

Summary
Protein C is a vitamin K-dependent serine protease zymogen in plasma which upon activation by thrombin in complex with thrombomodulin (TM) down-regulates the clotting cascade by a feedback loop inhibition mechanism. Activated protein C (APC) exerts its anticoagulant function through protein S-dependent degradation of factors Va and VIIIa. We recently identified a venous thrombosis patient whose plasma level of protein C antigen is normal, but its anticoagulant activity is only 34% of the normal level. Genetic analysis revealed that the proband and her younger brother carry a novel heterozygous mutation c.346G>A, p.Gly74Ser (G74S) in PROC. Thrombin generation assay indicated that the TM-dependent anticoagulant activity of the proband’s plasma has been significantly impaired. We expressed protein C-G74S in mammalian cells and characterized its properties in established coagulation assays. We demonstrate that the protein C variant can be normally activated by the thrombin-TM complex and the resulting APC mutant also exhibits normal amidolytic and proteolytic activities toward both FVa and FVIIIa. However, it was discovered the protein S-dependent catalytic activity of APC variant toward both procoagulant cofactors has been significantly impaired. Protein S concentration-dependence of FVa degradation revealed that the capacity of APC variant to interact with the cofactor has been markedly impaired. The same results were obtained for inactivation of FVa-Leiden suggesting that the protein S-dependent activity of APC variant toward cleavage of Arg-306 site has been adversely affected. These results provide insight into the mechanism through which G74S substitution in APC causes thrombosis in the proband carrying this mutation.
Keywords: Protein C, Protein S, Factor Va, Factor VIIIa, Thrombosis
Introduction
Protein C is a vitamin K-dependent serine protease zymogen in plasma which upon activation by thrombin in complex with thrombomodulin (TM) down-regulates thrombin generation by proteolytic degradation of factors Va and VIIIa (FVa and FVIIIa) (1–3). Protein C has a multi-domain structure composed of an N-terminal γ-carboxyglutamic acid (Gla) domain (residues 1–45), two epidermal growth factor (EGF)-like domains (residues 46–136), a linking peptide (residues 137–157) between light and heavy chains, an activation peptide (residues 158–169), and a C-terminal serine protease domain (residues 170–419) which contains the trypsin-like catalytic domain (4,5). Following removal of the activation peptide by thrombin and conversion of protein C to activated protein C (APC), the non-catalytic light chain of the protease remains covalently associated with its catalytic heavy chain by a single disulfide bond (4). The anticoagulant function of APC in degradation of both FVa and FVIIIa is stimulated by protein S bound to negatively charged membranes in the presence of calcium (6,7). In addition to its essential role in protein S-dependent regulation of thrombin generation, APC also possesses cytoprotective and anti-inflammatory properties when it binds to endothelial protein C receptor (EPCR) to activate protease-activated receptor 1 (PAR1) (8–11). The functional significance of individual domains of APC has been extensively studied. It has been demonstrated that the Gla-domain is involved in the Ca2+-dependent interaction of APC with both cofactors of the anticoagulant (protein S) and anti-inflammatory pathways (EPCR) (12,13). The N-terminal EGF domain is also believed to be required for the protein S-dependent anticoagulant function of APC (14–16). The role of the C-terminal EGF domain in the catalytic function of APC is not well known. Nevertheless, this domain is in intimate contact with the catalytic domain, thus the two domains likely constitute a single functional unit (17). Unlike its anticoagulant function, EPCR- and PAR1-dependent cytoprotective function of APC does not require interaction with protein S. The mechanism through which protein S augments the catalytic function of APC toward the procoagulant cofactors is poorly understood (18).
Protein C deficiency has an autosomal dominant pattern of inheritance and its heterozygous deficiency is associated with increased risk of venous thromboembolism (VTE) and its homozygous deficiency causes purpura fulminans, which is fatal unless treated by protein C replacement therapy (19,20). This is in agreement with the observation that complete deficiency of protein C in knockout mice is lethal (21). More than 200 natural variants of protein C with mutations scattered at all distinct functional domains (Gla, EGF1, EGF2 and catalytic domains) have been reported in the protein C database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PROC). In general, protein C deficiency is divided into type-I deficiency, which is characterized by equally low antigen (PC:Ag) and activity (PC:A) levels, and type-II deficiency which is characterized by only a lower activity level for APC (22). Type-II deficiency can be subdivided into type-IIa and type-IIb. Type-IIa variants show concordant reductions in PC amidolytic and anticoagulant activities because of abnormal function of the PC/APC serine protease domain, while type-IIb variants show reduced anticoagulant activity but normal amidolytic activity (23). In this study, we have identified a type-IIb protein C deficient VTE patient whose plasma PC:Ag level in ELISA and PC:A level in chromogenic assay are normal, but the PC:A level in clotting assay is 34% of that in normal plasma. By genetic analysis, we demonstrate the proband carries a heterozygous mutation (c.346G>A) in PROC, which leads to a Gly-74 to Ser substitution (p.Gly74Ser, G74S) on the N-terminal first EGF-like domain of protein C. We expressed this protein C variant in mammalian cells and after its characterization discovered the variant has normal amidolytic and catalytic activity toward both FVa and FVIIIa in the absence of protein S, but the anticoagulant activity of the variant was significantly impaired in the presence of the cofactor. The same results were obtained with the FVa Leiden protein, suggesting the mutation adversely affects the protein S-dependent recognition and cleavage of the Arg-306 site of FVa by APC. The results provide clinical evidence that the interaction of EGF1 of APC with protein S contributes to the anticoagulant function of the protease.
Materials and methods
Hemostasis assays
Routine coagulation screening assays including prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen (Fg), thrombin time (TT), d-dimer (DD) and fibrinogen/fibrin degradation products (FDP) were performed in all individuals under the study using an ACL-TOP automatic coagulometer (Instrumentation Laboratory, Bedford, MA, USA) according to manufacturer’s instructions. A detailed description of all hemostasis assays is presented as online Supplementary Materials.
Analysis of thrombin generation in plasma
Thrombin generation (TG) assay (Thermo Labsystems OY, Helsinki, Finland) was carried out with platelet-poor plasmas of the proband, her affected younger and normal older brothers according to manufacturer’s instructions. The reaction was initiated with 5pM tissue factor (TF), 4μM phospholipids, 16.7mM CaCl2 in the absence or presence of 5nM or 10nM soluble thrombomodulin (sTM) (Sekisui Diagnostics, LLC, Lexington) as described (24). The kinetics of thrombin generation was monitored by measuring the hydrolysis of a fluorogenic thrombin substrate as described (25). The lag time (LT, min), peak height (Peak, nM), and endogenous thrombin potential (ETP, nM*min) were deduced from thrombin generation curves plotted with Thrombinoscope Software, version 5.0.0.742 (Thrombinoscope BV) as described (24,25).
Genetic analysis
Genomic DNA was extracted from peripheral whole blood using the QIAamp DNA blood purification kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Detection of genetic defects of the PROC was carried out by directly sequencing on an ABI 3700 sequencer (Applied Biosystems, Foster City, CA).
Construction, expression, and purification of recombinant proteins
Both wild-type (WT) and Gly-74 to Ser (G74S) substitution mutant of protein C were expressed in human embryonic kidney (HEK-293) cells as described (26). Methodologies for isolation, activation and initial characterization of protein C derivatives and the source of plasma proteins and other reagents have been presented as Supplementary Materials.
