ ≅ 429 nm); however, a closer look reveals imperfect Gaussians above pH = 6 (Figure 3c). The transition observed likely corresponds to deprotonation of the two neutral tautomeric forms observed in the crystal structure and by 1H NMR in DMSO. The acidic points of the sigmoidal fit might hint to the presence of an event corresponding to the deprotonation of the protonated exocyclic amine. The slight difference in the pKa observed by fitting the wavelength maxima (pH = 7.6, Figure 3d) or by fitting the optical density values at specific wavelengths (pH = 7.0, Figure S4) is possibly due to a combination of tautomerization along with C-face deprotonation. Compound 8 exhibits a notable hypochromic effect with a large 19% intensity range as polarity changes (Figure S8). The sigmoidal fit of the relative intensities (Figure 4d) also likely corresponds to the tautomerization event and indicates the polarity at which the two tautomers are at a 1:1 ratio [ET(30) = 54.2 ± 0.1 kcal/mol]. The polarity of DMSO-d6 NMR sample and possibly a pure-solvent effect accounts for a roughly 7:3 ratio between the tautomers in the 1H NMR spectrum [ET(30) = 45.1 kcal/mol for pure DMSO].
The implementation of nucleodyes as visible-range probes ultimately depends on their ability to report microenvironmental changes. Nucleic acid binding and folding events are likely to result in local changes to polarity and possibly pH, which may be monitored by changes in the absorbance wavelength or intensity of the nucleodye probe. Both 5 and 8 are found to be sensitive to pH and polarity as reflected by changes in wavelength and/or intensity. Both compounds exhibit a notable hypochromic effect in water compared to solutions of reduced polarity with limited or no hydrogen bonding. However, the spectral changes induced by variations in polarity are relatively minor and changes in pH are clearly more significant. Importantly, the high extinction coefficients of the remote red-shifted maxima suggest that these single modifications can be conveniently detected (ABS = 0.05–0.1) at the low micro molar concentrations typical of biochemical assays.
Theoretical Calculations
Quantum chemical calculations may be used to approximate spectral properties and, thus, facilitate the synthesis of selected desired compounds that are likely to be highly absorbing visible dyes and useful acceptors in FRET pairs with fluorescent donor molecules.31 In general, calculations of absorptive properties of nucleobases have shown good correlation with experimental values.32 Such calculations featuring candidate nucleobase molecules have also been successfully used to predict fluorescence properties (kf)33. However, such predictions are strictly restricted to comparisons within a series of molecules built up from the same molecular scaffold and not introducing any moieties known to open up new excited-state deactivation pathways (knr) such as nitro groups. The potential of nucleodyes as sensitive solvatochromic probes and fluorescence quenchers renders prediction an attractive complementary tool for efficient design and synthesis of these compounds. Azo dyes are notorious for their high molar absorptivity; therefore, we merely used our calculations here to assess the lowest energy transition (S1–S0) wavelength and whether the 6-aminouracil nucleodyes are expected to have a high molar absorptivity.
The calculations predict that compounds 5 and 8a absorb at essentially the same wavelength (386 and 382 nm, respectively) and will have similar and high molar absorptivity (oscillator strengths are both close to 1). Previous calculations on azobenzene derivatives also predicted oscillator strengths around or slightly above 1.34 Generally, calculations like the ones we have used here overestimate the energy needed for the lowest energy transition, and correction factors of approximately 0.9 have previously been reported for nucleobases.32b Taking this correction into account, lowest energy peaks for 5 and 8a at wavelengths around or slightly above 400 nm can be expected. This is in good agreement with our experimental finding for 5 and 8 (λ = 397 and 411 nm, respectively; Figure 3). To be more exact, the wavelength value calculated for the dominant tautomer 8a should be compared to the absorption peak maximum of the form of 8 that exists at low pH26a in Figure 3c (λ = 405 nm). Moreover, the calculated high oscillator strengths (f) of 5 and 8a excellently predict the high molar absorptivity recorded for the two compounds (29000 and 35000 M−1 cm−1, respectively). Also, the hydrazo form of 8, 8b, that was calculated after it was identified in the solid state, is predicted to have a high molar absorptivity (f ≈ 1) and a lowest absorption peak slightly blue-shifted compared to 8a and 5 (λ = 363 nm). The mixture of 8a and 8b is expected to increase the complexity of the absorption spectra (Figure 3c), thus making a direct comparison of the calculated lowest absorption band of 8a and measurement even more challenging.
FRET-based techniques have become widely popular for studying biomolecule structure and function owing to the high sensitivity of the method to relative changes in distance and orientation of the FRET pair donor and acceptor.35 Much effort has been put forth to develop new fluorescent probes as well as fluorescent nucleoside analogues. However, with the exception of the FRET pair tC°/tCnitro,7 little effort has been made to develop FRET acceptor nucleosides to match existing FRET donors. The fundamental requirement of a FRET pair is spectral overlap of the donor emission and the acceptor absorption and with knowledge from that the Förster critical distance (R0) can be accurately calculated.
