

Abstract
Multiple binding and transport proteins facilitate many aspects of retinoid biology through effects on retinoid transport, cellular uptake, metabolism, and nuclear delivery. These include the serum retinol binding protein sRBP (aka Rbp4), the plasma membrane sRBP receptor Stra6, and the intracellular retinoid binding-proteins such as cellular retinol-binding proteins (CRBP) and cellular retinoic acid binding-proteins (CRABP). sRBP transports the highly lipophilic retinol through an aqueous medium. The major intracellular retinol-binding protein, CRBP1, likely enhances efficient retinoid use by providing a sink to facilitate retinol uptake from sRBP through the plasma membrane or via Stra6, delivering retinol or retinal to select enzymes that generate retinyl esters or retinoic acid, and protecting retinol/retinal from excess catabolism or opportunistic metabolism. Intracellular retinoic acid binding-proteins (CRABP1 and 2, and FABP5) seem to have more diverse functions distinctive to each, such as directing retinoic acid to catabolism, delivering retinoic acid to specific nuclear receptors, and generating non-canonical actions. Gene ablation of intracellular retinoid binding-proteins does not cause embryonic lethality or gross morphological defects. Metabolic and functional defects manifested in knockouts of CRBP1, CRBP2 and CRBP3, however, illustrate their essentiality to health, and in the case of CRBP2, to survival during limited dietary vitamin A. Future studies should continue to address the specific molecular interactions that occur between retinoid binding-proteins and their targets and their precise physiologic contributions to retinoid homeostasis and function.
Keywords: Acyl-CoA:retinol acyltransferase, Cellular retinol binding-protein, Cellularretinoicacidbinding-protein, CytochromeP-450, Acyl-CoA:diacylglycerol acyltransferase, Acyl-CoA:monoacylglycerol acyltransferase, Lecithin:retinol acyltransferase, Peroxisomal proliferator activated receptor δ/β, Retinal dehydrogenase, Retinol dehydrogenase, Retinal, Retinol, Retinoic acid, Retinoic acid receptor, Serum retinol binding-protein, Retinyl ester hydrolase
Introduction
Multiple binding-proteins display high specificity for retinoids. These binding-proteins apparently are involved in most facets of retinoid biology through mediating retinoid transport, metabolism and function. They belong to at least three gene families. The serum retinol binding-protein, sRBP (encoded by Rbp4), the main transporter of retinol through serum, belongs to the lipocalin gene family [3, 75]. Cellular retinaldehyde-binding protein, CRALBP (encoded by Rlbp1), which transports 11-cis-retinal in the eye, belongs to the CRAL_Trio gene family [240]. The intracellular retinoid-binding proteins, which transport and mediate metabolism and function of retinol, retinal and retinoic acid, belong to the fatty acid binding-protein gene family [263]. Data increasingly support occurrence of retinoid metabolons consisting of physiological interactions among intracellular retinoid binding-proteins and retinoid metabolizing enzymes that chaperone retinoid uptake, metabolism and action to enhance efficiency of retinoid use. This chapter focuses on intracellular retinoid binding-proteins of the fatty acid binding-protein gene family that have the most understood functions in retinoid transport and metabolism: cellular retinol binding-protein, type 1 (CRBP1), cellular retinol binding-protein, type 2 (CRBP2), cellular retinoic acid binding-protein, type 1 (CRABP1), cellular retinoic acid binding-protein, type 2 (CRABP2) and fatty acid binding-protein, type 5 (FABP5), and their interactions with enzymes that regulate retinoid homeostasis and nuclear receptors that mediate RA action.
History
Discovery of the Intracellular Retinoid Binding-Proteins
The existence of intracellular retinoid binding-proteins of the FABP gene family was established by the lab of Frank Chytil reporting what is currently known as CRBP1 (aka CRBP) [6]. Prompted by knowledge that steroids interact with specific intracellular receptors, Bashor et al. demonstrated the occurrence of a protein in cytosol of multiple rat tissues that binds retinol with high affinity and specificity. They noted that the 16 kDa molecular weight distinguished it from the steroid hormone receptors known at the time. CRBP1 expression was extended to additional tissues and to fetuses [5]. This was followed quickly by demonstration of two distinct retinoid binding-proteins in human uterus—one that binds retinol specifically (CRBP1) and another that binds RA specifically, currently known as CRABP1 [49, 185, 232, 241], and an RA binding-protein in skin that was likely CRABP2 [245]. Reviews of this early work with these two proteins have been published [46, 47, 232].
The next phase of work focused on purifying CRBP1 and CRABP1 [186, 187, 233, 234]. This was followed by determining their areas of expression in vertebrate tissues [67, 115, 188, 208]. Early work suggested that CRBP1 mediated uptake of retinol into cells and protected it from catabolism, whereas CRABP1 was postulated to serve as a potential mediator of RA action [48]. These deductions have been verified by work from multiple labs.
Approximately 10 years after the discovery of CRBP1 and CRABP1, a second retinol binding-protein, CRBP2, was discovered and purified [183]. Several tissues of the neonatal rat express CRBP2, with neonatal liver and intestine having ~100-fold higher expression than other tissues, and adult rat intestine having ~500-fold higher expression than other tissues. Expression of CRBP2 in adult tissues is more limited than that of CRBP1, both in location and in intensity, with the exception of intestinal mucosa, where CRBP2 levels are ~1000-fold higher than those of CRBP1. The earliest hypothesis for CRBP2 function suggested that it contributes to retinol absorption, which it does.
A second RA binding-protein was identified and cloned in 1990 and named CRABP2 [78], which likely is the same CRABP originally identified in skin [245]. Embryos and skin express CRABP2 most intensely, but low expression is widespread. RA induces the mRNA of CRABP2, suggesting that CRABP2 is involved in the mechanism of RA action.
In 1990, a third CRBP, CRBP3, was isolated from fish eyes [176]. In 2001, two groups reported additional retinol binding-proteins, also referred to as CRBP3 [71, 267]. The relationship of the CRBP3 reported by Nishiwaki et al. to the other two is not clear. It seems, however, that the proteins described by Vogel et al. and Foli et al. are not identical.
Mouse heart, muscle and epididymal white adipose express the CRBP3 reported by Vogel et al. most intensely. This protein binds retinol with a kd value of ~109 nM. It has similar affinities for 13-cis and 9-cis-retinol. This CRBP3 functions as a substrate for lecithin:retinol acyltransferase (LRAT), and seems necessary for optimal incorporation of RE into milk, as demonstrated by knocking out its gene [205]. Knocking out CRBP3 also results in reduced food intake, decreased adiposity and increased lean body mass. The knockout mice are more cold tolerant than WT when fed a high-fat diet, owing to increased mitochondrial fatty acid oxidation in brown adipose tissue. In contrast, kidney and liver express the human CRBP3 reported by Folli et al. most intensely: this CRBP3 has a kd value of ~60 nM for retinol. It is not the same protein as the CRBP3 reported by Vogel et al., and has no mouse ortholog. Its precise function remains unknown, as does the function and relationship of the fish CRBP3 to other retinoid binding-proteins.
Multiple reviews summarize the molecular characteristics, expression locations, and amino acid conservation of these binding proteins in detail. Readers are referred to these reviews for detailed information [134, 164, 174, 178, 184, 231].
Development of the Field
Affinity of CRBP for Retinoids
Even though initial purification of CRBP1 was slow, it still relied on monitoring fluorescence of bound retinol, despite opportunity for dissociation and distribution of retinol throughout membranes or other matrices. This established a high-affinity interaction with a slow off-rate between the ligand and the binding-protein (low dissociation constant) [182, 187, 233, 242]. Originally, a kd value of 16 nM was calculated using a standard fluorescence titration approach [187]. High affinity, however, presents a formidable problem for establishing a kd value, because a system must be at equilibrium to calculate an accurate equilibrium constant. Older fluorimeters were insufficiently sensitive to monitor CRBP1 titration with retinol using a protein concentration in the range of the anticipated kd value. Therefore, a CRBP1 concentration much higher than the kd value (~μM) had to be used. This determined the number of binding sites accurately, but allowed only an upper estimate of the kd. Initially, there was also disagreement whether CRBP1 and 2 could bind retinal, but ultimately, various studies demonstrated that both binding-proteins recognize retinal [133, 135, 145]. As fluorimeters improved, lower estimates of kd values were generated. In the 90s, CRBP1 kd values were reported to be 2 nM for retinol and 30 nM for retinal [149]. With further improvements in equipment and application of non-linear regression analyses to the raw data, the latest kd values for a variety of retinoids have been reported in the low nM range for CRBP1 and somewhat higher for CRBP2 (Table 2.1) [112]. These values also likely represent upper limits, because the fluorescence titrations were done with 150 nM CRBP1 for most retinoids, still well above the calculated kd values, owing to improved, yet not sufficiently sensitive fluorimeters. Reasonably, the true values may be an as much as an order of magnitude lower. Estimates of ~0.1 nM have been published [135].
Table 2.1.
Binding affinities (kd values, nM) of retinoid isomers for CRBP1 and 2. RA and its isomers do not bind with CRBP1 or 2
| Retinoid | CRBP1 | CRBP2 |
|---|---|---|
| all-trans-retinol | 3 ± 2 | 10 ± 3 |
| 13-cis-retinol | 3 ± 1 | 30 ± 12 |
| 9-cis-retinol | 11 ± 2 | 68 ± 7 |
| all-trans-retinal | 9 ± 4 | 11 ± 4 |
| 13-cis-retinal | – | – |
| 9-cis-retinal | 8 ± 4 | 5 ± 3 |
Data are from Kane et al. [112]
Affinities of CRABP1 and 2 for RA and Its Metabolites
CRABP1, purified to homogeneity from tissues, binds all-trans-RA (RA) with a kd value of 4 nM (rat testes) and 16 nM (rat liver) [186, 187]. Expression of bovine CRABP1 in E. coli provided an opportunity to extend binding analysis studies [68, 70]. Purified CRABP1 isolated from E. coli has ~threefold greater affinity for RA than E. coli –expressed CRABP2. CRABPs also bind other forms of RA. A retinoid active in skin, 3,4-didehyro-RA [215], binds to both CRABP1 and 2 with an affinity similar to RA. The RA metabolites, 18-OH-RA, 4-OH-RA, 4-oxo-RA and 16-OH-4-oxo-RA, also have affinity constants similar to RA. These values were established with CRABP 1 and 2 concentrations in the high nanomolar range. Similar to CRBP1, fluorescent titration was used to estimate kd values for CRABP 1 and 2, which provides only an upper limit kd value for high-affinity ligands when the protein concentration is much higher than the kd value, because the ligand saturates the protein, i.e. does not establish equilibrium. For lower affinity ligands, equilibrium can be established, allowing a more accurate value. Thus, the affinities of RA and its phase I trans metabolites could be much less, revealing greater differences relative to the cis isomers. This expectation was verified by re-visiting kd values of CRABP1 and 2 for RA, 9-cis-RA and 13-cis-RA, also generated by fluorescence titration, but with CRABP1 and 2 concentrations from 5 to 16 nM. This approach, along with non-linear regression analyses of the data, generated kd values of 0.4 and 2 nM RA for CRABP1 and CRABP2, respectively, and affirmed the order of binding: RA > 9-cis-RA > 13-cis-RA [177]. The kd values of 9-cis-RA for both RA binding proteins were ~200 nM, whereas 13-cis-RA showed little to no affinity.
Retinoids Have Low Solubility in Aqueous Media
Retinoids have a limited aqueous solubility. Aqueous media accommodate a maximum retinol concentration of ~60 nM [260]. Therefore, μM intracellular concentrations of retinol in solution in the aqueous phase are not possible unless retinol is esterified or “chaperoned” by binding proteins. The high-affinity association of retinol with binding-proteins provides background for appreciating a chaperone model.
In rat liver, 95 % of retinoids are esterified and most occur within lipid droplets. The remaining unesterified retinol [15] occurs predominantly in the microsomal or cytosolic fractions [91], where the concentrations of retinoid binding-proteins (sRBP in microsomes and CRBP1 in cytosol) match or exceed the amount of retinol, supporting the conclusion that retinol is binding-protein-bound. In retinal pigment epithelia cells, which harbor large amounts of retinoids for use by the visual cycle, 92 % of total retinol occurs as retinyl esters (RE) and 8 % as CRBP1-retinol. Thus, intracellular retinol is either esterified or associated with a binding protein.
Insights from Knocking Out Rbp1, the Gene that Encodes CRBP1
The CRBP1-null mouse, created by disrupting the Rbp1 gene though homologous recombination, illustrates the systemic physiological functions of CRBP1. Although Rbp1-null mice show no obvious morphological impairments, retinoid homeostasis is abnormal in multiple tissues [113].
Rbp1-null mice display severe vitamin A loss, but not deficiency, even when fed standard rodent chow, which contains copious vitamin A [77]. At the time of the studies, rodent chow supplied at least 25 IU vitamin A/g diet in the form of retinyl esters. Chow likely had a higher vitamin A equivalent value because of carotenoid content, as chow diets include plant material. Thus, the minimum vitamin A content was sixfold greater than the amount of vitamin A recommended for rodents [219, 220].
Despite copious vitamin A in the dam’s diet, livers of Rbp1-null mice had threefold decreased retinyl palmitate (the major RE) in day 16.5 and 18.5 fetuses relative to WT mice. Starting at 4 weeks of age, both retinyl palmitate and retinol were ~50 % lower. Hepatic stellate cells, which store most of the vitamin A in liver as RE and express CRBP1 and retinoid metabolizing enzymes, contained fewer and smaller cytoplasmic lipid droplets in 8-week-old knockouts relative to WT. The elimination half-life (t½) of total [3H]retinoids in liver was more rapid (10 days in Rbp1-null mice compared to 60 days in WT). Six hours after dosing [3H]retinol, livers of Rbp1-null mice retained 5 % of the dose and a retinyl palmitate/retinol ratio of 2, compared to WT, which retained 10 % of the dose with a retinyl palmitate/retinol ratio of 5. Differences in uptake, esterification, and mobilization were not determined. Rbp1 ablation resulted in 50–60 % lower liver RA concentrations, and allowed increased susceptibility to hepatic retinoid depletion upon dioxin treatment [97].
Retinoid homeostasis also is abnormal in mammary tissue of Rbp1-null mice. Relative to controls, RA is reduced 40 % in mammary tissue of Rbp1-null mice, a decrease attributed to reduced retinol dehydrogenase (RDH) activity [65, 124, 157, 207]. This aberration in retinoid metabolism is accompanied by morphologic abnormalities, such as epithelial hyperplasia and stromal hypercellularity, which promote tumor progression. These data are significant because ~ 25 % of human breast cancers silence Rbp1 epigenetically [123].