Interaction with EPCR
The interaction of protein C derivatives with EPCR was assessed by an ELISA-based binding assay using the HPC4-tagged recombinant soluble EPCR (sEPCR) as described (27). 96-well flat bottom microtiter plates were coated with the HPC4 monoclonal antibody in TBS containing 1mM CaCl2 overnight at 4 °C. Following washing and blocking of plates next day with 2% BSA in TBS/Ca2+, they were incubated with sEPCR (0.5μM in TBS/Ca2+ containing 0.1% BSA) for 1h. The plates were rinsed and then incubated with either wild-type or mutant APC (7.8–500 nM) for 1h. After washing, a goat anti-protein C polyclonal antibody (1μg/mL) was added and the plates were developed as described (27).
Endothelial cell permeability assay
The cytoprotective signaling activity of APC derivatives was evaluated by a permeability assay using transformed human umbilical vein endothelial cells (EA.hy926) as described (24,27). The cell permeability in response to thrombin (10nM for 10min) following treatment with APC derivatives (20nM for 3h) was quantitated by spectrophotometric measurement of the flux of Evans blue-bound albumin across functional cell monolayers using a modified 2-compartment chamber model as described (24,27). Results were expressed as mean ±SD and all experiments were repeated at least twice.
Anticoagulant assays
The anticoagulant activity of APC derivatives was monitored both in purified and plasma-based assay systems as described (24,26). In the purified system, degradation of both FVa and FVIIIa by APC was evaluated. In the case of FVa, the cofactor (2.5nM) was incubated with increasing concentrations of either APC-WT or APC-G74S (0–5 nM) on 25μM PC/PS vesicles in TBS/Ca2+. Following 10min incubation at room temperature, the remaining FVa activity was determined in a prothrombinase assay as described (24). Thrombin generation was monitored by an amidolytic activity assay using S2238 (100μM). The same assay was used to monitor the inactivation of FVa by increasing concentrations of APC in the presence of protein S (110nM) with the exception that incubation time was decreased to 1min. The same methods were used to measure the catalytic activity of APC derivatives toward FVa Leiden in both the absence and presence of protein S.
The inactivation of FVIIIa (10nM) by increasing concentrations of APC (0–20 nM) in the absence or presence of protein S (110nM) ± factor V (FV, 10nM) on PC/PS vesicles (50μM) was monitored in TBS/Ca2+ as described (24). Following 3–6 or 30min incubation at room temperature, the remaining FVIIIa activity was determined by an intrinsic Tenase assay as described (24). FXa generation was measured by an amidolytic activity assay using SpFXa (200μM).
The anticoagulant activities of APC derivatives were also evaluated in normal and protein S-deficient plasma by an aPTT assay using STart 4 fibrinometer (Diagnostica/Stago, Asnieres, France). In both cases, 0.05mL TBS containing 0–20nM APC was incubated with a mixture of 0.05mL of plasma plus 0.05mL aPTT reagent (Alexin) for 5min before initiating clotting by the addition of 0.05 mL CaCl2 (35mM) at 37 °C as described (24).
Molecular modeling
The structural model of the APC Gla-EGF1 domains was built based on the x-ray crystal structures of the Gla-domain of prothrombin and active-site inhibited Gla-domainless APC (14,17,28,29). The angle between EGF1 and Gla domains was taken from the x-ray structure of factor VIIa (30). In all cases, Gly-74 is fully solvent exposed and located in a loop structure in a region of the EGF1 domain involved in Ca2+ binding near the last helix of the Gla-domain.
Fragment mapping approach (FTMAP) was used to predict hotspot regions on the surface of the APC and protein S Gla-EGF1 regions (31). The model structures of protein S and APC were used as input for protein-protein docking experiments (14,17,29). Details of molecular modeling are given in Supplementary Materials.
Results
Clinical case
The proband (III-3) was referred to the hematology clinic for consultation because of mesenteric and portal vein thrombosis (Fig. 1A). Her younger brother (III-2), father (II-2) and aunt (II-3) had also experienced bilateral lower-limb deep vein thrombosis (DVT), but her older brother (III-1) was normal (Fig. 1A). Plasma levels of protein C obtained from the proband and her affected younger brother revealed a type-IIb protein C deficiency as evidenced by normal protein C antigen and activity levels based on ELISA and chromogenic assays, but a significantly lower activity level based on the aPTT clotting assay (Fig. 1C). Results of all other routine coagulation and thrombophilia screening assays were normal (data not shown). Genetic analysis identified a heterozygous missense mutation in PROC in both the proband and her younger brother, resulting in substitution of Gly-74 of protein C in EGF1 domain (exon 5 g.9698G›A) with Ser (Fig. 1B). This is a novel mutation in PROC which has not been reported before.
Figure 1.
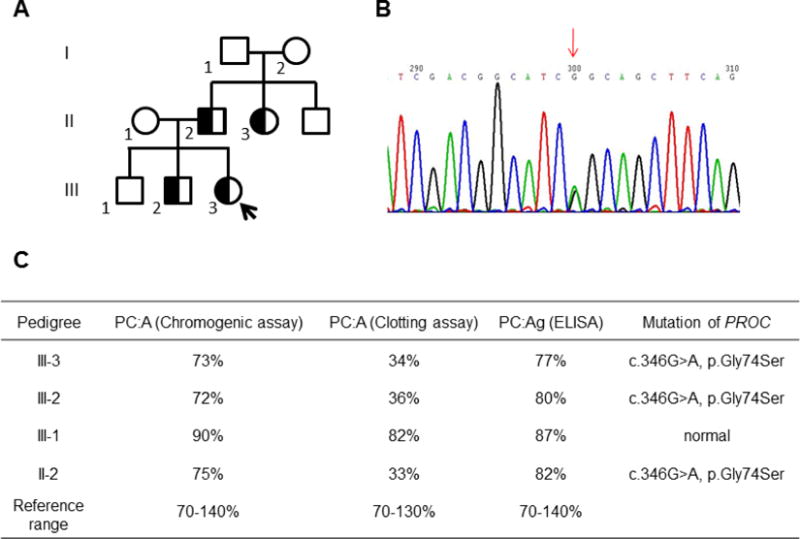
Phenotype and genotype analysis of the proband and family members. (A) The pedigree of the family members with thrombosis. The proband and her affected brother, father and aunt are shown in. (B) The genetic analysis showing the proband and her younger brother are carrying a heterozygous c.346G>A, p.Gly74Ser(G74S) mutation in PROC. (C) The phenotype and genotype data of the proband and her father and brothers. The samples of proband’s mother and grandparents were not available.
Thrombin generation assay
To evaluate the anticoagulant activity of protein C in the proband’s and her affected brother’s plasma, thrombin generation assay was conducted in both the absence and presence of sTM and utilizing a tissue factor concentration of 5 pM to initiate clotting. The results in the absence of sTM indicated near normal thrombin generation profiles for the proband and her younger brother (Fig. 2). However, in the presence of 5 nM or 10 nM sTM, plasma from the proband and her younger brother exhibited significantly higher values of Peak and ETP of thrombin generation (Fig. 2). Thus, in contrast to 90–95% sTM-mediated inhibition of thrombin generation in normal plasma, the inhibition ratio was decreased to 50–70% in both the proband’s (III-3) and her younger brother’s (III-2) plasma. These results suggest that the protein C anticoagulant activity of the mutant in plasma has been markedly impaired in both subjects carrying the Gly74Ser mutation.