2AP and PyC are two of the most commonly used fluorescent nucleoside analogues.6,36 2AP is of specific interest as it is known to pair with uridine and also with cytidine.37 The R0 calculations suggest that 5 and 8 are suitable FRET acceptors for 2AP and PyC, indicating their potential application in monitoring folding or hybridization events within oligonucleotides. Nucleodyes, owing to their large extinction coefficients, are excellent candidates for the design of FRET acceptors to match specific existing donors. We find the quantum chemical calculations of spectra of azo-compounds advantageous for the prediction of the position of the lowest energy band and its relative intensity, and thus, they represent a valuable tool in designing new FRET acceptor nucleodyes for existing FRET donors.
CONCLUSIONS
We present the concept of nucleodyes, visibly colored chromophoric nucleoside analogues in which the base-pairing face is part of the chromophore, thus influencing its photophysical properties through donor/acceptor electronic conjugation. We demonstrate that theoretical calculations can, in principle, facilitate the design of strongly absorbing nucleodyes, which overlap with the emission of known fluorophores.
Starting from 6-aminouracil, two pyrimidine nucleodyes were prepared and studied for their potential as visible-range probes. Crystal structure analysis indicates that both 6-amino-5-(4-nitrophenylazo)uridine (5) and 5-(4-nitrophenyl-hydrazono)-6-oxocytidine (8b) are good structural analogues of their native nucleoside counterparts. We recognize that the use of these nucleodyes as visible-range probes might be limited as the impact of pH and polarity on their photophysical features indicates that microenvironmental changes might be difficult to distinguish. We note, however, that the photophysical features of azo nucleodyes may be enhanced and/or fine-tuned by changing the substituent on the phenyl ring. The prospect of monitoring nucleic acid interactions through microenvironmental changes of the nucleodyes in the visible range remains appealing since UV–vis absorption spectrometers are widely common and affordable. In addition, such nucleodyes may become complementary to fluorescent probes since they can be custom designed as FRET acceptors to match existing donors.
EXPERIMENTAL SECTION
Materials and Methods
All reagents and solvents were purchased from commercial suppliers and used without further purification unless otherwise specified. Anhydrous solvents were purchased from commercial suppliers or dried by standard techniques. Spectroscopic-grade solvents and deuterated NMR solvents were purchased from commercial suppliers. All experiments involving air-and/or moisture-sensitive compounds were carried out under an argon atmosphere. All reactions were monitored with analytical TLC 60 F254. Column chromatography was carried out with silica gel particle size 40–63 μm. NMR spectra were recorded on 300 and 500 MHz spectrometers. Mass spectra were recorded on LR-ESI and HR-ESI-TOF mass spectrometers.
All photophysical values reflect the average of at least three independent measurements. Absorption spectra were measured with 1 nm resolution on a UV–vis spectrophotometer and corrected for the blank. A 1 cm four-sided Helma quartz cuvette was used, and the sample temperature was kept constant at 20 °C using a thermostat-controlled ethylene glycol–water bath fitted to a specially designed cell holder. The sensitivity of the nucleodyes to changes in pH was studied in aqueous phosphate buffers (10 mM phosphate, 100 mM NaCl) with pH values ranging from 2 to 12. The samples were prepared from concentrated DMSO stock solutions and contained up to 0.4 vol % DMSO. For 5-(4-nitrophenylazo)-6-oxocytidine 8 (Figure 3d), a triplicate pH titration was performed with unchanged solutions of different pH. The average absorption maxima value for every pH was taken, the values were fitted to a Boltzmann function, and the pKa value is reported. For 6-amino-5-(4-nitrophenylazo)uridine 5 (Figure 3b), a triplicate pH titration was performed with different sets of solutions of slightly variated pH values. Each data set of values was separately fitted to a Boltzmann function, and the average pKa value is reported.
Quantum mechanical calculations of electronic absorption spectra of the investigated compounds 5, 8a, and 8b were performed with the semiempirical ZINDO/S method as incorporated in the HyperChem program. All singly excited configurations using the 15 highest occupied and 15 lowest unoccupied orbitals were included in the configuration interaction (CI) calculation. The geometries used were obtained from AM1 optimizations as implemented in HyperChem.
R0 Calculations
The Förster critical distance (R0) was calculated in Å by the equation7,23
Where κ2 is the relative orientation of the donor and acceptor transition dipole moments, n is the refractive index of the medium, QD is the quantum yield of the donor in the absence of acceptor and J(λ) is the spectral overlap between the donor emission and acceptor absorption spectra calculated by the equation
where FD(λ) is the normalized fluorescence intensity of the donor in the wavelength range λ to λ + Δλ and εA(λ) is the is the extinction coefficient of the acceptor at λ.