The pancreas of the Rbp1-null mouse has increased retinol, intense ectopic expression of Rbp2 mRNA (encodes CRBP2), defective islet expression of glucose sensing and insulin secretion genes, α-cell infiltration into the β-cell interior of islets, diminished glucose-stimulated insulin secretion, high glucagon secretion, abnormally high gluconeogenesis, and hyperglycemia. Conversely, CRBP1 attenuates the negative impact of copious dietary retinol on glucose tolerance. Thus, glucose homeostasis and energy metabolism rely on CRBP1 moderating retinoid homeostasis.
Consistent with the foregoing observations, overexpression of Rbp1 in 3T3L1 pre-adipocytes resulted in a threefold increase in ability of RA to induce the expression of target genes [122]. This observation suggests irregular adipogenesis and lipid metabolism in the Rbp1-null mouse, and explains the more efficient adipocyte differentiation observed with embryonic fibroblasts from the Rbp1-null mouse [290]. The phenotype of the Rbp1-null mouse, the conservation of the CRBP1 primary amino acid sequence in vertebrates, and the widespread tissue distribution and expression of CRBP1 in multiple cell types [26, 115, 189, 190], indicate the survival value of CRBP1 and suggest its function as a chaperone that protects retinol from rapid degradation and efficient use of retinol.
Current State of the Field
CRBP1 Properties and Structure
The ability of select enzymes to recognize the CRBP1-retinol or CRBP1-retinal “cassette” and tease the retinoid from the binding protein into their own active sites provides a solution for regulating retinol flux into RE vs. RA and sparing bound retinol from metabolism by enzymes that do not recognize the holo-binding proteins. Enzyme interactions with holo-CRBP may occur through direct protein-protein contact or in a microenvironment of membrane lipids (cholesterol, phospholipids, ceramides, sphingosines, e.g.) that contribute to transfer of retinol by influencing protein conformations and/or interactions.
The structures of the fatty acid binding-protein gene family members, including CRBP1 and CRBP2, are similar but not identical. They fold similarly, but have different residues in the internal binding pockets that determine ligand specificity. This discussion will focus on CRBP1, but the major points apply to CRBP2.
The CRBP1 structure was solved first by X-ray crystallography [53]. Differences in the structures of apo-and holo-CRBP1 were apparent immediately. Apo-CRBP1 has a more flexible structure, relative to the more rigid holo-CRBP1. This difference was confirmed by limited proteolysis, which demonstrated resistance of holo-CRBP1 to multiple proteases, whereas apo-CRBP1 was digested by the endopeptidase Arg-C at R30 in α-helix II [100, 175]. Since the determination of the structure by X-ray, several NMR studies and a mass spectrometry-based study have confirmed and expanded insight into the flexibility and the ligand entry portal [38, 72, 73, 134, 135, 144, 158]. CRBP1 has a flattened β-barrel (aka β-clam) shape formed by two orthogonal β-sheets (Fig. 2.1). Each β-sheet consists of five anti-parallel β-strands. The N-terminus blocks one end of the barrel and a cap consisting of two short α-helices (helix-turn-helix) between βA and βB blocks the ligand entrance portal. The binding pocket exists as a closed cavity completely isolated from the external solvent. Retinol assumes a flattened (planar) conformation inside the binding pocket, as indicated by a 25 nm red shift in absorbance of bound retinol relative to retinol in solution [187]. The hydroxyl group points into the interior, hydrogen bonding with N108, which contributes to ligand specificity and affinity. Surprisingly, much of the CRBP1 binding cavity presents a hydrophilic environment with structured water molecules surrounding the isoprenoid side-chain and hydroxyl group. In contrast, the β-ionone ring exists in a hydrophobic region created by L29, I32, L36, F57 and I77. The helix-turn-helix “cap” has few interactions with the rest of the binding protein, supporting the likelihood that reduced flexibility of holo-CRBP1 stems from the β-ionone ring of retinol engaging with hydrophobic residues in αII (L29, I32, A33), the link between αII and βB (L36), the βC-βD hairpin turn (F57), and the βE-βF hairpin turn (I77). These areas have been identified by NMR as more disordered in apo-CRBP1, relative to the rest of the molecule. Thus, the β-ionone ring holds αII and the other disordered regions in place, imparting greater rigidity to holo-relative to apo-CRBP1. Retinol accesses apo-CRBP1 because of its relatively open conformation resulting from flexibility of αII and these hairpin turns.
Fig. 2.1.

CRBP1 structure. The “worm” diagram at the left shows the two α-helices (green) and the ten anti-parallel β-strands (tan) arranged in two orthogonal sets of five strands each. Light blue denotes links between the strands or strands and helices. Dark blue indicates retinol. Magenta shows selected exterior residues V27, K31 and L35 in αII. Some interior residues also are shown. Between interior residue R58 and the exterior residues are shown (unmarked) from top to bottom L29, F57 and L36. The space-filling diagram in the middle presents a similar perspective as the “worm” diagram, illustrating seclusion of retinol from the cellular milieu. The depiction of partial residues at the right shows a different perspective from that at the far left to reveal more clearly association or relativity close proximity of L29, I32, L36, F57, R58, and I77 with the β-ionone ring of retinol as they point into the interior of CRBP1, and the outward projections of V27, K31 and L35. Structures were generated with the program Cn3D (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml) with data downloaded from http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=24299
Only small sections of retinol are visible within CRBP1, when viewing a space-filling molecular model. Encapsulation explains observations made during purification of CRBP1, namely, after saturating crude preparations of CRBP1 from animal tissues with retinol, purification of holo-CRBP I by column chromatography could be monitored by following fluorescence of bound retinol in the eluates [182, 187, 233]. One purification even monitored fluorescence of endogenously bound retinol [242].
What would prompt CRBP1 to surrender its ligand? The hydrophilic environment deep in the binding pocket would exert repealing force on the isoprenoid side chain, which would “push” the β-ionone ring against CRBP1 “cap” residues, to force partial opening of the cap, and partial egress of retinol. The three larger residues on or near αII pointing out from CRBP1 (V27, K31, L35) also would contribute to retinol egress. Two of these are hydrophobic, posing the question of why hydrophobic residues projecting into the hydrophilic milieu occur on the surface of a soluble protein. Complementary surfaces on select enzymes might interact with these residues to enhance dissociation of the “disorderly” domains that close around retinol, to prompt retinoid release.
CRBP1 and Cellular Retinol Uptake
Ottonello et al. studied cellular retinol uptake in a cell-free model [195]. They chose plasma membrane-enriched fractions from RPE (retinal pigment epithelium), because RPE accumulates large quantities of retinol to support vision. Incubation with sRBP-retinol resulted in the membrane fractions absorbing and esterifying retinol in a concerted process. Inhibiting RE formation decreased retinol incorporation ~80 %, confirming coupling of retinol uptake and RE formation. They concluded that the overall process occurred without free retinol ever having to leave the binding-protein. They also showed that apo-CRBP1 promoted hydrolysis of membrane-bound RE, generating CRBP1-retinol. The latter point was developed further by the demonstration that apo-CRBP1 activates a bile salt-independent REH (bsiREH) that catalyzes hydrolysis of endogenous RE in liver microsomes, forming holo-CRBP1 [28].
The Ottonello data complemented conclusions of previous reports of a high-affinity (kd = 5 pM) membrane receptor in RPE and intestine that recognizes sRBP [94, 95, 217]. Supporting this conclusion were the observations that retinol uptake from sRBP is saturable and temperature dependent, and apo-sRBP and holo-sRBP compete with each other. This engendered the proposal that relative concentrations of apo- and holo-sRBP in blood control cellular retinol uptake. sRBP-mediated retinol uptake into Sertoli cells is also saturable, and labeled sRBP competes with unlabeled sRBP, again suggesting the presence of a membrane receptor [252]. The rate of uptake from sRBP by Sertoli cells was linear until the concentration of retinol reached the intracellular CRBP1 concentration, and then it decreased. Ultimately, cells contained both free retinol and RE, with RE biosynthesis presumably reflecting the second rate phase, which required discharge of CRBP1 to allow further retinol uptake.
Although efforts were made to isolate and characterize a membrane receptor for sRBP, many years passed before success was achieved. Discovery of an RA-induced gene, Stra6 (stimulated by RA gene 6), in an unbiased screen of RA-stimulated murine P19 embryonal carcinoma cells [34, 41] provided foundation for identification of an sRBP membrane receptor. The deduced amino acid sequence of STRA6 showed that it was not a known protein. STRA6 is expressed widely during mouse embryogenesis and has strong expression in adult brain, kidney, spleen, the female genital tract, and testis; weak expression in heart and lung; and little to no expression in liver. The research team concluded that Stra6 encoded a “new type” of membrane protein, expressed intensely in blood-organ barriers, which serves as a “component” of “transport machinery”. It wasn’t until 2007, however, that Stra6 was verified as an sRBP receptor [116]. Consistent with previous reports of retinol uptake, Kawaguchi et al. showed that LRAT-catalyzed formation of RE and enhanced retinol uptake via Stra6. This conclusion was reinforced by studies in an LRAT-null mouse, which also showed that retinol uptake via Stra6 was coupled with LRAT activity [4]. Wolf has published a detailed history of sRBP receptor studies and cellular retinol uptake [276].
Functions of Stra6 are complex and controversial [16, 258]. Although an original report of the Stra6-knockout concluded that it was embryonic lethal [99], subsequent reports showed this was not the case [22, 239]. It must be noted that the first Stra6-knockout retained a neoR cassette in the gene and that NeoR insertion into target genes can influence expression of neighboring genes, producing phenotypes related to nearby genes, rather than the target gene [204]. A second Stra6 knockout mouse was created that did not retain the neoR cassette. These later produced Stra6-null mice were born in Mendelian frequency and did show differences in the amounts of retinol and RE in multiple tissues relative to WT controls. When these Stra6-null mice that had been fed a diet copious in vitamin A for 12 weeks were switched to a vitamin A-deficient diet for another 4 weeks, total retinol plus RE levels decreased ~50 % in white adipose tissue and ~10–30 % in heart, kidney and testis. Absence of Stra6 marginally slowed the rate of retinol uptake into multiple, but not all, tissues. The only physiological dysfunction noted in these Stra6-null mice was in the eye, which experienced a hyperplastic primary vitreous body, distorted rod photoreceptor segments, and reduced numbers of cone cells. The newer work demonstrated that Stra6 does not protect cells from retinol toxicity by fostering egress of retinol as had been postulated by the Isken report [99]. Instead, the results were consistent with an essential function of Stra6 in retinoid uptake by the eye, but not for maintaining retinoid homeostasis in other tissues, at least in mice fed a diet copious in vitamin A. These conclusions were confirmed by the group that produced the original Stra6 knockout with a redeveloped knockout [4]. Together, available data indicate Stra6 is not the sole mechanism for retinol uptake by extra-ocular tissues in mice fed a diet with copious vitamin A.
Stra6 allows retinol to exit cells when presented with apo-sRBP, but cells that express CRBP1 and/or LRAT presented with holo-sRBP experience net uptake of retinol via STRA6. Inward flow is driven by CRBP1 sequestering retinol and LRAT accessing CRBP1-bound retinol for RE synthesis [21, 117, 118]. sRBP circulates bound with one of the thyroid hormone carriers transthyretin (TTR). This association prevents sRBP from rapid clearance by the kidney. But TTR blocks binding of sRBP with Stra6, indicating that sRBP functions only when its concentration exceeds that of TTR and/or with the free sRBP pool in equilibrium with TTR [19]. This suggests TTR regulates the impact of sRBP on Stra6.
Insulin-resistant mice and humans with obesity and type 2 diabetes have increased circulating sRBP. Overexpression of sRBP causes insulin resistance and ablation of Rbp4 (encodes sRBP) enhances insulin sensitivity [280]. The mechanism of these effects was unknown until Berry et al. reported that Stra6 functions as a signaling receptor activated by sRBP [18]. Binding of sRBP-retinol with Stra6 initiates a signaling cascade that ultimately induces STAT5 regulated genes. One of these, suppressor of cytokine signaling 3 (SOCS3), inhibits insulin signaling, providing a mechanistic connection between sRBP and insulin resistance. This work, along with previous studies, coupled retinol uptake with sRBP-stimulated cellular signaling. Notably, Stra6 signaling requires retinol flux through it to stimulate SOCS3, as revealed by protection from sRBP-induced insulin resistance in Lrat-null mice [152]. The cumulative evidence unites retinol transport by sRBP through blood, cellular retinol uptake, metabolism umpired by CRBP1, LRAT, and Stra6-mediated signaling functio ns [19, 21, 152].
CRBP and Retinoid-Metabolizing Enzymes
Intracellular CRBP1 Chaperoning of Retinol
Retinol and retinal in many cell types occur bound with CRBP, which therefore provides the most abundant substrates for retinoid metabolism, because rapid dissociation from CRBP does not occur through aqueous media [179]. Indeed, rapid dissociation of retinoid from CRBP would be self-defeating, because it would quickly deplete holo-CRBP of retinoids and expose the “free” retinoids to non-enzymatic degradation and to metabolism catalyzed by any enzyme that recognizes free retinoid as substrate, such as xenobiotic-metabolizing enzymes, which have evolved with low substrate specificity. As stated “…most enzymes are not perfectly specific for a single substrate…” [52]. Also, increased concentrations of “free” retinoids would increase RA biosynthesis by mass action, and a modest increase in RA underlies retinoid toxicity [51]. Enzymes that recognize holo-CRBP have access to relatively high substrate concentrations, while permitting CRBP to ensure efficient retinoid use.
Km values for RDH, retinal dehydrogenases, and LRAT with CRBP-bound retinoids are lower than their values with unbound retinoids [170, 172, 184]. An apparent exception is RDH10, for which a Km value was reported as 0.035 μM for unbound retinol, obtained with the lowest substrate concentration assayed greater than the extrapolated Km [12], which cannot generated a reliable value, because the substrate range did not include the curve’s inflection point. In contrast to this preparation of modified RDH10 (RDH10-His6) expressed as 10 % of total insect (Sf9) cell membrane protein, native human RDH10 assayed in membranes of COS-1 cells had an Km value of ~4 μM for unbound all-trans-retinol, indicating the importance of context concerning RDH activity [261].