Figure 2.
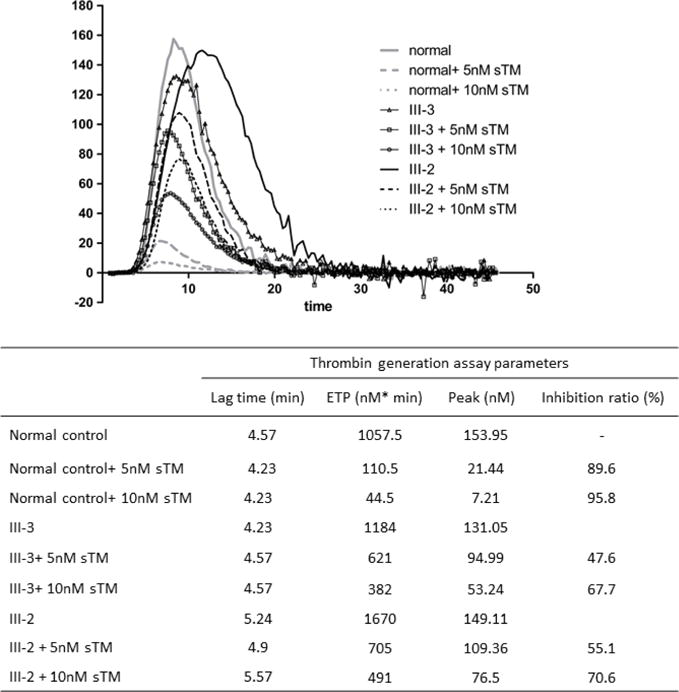
Assessment of thrombin generation in the absence and presence of sTM in normal plasma and plasmas derived from the proband and her younger brother. Citrated normal or test plasmas (80 μL) were incubated with 20 μL PPP reagent and the reaction was initiated with 5 pM TF in the absence or presence of 5 nM or 10 nM sTM. Each sample and the related calibrator were tested in duplicate. The kinetics of thrombin generation was monitored by measuring the hydrolysis of a fluorogenic thrombin substrate. The lag time (LT, min), peak height (Peak, nM), and endogenous thrombin potential (ETP, nM*min) were deduced from thrombin generation curves as described in “Materials and methods”.
Expression and characterization of recombinant protein C-Gly74Ser
Both wild-type and the Gly74Ser mutant of protein C were expressed in HEK-293 cells and following purification to homogeneity, activated by thrombin in TBS/ETDA buffer as described in Materials and methods. The amidolytic activity of APC-G74S toward the chromogenic substrate SpPCa was essentially identical to that observed with wild-type APC (Supplementary Materials, Fig. S1). The time course of the initial rate of activation of protein C by thrombin in both the absence and presence of sTM and calcium suggested a normal activation rate for the mutant zymogen (Fig. 3). The activation of the mutant protein C zymogen by thrombin in the presence of TM (Fig. 3A) and presence of EDTA (Fig. 3B) was similar to wildtype protein C. Interestingly, the activation of the mutant zymogen by thrombin alone (in the presence of calcium) was slightly, but reproducibly improved (Fig. 3C). These results suggest that the higher incidence of thrombosis observed in the proband is not caused by the slower rate of protein C activation by thrombin.
Figure 3.
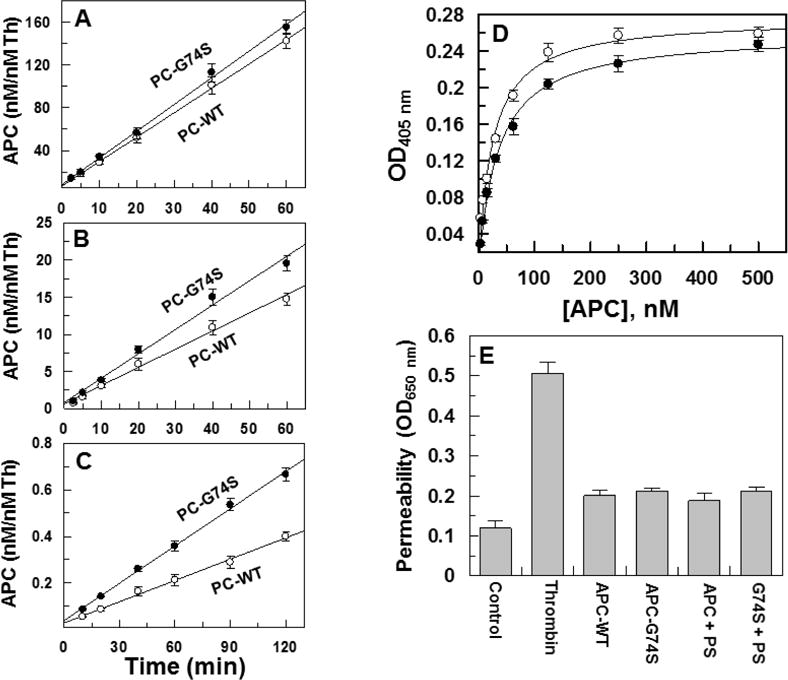
Initial rate of protein C activation by thrombin in the absence and presence of TM and interaction with EPCR. (A) Time course of activation of protein C zymogens (O, WT; ●, G74S) (1 μM) by thrombin (1 nM) in the presence of sTM (10 nM) was monitored in TBS/Ca2+. At indicated time intervals, the activity of thrombin was inhibited by antithrombin (20 nM) and heparin (100 nM) and the rate of APC generation was determined by an amidolytic activity using SpPCa as described in Materials and methods. (B) The same as A, except that in the absence of TM, the time course of protein C activation by 10 nM thrombin was carried out in TBS containing 1.0 mM EDTA. (C) The same as B, except that the zymogens were activated by thrombin (50 nM) in the absence of TM in TBS/Ca2+. (D) The affinity of APC-WT (O) and APC-G74S (●) for interaction with the HPC4-tagged soluble EPCR (0.5 μM) was measured by an ELISA-based binding assay using the monoclonal antibody, HPC4 (1 μg/mL), and a goat polyclonal anti-protein C antibody as described in Materials and methods. (E) The inhibition of thrombin-induced hyperpermeability (10 nM for 10 min) by APC derivatives (20 nM each) in the absence and presence of protein S (110 nM) was monitored from the flux of Evans blue-bound albumin across endothelial cells as described under Materials and methods. Data are derived from two independent measurements.
Interaction with EPCR and cytoprotective signaling activity
The effect of G74S substitution on the affinity of APC for interaction with EPCR was evaluated by an ELISA-based binding assay. The results suggest both APC-WT and APC-G74S interact with EPCR with similar affinities thus yielding a dissociation constant of ~30 nM for both proteases (Fig. 3D). A similar affinity for the interaction of APC with EPCR has been reported in other studies (12). The results further suggested that the EPCR-dependent signaling activity of APC is not adversely affected by the G74S mutation since both APC-WT and APC-G74S exhibited similar cytoprotective activity in a thrombin-mediated permeability assay (Fig. 3E). The cytoprotective activity of APC was independent of protein S for both proteases (Fig. 3E).