For the R0 calculations, a value of 2/3 was taken for the orientation factor κ2 corresponding to free rotation and a value of 1.333 for the refractive index n of water.
Synthesis
N6-DMF-6-aminouracil (2)
To a cooled to 0 °C suspension of 6-aminouracil (300 mg, 2.36 mmol) in dry DMF (50 mL, filtered over silica gel) was dropwise added N,N-dimethylformamide dimethyl acetal (0.627 mL, 4.72 mmol). The mixture was slowly heated and stirred o.n. at 60 °C. The solvent and excess DMF–DMA were evaporated, and the residue was coevaporated with dry DMF (2 × 20 mL). The solid was used in the next reaction without further purification. To obtain NMR spectra, a small amount of the solid was washed with MeOH and filtered: white solid; mp 245 °C dec; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 10.38 (s, 1H), 8.08 (s, 1H), 4.89 (d, J = 1.5 Hz, 1H), 3.07 (s, 1H), 2.94 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 165.0, 160.5, 156.7, 151.7, 81.5, 40.3, 34.3; ESI-HRMS calcd for [C7H10N4O2Na]+ [M + Na]+ 205.0696, found 205.0698.
N6-DMF-2′,3′,5′-tri-O-benzoyl-6-aminouridine (3)
To the suspension of N6-DMF-6-aminouracil (425 mg, 2.33 mmol) in dry DCE (25 mL) was dropwise added N,O-bis(trimethylsilyl)acetamide (1.43 mL, 5.83 mmol). The mixture was stirred at rt for 2 h until the suspension became clear. β-D-Ribofuranose 1-acetate 2,3,5-tribenzoate (1.18 g, 2.33 mmol) and TMSOTf (0.675 mL, 3.73 mmol) were added successively. The reaction mixture was heated to reflux for 1 h, cooled to rt, and then evaporated. The residue was dissolved in CH2Cl2, and the solution was washed with saturated aqueous NaHCO3 solution and brine, dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by column chromatography with CH2Cl2/MeOH = 100:0–95:5 to afford a white solid (790 mg, 54%): mp 105–107 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.03 (s, 1H), 8.08 (s, 1H), 7.99 (d, J = 5.5 Hz, 2H), 7.88 (dd, J = 8.3, 1.1 Hz, 2H), 7.83 (d, J = 7.5 Hz, 2H), 7.67–7.58 (m, 3H), 7.50–7.42 (m, 4H), 7.41–7.36 (m, 2H), 6.85 (s, 1H), 6.19–5.99 (m, 2H), 5.09 (s, 1H), 4.68–4.59 (m, 2H), 4.56–4.48 (m, 1H), 3.06 (s, 3H), 2.84 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 164.8, 164.7, 163.1, 159.1, 156.2, 150.8, 133.9, 133.8, 133.5, 129.4, 129.3, 129.2, 128.8, 128.74, 128.70, 128.64, 128.60, 86.7, 82.9, 77.9, 74.1, 70.6, 63.6, 40.4, 34.4; ESI-HRMS calculated for [C33H31N4O9]+ [M + H]+ 627.2086, found 627.2083.
6-Aminouridine (4)
A solution of N6-DMF-2′,3′,5′-tri-O-benzoyl-6-aminouridine (500 mg, 0.8 mmol) in saturated methanolic ammonia (38 mL) was heated overnight at 60 °C in a pressure vessel. All volatiles were evaporated, and the residue was coevaporated with methanol (2 × 50 mL). The residue was dissolved in MeOH (50 mL), treated with silica gel, and evaporated to dryness. The residue was loaded onto a silica gel chromatography column and eluted with CH2Cl2/MeOH = 100:0 to 75:25 to afford a white solid (174 mg, 84%): mp 197 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 10.47 (d, J = 1.7 Hz, 1H), 6.87 (s, 2H), 6.18 (d, J = 7.4 Hz, 1H), 5.50 (t, J = 4.5 Hz, 1H), 5.24 (d, J = 6.4 Hz, 1H), 5.03 (d, J = 5.0 Hz, 1H), 4.58 (d, J = 2.1 Hz, 1H), 4.37 (dd, J = 13.6, 6.5 Hz, 1H), 4.04–3.98 (m, 1H), 3.83–3.79 (m, 1H), 3.64–3.55 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 162.4, 155.7, 151.3, 87.7, 85.0, 76.9, 69.6, 69.5, 60.4; ESI-HRMS calcd for [C9H13N3O6Na]+ [M + Na]+ 282.0697, found 282.0698.