RE Biosynthesis in the Presence and Absence of CRBP
Early efforts to characterize the enzymology of RE formation identified activity in liver and mammary gland microsomes that relied on acyl-CoA as co-substrate, which was referred to as acyl-CoA:retinol acyltransferase (ARAT) [229, 230]. Liver ARAT activity has a Km value of ~25 μM for retinol and a Vm ranging from 450 to 675 pmol/min/mg of rat liver microsomal protein. The reaction catalyzed by microsomes from the lactating rat mammary has a Km value of ~39 μM for retinol and a Vm ~270 pmol/min/mg protein. ARAT does not recognize CRBP1 as substrate. CRBP1 levels are very low in the mammary gland, consistent with the conclusion that ARAT would contribute to RE formation for milk [216]. Knockout of LRAT, which recognizes CRBP1 and 2 as substrates, however, resulted in a transition from mostly RE to mostly unesterified retinol in milk, supporting the conclusion that LRAT generates RE for milk.
ARAT activity also was reported in microsomes of rat small intestine with a Km value ~5 μM and a Vm of ~300 pmol/mg microsomal protein/mg [93]. Subsequently, ARAT activity was confirmed in intact rat liver cells, which had a combined retinol uptake and esterification maximum rate ~35 pmol/min/mg protein, and a retinol concentration that supported a half-maximal combined rate of uptake and esterification that occurred at ~50 μM [59]. Based on rat liver retinol concentrations, the Km values of liver ARAT suggest that ARAT works most effectively at higher retinol concentrations. One measurement reported retinol in rat liver as ~6 μM [247]. But other measurements place retinol in the range of 13–30 μM in rat liver [63]. Measurements in mouse liver yielded values from 5 to 50 μM [108, 141, 181, 247]. The reasons for these differences are unclear, as all measurements were done on rodents fed chow diets. The point remains, the Km values seem reasonable in view of total unesterified retinol, but not in terms of the physiological form of unesterified retinol, which occurs predominantly as CRBP1 or 2-bound. This suggested that other retinol esterifying enzymes function as a major source of RE in CRBP-expressing cells--a prediction that was validated by the discovery of LRAT.
Although LRAT catalyzes most RE biosynthesis in most tissues, a physiological contribution of ARAT activity to RE formation has been revealed by the presence of RE in tissues of Lrat-null mice fed a vitamin A-deficient diet [141]. RE levels were normal in kidney and brain, increased in adipose, but <1 to ~5 % in liver, lung and eye. The ARAT activities in these tissues were identified as acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) and acyl-CoA:monoacylglycerol acyltransferase 1 (MGAT1), with the former having the higher specific enzymatic activity in the human intestine CACO2 cell line and in mouse liver, testis, kidney, and intestine, and in substrate studies with recombinant DGAT1 [194, 277, 281]. It is now accepted that ARAT activity is produced by these two enzymes, especially DGAT1.
The importance of DGAT1 to RE formation in skin was demonstrated using the Dgat1-null mouse, which showed that DGAT1 accounts for most ARAT activity [251]. In skin of Dgat1-null mice fed a vitamin A-sufficient diet (4 IU vitamin A/g), unesterified retinol increased ~22 % and RA increased ~40 %, but RE levels remained similar to WT, which likely reflects LRAT activity. In contrast, liver retinol, RE and RA levels were similar to WT, consistent with local events driving the changes in skin retinoid homeostasis. When mice were fed a vitamin A-deficient diet, skin retinoids did not differ between null and WT. When mice were fed a diet copious in vitamin A (i.e. standard lab chow, which currently has ~15 IU vitamin A/g), Dgat1-deficiency induced a multifold increase in RA-regulated gene expression in skin, consistent with increased RA levels. Therefore, DGAT1 functions as an ARAT in skin to maintain retinoid homeostasis and prevent toxicity, but makes a minimal contribution in liver. These data generated with the Lrat and Dgat1-null mice demonstrate that LRAT and DGAT1 produce tissue-specific effects on retinoid homeostasis that are influenced by the amount of dietary retinol.
CRBP1 and CRBP2 Contribute to RE Biosynthesis via LRAT
Work on retinol esterification in the mid 1980’s established the existence of an activity in intestine and liver that is independent of ARAT (acyl-CoA) activity [74, 148]. Subsequently, CRBP1 and 2 were evaluated as substrates to test the hypothesis that the two binding-proteins serve as the actual substrates for RE biosynthesis and to determine the relative importance of acyl-CoA dependent and independent activities [96, 146, 147, 192, 253, 282]. With holo-CRBP as substrate, retinol esterification by rat and human liver, rat Sertoli cells, and rat intestinal microsomes proceeded in an acyl-CoA independent reaction that relied on the SN1 position of phosphotidyl choline (lecithin) as fatty acid donor: i.e. a lecithin:retinol acyltransferase or LRAT. The acyl-compositions of RE generated by LRAT were similar to the acyl-composition of RE that occurs in tissues. Specifically inhibiting LRAT reduced RE formation in cultured cells (Caco-2, Sertoli) by as much as 90 %, even though acyl-CoA supported activity was not affected [214, 253].
Reaction kinetics with CRBP1 and 2 as substrates using microsomes and a 1:1 ratio of CRBP/retinol produced Km values lower than ARAT activity. The Km values were below the concentrations of holo-CRBP1 in liver and holo-CRBP2 in intestine (Fig. 2.2, Table 2.2) and with both CRBP1 and 2, were close to those of free retinol, indicating that dissociation of retinol from CRBP is not required to supply LRAT with retinol. This conclusion ensues because the extreme reduction in unbound retinol in the presence of CRBP would profoundly increase the apparent Km and lower the Vm values if only free retinol served as substrate.
Fig. 2.2.
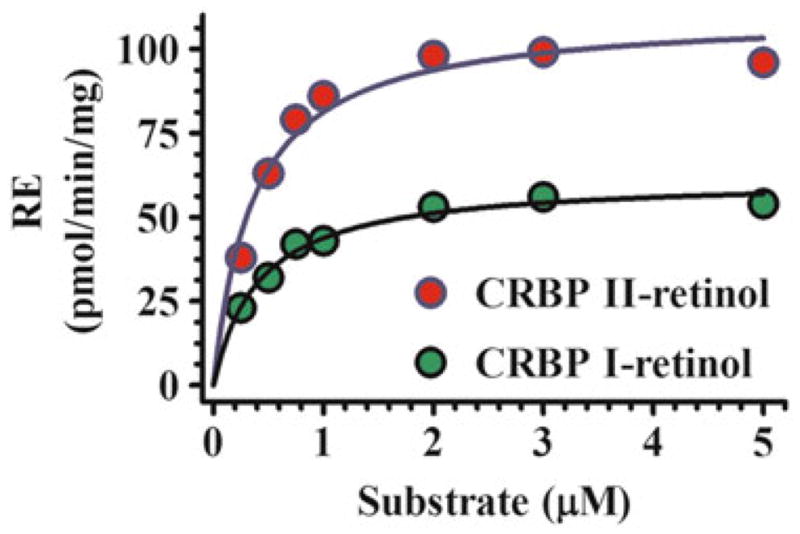
Kinetics of RE formation by LRAT catalyzed by rat liver microsomes with holo-CRBP substrates. Each reaction was run with CRBP1 in a 1:1 ratio with retinol [192]. The Km values were ~1 and 0.7 μM for holo-CRBP1 and holo-CRBP2, respectively, with adult rat liver microsomes. Use of holo-CRBP did not support RE formation from ARAT activity
Table 2.2.
Kinetic values of LRAT for retinol vs. CRBP-bound retinol (holo-CRBP). Km values in μM; Vm values in pmol/min/mg protein
| Substrate | Intestinal microsomes Km; Vm | Liver microsomes Km; Vm |
|---|---|---|
| Retinol | 0.6 ± 0.3; 54 ± 13 | 0.4 ± 0.2; 80 ± 15 |
| CRBP1-retinol | 0.2 ± 0.1; 33 ± 12 | 0.8 ± 0.1; 76 ± 6 |
| CRBP2-retinol | 0.2 ± 0.04; 47 ± 3 | 0.3 ± 0.02; 61 ± 5 |
LRAT and CRBP1 Associate with Lipid Droplets
When cells are not undergoing lipid droplet formation, LRAT embeds in the endoplasmic reticulum (microsomal fraction) via a C-terminal transmembrane domain, with most of the enzyme, including its active site, projecting into the cytoplasm [159, 192, 282]. LRAT surrounds CRBP1, which co-localizes with mitotracker, suggesting association with the outer mitochondrial membrane or mitochondria associated membranes [102]. During lipid droplet formation stimulated by oleate or retinol, LRAT surrounds growing lipid droplets, colocalizing with the lipid droplet surface proteins, desnutrin/adipose triglyceride lipase and perilipin 2, and CRBP1 co-localizes with LRAT. Association of LRAT with lipid droplets is specific as shown by (1) a requirement for two amino acids residues (K36 and R38) in its N-terminus for lipid droplet association and (2) the enhancement of LRAT specific activity upon lipid droplet association [102]. These data place both LRAT and CRBP1 at the surfaces of lipid droplets during acyl ester formation, which is consistent with the kinetic data that reveals a substrate-enzyme relationship between the two proteins. The data also illustrate the mobility of the proteins, which do not remain fixed but migrate during metabolism.
Genetic Ablation of LRAT
Knockout of Lrat revealed that LRAT serves as the major, but not sole retinol esterifying enzyme in several mouse tissues [8, 121, 141, 143, 180, 238]. Lrat-null mice fed chow diets (copious vitamin A) have severely depleted RE levels in liver, lung, eye and testes, but not in kidney, adipose, pancreas or brain. Despite reduced RE, chow-fed Lrat--null mice develop normally, but have degraded rod and cone functions of the neural retina because of a need for profuse vitamin A flux into the eye to support vision. The Lrat-null mouse also responds to excess dietary retinol by increasing retinol excretion, RE deposition in adipose, and induction of Cyp26A1, which accelerates RA catabolism [143]. Thus, LRAT contributes to regulating vitamin A homeostasis in multiple tissues during copious vitamin A intake.
When Lrat-null mice are fed a vitamin A-deficient diet for 6 weeks, retinol is undetectable <0.007 pmol/mg) in most tissues. Liver and kidney have detectable retinol, albeit <6 % of WT. This observation should be evaluated in context of the well-established phenomenon that mice bred from dams fed a chow diet do not become vitamin A-deficient during 36 weeks of feeding a vitamin A-deficient diet from weaning (Napoli, unpublished data). These data demonstrate that LRAT is essential for maintaining retinoid levels in select tissues during restricted dietary vitamin A—the normal situation throughout evolution.
Recent work has confirmed the primacy of LRAT as the RE forming enzyme in liver and its function in preventing retinol loss [278]. Interestingly, although low, RE was 50-fold higher in livers of the Lrat/Rbp1double knock-out mice compared to the single Lrat knockout (Table 1 of [278]), whereas unesterified retinol was ~90 % lower (Fig. 3 of [278]), indicating CRBP1 restricts access of acyl transferases other than LRAT to retinol in vivo. Because a chow diet was fed, the impact of this partnership during low amounts of retinol, a situation of potentially greater CRBP1 impact, remains unclear. Regardless, these data indicate that the CRBP1/LRAT partnership promotes retinol sequestration by creating a gradient driven by RE biosynthesis. Although this channeling would protect retinol from ARAT activity, RE formation in the absence of LRAT and CRBP1 is quantitatively minor relative to WT, reinforcing the model that the channeling partnership promotes efficient intracellular retinol sequestration.
Retinyl Ester Hydrolysis
Initial studies of retinyl ester hydrolysis (REH) in vitro focused on activity that required cholate [86]. This bile salt-dependent activity has a specific requirement for ≥ 10 mM cholate or taurocholate, hydrolyzes triacylglycerol and cholesteryl oleate at higher rates than RE, and is remarkably similar to pancreatic carboxyester hydrolases [85]. The specific activity varies radically from animal to animal, suggesting confounding influences, such as erratic contamination by secreted and/or plasma hydrolases. Because most tissues that store and mobilize RE and generate RA are unlikely to contain millimolar bile salts, attention turned to alternative enzymes. In 1989 two labs independently demonstrated occurrence of a bile-salt independent microsomal REH (bsiREH) activity in liver and in other vitamin A target tissues, including lung, intestine, kidney and testes [89, 90, 169]. The bsiREH did not hydrolyze cholesterol esters and had higher activity with RE than triacylglycerol.
Several intracellular REH activities have been isolated from the intestinal brush border [226–228], liver and other tissues [140, 259]. They belong to the ES or CES family (i.e. ES-2, and ES-10). In adipocytes hormone-sensitive lipase has higher specific activity with RE compared to diacylglycerol, and its ablation reduces RA signaling, indicating its importance to generation of adipocyte RA [257]. Likely, multiple membrane-associated enzymes function as intracellular REH with tissue-specific expression [87].
The Ratio of Holo-CRBP1 to Apo-CRBP1 Influences Retinol Metabolism
Apo-CRBP1 activates bsiREH-catalyzed hydrolysis of resident RE in liver microsomes and displays Michaelis-Menten kinetics [28]. RE hydrolysis occurs with 2.6 μM apo-CRBP1 stimulating a half-maximum rate (Fig. 2.3). The effect is specific because other proteins capable of sequestering retinol are ineffective, such as (bovine serum albumin and β-lactoglobulin). apo-CRBP1 functions as a competitive inhibitor of LRAT, with a Ki value ~0.2 μM [96]. These actions suggest that the ratio of apo-CRBP1 to holo-CRBP1 senses intracellular retinol status and modifies retinol channeling. Increasing apo-CRBP1 during declining retinol concentrations would stimulate RE hydrolysis to prevent total retinol sequestration as RE and maintain sufficient holo-CRBP1 to support RA biosynthesis.
Fig. 2.3.