Anticoagulant activity
The anticoagulant activity of APC-WT and APC-G74S was evaluated in both the absence and presence of protein S. The APC concentration dependence of FVa inactivation indicated that APC-G74S has normal anticoagulant activity toward FVa in the absence of protein S and that the rate of cofactor degradation is essentially identical to that observed with APC-WT (Fig. 4A). However, the same assay indicated that the anticoagulant activity of the APC mutant toward FVa has been significantly impaired in the presence of protein S (Fig. 4B). Essentially identical results were obtained in reaction with FVIIIa. Thus, the APC mutant exhibited a normal anticoagulant activity toward FVIIIa in the absence of protein S (Fig. 4C), but its activity toward the cofactor was markedly impaired in the presence of the cofactor (Fig. 4D). Further FVa and FVIIIa inactivation studies in the presence of increasing concentrations of protein S suggested the capacity of APC-G74S to interact with protein S has been significantly (2–3-fold) impaired (Fig. 5A,B). The PC/PS-concentration dependence of FVa inactivation in the absence (Fig. 5C) and presence of protein S (Fig. 5D) suggested the impaired protein S-dependent inactivation of FVa by the APC-G74S is not due to the loss affinity of the mutant APC for interaction with negatively charged membrane surfaces since both APC-WT and APC-G74S exhibited identical apparent dissociation constant of 1 μM for interaction with the PC/PS vesicles (Fig. 5C,D).
Figure 4.
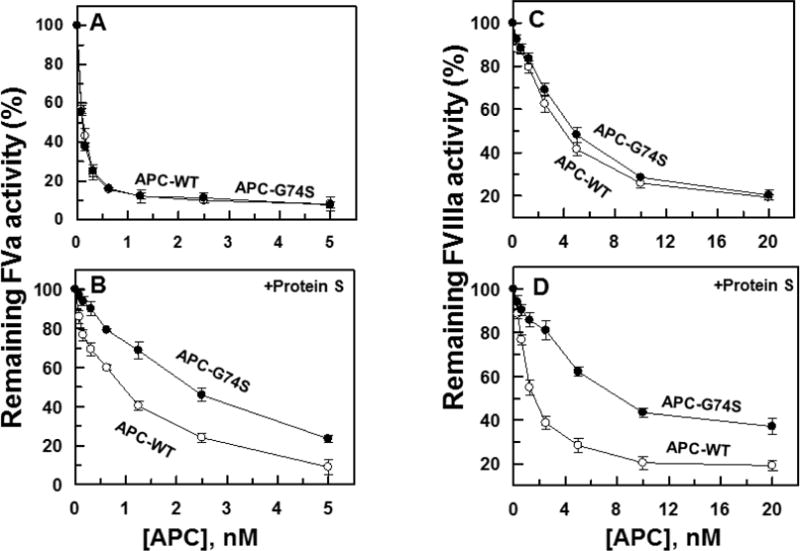
Factors Va and VIIIa degradation by APC derivatives in the absence and presence of protein S. (A) The degradation of human FVa (2.5 nM) by increasing concentrations of APC-WT (O) and APC-G74S (●) was carried out on PC/PS vesicles (25 μM) in TBS/Ca2+ in a 96-well assay plate. Following 10 min incubation at room temperature, the remaining cofactor activity of FVa was determined by a prothrombinase assay (0.5 nM FXa and 500 nM prothrombin for 1 min) as described in Materials and methods. (B) The same as A, except that the APC concentration dependence of FVa degradation was carried out in the presence of protein S (110 nM) for 1min. (C,D) The degradation of FVIIIa (20 nM) by APC-WT (O) and APC-G74S (●) in the absence and presence of protein S (110 nM) was analyzed by incubating increasing concentrations of each protease with the cofactor on PC/PS vesicles (50 μM) in TBS/Ca2+ in a 96-well assay plate for 30 min and 6min, respectively. The remaining cofactor activity of FVIIIa was determined by an intrinsic Tenase (1 nM FIXa and 100 nM FX for 1 min) as described in Materials and methods.
Figure 5.
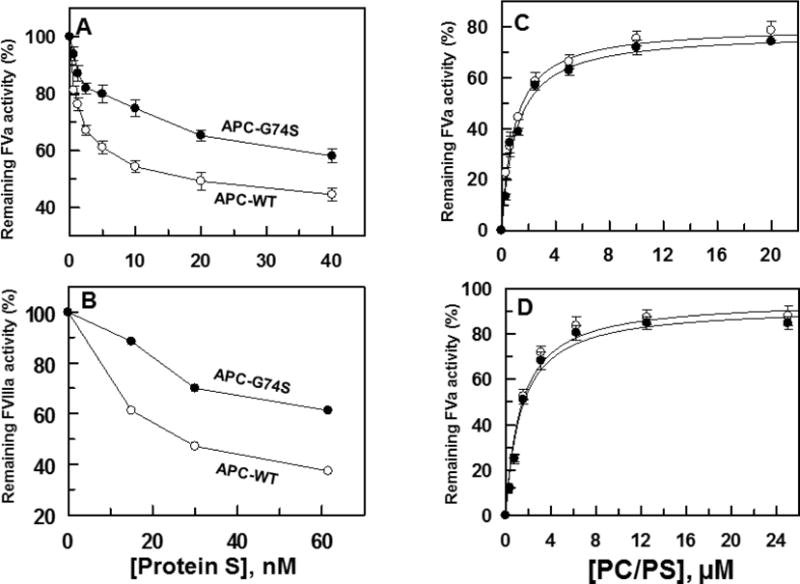
Protein S-concentration dependence of FVa and FVIIIa degradation by APC derivatives. (A) FVa (2.5 nM) degradation by APC-WT (O) and APC-G74S (●) (1 nM each) in the presence of increasing concentrations of protein S (from 0 to 50 nM) was analyzed on PC/PS vesicles (25 μM) in TBS/Ca2+ for 1min. The remaining cofactor activity of FVa was determined by a prothrombinase assay (1nM FXa and 1μM prothrombin for 1 min). (B) Similar to A, except that FVIIIa (10 nM) degradation by APC-WT and APC-G74S (20 nM each) in the presence of increasing concentrations of protein S (from 0 to 500 nM) was analyzed on PC/PS vesicles (40 μM) in TBS/Ca2+ for 6 min. The remaining cofactor activity of FVIIIa was determined by an intrinsic Tenase (1 nM FIXa and 100 nM FX for 1 min) as described in Materials and methods. (C) The same as panel A, except that the PC/PS concentration-dependence of FVa (2.5 nM) degradation by APC-WT and APC-G74S (1 nM each) in the absence and presence of protein S (110 nM) was monitored.
Factor V is known to act as a cofactor and make additional contribution to degradation of FVIIIa by the APC/protein S complex in the intrinsic Tenase assay (32). While the addition of FV to the FVIIIa degradation assay improved the anticoagulant activity of both APC-WT and APC-G74S, nevertheless FV did not restore the defective cofactor function of protein S, suggesting that the protein S cofactor defect with the mutant protease is independent of the cofactor effect of FV (Fig. 6A,B).
Figure 6.