6-Amino-5-(4-nitrophenylazo)uridine (5)
4-Nitroaniline (40 mg, 0.29 mmol) was dissolved in 1 M HCl (5 mL) and cooled to 0 °C. Sodium nitrite (1 M, 290 μL, 0.29 mmol) was added, and the reaction was kept at 0 °C for 1 h. A mixture of 6-aminouridine (50 mg, 0.193 mmol) predissolved in aqueous NaHCO3 solution (1 M, 5 mL) was slowly added at which a yellowish-orange precipitation formed. The ice bath was removed, and the reaction was stirred for 2 h at rt to give a deep orange-red slurry. The slurry was filtered on a Buchner funnel, washed twice with water, and dried thoroughly over vacuum to give an orange solid (78 mg, 99%): mp >300 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 11.50 (s, 1H), 9.32 (s, 1H), 8.32 (d, J = 9.1 Hz, 2H), 7.82 (d, J = 9.1 Hz, 2H), 6.36 (d, J = 8.3 Hz, 1H), 5.95 (s, 1H), 5.46 (d, J = 5.7 Hz, 1H), 5.19 (d, J = 4.3 Hz, 1H), 4.51–4.43 (m, 1H), 4.10–4.01 (m, 1H), 4.02–3.97 (m, 1H), 3.73–3.66, (m, 1H), 3.65–3.58 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 159.3, 156.4, 149.6, 149.1, 145.6, 125.2, 121.3, 112.2, 88.1, 86.1, 70.4, 68.4, 60.8; ESI-HRMS calcd for [C15H15N6O8]− [M – H]− 407.0957, found 407.0953.
2′,3′,5′-Tri-O-benzoyl-6-oxocytidine (6)
To a suspension of 6-aminouracil (1.00 g, 7.87 mmol) in dry DCE (35 mL) was dropwise added N,O-bis(trimethylsilyl)acetamide (9.62 mL, 39.34 mmol). The mixture was heated to 50 °C and stirred for 5 h until the suspension became clear. The reaction was cooled to rt, and β-D-ribofuranose 1-acetate 2,3,5-tribenzoate (3.97 g, 7.87 mmol) and TMSOTf (2.28 mL, 12.59 mmol) were added successively. The reaction mixture was heated to reflux for 1 h, cooled to rt, and then evaporated. The residue was dissolved in CH2Cl2, and the solution was washed with saturated aqueous NaHCO3 solution and brine, dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by column chromatography with CH2Cl2/MeOH = 100:0–95:5 to afford a white solid (3.15 g, 70%): mp 201–203 °C; 1H NMR (500 MHz, CDCl3) δ 11.06 (s, 1H), 7.99 (d, J = 7.2 Hz, 2H), 7.92 (d, J = 7.3 Hz, 2H), 7.85 (d, J = 7.5 Hz, 2H), 7.55–7.47 (m, 3H), 7.39–7.26 (m, 6H), 6.75 (s, 1H), 6.28 (dd, J = 8.7, 6.1 Hz, 1H), 6.22 (dd, J = 5.9, 1.3 Hz, 1H), 5.23 (s, 2H), 4.88 (d, J = 1.9 Hz, 1H), 4.78–4.66 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 166.6, 165.9, 163.4, 153.8, 152.0, 133.6, 133.5, 133.4, 129.9, 129.8, 129.5, 129.1, 128.8, 128.5, 128.4, 85.1, 78.1, 76.2, 74.5, 70.9, 63.3; ESI-HRMS calcd for [C30H25N3O9Na]+ [M + Na]+ 594.1483, found 594.1481.
6-Oxocytidine (7)
A solution of 2′,3′,5′-tri-O-benzoyl-6-oxocyti-dine (600 mg, 1.05 mmol) in saturated methanolic ammonia (38 mL) was heated overnight at 60 °C in a pressure vessel. All volatiles were evaporated, and the residue was coevaporated with methanol (2 × 50 mL). The residue was dissolved in MeOH (50 mL), treated with silica gel, and evaporated to dryness. The residue was loaded onto a silica gel chromatography column and eluted with CH2Cl2/MeOH = 100:0–75:25 to afford a white solid (268 mg, 98%): mp 198–200 °C;1H NMR (500 MHz, DMSO-d6) δ 10.45 (s, 1H), 6.36 (s, 2H), 6.00 (d, J = 2.6 Hz, 1H), 4.96 (d, J = 5.3 Hz, 1H), 4.79 (d, J = 6.5 Hz, 1H), 4.61 (t, J = 5.7 Hz, 1H), 4.53 (s, 1H), 4.44 (dd, J = 9.6, 5.3 Hz, 1H), 4.04 (q, J = 6.3 Hz, 1H), 3.63 (td, J = 6.0, 3.1 Hz, 1H), 3.56 (ddd, J = 11.6, 4.6, 3.2 Hz, 1H), 3.41–3.36 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.8, 154.1, 150.7, 86.8, 84.1, 74.0, 71.1, 70.3, 62.5; ESI-HRMS calcd for [C9H12N3O6]− [M – H]− 258.0732, found 258.0736.