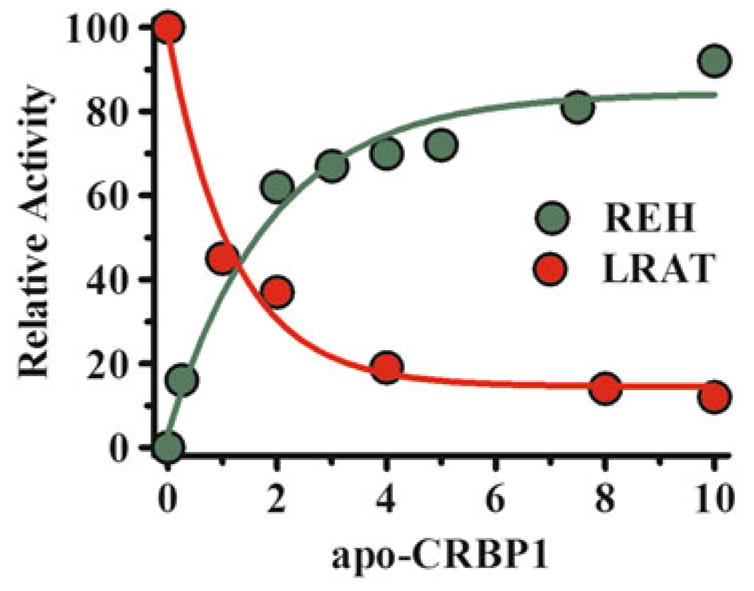
Apo-CRBP1 effects on retinol esterification and RE mobilization. Increasing concentrations of apo-CRBP1 increase the hydrolysis of resident RE in liver microsomes, whereas titrating apo-CRBP1 into an LRAT reaction using holo-CRBP1 as substrate inhibits RE formation. These data indicate that the ratio holo-CRBP1/apo-CRBP1 influence flux between retinol and RE. Fully-charged CRBP1 would favor RE formation, while still allowing retinal formation for RA biosynthesis; whereas appreciable apo-CRBP1 would stimulate RE hydrolysis to maintain holo-CRBP1 as substrate for RA biosynthesis
CRBP2 Favors Retinal Reduction and Retinol Esterification in the Intestine
The first demonstration that a retinoid binding-protein was involved in retinol metabolism was reported with CRBP2 [191]. The high concentration of CRBP2 (~1 % of total soluble protein in the intestinal enterocyte) predicts a vital contribution for managing the efficiency of dietary retinoid absorption. This prediction was verified by demonstrating that LRAT in rat intestinal microsomes converts retinol bound to CRBP2 into RE with a fatty-acyl composition that reflects the composition of RE in chylomicrons. The apparent Km for the CRBP2-retinol complex (~0.2 μM) is likely well below the concentration of the complex generated from retinol absorption or β-carotene metabolism.
Carotenoids provide most dietary vitamin A. A dioxygenase centrally cleaves β-carotene, the prototypical dietary provitamin A carotenoid, into two molecules of retinal [55, 88]. Retinal can undergo reduction into retinol for esterification and distribution as RE in chylomicrons, or conversion into RA. What prevents generation of large amounts of RA rather than predominantly RE? The answer was provided by the demonstration that CRBP2-bound retinal serves as substrate for retinal reduction, catalyzed by rat intestinal microsomes with a Km value ~0.5 μM [106]. Retinol generated by the reductase remains bound to CRBP2, available for esterification without diffusion into the aqueous medium. Significantly, CRBP2-bound retinol is not oxidized efficiently (<0.3 pmol/min/mg protein). compared to the reduction of CRBP2-bound retinal (~300 pmol/min/mg protein). These data reveal the mechanism that directs most intestinal carotenoid metabolites into RE, while avoiding generating large amounts of RA (Fig. 2.4). The phenotype of the Rbp2-null mouse (RBP2 encodes CRBP2) supports this conclusion. Rbp2-null mice fed a chow diet (copious vitamin A) have RE levels that are 40 % of the levels in WT mice, but show no symptoms of vitamin A-deficiency. When fed marginal dietary vitamin A (0.6 IU/g), Rbp2-null pups suffer from 100 % mortality within 24 h after birth vs 0 % mortality for WT. These results illustrate two points: (1) cellular retinoid-binding proteins are essential for efficient use of vitamin A; (2) contributions of cellular retinoid-binding proteins to retinoid homeostasis and function can be overwhelmed by diets copious in vitamin A. Table 2.3 provides a summary of rationale supporting interactions of CRBP with retinoid-metabolizing enzymes.
Fig. 2.4.

Channeling of intestinal retinal metabolism to direct pro-retinoid and retinoid flux into RE. β-Carotene and pro-vitamin A carotenoids provide the major sources of dietary vitamin A. A dioxygenase (BCO1) generates retinal from β-carotene. Retinal can then be reduced by retinal reductase or dehydrogenated into RA. The dehydrogenation rate of retinal bound to CRBP2 is <300-fold the rate of reduction into retinol. β-Carotene is not toxic, whereas RA is toxic. Moreover, large scale conversion of retinal in the intestine into RA likely would not serve the vitamin A needs of the animal. CRBP2-directed metabolism of retinal through retinol into RE for incorporation into chylomicrons provides a mechanism for efficient conservation of vitamin A as its ester, systemic delivery of RE via chylomicrons, and limiting RA biosynthesis
Table 2.3.
Rationale supporting substrate-product relationships between CRBP and retinoid-metabolizing enzymes. These observations have been developed mostly with CRBP1, but several pertain to CRBP2, CRABP1, and CRABP2
| High affinity | CRBP binds retinoids with affinities near those of nuclear hormone receptors and engulfs retinoids in an interior pocket protected from the cellular milieu. |
| Abundance | Holo-CRBP embodies the major form of non-esterified retinol in many tissues and therefore the substrate of highest concentration. |
| Regulation | By allowing only specific enzymes access to its tightly-bound retinoids, CRBP would control retinoid metabolism (see CRBP-null below). |
| Security | CRBP sheltering enhances retinol stability and protects retinal from forming adventitious Schiff’s bases with amine residues. |
| Kinetics | Prototypical enzyme-substrate kinetic relationships occur between holo-CRBP (retinol or retinal) concentrations and rates of retinoid metabolism. |
| Crosslinking | Protein crosslinking reagents form covalent bonds between holo-CRBP1 and RDH and LRAT in microsomes (many possible targets, only two interact). |
| CRBP mutants | A single exterior amino acid mutation (L35A) of holo-CRBP1 decreases the rate of retinal formation without affecting retinol binding affinity. |
| Gene knockouts | Rbp1 and Rbp2-null mice experience depleted retinol uptake and/or enhanced depletion and metabolic disease, related to changes in retinoid metabolism. |
RA Biosynthesis Independent of CRBP: ADH
The earliest studies demonstrating conversion of retinol into retinal outside of the visual cycle were done before RA was appreciated as essential for the systemic functions of vitamin A. Lacking this insight, early studies did not focus on investigating de novo pathway(s) that generate RA from physiological substrates, but rather, focused on catalysis by members of the medium-chain alcohol dehydrogenase gene family (ADH) [27, 223]. Later work, which concluded that ethanol inhibits generation of “active retinal”, relied on 60 μM retinol (exceeds the testis concentration of retinol by 750-fold) in a crude cytosolic fraction [265]. The latter report hypothesized that competitive inhibition by ethanol reduced retinal formation and caused the testicular atrophy and aspermatogenesis experienced by alcoholics. These studies were done before: (1) quantification of retinol concentrations in tissues; (2) identification of specific high-affinity retinoid binding-proteins; (3) there was an appreciation for RA’s occurrence and importance. Therefore, there was no background to question whether xenobiotic clearing enzymes would represent credible candidates for maintaining RA homeostasis. Also, the short-chain dehydrogenase/reductase (SDR) gene family was not known. If it were, earlier investigators might have inquired about its involvement in RA biosynthesis, because SDR metabolize hormones and autocoids, such as steroids and prostanoids [193].
The presumption that ADH catalyze retinol metabolism spawned a hypothesis that ethanol interferes with retinoid metabolism by competitive inhibition. This hypothesis has been long-lived, despite not having been tested by analytically rigorous assays of ethanol effects on RA levels in vivo, and despite much evidence to the contrary [165]. For example, a spontaneous mutation in ADH1 in peromyscus (aka deermouse) was reported in 1978 [37]. Adh1-null deermice have normal testes histology and reproduction, which indicate the animals have normal vitamin A-supported processes [131]. Cytosol of the Adh1-null deermouse catalyzes RA biosynthesis from physiological concentrations of retinol with rates from 0.3 to 0.7 nmol/h/mg of protein, more than sufficient to fulfill cellular needs [209, 210]. Finally, ADH1 expression in vivo does not correlate with sites of RA biosynthesis [268].
Further, a concentration of ethanol (1000 mM) that far exceeds the blood alcohol concentration of chronic alcoholics (~25 mM) did not inhibit RA synthesis in kidney cytosol from the Adh1-null deermouse. Analytically robust assays also demonstrated that ethanol does not decrease RA concentrations in vivo in multiple tissues of WT mice [111, 155, 165], findings that support earlier observations showing that a 10,000–30,000-fold excess of ethanol (100–300 mM) relative to retinol only partially inhibits (54–69 %) RA biosynthesis from 10 μM retinol in rat liver cytosol [168]. Nor is RA biosynthesis from retinol inhibited by ethanol in intact cells in culture [111, 155, 162, 165, 254]. In fact, ethanol causes massive loss of hepatic RE, a phenomenon established earlier, but for which a mechanism has not been determined [127, 246]. One potential mechanism seems to be RA catabolism by Cyp2e1, a xenobiotic clearing enzyme with over 85 substrates [136, 142, 270]. Ethanol-induced RA catabolism causes a compensatory mechanism to replenish RA by drawing on RE stores [111]. Ethanol and its metabolite acetaldehyde, however, affect multiple binding proteins and receptors that participate in generating RA and mediating vitamin A function, and undoubtedly many related processes. Further discussions of ethanol effects on retinoid metabolism are available [165, 270, 284].
ADH-null house mice have not been informative concerning whether they contribute to RA biosynthesis under physiological conditions. Studies of ADH knockout mice have not reported essential experiments, such as RA levels during normal vitamin A nutriture, pathology resulting from reduced RA, complete rescue with reasonable amounts of RA, or gene expression changes indicating changes in RA signaling and/or compensation for decreased RA [56]. Thus, claims of ADH involvement in the physiological metabolism of retinol remain unsubstantiated, despite the long-term availability of ADH knockout mice.
A revised hypothesis proposed that ADH1 and ADH3 catabolize “excess” retinol to alleviate retinol toxicity. This hypothesis overlooks a large body of data and contradicts the original hypothesis by the same group that ADH produce RA to serve physiological functions [160]. Throughout evolution, vitamin A has been a scarce but essential nutrient—retinol insufficiency remains the third most prevalent nutrient deficiency worldwide. Mammals evolved to sequester retinol efficiently, as shown by the Crbp2-null mouse, not to eliminate an “excess”. There was no excess and therefore no evolutionary pressure to catabolize an excess. Abundant dietary retinol is stored as RE in liver, which can achieve concentrations exceeding 1 mM. Other tissues, such as adipose, also can accumulate relatively high RE concentrations. It is unlikely that ADH evolved to “detoxify” an essential nutrient that can be stored in substantial amounts, and do so by sending it down a path that generates a metabolite (RA) with greater toxicity.
A single 3 mg retinol/kg dose induces 71 % incidence of cleft palate and a 39 mg/kg dose induces 76 % incidence of neural tube defects [23]. The data used to support the contention that ADH detoxifies retinol were generated by measuring RA in Adh1-null or Adh3-null mice after administering a retinol dose of 50 mg/kg or 100 mg/kg. The 50 mg/kg dose delivers ~300-fold more vitamin A than the recommended daily intake for mice, and was given to mice fed a diet copious in vitamin A. This dose produces serum RA concentrations ~1600–2330-fold greater than the steady-state value (~2 nM) in WT mice, ~100–200-fold greater than normal in the Adh1-null mouse, and ~650-fold greater than normal in the Adh3-null mouse. These data do not support ADH1 or 3 as detoxifying retinol because prolonged exposure to even modestly increased concentrations of RA causes teratology [50, 173, 264]. Generation of RA from toxic amounts of retinol likely contributes to retinoid toxicity. Figure 2.5 summarizes the distinction between metabolism of low amounts of dietary retinol involving binding-proteins vs. copious or toxic amounts of retinol that overwhelm binding-proteins and become accessible to xenobiotic-clearing enzymes.
Fig. 2.5.

Disposition of low amounts of dietary retinol vs. copious or toxic dietary retinol. This figure presents a model for retinol toxicity consistent with saturation of retinoid binding-proteins by copious dietary retinol resulting in unmetered generation of RA by xenobiotic clearing enzymes. This path is in contrast to channeling of dietary sufficient amounts of vitamin A by retinoid binding-proteins, such as CRBP1. This path complements the path imposed upon the intestine by CRBP2, which directs dietary retinol into RE, rather than RA
A De Novo Approach to RA Biosynthesis
Study of retinol metabolism, using analytically validated retinoid quantification by HPLC and GC/MS, established that conversion of retinol into retinal is rate-limiting in intact cells (pig kidney cell line LLC-PK1) [162]. Reduction of retinal into retinol and dehydrogenation of retinal into RA occur at rates four–eightfold and 30–60-fold faster, respectively, than retinol dehydrogenation. Identification of the rate-limiting step was reinforced in studies of RA biosynthesis by primary hippocampus astrocytes [273].
The work with LLC-PK1 cells also showed, using the ADH inhibitor 4-methylpyrazole, that ethanol metabolizing enzymes do not catalyze RA formation in intact cells in the physiological range of retinol concentrations. Cultured human keratinocytes provided the same insights: conversion of retinol into retinal is rate-limiting and ADH are not involved in the RA biosynthesis in intact cells [254].
CRBP1 as Bait to Identify Physiologically Relevant Retinol Dehydrogenases (RDH)
Many cell types, subcellular fractions, and enzyme types convert free retinol into retinal [163, 168]. The issue was identifying the enzyme(s) that catalyze the rate-limiting step in vivo under physiological conditions. Based on awareness that holo-CRBP1 provides the major potential substrate for retinol metabolism, holo-CRBP1 was used as bait to identify physiologically significant RDH [29, 211].
Technical Issues with CRBP and Retinol as Substrates
Experiments that evaluate free vs. bound retinol as substrate require careful attention to preparing holo-CRBP. It is important to prepare holo-CRBP1 frequently and to pass it through a Sephadex column to insure a 1:1 ratio of CRBP1:retinol. Only this assures a true 1:1 ratio. Apo-CRBP1 is then added to give the most accurate ratio of apo/holo. Storing holo-CRBP1 for longer times, even at −80 °C, allows conversion of some all-trans-retinol into 9-cis-retinol (Napoli, unpublished data). This became obvious because reactions with “old” holo-CRBP1 produced some 9-cis-RA, which our HPLC system distinguishes from RA. Freshly prepared holo-CRBP1 does not produce 9-cis-RA. Experiments done in my lab used CRBP1 prepared as just described.