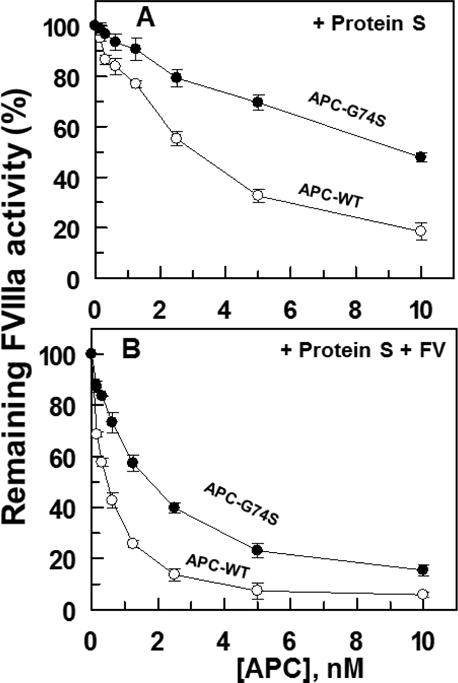
Factor VIIIa degradation by APC derivatives in complex with protein S in the absence and presence of factor V. (A) The degradation of FVIIIa (10 nM) by APC-WT (O) and APC-G74S (●) in complex with protein S (110 nM) in the absence of FV was analyzed by incubating increasing concentrations of each protease with FVIIIa on PC/PS vesicles (50 μM) in TBS/Ca2+ in a 96-well assay plate for 3 min. The remaining cofactor activity of FVIIIa was determined by an intrinsic Tenase (1 nM FIXa and 100 nM FX for 1 min) as described in Materials and methods. (B) The same as panel A except that the reaction was carried out in the presence of human FV (10 nM).
The anticoagulant activity of APC derivatives toward FVa Leiden was also evaluated in both the absence and presence of protein S. The results indicate both APC-WT and APC-G74S exhibit similar anticoagulant activities toward FVa Leiden in the absence of protein S (Fig. 7A). However, similar to degradation of wild-type FVa, the anticoagulant activity of the APC mutant toward FVa Leiden was significantly impaired in the presence of protein S (Fig. 7B). In FVa Leiden, the APC recognition site, Arg-506, is mutated to Gln (33,34). Thus, these results suggest that the cofactor function of protein S in promoting the catalytic efficiency of the APC mutant toward the FVa Arg-306 site has been impaired.
Figure 7.
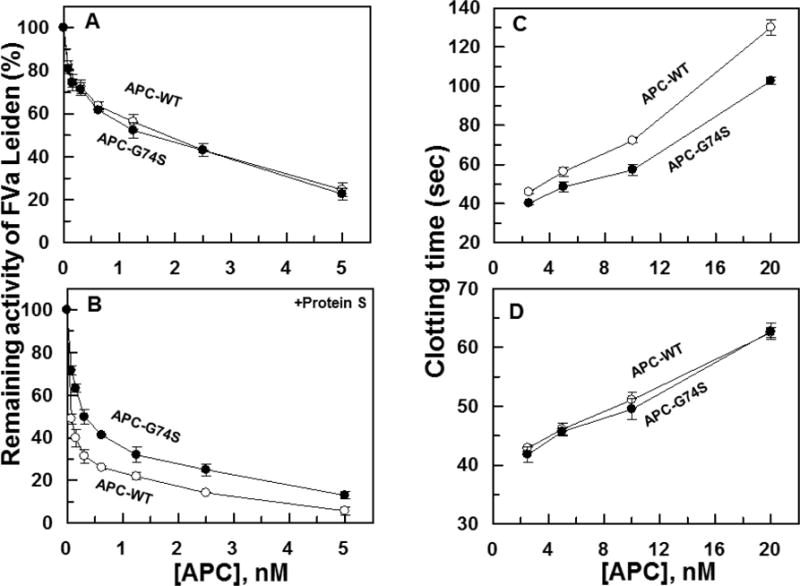
Purified FVa Leiden degradation and clotting activity of APC derivatives in protein S-deficient plasma. (A) The degradation of FVa Leiden (2.5 nM) by increasing concentrations of APC-WT (O) and APC-G74S (●) was carried out on PC/PS vesicles (25 μM) in TBS/Ca2+ in a 96-well assay plate. Following 10 min incubation at room temperature, the remaining cofactor activity of FVa Leiden was determined by a prothrombinase assay (0.5 nM FXa and 500 nM prothrombin for 1 min) as described in Materials and methods. (B) The same as A, except that the APC concentration dependence of FVa Leiden degradation was carried out in the presence of protein S (110 nM) for 1 min. (C) The anti-clotting activities of APC-WT (O) and APC-G74S (●) were determined in normal plasma by an aPTT assay as a function of increasing concentrations of APC at 37 °C as described in Materials and methods. (D) The same as panel C except that the anticoagulant activities of the proteases were evaluated in protein S-deficient plasma.
Consistent with results in the purified system, APC-G74S exhibited defective anticoagulation activity in the plasma-based aPTT assay, thus prolonging the clotting time of normal plasma with an efficiency that was significantly lower than that observed with wild-type APC (Fig. 7C). In agreement with the hypothesis that the G74S mutation has specifically affected the protein S-dependent function of APC the mutant exhibited a normal anticlotting activity in protein S-deficient plasma (Fig. 7D).
The reactivity of APC-WT and APC-G74S with plasma inhibitors was evaluated by incubating the proteases with plasma at a final concentration of 20 nM followed by monitoring their residual amidolytic activities toward the chromogenic substrate SpPCa. Time course analysis indicated both APC-WT and the variant exhibit identical reactivity with plasma inhibitors at several time points examined (data not presented).
Discussion
We demonstrated in this study that heterozygous G74S mutation in PROC is associated with DVT in the proband and two of her family members. To determine whether a molecular defect in the anticoagulant function of the protein C mutant may be responsible for the clotting defect in the proband and her family members, we expressed the protein C mutant in mammalian cells and characterized its properties in established protein C/APC assay systems. The results indicated the protein C mutant is activated normally by the thrombin-TM complex and the resulting APC mutant has normal amidolytic and proteolytic activities in all assays examined in the absence of a cofactor. However, the results in the presence of protein S indicated that the APC mutant has lost its high-affinity interaction with its anticoagulant cofactor. Thus, the protein S concentration-dependence of FVa and FVIIIa degradation by APC in the presence of increasing concentrations of PC/PS vesicles suggested that the G74S mutation weakens the affinity of APC for protein S ~2–3-fold without adversely affecting the affinity of the Gla-domain of the APC mutant for interaction with negatively charged membrane surfaces. Further support for this hypothesis was provided by the observation that both APC-WT and APC-G74S exhibited identical anticoagulant activities in protein S-deficient plasma but APC-G74S had reduced activity in normal plasma. The G74S mutation did not adversely affect the interaction of APC with EPCR and/or its EPCR-dependent cytoprotective signaling function. Noting that the Gla-domain dependent interaction of APC with either negatively charged membrane surfaces or endothelial cell surface receptor, EPCR, is required for the anticoagulant and cytoprotective function of APC (8–16), respectively, the results suggest that G74S mutation does not adversely affect the folding and/or the conformation of the Gla-domain of APC.
The mechanism by which G74S mutation impairs the protein S-dependent anticoagulant function of APC is not known. However, it is known that APC sequentially cleaves at least two bonds after Arg-506 and Arg-306 sites to inactivate FVa (35–37). It has been demonstrated that the APC cleavage of FVa at the Arg-306 site is membrane dependent (35–37). By contrast, the APC cleavage of the Arg-506 is membrane independent, but APC cleaves this site faster than that of the Arg-306 site. The slower activity of APC toward the Arg-306 site is compensated by the cofactor function of protein S which has been shown to preferentially improve the cleavage rate of Arg-306 site to a greater extent than that of the Arg-506 site (36,37). To evaluate whether or not the defective protein S binding property of APC-G74S affects the cleavage rate of Arg-306 site, the capacity of APC derivatives to inactivate FVa Leiden was examined. In this natural FVa variant, the Arg-506 site is mutated to Gln so that the inactivation reaction is only monitoring the cleavage of Arg-306 site by APC on the membrane surface. The observation that both APC-WT and APC-G74S exhibited identical activity toward the cleavage of Arg-306 in the absence of protein S, but the activity of APC-G74S in the presence of protein S was impaired to a similar extent as with wild-type FVa suggests that the cofactor function of protein S in promoting the catalytic efficiency of the APC mutant toward cleavage of Arg-306 site has been impaired. It should be noted that these results do not exclude the possibility that the cofactor activity of protein S toward recognition of Arg-506 has also been impaired in the APC mutant.