5-(4-Nitrophenylazo)-6-oxocytidine (8)
4-Nitroaniline (40 mg, 0.29 mmol) was dissolved in 1 M HCl (3 mL) and cooled to 0 °C. Sodium nitrite (1 M, 290 μL, 0.29 mmol) was added, and the reaction was kept at 0 °C for 1 h. A mixture of 6-aminouridine (50 mg, 0.193 mmol) predissolved in aqueous NaOAc solution (1 M, 3 mL) was slowly added at which a yellowish-orange precipitation formed. The ice bath was removed, and the reaction was stirred for 2 h at rt to give a deep orange-red slurry. The solution was slightly acidified with a few drops of 1 M HCl and evaporated to dryness. The crude was taken up in MeOH (50 mL) and sonicated for 5 min, and the salts were filtered off. The solution was treated with silica gel and evaporated to dryness. The residue was loaded onto a silica gel chromatography column and eluted with CH2Cl2/MeOH = 100:0 to 75:25 to afford an orange solid (71 mg, 90%): mp 237 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 13.94 (s, 0.3H), 11.17 (s, 0.7H), 10.67 (s, 0.7H), 8.32 (d, J = 8.6 Hz, 1.4H), 8.27(s, 0.6H), 8.26 (d, J = 5.3 Hz, 0.6H), 8.08 (d, J = 6.5 Hz, 0.6H), 7.93 (s, 0.7), 7.86 (d, J = 8.5 Hz, 1.4H), 6.12 (s, 0.7H), 6.03 (s, 0.3H), 5.03 (d, J = 4.4 Hz, 1H), 4.87 (d, J = 6.2 Hz, 1H), 4.58 (t, J = 4.8 Hz, 1H), 4.52 (s, 0.7H), 4.47 (s, 0.3H), 4.15 (dd, J = 12.5, 6.2 Hz, 1H), 3.74–3.66 (m, 1H), 3.66–3.59 (m, 1H), 3.49–3.41 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.2, 160.6, 159.8, 156.7, 154.6, 148.9, 148.1, 146.8, 145.7, 144.0, 125.1, 121.4, 117.4, 116.6, 111.6, 87.9, 84.3, 71.3, 70.2, 62.5; ESI-HRMS calcd for [C15H15N6O8]− [M – H]− 407.0957, found 407.0964.
5-(4-Nitrophenylhydrazono)-6-oxouridine (9)
Attempts to obtain crystals of 5-(4-nitrophenylazo)-6-oxocytidine (8) in strongly acidic conditions resulted in isolation of yellow crystals of the title product. Method A, slow evaporation: 8 (minuscule amount) was suspended in a few drops of acetonitrile, and a drop of HCl 4 M in EtOH was added. The clear solution obtained was allowed to evaporate at room temperature to give yellow crystals. Method B, vapor diffusion: 8 (minuscule amount) in a crystallization tube was dissolved in TFA (≅0.5 mL) and was set in a sealed vial with water (≅2.5 mL). Mass spectra analysis of the yellow crystals obtained displayed one compound of one mass unit more than the original mass previously obtained for the desired product 8. Mass spectra analysis of the crystallization solution displayed a mixture of both masses attesting to the transformation occurring in acidic conditions (Figure S12). The amount of 9 obtained was insufficient for NMR analysis: HRMS calcd for [C15H14N5O9]− [M – H]− 408.0797, found 408.0799.
Supplementary Material
Acknowledgments
We thank Drs. A. Fin and E. Wexselblatt (Chemistry and Biochemistry, UCSD) for technical help and valuable discussions. We also thank the UCSD Crystallography Facility and the UCSD Chemistry and Biochemistry NMR Facility and the Molecular Mass Spectrometry Facility. We are grateful to the National Institutes of Health for generous support (GM 069773).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b00310.