Other issues arise from the lability of retinol in aqueous media and impurities that contaminate commercially available retinol, including retinal. In the absence of CRBP1, 60 % of the total retinal generated in vitro was artifactual (did not rely on presence of cofactor or enzyme), but the retinal generated from CRBP1-retinol exceeded background by >tenfold. Thus, it is imperative to purify retinol through HPLC or by binding it to CRBP and purifying the holo-CRBP.
CRBP1 and Retinal Biosynthesis
Multiple experimental approaches indicate that RDH recognize holo-CRBP1 as substrate. Typical Michaelis-Menten enzyme kinetics are observed between concentrations of holo-CRBP1 and rates of retinal formation catalyzed by liver microsomes [211]. (Liver microsomes do not produce RA). Rates of retinal formation tested with two different ratios of total CRBP1 to retinol, 1:1 and 1.5:1, each produced the same Km and Vm values, which could not occur if only free retinol were substrate (Fig. 2.6). The Vm would have decreased markedly with the nearly five-fold decrease in free retinol after adding the additional apo-CRBP1. Furthermore, the nanomolar concentrations of free retinol present with the micromolar amounts of holo-CRBP1 were not sufficient to drive the observed rates of retinal biosynthesis. N-ethylmaleamide preincubated with RDH inhibited retinal generation from holo-CRBP1, but did not prevent RDH from converting unbound retinol into retinal, consistent with obstructing access of CRBP1 to RDH without affecting RDH catalytic activity [30]. In contrast, N-ethylmaleamide preincubated with holo-CRBP1 did not prevent retinal biosynthesis. With free retinol in the absence of cofactor, retinal was produced at ~75 % of the cofactor-supported rate owing to non-enzymatic oxidation. With holo-CRBP1 in the absence of cofactor, retinal generation was negligible, indicating protection of retinol from non-enzymatic oxidation.oxidation. Velocities, however, were lower with CRBP1-retinol than with unbound retinol, possibly because transfer of retinol from CRBP1 to RDH became rate limiting [30, 171]. These data indicate that RDH must recognize holo-CRBP1, in addition to free retinol.
Fig. 2.6.

Kinetic relationships between RDH and CRBP1-retinol. The top panel shows the Michaelis-Menten relationship (substrate-concentration dependent, saturable kinetics, initial velocity conditions) between holo-CRBP1 and the rate of retinal formation catalyzed by microsomes. The kinetic constants generated were independent of the ratio total CRBP1/retinol, indicating ability of CRBP1 to deliver retinol without diffusion. The bottom panel shows the impact of mutating external residues of CRBP1 on kinetics of retinal formation using microsomes as source of RDH. The sensitivity of the kinetics to changes in one external CRBP amino acid residue, without affecting affinity for retinol, corroborates an interaction between RDH and CRBP1
A similar approach, also using two different ratios of total CRBP1 to retinol (1:1 and 2:1) demonstrated a Michaelis-Menten relationship with a Km value of ~0.8 μM between holo-CRBP1 and RDH activity in calf liver cytosol [196]. Cytosol contains RALDH isoforms, so cytosol can convert retinal generated from retinol into RA. In this preparation, increasing the ratio of CRBP1/retinol to 2 decreased the Vm ~50 %. Because increasing the ratio decreased the free retinol concentration from 120 to 3 nM (40-fold) at a nominal CRBP1-retinol concentration of 5 μM, this result confirmed that holo-CRBP1 acts as substrate and that apo-CRBP1 competes with holo-CRBP1. The Km value for CRBP1-bound retinol was lower than that of free retinol, but was greater than the kd value for retinol dissociation from CRBP1. These phenomena cannot occur if retinol must dissociate from CRBP1 prior to association with RDH. This work also showed that apo-CRBP1 captured the retinal generated from CRBP1-retinol.
To resolve the relative contributions of microsomal vs cytosolic RDH, both subcellular fractions from liver, kidney, testes, and lung were assayed for activity with 5 μM holo-CRBP1 and with 5 μM holo-CRBP1 plus 2 μM apo-CRBP1 [31]. The latter was done to model RA biosynthesis during states of moderate vitamin A-status in which CRBP1 would not be saturated with retinol, because apo-CRBP1 is a potent inhibitor of cytosolic RDH (IC50 ~ 1 μM). In the absence of apo-CRBP1, microsomes accounted for 60–83 % of the RDH activity (total enzyme units). In the presence of apo-CRBP1, microsomes accounted for 80–94 % of the combined microsomal plus cytosolic activity. Specific activities of microsomal RDH in presence of 5 μM holo-CRBP1 plus 2 μM apo-CRBP1 ranged from 13 to 48-fold higher than those in cytosol, depending on the tissue. This showed that microsomal RDH generates the quantitatively major share of retinal for RA biogenesis from the predominant physiological substrate, holo-CRBP1. The SDR inhibitor carbenoxolone and apo-CRBP1 confirmed the cytosolic activity as resulting from an SDR. Possibly, cytosolic RDH that recognize holo-CRBP1 function as reductases (see below). If so, inhibition by apo-CRBP1 would allow continued RA biosynthesis during restricted cellular retinol. Thus, work reported in 1986 and continued through 1996 established that RDHs serve as physiological generators of retinal for RA biosynthesis in multiple species and tissues, and excluded ADH participation in RA biosynthesis under normal conditions.
External Residue Mutation Affects CRBP1 as Substrate Without Altering Affinity for Retinol
Specific CRBP1 residues were mutated to assess ligand binding interactions, conformation flexibility, and the kinetics of retinal formation catalyzed by microsomes [198]. Data generated from these mutants show that residues L29, I32, and R58 do not affect substrate efficiency, but contribute to ligand binding and protein rigidity. In contrast, mutation of L36 (L36A) increased substrate efficiency, but the mutant also was more flexible and had a lower ligand binding affinity (Fig. 2.1).
Surface residue L35 projects from the body of CRBP1. Converting it into an alanine residue (L35A) did not change affinity for retinol, but decreased the Vm of retinal formation by 50 % (Fig. 2.6). This suggests RDH interacts with the L35 side-chain to counteract interactions of interior residues with the β-ionone ring of retinol. Enzyme binding to L35 apparently alters the position of the cap to allow transfer of retinol from CRBP1. Residues V27 and K31 in αII may also contribute to interactions with RDH, because they are bulky and project from the CRBP1 surface. Unfortunately, these residues were not mutated. Individual mutations of V27 and K31, as well as triple mutation of V27, K31, and L35 would test this hypothesis further. Mutant L35A was somewhat less rigid than WT, as judged by Arg-C digestion of holo-CRBP1, which cleaves at R30 in α-helix II (αII). Charged mutations L35E and L35R had the same efficiency for retinal formation as WT, consistent with a van der Waals interaction between L35 and RDH. Each charged L35 mutation has affinity for retinol equivalent to WT, but each had much more flexibility than L35A, presumably because of charge repulsion from nearby residues. These are remarkable kinetic results for mutation of a single exterior residue that does not determine affinity of ligand binding.
In contrast to L35A, CRBP1 mutants (L29A, I32A, L36A, F57A, F58A, R58E) each had ~two–fivefold reduced affinity for retinol and were proteolyzed as much as ~fivefold more rapidly than WT. These data connect affinity of retinol binding with protein flexibility and are consistent with the greater flexibility of apo-vis-à-vis holo-CRBP1. None of these mutations, however, had a Vm for retinal formation lower than WT. In fact, mutants L36A and R58A had 50 % increases in Vm values and >tenfold increases in efficiency relative to WT, likely reflecting increased ease of portal opening and retinol release. Taken together, these data suggest that the β-ionone ring grasps the “handle” provided by the interior residues discussed, offset by the hydrophilic cavity exerting repulsive forces on the isoprene side chain. Enzyme interaction with the αII exterior residues would contribute sufficient force to promote retinol egress. Thus, pressure for the conservation of the external residues of CRBP seems to reflect a need for interaction with enzymes that rely on CRBP1 to provide retinol, not merely for protein conformation and/or ligand binding.
CRBPI Identified SDR as RDH
Microsomal RDH that recognize holo-CRBP1 occur in at least two forms: integral and peripheral membrane [29]. RDH1 has been identified as an integral microsomal enzyme that anchors in the membrane facing the cytoplasm [272, 288]. The more stable peripheral form was purified to two major bands on SDS-PGE, one ~35 kDa and the other ~54 kDa [29]. To determine which might represent RDH, either microsomes or a glycerol extract of microsomes was presented with CRBP1 covalently bound with a radioiodinated, heterobifunctional, cleavable crosslinking arm (Fig. 2.7). This allowed transfer of radioiodine from the donor (CRBP1) to specific acceptors. Only two molecules cross-linked. One was ~35 k Da and the other was ~25 kDa. Crosslinking the ~35 kDa protein required pyridine nucleotide cofactor, consistent with the ordered bisubstrate reaction mechanism of a NAD(P)+-supported dehydrogenase, which must bind cofactor before accepting substrate. The ~25 kDa protein did not require cofactor to crosslink, and likely represented LRAT, as purification of LRAT showed that its molecular weight was ~25 kDa [237]. Crosslinking was specific, because only these two proteins in microsomes cross-linked, and crosslinking occurred with each protein based on their individual cofactor requirements.
Fig. 2.7.

Holo-CRBP1 crosslinks with RDH. A. CRBP1 was covalently modified with a heterobi-functional, cleavable crosslinking reagent. The crosslinker was radioiodinated. B. UV irradiation activated the azide of the crosslinker to a nitrene residue that formed a covalent bond between CRBP1 and closely associating RDH (and LRAT). C) Cleavage of the crosslinking reagent left the radioiodine on the target protein, effectively resulting in a transfer of radioiodine from holo-CRBP1 to RDH and LRAT. CRBP1 cross-linked only with two proteins in microsomes, RDH and LRAT, indicating specificity of the interaction. Crosslinking with RDH required presence of NAD(P)+, consistent with the ordered bisubstrate reaction mechanism of a dehydrogenase
The 35 kDa protein had a molecular weight and other attributes of an SDR, including the highly conserved sequence WXLVNNAG, Zn 2+ independence, inhibition by carbenoxolone (IC50 = 55 pM), and insensitivity to inhibition by ethanol and the ADH inhibitor 4-methylpyrazole. Multiple different cDNA were identified. One of these was cloned first and referred to as RODHI. The cDNA validated it as an SDR that recognized holo-CRBP1 (1.4:1 ratio total CRBP1/retinol, to ensure virtually no free retinol) with a Michaelis-Menten relationship with a Km value <1 μM [39]. Soon, other RDH/SDR were cloned based on the data generated from RODHI [39, 40, 43, 256, 287]. Subsequently, human orthologs were cloned [25, 80, 105, 150] (Table 2.4). Of these, RDH1, RDH10 and DHRS9 have been associated with vitamin A-dependent processes as indicated by studies of gene ablation in the mouse or in zebrafish, or overexpression in cells.
Table 2.4.
Dehydrogenases and reductases likely to contribute physiologically to RA homeostasis1
| Mouse | Rat | Human | Function |
|---|---|---|---|
| Rdh1 (9C17) | Rdh2, Rdh7 (9C28, 9C29) (RodhII, RoDHIII) | RDH16 (9C8) (RODH4, RDH-E) | Dehydrogenase |
| Rdh10 (16C15) | RDH10 (16C4) | Dehydrogenase | |
| Rdh10 (16C10) | Dhrs9 (9C26) (eRolDH, Rdhl) | DHRS9 (9C4) (retSDR8, RDHL, Rdh-TBE, RoDH-E2, 3α-HSD) | Dehydrogenase |
| SDR16C5 (16C114) | RDHE2 (16C5) | Dehydrogenase | |
| Dhrs9 (9C12) (Rdh15) | SDR16c6 (16C113) | a SDR (16C6P) | Dehydrogenase |
| Rdhe2 (16C11) (Scdr9, Sdr16c5) | – | RDH14 (7C4) | Dehydrogenase |
| Rdhe2S (16C12) (SDR16c6) | (PAN2) | Reductase | |
| Rdh14 (7C12) | Dhrs3 (16C16) | DHRS3 (16C1) (RDH17, SDR1, SDR16C1, retSDR1, Rsdr1) | |
| Dhrs3 (16C9) | Dhrs4 (25C7) (PSCD) | DHRS4 (25C2) (SCAD-SRL, SDR-SRL) | Reductase |
| (retSDR1, Rsdr1) | Rdh11 (7C14) | Rdh11 (7C1) (PSDR1, RalR1) | Reductase |
| Dhrs4 (25C5) (RRD) | – | RDH13 (7C3) | Reductase |
| Rdh11 (7C9) (SCALD, Psdr1) | |||
| Rdh13 (7C11) |
All belong to the SDR gene family. Orthologs are aligned in rows. “Family designations” are given in parentheses next to each human gene, each is preceded by SDR, e.g. SDR9C8 [202, 203]. Alternative names are given in parentheses below. Several genes were cloned multiple times and named differently. Various nomenclature initiatives introduced further confusion by assigning different common names to orthologs (e.g. mRDH1, rRDH2, hRDH16)
This is a pseudo gene in human. –, no entry for rat
RDH10 is one of multiple members of the SDR gene family that catalyze inter-conversion of retinol and retinal in the visual cycle, but also contributes to RA biosynthesis in multiple tissues [197, 206]. RDH10 was first cloned from the RPE and identified as important to the visual cycle [279], and recognizes two other proteins that mediate retinoid metabolism in the eye, RPE65 and CRALBP [66]. RDH10 immunoprecipitates with both proteins and catalyzes the conversion of retinol bound to a twofold molar excess of CRALBP into retinal at the same rate as with free retinol. Outside the eye, RDH10 is broadly distributed in the cell in microsomes and mitochondria and mitochondrial associated membranes, but during acyl ester and lipid droplet biosynthesis, it translocates to lipid droplets and co-localizes with LRAT and CRBP1 [103]. This suggests that lipid droplets, which store RE, also serve as a source of RA, and again illustrates the intracellular mobility of retinoid binding-proteins and enzymes.