The structural basis for the weaker affinity of APC-G74S for protein S is not known. We investigated this question using several computational approaches and structural analyses of both x-ray structures and homology models. Structural data shows that Gly-74 is located near Ca2+-binding residues of EGF1 of APC (5,17,29). The binding of Ca2+ to this site of APC-EGF1, immediately outside of the Gla-domain is required for the normal anticoagulant function of APC and its interaction with protein S (38). Thus, it is possible that the G74S mutation alters the Ca2+-dependent affinity of APC for protein S. This site has a much higher affinity for Ca2+ than the Gla-domain of APC, thus the evaluation of the effect of mutagenesis on the affinity of EGF1 for Ca2+ was not feasible by functional assays. However, molecular modeling of Gla and EGF1 domains of APC (Fig. 8A) predicts that the Gly to Ser substitution at this position should be structurally tolerated. Moreover, based on simulation data, the Ser side chain in the APC variant is expected to point away from the Ca2+-binding site without affecting the interaction of EGF1 with the metal ion. Short simulations indeed suggested that a Ser at this position could be inserted without causing steric clashes or folding problems. A Gly at this site is found in APC sequences from different species but this residue is not conserved in the same position of other vitamin K-dependent coagulation proteins such as FIX and FVII which also possess a similar high-affinity Ca2+-binding site on EGF1 domain (5). Further, this residue is Ser in FIX and Lys in FVII, yet the EGF1 domains of both coagulation factors harbor a high-affinity Ca2+-binding site in the same position as in APC (5). The prediction of the stability change (ΔΔG value) upon the amino acid substitution of Gly to Ser, as computed with two different software programs (39,40), indicated the mutation is neither stabilizing nor destabilizing and, as such, it should be tolerated structurally. Thus, the molecular modeling data does not predict a disruptive role for Ser at this position for the ability of APC-G74S to interact with Ca2+.
Figure 8.
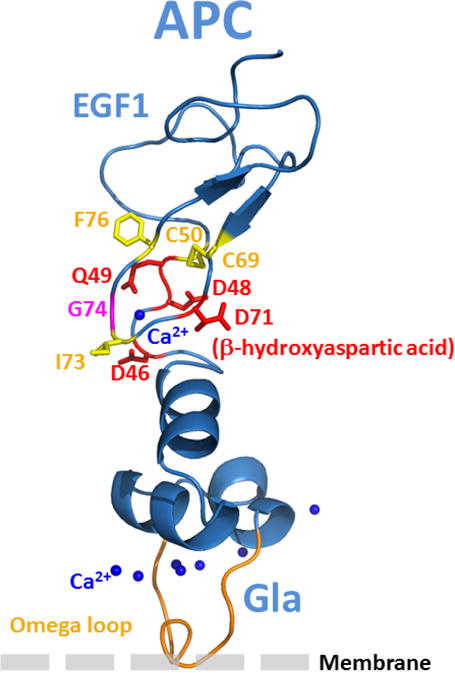
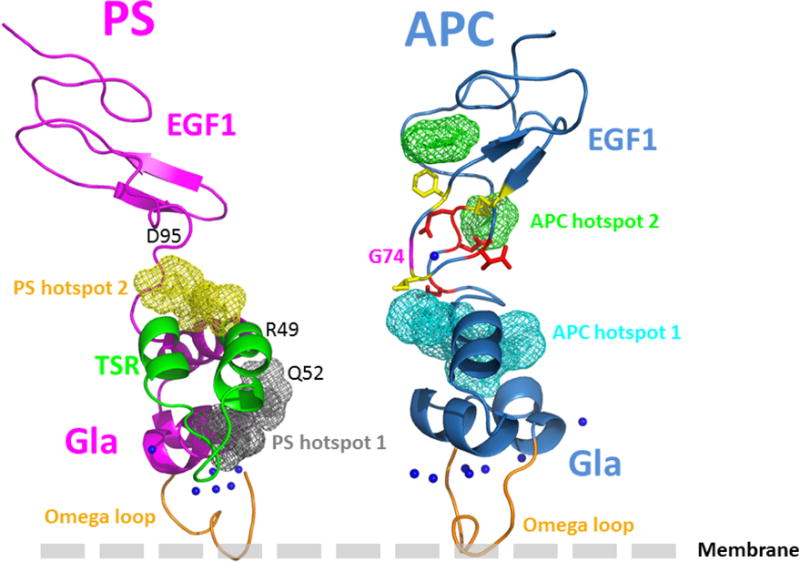
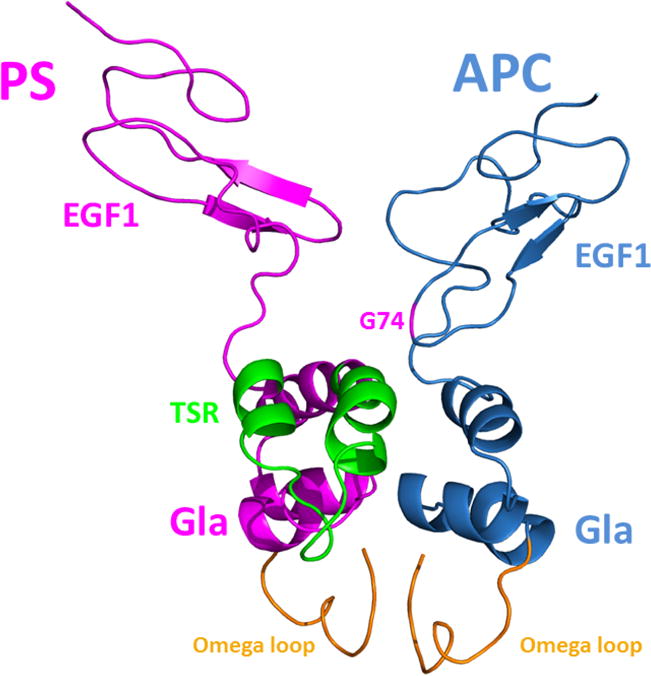
Structural models of the APC and protein S Gla-EGF1 domains. (A) The model of APC Gla-EGF1 domains was built based the x-ray structures of prothrombin, Gla-domainless APC and FVIIa as described in Materials and methods. The side chains of several residues near the Ca2+-binding residue, β-hydroxylated Asp-71, in the vicinity of Gly-74 are displayed. Some polar and negatively charged residues are shown in red while some surrounding hydrophobic and/or aromatic residues are shown in yellow. (B) The fragment mapping approach, FTMAP, was used to predict protein-protein interaction sites and hotspot regions as described in Materials and methods. Two main hotspots in these regions of both proteins were identified (C) The pyDock protein-protein docking algorithm was used to propose about 100 different models of the APC-protein S complex as described in Materials and methods. Structural analysis and integration of previously reported mutagenesis data, predicted hotspot data and results of the present analysis of the G74S variant led to the shown structural model. The omega loops (in orange) of the two proteins are shown pointing toward a membrane surface.