Supporting figures and NMR spectra (PDF)
X-ray crystallographic data (ZIP)
References
- 1.(a) Tanpure AA, Pawar MG, Srivatsan SG. Isr J Chem. 2013;53:366–378. [Google Scholar]; (b) Noe MS, Xie Y, Tor Y. Methods for Studying Nucleic Acid/Drug Interactions. 2011:159–182. [Google Scholar]; (c) Srivatsan SG, Sawant AA. Pure Appl Chem. 2010;83:213–232. [Google Scholar]; (d) Wilhelmsson LM. Q Rev Biophys. 2010;43:159–183. doi: 10.1017/S0033583510000090. [DOI] [PubMed] [Google Scholar]; (e) Sinkeldam RW, Greco NJ, Tor Y. Chem Rev. 2010;110:2579–2619. doi: 10.1021/cr900301e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dodd DW, Hudson RHE. Mini-Rev Org Chem. 2009;6:378–391. [Google Scholar]; (g) Wilson JN, Kool ET. Org Biomol Chem. 2006;4:4265–4274. doi: 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]; (h) Okamoto A, Saito Y, Saito I. J Photochem Photobiol, C. 2005;6:108–122. [Google Scholar]; (i) Rist MJ, Marino JP. Curr Org Chem. 2002;6:775–793. [Google Scholar]
- 2.Asseline U. Curr Org Chem. 2006;10:491–518. [Google Scholar]
- 3.(a) Rovira AR, Fin A, Tor Y. J Am Chem Soc. 2015;137:14602–14605. doi: 10.1021/jacs.5b10420. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hopkins PA, Sinkeldam RW, Tor Y. Org Lett. 2014;16:5290–5293. doi: 10.1021/ol502435d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sinkeldam RW, Hopkins PA, Tor Y. Chem Phys Chem. 2012;13:3350–3356. doi: 10.1002/cphc.201200375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Noe MS, Rios AC, Tor Y. Org Lett. 2012;14:3150–3153. doi: 10.1021/ol3012327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sinkeldam RW, Marcus P, Uchenik D, Tor Y. Chem Phys Chem. 2011;12:2260–2265. doi: 10.1002/cphc.201100315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shin D, Sinkeldam RW, Tor Y. J Am Chem Soc. 2011;133:14912–14915. doi: 10.1021/ja206095a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Mata G, Luedtke NW. J Am Chem Soc. 2015;137:699–707. doi: 10.1021/ja508741u. [DOI] [PubMed] [Google Scholar]; (b) Barthes NPF, Karpenko IA, Dziuba D, Spadafora M, Auffret J, Demchenko AP, Mely Y, Benhida R, Michel BY, Burger A. RSC Adv. 2015;5:33536–33545. [Google Scholar]
- 5.(a) Sholokh M, Sharma R, Shin D, Das R, Zaporozhets OA, Tor Y, Mely Y. J Am Chem Soc. 2015;137:3185–3188. doi: 10.1021/ja513107r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wierzchowski J. Nucleosides, Nucleotides Nucleic Acids. 2014;33:626–644. doi: 10.1080/15257770.2014.913065. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hall KB. Methods Enzymol. 2009;469:269–285. doi: 10.1016/S0076-6879(09)69013-3. [DOI] [PubMed] [Google Scholar]; (b) Tinsley RA, Walter NG. RNA. 2006;12:522–529. doi: 10.1261/rna.2165806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borjesson K, Preus S, El-Sagheer AH, Brown T, Albinsson B, Wilhelmsson LM. J Am Chem Soc. 2009;131:4288–4293. doi: 10.1021/ja806944w. [DOI] [PubMed] [Google Scholar]
- 8.(a) Dong M, Babalhavaeji A, Samanta S, Beharry AA, Woolley GA. Acc Chem Res. 2015;48:2662–2670. doi: 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]; (b) Beharry AA, Woolley GA. Chem Soc Rev. 2011;40:4422–4437. doi: 10.1039/c1cs15023e. [DOI] [PubMed] [Google Scholar]; (c) Hamon F, Djedaini-Pilard F, Barbot F, Len C. Tetrahedron. 2009;65:10105–10123. [Google Scholar]; (d) Zollinger H. Color Chemistry: Syntheses, Properties and Applications of Organic Dyes and Pigments. 3. Wiley–VCH; Weinheim: 2003. [Google Scholar]; (e) Griffiths J. Chem Soc Rev. 1972;1:481–493. [Google Scholar]
- 9.Li J, Wang XY, Liang XG. Chem - Asian J. 2014;9:3344–3358. doi: 10.1002/asia.201402758. [DOI] [PubMed] [Google Scholar]