Retinal Reductases Contribute to Retinol and RA Homeostasis Outside of Intestine and Eye
Reductases of the SDR gene family have been identified that convert retinal into retinol [9, 10, 84, 114, 120, 130] (Table 2.4). Most have not been tested for recognition of CRBP1-retinal as substrate, with the exception of DHRS4 (originally RRD), a peroxisomal reductase that leaks into cytosol. DHRS4 recognizes CRBP1-bound retinal as evidenced by a Michaelis-Menten relationship with CRBP1-retinal (twofold molar excess of total CRBP1 to retinal), but with a Vm ~25 % of the rate of unbound retinal [130]. To provide insight into whether competitive inhibition between apo- and holo-CRBP1 produced these results, apo-CRBP1 was titrated into a reaction with unbound retinal as substrate. The rate of reduction did not decrease markedly as long as the total CRBP1 concentration was less than the retinal concentration, i.e. as long as no apo-CRBP1 was present. Adding apo-CRBP1 concentrations greater than the retinal concentration decreased reduction rates with an IC50 ~ 0.6 μM CRBP1, indicating competitive inhibition between CRBP1-retinal and apo-CRBP1.
Of the reductases, only DHRS3 has been knocked out in mice [24]. Ablation caused a 40 % increase in RA and 55–60 % decreases in retinol and RE during embryogenesis. These results show that regulation of RA homeostasis requires both retinol dehydrogenation and retinal reduction, achieved through the actions of SDRs.
Retinoid Metabolizing Enzymes Have Interactions Beyond Binding-Proteins
Knockdown of Dhrs9 in primary astrocytes increased RA biosynthesis ~ 40 % by increasing RALDH1 expression [273]. It may seem odd that increasing RALDH1 would increase RA biosynthesis, because RDH catalyze the rate-limiting reaction. But the Vm of the slowest enzyme in a branched pathway is not necessarily rate determining for any one final product, and the pathway from retinol to RA is non-linear. Retinal can be fed into the path from retinol dehydrogenation or carotene cleavage, and can be removed by reduction or dehydrogenation. Therefore, at least four reactions determine the concentration of the retinal pool and the rate of RA biosynthesis. Increasing the concentration of RALDH1 would allow more efficient competition for the retinal pool (reaction rate ∝ [enzyme] x kcat x [substrate]), especially because the reaction is irreversible.
Repression of RALDH1 expression complements the observation that DHRS9 “moonlights” as a transcriptional repressor [151]. Another form of interaction may be through direct binding to other proteins. During purification, rat RDH activity required binding to a 54 kDa protein, identified as CYP2D1 [29, 98]. In another example, recombinant tagged RDH10 and DHRS3 expressed in HEK293 cells form a heterodimer that enhances activities of both [1]. These data indicate the complexity of regulating RA biosynthesis and maintaining RDH structure to retain enzymatic activity and interactions of enzymes with retinoid binding-proteins.
Features of RDH Knockouts
The Rdh1-knockout produced the unexpected results of no apparent developmental or morphological phenotype, and no decrease in liver RA, even though Rdh1 expression correlates with RA targets. The Rdh1-null mouse, however, has altered retinoid homeostasis [285, 286, 289]. Decreases in enzymes that catabolize RA, i.e. liver Cyp26A1 and brown adipose Cyp26B1, and increases in liver RDH10 and brown adipose DHRS9 and RALDH1 modify the phenotype of the Rdh1-null mouse. When fed low-fat diets with modest amounts of vitamin A, the Rdh1-null mouse gains up to 37 % more weight than WT, due to increased adiposity. Rescue of the phenotype with diets high in vitamin A, and the ameliorating effect of pharmacological RA doses on weight gain in mice challenged with a high-fat diet corroborate the phenotype of the Rdh1-null mice as RA related [20]. In another model, RA was reduced 50 % in the kidney cortex of Tg26 mice (HIV-1 transgenic) and 70 % in their glomeruli, associated with a 70 % reduction in Rdh1 mRNA and ~twofold increase in Cyp26A1 mRNA [218].
Although Rdh10 ablation is embryonic lethal, it does not produce complete vitamin A-deficiency, indicating essentiality of additional RDH during development [224]. This activity may be supported by either RDH1 or DHRS9.
Both free- and CRBP1-bound retinol serve as substrates for DHRS9 [150]. Transfection of Dhrs9 into COS cells increases their ability to biosynthesize RA [221, 273]. Dhrs9 expression seems to be estrogen regulated, as the rat uterine lining epithelium expresses Dhrs9 only during estrus and co-expresses it with CRBP1 and CRABP2 [64, 221]. Dhrs9 has not been knocked out in mice, but zebra fish require Dhsr9 for normal gut development and differentiation, both RA dependent processes [161]. Introduction of the tumor suppressor adenomatous polyposis coli into the colon carcinoma cell line HT29 induces Dhrs9 mRNA fivefold and increases RA biosynthesis ~twofold [101].
Controversies and Confounding Factors in Evaluating CRBP1 Function and Retinol Metabolism
One concern is the tendency of endogenous RDH in their native mammalian membrane environments generating retinal from holo-CRBP1 at lower rates than from unbound retinol, possibly because release of retinol from holo-CRBP1 imposes a rate-limiting event slower than the release of cofactor from RDH, the normal rate-limiting event with dehydrogenases [150, 196, 211]. Nevertheless, holo-CRBP1 supports retinal formation in vitro at rates sufficient to provide the 1–50 nmol/g RA observed in serum and tissues [107].
Because the Vm values in vitro supported by holo-CRBP1 are lower than the values supported by unbound retinol, an alternative perspective proposes that RDH recognize only unbound (free) retinol [119]. This contention does not address the rate of RDH catalysis possible in vivo from the low intracellular concentrations of free retinol established by CRBP1 and other modes of sequestering retinol (Tables 2.1 and 2.2). A practical assessment requires comparing the rate that could be supported by the fraction of intracellular free retinol actually available to the rate that holo-CRBP1 would support. In addition, arguments that RDH recognize only unbound retinol should address all data supporting participation of retinoid binding-proteins in the RDH reaction. It also would be prudent to address the data that support participation of retinoid binding-proteins in the entire pathway of retinol/RA metabolism; otherwise the inference seems that the rate-limiting step is the only one that does not involve a binding-protein.
Some rate evidence used to challenge participation of holo-CRBP1 actually may support participation, or leave the question unresolved. Reduction of 2 μM retinal into retinol by Sf9-generated RDH12 with a C-terminal His6-tag was monitored with increasing concentrations of apo-CRBP1 from 0 to 40 μM [11]. Increasing apo-CRBP1 up to 10 μM decreased the rate of retinol formation to ~25 % of the rate observed in the absence of CRBP1. Based on a 50 mM kd of retinal with CRBP1 (one of the values in the literature at the time, the other being 30 nM), unbound retinal in the solution of 2 μM retinal and 10 μM CRBP1 was calculated as sufficient to account for product generation. The recently revised kd value of ~9 nM (Table 2.1, undoubtedly still an underestimate of affinity) indicates that the rate observed actually was ~fivefold greater than could be supported by the free retinal in this experiment. The decrease in rate between 0 and 10 μM CRBP1 might result from two substrates contributing to retinol formation—bound and free retinal, each supporting a different Vm. A Michaelis-Menten experiment with varying ratios of CRBP1 at each concentration of retinal would have addressed this possibility (assuming that an SDR over expressed in an insect cell would behave identically to one in its native mammalian cell environment). Significantly, the rate did not decrease further when total CRBP1 was increased from 10 to 20 to 40 μM (5–20-fold excess over retinal). This fourfold increase in the ratio CRBP1 to retinal would have decreased unbound retinal by fivefold, which would have decreased the reaction rate by a little over fourfold—based on the Km of RDH12 with free retinal—if free retinal were the only substrate. These experimental conditions and data generated do not support the paper’s conclusion that only free retinal functioned as substrate.
Lower rates of retinal production catalyzed by RDH16 (human ortholog of RDH1) and RDH10 from holo-CRBP1 relative to unbound retinol also have prompted arguments that these enzymes recognize only free retinol. In both cases possible Michaelis-Menten relationships between holo-CRBP1 and product were not reported. In fact, the RDH16 assayed had a kcat of 1.1 min−1 with free reinol, i.e. generated one mole of retinal per mole of enzyme in 50 s, indicating an enzyme with severely impaired activity. RDH with impaired activity may not appear to interact with holo-CRBP1 because retinal generation falls below limits of detection, or may not interact because the changes that impaired activity may have impaired interaction.
Another confounding factor was overexpression of RDH16 and RDH10 in Sf9 cells with RDH10 representing 10 % of total insect cell protein. Over expression of DHRS9 in Sf9 cells also resulted in low enzymatic activity for retinol relative to steroids, and relative to activity of endogenous enzyme in its natural membrane environment [221]. Membrane-associated proteins depend on membrane composition and their stoichiometry with membrane lipids and proteins for activity and function [2, 35, 92, 126]. Rat RODH activity during purification was maintained only with phosphatidylcholine (no other phospholipids were as effective), and required binding to the membrane protein CYP2D1, illustrating the importance of the membrane environment to activity [29, 30, 98]. This suggests that RDH expressed in insect cells might have inherently low activity because of the unnatural environment and/or stoichiometry.
Modification of RDH and/or CRBP1 introduced further confounding factors. RDH16 was reacted with an N-terminal fusion of CRBP1 with glutathione S-transferase or with a C-terminal fusion of CRBP1 with the chitin binding domain. Mutating single exterior residues of CRBP1 altered ability to serve as substrate (see above). A His6 construct was used with RDH10. Modifying microsomal RDH activity with N-ethylmaleimide inhibited holo-CRBP1-supported retinal biosynthesis by 90 %, but did not inhibit RDH activity with unbound retinol, consistent with obstructing access of CRBP1 to RDH without affecting RDH catalytic activity [30]. This latter observation provides further support for RDH recognizing holo-CRBP, and indicates caution when testing the premise with modified RDH or CRBP1.
Lower Vm values are difficult to interpret without a context of supporting data, including assaying for a Michaelis-Menten relationship, determining whether apo-binding proteins inhibit the reaction, allowing for the possibility of rate-limiting transfer of retinoid from binding-protein to enzyme, and attention to the environment of the enzyme.
CRBP1 Participates in Irreversible RA Biosynthesis Catalyzed by RALDH
Three cytosolic RALDH catalyze irreversible conversion of retinal into RA: RALDH1 (Aldh1A1); RALDH2 (Aldh1A2); RALDH3 (Aldh1A3) [166]. RALDH1 and 2 recognize CRBP1-retinal as substrate, whereas RALDH3 has not been tested with CRBP1 [199, 200, 212, 271]. A fourth, RALDH4 (ALDH8A1) recognizes only 9-cis-retinal [137, 139]. Though RALDH4 is widely expressed and has especially intense expression in liver, its function is uncertain because 9-cis-RA has not been detected outside pancreas [110].
Multiple RALDH candidates occur in cytosol using free retinal as substrate [125, 128, 154, 212]. To identify physiologically relevant candidates RALDH, CRBP1-retinol was incubated with liver microsomes (as a source of RDH) to generate retinal in situ. Liver cytosolic fractions, resolved via anion-exchange chromatography, were added and the combination was assayed for RALDH activity [212]. RA biosynthesis from CRBP1-retinol required both microsomes and the cytosolic fraction P1 (Table 2.5). Adding pyridine nucleotide cofactors generated maximum RA. This experiment achieved the dual purposes of verifying that the CRBP1-retinol/microsomal RDH partnership generates retinal, and showing that retinal generated by microsomes supports RA biosynthesis by cytosolic RALDH. These data suggested that CRBP1 acts as conduit of retinal from microsomes to cytosol to overcome low solubility of retinal in aqueous media. These data were complemented by the observation that retinal generated from CRBP1-retinol occurs bound with CRBP1 [196]. The ability of microsomes and CRBP1-retinol to provide retinal for RA biosynthesis was revisited with purified recombinant RALDH1 [199, 200]. Data very similar to those in Table 2.5 were generated, reinforcing the notion of an RA-generating metabolon consisting of CRBP1, microsomal RDH and cytosolic RALDH1.
Table 2.5.
RA biosynthesis catalyzed by microsomes (RDH activity) and a cytosolic fraction (RALDH activity) using CRBP1-retinol as substrate1
| RA generated (pmol ± SD)
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Addition | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ~2 | 5±4 | 9±5 | 20±2 | 49±4 |
| μsomes | + | + | + | − | − | − | + | − | + | + | + | + |
| P1 | − | − | − | + | + | + | + | + | − | + | + | + |
| NAD+ | − | + | − | − | + | − | − | + | + | + | − | + |
| NADP+ | − | − | + | − | − | + | − | + | + | − | + | + |
A total of 7.5 μM CRBP1 was used with 5 μM retinol to insure minimal free retinol [212]. With a kd of 3 nM, the unbound retinol concentration was 6 nM, whereas the holo-CRBP1 concentration was 4994 nM. The liver cytosol fraction P1 with RALDH activity was isolated by anion-exchange chromatography
Kinetics of CRBP1-delivered retinal with RALDH1 and 2 are consistent with the binding protein “cassette” serving as substrate (Fig. 2.8). CRBP1-retinal produces a Michaelis-Menten relationship with RALDH1-catalyzed RA formation, with a K0.5 value less than half that obtained with unbound retinal (Table 2.6). The Vm using CRBP1-retinal was much lower than with unbound retinal under the conditions used, which consisted of a twofold molar excess of CRBP1 at each retinal concentration [199]. This ratio practically eliminates any free retinal. For example, at 4 μM retinal, the 8 μM total CRBP1 would reduce free retinal >130-fold, but resulted in only a fourfold decrease in the rate of RA production relative to the reaction with 4000 nM free retinal, which indicates that CRBP1 acts as substrate. The decrease in Vm was caused by the ~4000 nM apo-CRBP1 inhibiting RALDH1, as revealed by a Dixon plot (Fig. 2.8). A Dixon plot allows calculating the rate that occurs at a precise 1:1 ratio of CRBP1 to retinal, and can be more reliable than trying to achieve a precise 1:1 ratio experimentally. The Dixon plot revealed that the Vm value for 1:1 ratio CRBP1-retinal (i.e. no apo-CRBP1) was 89 % of the value with unbound retinal. Thus, delivering retinal bound to CRBP1 allowed RALDH1 to function ~twofold more efficiently than with unbound retinal (by comparing Vm/Km). This outcome illustrates the problem encountered when interpreting lower rates produced by titrating CRBP1 into a fixed concentration of retinoid.
Fig. 2.8.