Prediction of protein-protein interaction sites
Mutations in the protein-protein interaction sites are known to be enriched in disease-causing missense mutations compared to other protein surface regions (41). Since Gly-74 of APC is in a fully exposed region and its substitution to Ser is predicted to be structurally tolerated, we hypothesize that this residue should be within a protein-protein interaction site area, and according to the experimental data, possibly in a region interacting with protein S. Numerous computational approaches can be used to predict protein-protein interaction sites and hotspot regions (42,43). Here we used the fragment mapping approach, FTMAP (31), as a tool to identify hotspot regions potentially present in the interface regions between the Gla-EGF1 domains of APC and the Gla thrombin-sensitive region (TSR)-EGF1 domains of protein S (14). Two main binding hotspot regions were identified on the surface of human protein S: hotspot 1 involves a region of the Gla-domain and several residues of the TSR (Arg-49 and/or Gln-52) that have been hypothesized to be involved in APC binding (44–46); hotspot 2 involves a region at the interface of EGF1 domain (i.e., Asp-95) that has been shown to play a role in binding to APC (Fig. 8B) (47). Of interest, these predicted hotspot regions are supported by results of previously reported mutagenesis studies (44–47). Using the same orientation as in Fig. 8A, the two main computed APC hotspot regions are shown in Fig. 8B, hotspot 1 is at the Gla-EGF1 interface near Gly-74 and hotspot 2 is located in the EGF1 domain. The protein-protein docking experiments employing the pyDock computational modeling package predicts that Gly-74 is located in the vicinity of several residues of the Gla-TSR-EGF1 regions of protein S and expected to be in the interface of the APC-protein S interaction sites (Fig. 8C). This structural model is still a “low resolution” structure at this time, thus it is not possible to predict whether the Gly to Ser substitution would directly clash against some nearby protein S residues or if the substitution induces some local dynamic structural changes in this region of APC that impedes the proper interaction of APC with this region of protein S (e.g., perturb stabilizing hydrogen bond networks including water molecules commonly found at protein-protein interfaces).
In summary, we have demonstrated that EGF1 Gly-74 of APC plays a key role in the protein S-dependent anticoagulant function of APC. The heterozygous Ser substitution of this residue was determined to be responsible for VTE in the proband and her affected family members. The results suggest that the G74S mutation would be most harmful under conditions where protein S levels are low (i.e., pregnancy, oral contraceptive use, etc.) (48,49). Molecular modeling of the APC-protein S complex predicts the lack of a side chain at position 74 of the APC EGF1 domain facilitates its optimal interaction with a region in the TSR-EGF1 domain of protein S on the membrane surface. This interaction appears to be important for maintaining the active-site of APC in a topographical orientation that is optimal for efficient recognition and degradation of its procoagulant substrates, FVa and FVIIIa, on the membrane surface.
Supplementary Material
Acknowledgments
This study was supported by the General Program of National Natural Science Foundation of China (81570114), Shanghai Natural Science Foundation (15ZR1426000) and by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health HL101917 and HL062565 to ARR. We thank ccontinuous financial supports from the Inserm and University Paris Diderot to BOV. The characterization of the recombinant protein C-G74S mutant was partially conducted by C. Chen when he was a visiting student in the A. R. Rezaie’s laboratory in St. Louis University Medical School. The authors also thank Audrey Rezaie for editorial work on the manuscript.
Footnotes
Authorship contributions
C.C., L.Y. designed experiments and performed research; B.O.V. performed molecular modeling; X.W. and Q.D. designed experiments, supervised studies conducted in Ruijin Hospital with the subjects’ plasma and contributed to writing of the manuscript; and A.R.R. designed experiments, analyzed data, wrote the manuscript and supervised the project.
All authors approved the final version of this manuscript.
Disclosure of Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70:29–35. [PubMed] [Google Scholar]
- 2.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Ann Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 3.Walker FJ, Fay PJ. Regulation of blood coagulation by the protein C system. FASEB J. 1992;6:2561–2567. doi: 10.1096/fasebj.6.8.1317308. [DOI] [PubMed] [Google Scholar]
- 4.Foster D, Davie EW. Characterization of a cDNA coding for human protein C. Proc Natl Acad Sci (USA) 1984;81:4766–4770. doi: 10.1073/pnas.81.15.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stenflo J. Structure-function relationships of epidermal growth factor modules in vitamin K-dependent clotting factors. Blood. 1991;78:1637–1651. [PubMed] [Google Scholar]
- 6.Heeb MJ, Griffin JH. Activated protein C-dependent and —independent anticoagulant activities of protein S have different structural requirements. Blood Cells Mol Dis. 2002;29:190–199. doi: 10.1006/bcmd.2002.0558. [DOI] [PubMed] [Google Scholar]
- 7.Norstrom EA, Steen M, Tran S, et al. Importance of protein S and phospholipid for activated protein C-mediated cleavage in factor Va. J Biol Chem. 2003;278:24904–24911. doi: 10.1074/jbc.M303829200. [DOI] [PubMed] [Google Scholar]
- 8.Fukudome K, Esmon CT. Identification, cloning and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. 1994;269:26486–26491. [PubMed] [Google Scholar]
- 9.Mosnier LO, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 10.Ruf W, Dorfleutner A, Riewald M. Specificity of coagulation factor signaling. J Thromb Haemost. 2003;1:1495–1503. doi: 10.1046/j.1538-7836.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 11.Joyce DE, Grinnell BW. Recombinant human activated protein C attenuates the inflammatory response in endothelium and monocytes by modulating nuclear factor-kB. Crit Care Med. 2002;30:S288–293. doi: 10.1097/00003246-200205001-00019. [DOI] [PubMed] [Google Scholar]
- 12.Regan LM, Mollica JS, Rezaie AR, et al. The interaction between the endothelial cell protein C receptor and protein C is dictated by the gamma-carboxyglutamic acid domain of protein C. J Biol Chem. 1997;272:26279–26284. doi: 10.1074/jbc.272.42.26279. [DOI] [PubMed] [Google Scholar]
- 13.Ahnström J, Andersson HM, Canis K, et al. Activated protein C cofactor function of protein S: a novel role for a γ-carboxyglutamic acid residue. Blood. 2011;117:6685–6693. doi: 10.1182/blood-2010-11-317099. [DOI] [PubMed] [Google Scholar]
- 14.Villoutreix BO, Teleman O, Dahlbäck B. A theoretical model for the Gla-TSR-EGF-1 region of the anticoagulant cofactor protein S: from biostructural pathology to species-specific cofactor activity. J Comput Aided Mol Des. 1997;11:293–304. doi: 10.1023/a:1007912929828. [DOI] [PubMed] [Google Scholar]
- 15.He X, Shen L, Dahlbäck B. Expression and functional characterization of chimeras between human and bovine vitamin-K-dependent protein-S-defining modules important for the species specificity of the activated protein C cofactor activity. Eur J Biochem. 1995;227:433–440. doi: 10.1111/j.1432-1033.1995.tb20406.x. [DOI] [PubMed] [Google Scholar]