- 10.(a) Goldau T, Murayama K, Brieke C, Steinwand S, Mondal P, Biswas M, Burghardt I, Wachtveitl J, Asanuma H, Heckel A. Chem - Eur J. 2015;21:2845–2854. doi: 10.1002/chem.201405840. [DOI] [PubMed] [Google Scholar]; (b) Kou B, Guo X, Xiao SJ, Liang X. Small. 2013;9:3939–3943. doi: 10.1002/smll.201301134. [DOI] [PubMed] [Google Scholar]; (c) Asanuma H, Takarada T, Yoshida T, Tamaru D, Liang X, Komiyama M. Angew Chem, Int Ed. 2001;40:2671–2673. [PubMed] [Google Scholar]; (d) Asanuma H, Ito T, Komiyama M. Tetrahedron Lett. 1998;39:9015–9018. [Google Scholar]
- 11.(a) McKeen CM, Brown LJ, Nicol JT, Mellor JM, Brown T. Org Biomol Chem. 2003;1:2267–2275. doi: 10.1039/b301859h. [DOI] [PubMed] [Google Scholar]; (b) Gunnlaugsson T, Kelly JM, Nieuwenhuyzen M, O’Brien AMK. Tetrahedron Lett. 2003;44:8571–8575. [Google Scholar]; (c) Asanuma H, Yoshida T, Ito T, Komiyama M. Tetrahedron Lett. 1999;40:7995–7998. [Google Scholar]
- 12.(a) Katritzky AR, Khelashvili L, Kovacs J, Shanab K. Chem Biol Drug Des. 2009;73:396–402. doi: 10.1111/j.1747-0285.2009.00792.x. [DOI] [PubMed] [Google Scholar]; (b) Adamczyk M, Akireddy SR, Mattingly PG, Reddy RE. Tetrahedron. 2003;59:5749–5761. [Google Scholar]
- 13.(a) Mori S, Morihiro K, Obika S. Molecules. 2014;19:5109–5118. doi: 10.3390/molecules19045109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kovaliov M, Wachtel C, Yavin E, Fischer B. Org Biomol Chem. 2014;12:7844–7858. doi: 10.1039/c4ob01185f. [DOI] [PubMed] [Google Scholar]; (c) Moustafa ME. PhD Thesis. University of Western Ontario; 2011. Design and Syntheses of Novel Quenchers for Fluorescent Hybridization Probes. [Google Scholar]; (d) Ogasawara S, Ito S, Miyasaka H, Maeda M. Chem Lett. 2010;39:956–957. [Google Scholar]; (e) Boge N, Schroder M, Meier C. Synlett. 2008;2008:1066–1070. [Google Scholar]; (f) Ikeda K, Sumi T, Yokoi K, Mizuno Y. Chem Pharm Bull. 1973;21:1327–1332. [Google Scholar]; (g) Ikeda K, Mizuno Y. Chem Pharm Bull. 1971;19:564–570. [Google Scholar]; (h) Lohrmann R, Lagowski JM, Forrest HS. J Chem Soc. 1964:451–459. [Google Scholar]
- 14.Nishimura N, Sueyoshi T, Yamanaka H, Imai E, Yamamoto S, Hasegawa S. Bull Chem Soc Jpn. 1976;49:1381–1387. [Google Scholar]
- 15.Birnbaum PP, Linford JH, Style DWG. Trans Faraday Soc. 1953;49:735–744. [Google Scholar]
- 16.The sensitivity of the nucleodyes to polarity changes was studied in dioxane [ET(30) = 36.0 kcal/mol], methanol [ET(30) = 55.4 kcal/mol], and mixtures thereof. MeOH–dioxane mixtures were chosen for this study over water–dioxane solutions to eliminate pH-related effects.
- 17.Alternative fitting of the optical density values at specific wavelengths gives a slightly lower value. The independent fitting of five wavelengths (λ = 370, 437, 460, and 475 nm) which include both Gaussian maxima as well as the Gaussian base indicate a transition at neutral pH (pKa1 = 7.0 ± 0.1) (Figure S4).
- 18.Hardman SJ, Botchway SW, Thompson KC. Photochem Photobiol. 2008;84:1473–1479. doi: 10.1111/j.1751-1097.2008.00368.x. [DOI] [PubMed] [Google Scholar]
- 19.Stock LM, Hung MH. Heterocycles. 1982;18:67–75. [Google Scholar]
- 20.Bogert MT, Davidson D. J Am Chem Soc. 1933;55:1667–1668. [Google Scholar]
- 21.(a) Yazdanbakhsh MR, Abbasnia M, Sheykhan M, Ma’mani L. J Mol Struct. 2010;977:266–273. [Google Scholar]; (b) Yazdanbakhsh MR, Moradi-e-Rufchahi E. Orient J Chem. 2009;25:41–48. [Google Scholar]; (c) Seferoglu Z, Ertan N. Cent Eur J Chem. 2008;6:81–88. [Google Scholar]
- 22.Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]
- 23.(a) Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; New York: 2006. [Google Scholar]; (b) Hink MA, Visser NV, Borst JW, van Hoek A, Visser AJWG. J Fluoresc. 2003;13:185–188. [Google Scholar]
- 24.(a) Parsch U, Engels JW. Chem - Eur J. 2000;6:2409–2424. doi: 10.1002/1521-3765(20000703)6:13<2409::aid-chem2409>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; (b) Berressem R, Engels JW. Nucleic Acids Res. 1995;23:3465–3472. doi: 10.1093/nar/23.17.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Muller CE. Tetrahedron Lett. 1991;32:6539–6540. [Google Scholar]