Kinetic relationships between CRBP1 and recombinant RALDH1 or RALDH2. Top: conversion of retinal into RA by RALDH1 using CRBP1-retinal (2:1 ratio) or unbound retinal. Middle: Dixon plot of the effect of titrating apo-CRBP1 into 2 μM retinal on generation of RA by RALDH1 illustrating inhibition by apo-CRBP1. These data allow calculating the uninhibited Vm of RALDH1 with CRBP1-retinal, which was ~90 % of the Vm with unbound retinal. The reaction with RAlDH1 was run at 25 °C to reduce its rate to preserve initial velocity conditions. The IC50 of apo-CRBP1 for inhibiting RALDH1 was 1.4 μM. Bottom: conversion of retinal into RA by RALDH2. A twofold excess of binding protein to retinal did not affect the reaction rate, indicating that RALDH2 interacts with CRBP1 [199]
Table 2.6.
Kinetic values of RALDH substrates. The Vm/Km values for RALDH1 and 2 are each twofold more efficient with CRBP1-retinal as substrate compared to free retinal
In contrast to the impact on RALDH1 activity, adding a twofold molar ratio of CRBP1 to each retinal concentration produced RADLH2 kinetics similar to those obtained with unbound retinal. The Vm/Km for RALDH2 was twofold higher with CRBP1-retinal, again indicating a more efficient reaction. The ability of apo-CRBP1 to inhibit RALDH1, but not RALDH2, suggests each operates at different levels of vitamin A nutriture, and reveals the complexity of apo- and holo-CRBP1nteractions with retinoid metabolizing enzymes.
CRABPs Offer RA for Catabolism
Since the demonstrations of CRABP1 and 2 expression in multiple species and tissues, much effort has gone into understanding their functions [49, 185, 241, 245]. One purpose seems to be regulating RA concentrations.
Both unbound RA and CRABP1-bound RA have a Michaelis-Menten relationship with the rate of RA metabolism catalyzed by testes microsomes (Fig. 2.9). Kinetic constants for total metabolite formation are Km ~50 nM and Vm ~10 pmol/min/mg protein for unbound RA, compared to 1.8 nM and 2.6 pmol/min/mg protein, respectively, for CRABP1-RA. Efficiency of the reaction with CRABP1-RA (3/1 ratio at all substrate concentrations) is ~sevenfold higher than with unbound RA, based on values of Vm/Km = 1.4 vs. 0.2 for bound vs. free, respectively. Titrating apo-CRABP1 into a solution of 120 nM CRABP1-RA (1:1) reduces the rate of reaction by 35 % at a fivefold ratio CRABP1/RA (total CRABP1 = 600 nM). Using a Kd value of 0.4 nM, the decrease in unbound RA would have reduced the rate 60-fold if only unbound RA were substrate (calculating from a Km value for unbound RA of 50 nM and a concentration of unbound RA decreasing from 7 nM (1:1 ratio CRABP:RA) to 0.1 nM (5:1 ratio CRABP1:RA). Overall, three criteria indicate that holo-CRABP1 serves as substrate: (1) the rate of RA metabolism in the presence of CRABP1 is faster than the rate supported by any unbound RA present; (2) increasing the concentration of apo-CRABP1 does not decrease the rate of metabolism to the extent predicted if only free RA were substrate; (3) the CRABP1 reaction is more efficient (Vm/Km) than the reaction with unbound RA. These results were also observed with CRABP2 (Fiorella and Napoli, unpublished).
Fig. 2.9.

Kinetic relationship between CRABP1 and RA metabolism. Testis microsomes were used as source of RA catabolic activity, because testis has one of the highest intracellular RA concentrations [181]. Kinetics with CRABP1 were generated with a threefold molar excess of CRABP1 to RA to reduce free RA to negligible amounts [68]. Note that the reaction with CRABP1-RA is more efficient than with unbound RA (higher Vm/Km). Note also that the elimination half-life of RA does not change between unbound and CRABP1-bound catabolism, again suggesting interaction between enzyme and binding protein. The rates of RA oxidized at C4 do change, however, suggesting that these bound retinoids would inhibit RA catabolism if in sufficiently high concentrations. With physiological amounts of RA, the 4-oxidized derivatives have very low concentrations, but with toxic amounts of RA, the concentrations of 4-oxidized derivatives increase and contribute to RA toxicity
CRABP1 also serves as substrate for RA metabolism with microsomes from other tissues that express the binding-protein intensely, such as kidney and lung [69]. The composition of RA metabolites reflected the tissue source of microsomes, consistent with a previous report of tissue specificity in RA metabolism [167]. 4-OH-RA had a much higher concentration in the RA metabolite pool generated by testes when RA was presented bound to CRABP1 than when RA was presented unbound. This occurred because RA metabolites, such as 4-OH-RA and 4-oxo-RA, bind with CRABP1 upon formation in the incubation medium, and are not accessible to further metabolism when bound. The nearly complete arrest of 4-OH-RA and 4-oxo-RA degradation by CRABP1 contrasts with CRABP’s lack of interference in the overall rate of RA catabolism, indicating that pathways other than those initiated by C4 oxidation occur, such as 18-hydroxylation and epoxidation.
Similar results have been demonstrated in the murine embryonic stem cell line, AB1, and a mutant AB1 cell line with CRABP1-ablated [44]. Deletion of CRABP1 caused a 50 % decrease in intracellular RA when medium RA concentrations were in the range 1–10 nM (serum concentrations are ~1–4 nM) indicating in this experiment CRABP1 served as a reservoir to draw RA into cells. Deletion of CRABP1 did not change the elimination t½ of RA, however. Loss of CRABP1 increased the polar metabolites in cells ~30 % (1 nM RA treatment) relative to RA, but with no prior RA treatment or prior treatment with 1 nM RA, RA remained the quantitatively major intracellular retinoid. These data should be considered in context of the demonstration that F9 cells incubated with 50 nM RA has 99 % of the total retinoids in the medium, but only 44 % of the 99 % occurs as unmetabolized RA, whereas unmetabolized RA accounts for 76 % of the retinoids in cells, and 94 % of the retinoids in nuclei [274]. In other words, despite metabolism, RA accounts for most of the retinoids in the nucleus. Thus, the importance of increased 4-OH-RA and 4-oxo-RA is not certain. Their kd values for RAR rival that of RA, but their low concentrations under physiological conditions (as opposed to during toxicity) do not suggest a major role in nuclear retinoid signaling. These data suggest that CRABP1 modulates the intracellular concentrations of RA and increases levels of some polar metabolites, but the physiological impact is uncertain. CRABP1-binding could prevent cell membranes from absorbing RA and diminishing its functional availability.
The CRABP1-null mouse lacks an obvious morphological phenotype, as tested in mice fed a diet containing copious vitamin A [79], which rescues phenotypes of the Rbp4-null mouse (Rbp4 encodes sRBP), the Rbp2 null mouse (Rbp2 encodes CRBP2), and the Rdh1-null mouse [60, 213, 289]. Moreover, no other phenotypes related to retinoid action were studied, such as immune dysfunction, cancer, or short-term memory.
13-cis-RA (isotretinoin) exhibits about tenfold lower teratogenicity than RA (aka, tretinoin) [236]. To examine mechanisms, teratogenic amounts of RA and 13-cis-RA were dosed to mice during gestation, and retinoid distribution was monitored in embryos. More 13-cis-RA localized to the nucleus than RA, and nuclear 13-cis-RA gave rise to nuclear RA. Two conclusions were reached from these data: (1) the low affinity of 13-cis-RA for CRABP1 allowed greater nuclear localization; (2) conversion of 13-cis-RA into RA allowed RAR binding, thereby inducing teratogenic effects. These data are consistent with CRABP1 regulating access of RA to the nucleus.
Although 4-OH-RA and 4-oxo-RA have similar affinity for RAR as RA, they do not seem to affect RAR-gene regulation under normal circumstances because their concentrations are very low relative to RA. But with high doses of retinol or RA, the concentrations of 4-oxidized RA metabolites increase to the point where they likely contribute to retinoid toxicity [54, 61]. This suggests that during toxic vitamin A exposure, the ability of CRABP1 to repress metabolism of 4-OH-RA and 4-oxo-RA would contribute to toxicity, which could be augmented by the ability of CRABP1 to deliver retinoids to the nucleus.
Ketoconazole, an imidazole antimycotic and a potent inhibitor of CYP, inhibits metabolism of unbound and CRABP1-bound RA catalyzed by testes microsomes with IC50 values of 2 and 0.7 μM, respectively [162, 274, 275]. When this work was reported, the specific CYPs responsible for catalyzing RA metabolism were not known. Inhibition of RA metabolism by imidazole antimycotics, however, established the principle that arresting RA metabolism increased its potency. This principle was the foundation for using RA metabolism blocking agents (RAMBA) to treat RA-responsive diseases, as an alternative to dosing with RA or synthetic retinoids [156].
CRABPs Direct RA to Nuclear Receptors and/or Mediate Non-canonical Actions
Additional functions of RA binding-proteins involve delivering RA to the nucleus and mediating non-genomic mechanisms of RA action. Several labs have verified that CRABP1 localizes in nuclei as well in extra-nuclear structures [76, 83, 235, 243, 244, 262]. Notably, the precise subcellular locus of CRABP1 depends to a large extent on the fixation procedure used [76, 235]. The significance of CRABP1’s nuclear localization (in terms of activating receptors) was cast into doubt with the demonstration that CRABP1 did not enhance interaction of RARα with RAR response-elements, nor affect transcriptional activation by RA. these data supported a conclusion that CRABP1 did not directly deliver RA to RAR [266]. This observation was taken a step further with the demonstration that RA was transferred from CRABP1 to RARα only through diffusion in a cell-free system—a report that also confirmed the inability of CRABP1 to stimulate RA-induced transcription [58]. It is likely there is more to this story.
RA does not function exclusively through transcriptional activation, however. In hippocampus neurons, RA stimulates dendritic growth within minutes by activating translation through MAPK, ERK1/2, mTOR and their targets 4E-BP, p70S6K, and ribosome protein S6 [42, 45]. RARα, within dendritic RNA transport granules, binds the RNA-binding protein PURα. RA binding to RARα frees PURα to allow translation. Another mechanism of non-canonical (non-transcriptional) regulation by RA involves the CRABP1-RA complex activating ERK1/2 in embryonic stem cells [201].
In conclusion, non-nuclear CRABP1 serves to control RA concentrations, deliver RA for catabolism, affect the intracellular steady-state composition of RA metabolites, mediate rapid non-canonical actions of RA, and may contribute to toxicity during vitamin A excess by impeding clearance of 4-oxidized RA metabolites.
CRABP2 Functions Differently from CRABP1 and Delivers RA to RARs in the Nucleus
Analysis of CRABP2 mRNA expression in 12 breast cell lines revealed a correlation with ability of RA to inhibit growth. Overexpression of CRABP2 enhanced transcriptional activity of RA in these cells [104]. These observations are consistent with nuclear detection of CRABP2 in transfected COS cells, mouse embryos and other cell lines [76]. Further study led to the conclusion that nuclear CRABP2 in its unliganded state associates with RAR and confirmed the observation of CRABP2 enhancing transcription potency of RA [7, 57]. Remembering that most CRABP2 resides in cytosol, a plausible model for its dual location is provided by the observation that after binding with RA, the CRABP2-RA complex translocates from cytosol to the nucleus and channels RA to RAR through protein-protein interactions [36, 58]. The CRABP2-RAR interaction was reported to be short-lived, allowing recycling of CRABP2 back to the cytosol. As with previous reports, these studies also demonstrated that overexpression of CRABP2 enhanced cell sensitivity to RA-induced growth inhibition in MCF7 mammary carcinoma cells, whereas reducing CRABP2 expression reduced RA action.
FABP5 and Not CRABP2 Delivers RA to PPARδ/β
RA has pleiotropic actions, including diverse actions in different cell types. One mechanism for these diverse actions could depend on expression of dissimilar nuclear coactivators. Another could include differential activation of nuclear hormone receptors. The demonstration that RA binds and activates PPARδ/β (hereafter referred to as PPARδ) provided insight into mechanisms underlying these diverse cell responses. RA binds to PPARδ with high-affinity (kd = 17 nM; an affinity likely underestimated because fluorescent titration was done with 0.77 μM PPARδ [250]. RA selectively transcriptionally activates PPARδ (vs. PPARα and PPARγ) with an EC50 between 0.2 and 0.3 μM in COS-7 cells transfected with PPRE-driven luciferase reporters. The EC50 is higher than typical concentrations of RA in tissues, which fall between 1 and 50 nM [107, 109], which may be the result of RA insolubility in aqueous media and its sequestration by medium components and in cell membranes.
Binding of RA to PPARδ prompted a model for two intracellular RA binding-proteins, CRABP2 and FABP5, delivering RA to either RAR or PPARδ, respectively [248]. In cells with high CRABP2/FABP5, RA would function mainly through RAR as a proapoptotic agent, but in cells with relatively low CRABP2/FABP5, RA would function through PPARδ to promote cell survival and proliferation. In HaCaT keratinocytes, 100 nM RA causes fivefold activation of a PPAR response element, but the synthetic RAR-specific ligand TTNPB does not. Both the PPARδ-specific agonist GW0742 and RA induce expression of three PPARδ-regulated genes from 1.5 to ~3-fold: FIAF, ADRP and PDK-1. Notably, one of these, the PPARδ-target PDK-1, contributes to the anti-apoptotic activity of PPARδ. This work also demonstrated that RA serves as a ligand for FABP5 (kd ~35 nM) and endogenous FABP5 is predominantly cytosol, until RA exposure, which causes translocation to the nucleus. Remarkably, increasing the ratio CRABP2/FABP5 converts RA from a survival to a proapoptotic agent in HaCaT and NaF (mouse mammary tumor) cells. These results support the hypothesis that CRABP2 delivers RA to RAR, which causes cell cycle arrest, apoptosis and growth inhibition, whereas FABP5 delivers RA to PPARδ, which stimulates survival paths to enhance proliferation.
This model was extended in the RA-resistant mouse model of breast cancer, MMTV-neu, to show that RA activates PPARδ through an “aberrantly” high ratio of FABP5/CRABP2 and changing this ratio reduced RA-resistance [248, 249]. A switch of RA action mediated by binding proteins and their associated receptors also seems to function during neuronal [283] and preadipocyte differentiation [17]. This was verified in the P19 neuronal stem cell model, where RA induced neurogenesis via CRABP2/RAR, whereas FABP5/PPARδ promotes the development of mature neurons as differentiation proceeds. In contrast, in NIH3T3 L1 preadipocytes, down regulation of CRABP2 impairs ability of RA to prevent differentiation into mature adipocytes.