- 16.Hackeng TM, Yegneswaran S, Johnson AE, et al. Conformational changes in activated protein C caused by binding of the first epidermal growth factor-like module of protein S. Biochem J. 2000;349:757–764. doi: 10.1042/bj3490757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mather T, Oganessyan V, Hof P, et al. The 2.8 Å crystal structure of Gla-domainless activated protein C. EMBO J. 1996;15:6822–6831. [PMC free article] [PubMed] [Google Scholar]
- 18.Preston RJ, Ajzner E, Razzari C, et al. Multifunctional specificity of the protein C/activated protein C Gla domain. J Biol Chem. 2006;281:28850–28857. doi: 10.1074/jbc.M604966200. [DOI] [PubMed] [Google Scholar]
- 19.Dahlbäck B. The protein C anticoagulant system: inherited defects as basis for venous thrombosis. Thromb Res. 1995;77:1–43. doi: 10.1016/0049-3848(94)00138-4. [DOI] [PubMed] [Google Scholar]
- 20.Griffin JH, Evatt B, Zimmerman TS, et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–1373. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jalbert LR, Rosen ED, Moons L, et al. Inactivation of the gene for anticoagulant protein C causes lethal perinatal consumptive coagulopathy in mice. J Clin Invest. 1998;102:1481–1488. doi: 10.1172/JCI3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reitsma PH, Bernardi F, Doig RG, et al. Protein C deficiency: a database of mutations, 1995 update. On behalf of the Subcommittee on Plasma Coagulation Inhibitors of the Scientific and Standardization Committee of the ISTH. Thromb Haemost. 1995;73:876–889. [PubMed] [Google Scholar]
- 23.Mackie I, Cooper P, Lawrie A, Kitchen S, Gray E, Laffan M, British Committee for Standards in Haematology Guidelines on the laboratory aspects of assays used in haemostasis and thrombosis. Int J Lab Hematol. 2013;35:1–13. doi: 10.1111/ijlh.12004. [DOI] [PubMed] [Google Scholar]
- 24.Ding Q, Yang L, Dinarvand P, et al. Protein C Thr315Ala variant results in gain of function but manifests as type II deficiency in diagnostic assays. Blood. 2015;125:2428–2434. doi: 10.1182/blood-2014-12-617274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Manithody C, Rezaie AR. Contribution of basic residues of the 70–80-loop to heparin binding and anticoagulant function of activated protein C. Biochemistry. 2002;41:6149–6157. doi: 10.1021/bi015899r. [DOI] [PubMed] [Google Scholar]
- 27.Ding Q, Yang L, Hassanian SM, et al. Expression and functional characterization of natural R147W and K150del variants of protein C in the Chinese population. Thromb Haemost. 2013;109:614–624. doi: 10.1160/TH12-10-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soriano-Garcia M, Padmanabhan K, de Vos AM, et al. The Ca2+ ion and membrane binding structure of the Gla domain of Ca-prothrombin fragment 1. Biochemistry. 1992;31:2554–2566. doi: 10.1021/bi00124a016. [DOI] [PubMed] [Google Scholar]
- 29.Villoutreix BO, Covell DG, Blom AM, et al. Screening the molecular surface of human anticoagulant protein C: a search for interaction sites. J Comput Aided Mol Des. 2001;15:13–27. doi: 10.1023/a:1011158717139. [DOI] [PubMed] [Google Scholar]
- 30.Banner DW, D’Arcy A, Chène C, et al. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 31.Kozakov D, Grove LE, Hall DR, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10:733–755. doi: 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen L, Dahlbäck B. Factor V and protein S as synergistic cofactors to activated protein C in degradation of factor VIIIa. J Biol Chem. 1994;269:18735–18738. [PubMed] [Google Scholar]
- 33.Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc Natl Acad Sci (USA) 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 35.Kalafatis M, Mann KG. Role of the membrane in the inactivation of factor Va by activated protein C. J Biol Chem. 1993;268:27246–27257. [PubMed] [Google Scholar]
- 36.Nicolaes GA, Tans G, Thomassen MC, et al. Peptide bond cleavages and loss of functional activity during inactivation of factor Va and Factor VaR506Q by activated protein C. J Biol Chem. 1995;270:21158–21166. doi: 10.1074/jbc.270.36.21158. [DOI] [PubMed] [Google Scholar]
- 37.Rosing J, Hoekema L, Nicolaes GA, et al. Effects of protein S and factor Xa on peptide bond cleavages during inactivation of factor Va and factor VaR506Q by activated protein C. J Biol Chem. 1995;270:27852–27858. doi: 10.1074/jbc.270.46.27852. [DOI] [PubMed] [Google Scholar]
- 38.Ohlin AK, Landes G, Bourdon P, et al. Beta-hydroxyaspartic acid in the first epidermal growth factor-like domain of protein C. Its role in Ca2+ binding and biological activity. J Biol Chem. 1988;263:19240–19248. [PubMed] [Google Scholar]
- 39.Laimer J, Hiebl-Flach J, Lengauer D, et al. MAESTROweb: a web server for structure-based protein stability prediction. Bioinformatics. 2016;32:1414–1416. doi: 10.1093/bioinformatics/btv769. [DOI] [PubMed] [Google Scholar]
- 40.Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nat Methods. 2007;4:466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 41.David A, Razali R, Wass MN, et al. Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum Mutat. 2012;33:359–363. doi: 10.1002/humu.21656. [DOI] [PubMed] [Google Scholar]
- 42.Villoutreix BO, Lagorce D, Labbé CM, et al. One hundred thousand mouse clicks down the road: selected online resources supporting drug discovery collected over a decade. Drug Discov Today. 2013;18:1081–1089. doi: 10.1016/j.drudis.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 43.Villoutreix BO, Kuenemann MA, Poyet JL, et al. Drug-Like Protein-Protein Interaction Modulators: Challenges and Opportunities for Drug Discovery and Chemical Biology. Mol Inform. 2014;33:414–437. doi: 10.1002/minf.201400040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saller F, Villoutreix BO, Amelot A, et al. The gamma-carboxyglutamic acid domain of anticoagulant protein S is involved in activated protein C cofactor activity, independently of phospholipid binding. Blood. 2005;105:122–130. doi: 10.1182/blood-2004-06-2176. [DOI] [PubMed] [Google Scholar]
- 45.Giri TK, Villoutreix BO, Wallqvist A, et al. Topological studies of the amino terminal modules of vitamin K-dependent protein S using monoclonal antibody epitope mapping and molecular modeling. Thromb Haemost. 1998;80:798–804. [PubMed] [Google Scholar]
- 46.He X, Shen L, Villoutreix BO, Dahlbäck B. Amino acid residues in thrombin-sensitive region and first epidermal growth factor domain of vitamin K-dependent protein S determining specificity of the activated protein C cofactor function. J Biol Chem. 1998;273:27449–27458. doi: 10.1074/jbc.273.42.27449. [DOI] [PubMed] [Google Scholar]
- 47.Andersson HM, Arantes MJ, Crawley JT, et al. Activated protein C cofactor function of protein S: a critical role for Asp95 in the EGF1-like domain. Blood. 2010;115:4878–4885. doi: 10.1182/blood-2009-11-256610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boerger LM, Morris PC, Thurnau GR, et al. Oral contraceptives and gender affect protein S status. Blood. 1987;69:692–694. [PubMed] [Google Scholar]
- 49.Malm J, Laurell M, Dahlbäck B. Changes in the plasma levels of vitamin K-dependent proteins C and S and of C4b-binding protein during pregnancy and oral contraception. Br J Haematol. 1988;68:437–443. doi: 10.1111/j.1365-2141.1988.tb04232.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.