- 25.(a) Hirano T, Kuroda K, Kodama H, Kataoka M, Hayakawa Y. Lett Org Chem. 2007;4:530–534. [Google Scholar]; (b) Kuroda K, Kodama H, Kataoka M, Hayakawa Y. Nucleic Acids Symp Ser. 2006;50:17–18. doi: 10.1093/nass/nrl009. [DOI] [PubMed] [Google Scholar]
- 26.(a) Debnath D, Roy S, Li BH, Lin CH, Misra TK. Spectrochim Acta, Part A. 2015;140:185–197. doi: 10.1016/j.saa.2014.12.027. [DOI] [PubMed] [Google Scholar]; (b) Gaballa AS, Teleb SM, Asker MS, Yalcin E, Seferoglu Z. J Coord Chem. 2011;64:4225–4243. [Google Scholar]; (c) Seferoglu Z. ARKIVOC. 2009:42–57. [Google Scholar]; (d) Masoud MS, Abou El-Enein SA, Ayad ME, Goher AS. Spectrochim Acta, Part A. 2004;60:77–87. doi: 10.1016/s1386-1425(03)00226-9. [DOI] [PubMed] [Google Scholar]; (e) Masoud MS, Abou El Enein S, Kamel HM. Indian J Chem A. 2002;41:297–303. [Google Scholar]
- 27.Moradi-e-Rufchahi EO, Ghanadzadeh A. J Mol Liq. 2011;160:160–165. [Google Scholar]
- 28.(a) Portalone G, Colapietro M. Acta Crystallogr, Sect E: Struct Rep Online. 2007;63:O1869–O1871. doi: 10.1107/S1600536807066561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharma BD, McConnell JF. Acta Crystallogr. 1965;19:797–806. doi: 10.1107/s0365110x65004371. [DOI] [PubMed] [Google Scholar]; (c) Mcconnell JF, Sharma BD, Marsh RE. Nature. 1964;203:399–400. [Google Scholar]
- 29.Wagner T, Han B, Koch G, Krishnamurthy R, Eschenmoser A. Helv Chim Acta. 2005;88:1960–1968. [Google Scholar]
- 30.Dauber P, Hagler AT. Acc Chem Res. 1980;13:105–112. [Google Scholar]
- 31.(a) Luan F, Xu X, Liu HT, Cordeiro MNDS. Color Technol. 2013;129:173–186. [Google Scholar]; (b) El-Shafei A, Hinks D, Freeman HS. Handbook of Textile and Industrial Dyeing, Vol 1: Principles, Processes and Types of Dyes. 2011:225–244. [Google Scholar]
- 32.(a) Dierckx A, Miannay FA, Ben Gaied N, Preus S, Bjorck M, Brown T, Wilhelmsson LM. Chem - Eur J. 2012;18:5987–5997. doi: 10.1002/chem.201103419. [DOI] [PubMed] [Google Scholar]; (b) Sandin P, Borjesson K, Li H, Martensson J, Brown T, Wilhelmsson LM, Albinsson B. Nucleic Acids Res. 2007;36:157–167. doi: 10.1093/nar/gkm1006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wilhelmsson LM, Sandin P, Holmen A, Albinsson B, Lincoln P, Norden B. J Phys Chem B. 2003;107:9094–9101. [Google Scholar]
- 33.(a) Foller Larsen A, Dumat B, Wranne MS, Lawson CP, Preus S, Bood M, Graden H, Wilhelmsson LM, Grotli M. Sci Rep. 2015;5:12653. doi: 10.1038/srep12653. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dumat B, Bood M, Wranne MS, Lawson CP, Larsen AF, Preus S, Streling J, Graden H, Wellner E, Grotli M, Wilhelmsson LM. Chem - Eur J. 2015;21:4039–4048. doi: 10.1002/chem.201405759. [DOI] [PubMed] [Google Scholar]
- 34.Chen PC, Chieh YC, Wu JC. J Mol Struct: THEOCHEM. 2005;715:183–189. [Google Scholar]
- 35.(a) Ma L, Yang F, Zheng J. J Mol Struct. 2014;1077:87–100. doi: 10.1016/j.molstruc.2013.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuan L, Lin W, Zheng K, Zhu S. Acc Chem Res. 2013;46:1462–1473. doi: 10.1021/ar300273v. [DOI] [PubMed] [Google Scholar]; (c) Preus S, Wilhelmsson LM. Chem Bio Chem. 2012;13:1990–2001. doi: 10.1002/cbic.201200400. [DOI] [PubMed] [Google Scholar]; (d) Sahoo H. J Photochem Photobiol, C. 2011;12:20–30. [Google Scholar]; (e) Shanker N, Bane SL. Methods Cell Biol. 2008;84:213–242. doi: 10.1016/S0091-679X(07)84008-8. [DOI] [PubMed] [Google Scholar]; (f) Sapsford KE, Berti L, Medintz IL. Angew Chem, Int Ed. 2006;45:4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 36.Jones AC, Neely RK. Q Rev Biophys. 2015;48:244–279. doi: 10.1017/S0033583514000158. [DOI] [PubMed] [Google Scholar]
- 37.(a) Sowers LC, Boulard Y, Fazakerley GV. Biochemistry. 2000;39:7613–7620. doi: 10.1021/bi992388k. [DOI] [PubMed] [Google Scholar]; (b) Sowers LC, Fazakerley GV, Eritja R, Kaplan BE, Goodman MF. Proc Natl Acad Sci U S A. 1986;83:5434–5438. doi: 10.1073/pnas.83.15.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.