Collectively, these data support a model of CRABP2 and FABP5 influencing RA action by sorting delivery to either RAR or PPARδ, which in turn prompts selective actions. Not everyone embraces the aspect of the model that includes RA binding FABP5 and activating PPARδ, however. One report challenged the notions that RA activates PPARδ and that PPARδ stimulates proliferation in HaCaT keratinocytes [32]. Both RA and the “highly specific” PPARδ ligands (GW0742; GW501516) were observed to inhibit proliferation, but only the PPARδ ligands increased expression of PPARδ-target genes. Another group reported that concentrations of GW501516 from 0.1 to 1 μM induced transcription ~17-fold of an PPARδ-activated luciferase reporter driven by a PPRE in NIH3T3 cells, but the same concentration range of RA had no effect [225]. In HaCaT cells, this report concluded that GW5051516 induced expression of the PPARδ-target genes ADRP (adipocyte differentiation-related protein) and ANGPTL4 (angiopoietin-like protein 4), but RA did not, even though RA induced the RAR-target gene Cyp26A1. This report found no binding of RA to PPARδ in vitro by fluorescence assay in concentrations to 100 μM, in contrast to GW501516, which saturated PPARδ at 10 nM. A third report observed enhanced ligand-induced expression of ADRP and ANGPTL4 in HaCaT cells that over-expressed PPARδ [33], but reported that RA did not increase expression of ANGPTL4, even though cells expressed a high FABP5/CRABP2 ratio. The reasons for disparate results produced by different labs with the (nominally) same cell lines are not clear.
The lab that originally reported RA binding to FABP5 and delivery by FABP5 to PPARδ revisited the model [132]. They again showed that RA binds recombinant FABP5 with a kd ~42 nM by fluorescence assay. They also crossbred FABP5-null mice with MMTV-ErbB2/HER2 oncomice. The latter strain develops mammary tumors spontaneously. The double genetically modified model developed tumors in only 6 of 14 mice, whereas 100 % of the MMTV-ErbB2/HER2 mice developed tumors. Survival time of the latter did not exceed 39 weeks, but mice with the double mutation lived until at least 64 weeks, and had tumor volumes ~3.5-fold less than the control group. These data attest to the oncogenic (proliferative) actions of FABP5 in vivo. This work did not test the impact of RA on tumor growth in either strain of mouse, but showed that RA and FABP5 promote oncogenic metrics in NIH3T3 cells. Resolving the role of FABP5 and PPARδ in RA action is crucial to understanding mechanisms of RA action, and will have essential impact on therapeutic use of retinoids and PPARδ agonists.
CRABP2 Stabilizes mRNA
Apo-CRABP2 also binds with the RNA binding protein HuR and increases its affinity for the 3′-untranslated regions of mRNA, which delays degradation. This suggests that RA binding would cause CRABP2 release from HuR, allowing translocation to the nucleus [269].
Relevance
Data from multiple labs generated during the last 40 years indicate participation of retinoid binding-proteins during each step from retinol uptake into cells to its metabolism into RE or RA, with binding proteins also participating in RA delivery to nuclei, non-genomic (non-canonical) retinoid actions, and RA catabolism (Fig. 2.10). A number of aspects of this model and the data that support it are important to recognize. Neither the model nor the data indicate obligatory participation of binding-proteins in retinoid metabolism (as shown by knockouts). Rather, binding proteins enhance efficiency of metabolism to spare essential and rare (during evolution) retinoids from excess catabolism and to chaperone lipophilic retinoids through aqueous media. The model proposed does not address vitamin A metabolism during intake of copious or toxic amounts of vitamin A. With copious and/or toxic amounts of dietary or dosed vitamin A, enzymes that did not evolve to participate in vitamin A metabolism are presented with substrate as a result of saturated retinoid binding-proteins and high intracellular retinoid concentrations. This conclusion is supported by rescue of at least three knockouts (Rbp4, Rdh1, Rbp2) by chow diets (copious in vitamin A). It would seem important, therefore, when testing the contribution of a binding protein or enzyme, to avoid copious dietary vitamin A to reveal their functions (unless necessary for breeding, but returning to restricted amounts afterwards). Also, in light of the complexity of RA generation and homeostasis, and potential “moonlighting” by retinoid-metabolizing enzymes, it would seem important to assess multiple components of the retinoid metabolon before postulating sites that generate RA.
Fig. 2.10.

Model of retinoid homeostasis illustrating roles of retinoid binding-proteins. This model illustrates contributions of retinoid binding-proteins to multiple aspects of retinol uptake and metabolism, and RA catabolism and action. The complexity of storing and activating retinol undoubtedly contributes to the multiplicity of retinoid actions. This chaperoning increases the efficiency of retinoid metabolism, but is not obligatory for life. Note, however, that the CRBP1-null mouse suffers from disrupted retinol metabolism, retinol wasting, and dysfunctions in intermediary metabolism. The dotted lines represent regulatory points
Future Directions
Much work remains to be done concerning specific interactions among retinoid binding-proteins and the enzymes they engage. In reality, functions of the binding-proteins and the enzymes RDH and RALDH are known only in “outline”. Not entirely clear are the purposes of multiple CRBP, CRABP, RDH, RALDH and CYP isoforms. Finally, the different phenotypes of the multiple orthologs of binding-proteins, RDH, and RALDH knockouts suggest specialized metabolons generating RA for specific functions. This hypothesis and systemic as well as mechanistic regulation of retinoid homeostasis remain to be explored.
Acknowledgments
The author is grateful for thought-provoking suggestions from David Ong, especially concerning but not limited to, the structures and binding characteristics of retinoid binding-proteins.
Abbreviations
- ADH
alcohol dehydrogenase(s) (medium chain ADH gene family)
- ARAT, acyl-CoA
retinol acyltransferase
- bsiREH
bile-salt independent REH
- CRBP1
cellular retinol binding-protein type 1
- CRBP2
cellular retinol binding-protein type 2
- CRABP1
cellular retinoic acid binding-protein type 1
- CRABP2
cellular retinoic acid binding-protein type 2
- CYP
cytochrome P-450
- DGAT1, acyl-CoA
diacylglycerol acyltransferase 1
- FABP5
fatty acid binding-protein type 5
- kcat
catalytic constant (defines the efficiency of an enzyme for product formation)
- kd
equilibrium dissociation constant
- LRAT
lecithin: retinol acyltransferase
- MGAT
acyl-CoA:monoacylglycerol acyltransferase
- PPARδ/β
peroxisomal proliferator activated receptor δ/β
- RA
all-trans-retinoic acid
- RALDH
retinal dehydrogenase encoded by Aldh gene family 1A
- RAR
RA receptor
- Rbp1
gene that encodes CRBP1
- Rbp2
gene that encodes CRBP2
- Rbp4
gene that encodes serum retinol binding-protein (sRBP)
- RDH
retinol dehydrogenase(s) (SDR gene family)
- RE
retinyl ester(s)
- REH
retinyl ester hydrolase(s)
- SDR
short-chain dehydrogenase/reductase
- sRBP
serum retinol binding-protein
- TTR
transthyretin
- WT
wild-type
References
- 1.Adams MK, Belyaeva OV, Wu L, Kedishvili NY. The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J Biol Chem. 2014;289:14868–14880. doi: 10.1074/jbc.M114.552257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn T, Kim M, Yun C-H, Chae H-J. Functional regulation of hepatic cytochrome p450 enzymes by physicochemical properties of phospholipids in biological membranes. Curr Protein Pept Sci. 2007;8:496–505. doi: 10.2174/138920307782411392. [DOI] [PubMed] [Google Scholar]
- 3.Akerstrom B, Flower DR, Salier JP. Lipocalins: unity in diversity. Biochim Biophys Acta. 2000;1482:1–8. doi: 10.1016/s0167-4838(00)00137-0. [DOI] [PubMed] [Google Scholar]
- 4.Amengual J, Zhang N, Kemerer M, Maeda T, Palczewski K, Von Lintig J. STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bashor MM, Chytil F. Cellular retinol-binding protein. Biochim Biophys Acta. 1975;411:87–96. doi: 10.1016/0304-4165(75)90287-1. [DOI] [PubMed] [Google Scholar]
- 6.Bashor MM, Toft DO, Chytil F. In vitro binding of retinol to rat-tissue components. Proc Natl Acad Sci U S A. 1973;70:3483–3487. doi: 10.1073/pnas.70.12.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bastie JN, Despouy G, Balitrand N, Rochette-Egly C, Chomienne C, Delva L. The novel co-activator CRABPII binds to RARalpha and RXRalpha via two nuclear receptor interacting domains and does not require the AF-2 “core”. FEBS Lett. 2001;507:67–73. doi: 10.1016/s0014-5793(01)02938-6. [DOI] [PubMed] [Google Scholar]
- 8.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, et al. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belyaeva OV, Kedishvili NY. Human pancreas protein 2 (PAN2) has a retinal reductase activity and is ubiquitously expressed in human tissues. FEBS Lett. 2002;531:489–493. doi: 10.1016/s0014-5793(02)03588-3. [DOI] [PubMed] [Google Scholar]
- 10.Belyaeva OV, Stetsenko AV, Nelson P, Kedishvili NY. Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry (Mosc) 2003;42:14838–14845. doi: 10.1021/bi035288u. [DOI] [PubMed] [Google Scholar]
- 11.Belyaeva OV, Korkina OV, Stetsenko AV, Kim T, Nelson PS, Kedishvili NY. Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry (Mosc) 2005;44:7035–7047. doi: 10.1021/bi050226k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belyaeva OV, Johnson MP, Kedishvili NY. Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J Biol Chem. 2008;283:20299–20308. doi: 10.1074/jbc.M800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belyaeva OV, Korkina OV, Stetsenko AV, Kedishvili NY. Human retinol dehydrogenase 13 (RDH13) is a mitochondrial short-chain dehydrogenase/reductase with a retinaldehyde reductase activity. FEBS J. 2008;275:138–147. doi: 10.1111/j.1742-4658.2007.06184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belyaeva OV, Lee S-A, Adams MK, Chang C, Kedishvili NY. Short chain dehydrogenase/reductase rdhe2 is a novel retinol dehydrogenase essential for frog embryonic development. J Biol Chem. 2012;287:9061–9071. doi: 10.1074/jbc.M111.336727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berman ER, Segal N, Feeney L. Subcellular distribution of free and esterified forms of vitamin A in the pigment epithelium of the retina and in liver. Biochim Biophys Acta. 1979;572:167–177. doi: 10.1016/0005-2760(79)90212-1. [DOI] [PubMed] [Google Scholar]
- 16.Berry DC, Noy N. Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim Biophys Acta. 2012;1821:168–176. doi: 10.1016/j.bbalip.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berry DC, Soltanian H, Noy N. Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. J Biol Chem. 2010;285:15324–15332. doi: 10.1074/jbc.M110.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berry DC, Jin H, Majumdar A, Noy N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc Natl Acad Sci U S A. 2011;108:4340–4345. doi: 10.1073/pnas.1011115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry DC, Croniger CM, Ghyselinck NB, Noy N. Transthyretin blocks retinol uptake and cell signaling by the holo-retinol-binding protein receptor STRA6. Mol Cell Biol. 2012;32:3851–3859. doi: 10.1128/MCB.00775-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berry DC, DeSantis D, Soltanian H, Croniger CM, Noy N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes. 2012;61:1112–1121. doi: 10.2337/db11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry DC, O’Byrne SM, Vreeland AC, Blaner WS, Noy N. Cross talk between signaling and vitamin A transport by the retinol-binding protein receptor STRA6. Mol Cell Biol. 2012;32:3164–3175. doi: 10.1128/MCB.00505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berry DC, Jacobs H, Marwarha G, Gely-Pernot A, O’Byrne SM, DeSantis D, et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J Biol Chem. 2013;288:24528–24539. doi: 10.1074/jbc.M113.484014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biesalski HK. Comparative assessment of the toxicology of vitamin A and retinoids in man. Toxicology. 1989;57:117–161. doi: 10.1016/0300-483x(89)90161-3. [DOI] [PubMed] [Google Scholar]
- 24.Billings SE, Pierzchalski K, Butler Tjaden NE, Pang X-Y, Trainor PA, Kane MA, et al. The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27:4877–4889. doi: 10.1096/fj.13-227967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas MG, Russell DW. Expression cloning and characterization of oxidative 17beta- and 3alpha-hydroxysteroid dehydrogenases from rat and human prostate. J Biol Chem. 1997;272:15959–15966. doi: 10.1074/jbc.272.25.15959. [DOI] [PubMed] [Google Scholar]
- 26.Blaner WS, Das K, Mertz JR, Das SR, Goodman DS. Effects of dietary retinoic acid on cellular retinol- and retinoic acid-binding protein levels in various rat tissues. J Lipid Res. 1986;27:1084–1088. [PubMed] [Google Scholar]
- 27.Bliss AF. The equilibrium between vitamin A alcohol and aldehyde in the presence of alcohol dehydrogenase. Arch Biochem Biophys. 1951;31:197–204. doi: 10.1016/0003-9861(51)90206-8. [DOI] [PubMed] [Google Scholar]
- 28.Boerman MH, Napoli JL. Cholate-independent retinyl ester hydrolysis. Stimulation by Apo-cellular retinol-binding protein. J Biol Chem. 1991;266:22273–22278. [PubMed] [Google Scholar]
- 29.Boerman MH, Napoli JL. Characterization of a microsomal retinol dehydrogenase: a short-chain alcohol dehydrogenase with integral and peripheral membrane forms that interacts with holo-CRBP (type I) Biochemistry (Mosc) 1995;34:7027–7037. doi: 10.1021/bi00021a014. [DOI] [PubMed] [Google Scholar]
- 30.Boerman MH, Napoli JL. Effects of sulfhydryl reagents, retinoids, and solubilization on the activity of microsomal retinol dehydrogenase. Arch Biochem Biophys. 1995;321:434–441. doi: 10.1006/abbi.1995.1415. [DOI] [PubMed] [Google Scholar]
- 31.Boerman MH, Napoli JL. Cellular retinol-binding protein-supported retinoic acid synthesis. Relative roles of microsomes and cytosol. J Biol Chem. 1996;271:5610–5616. doi: 10.1074/jbc.271.10.5610. [DOI] [PubMed] [Google Scholar]
- 32.Borland MG, Foreman JE, Girroir EE, Zolfaghari R, Sharma AK, Amin S, et al. Ligand activation of peroxisome proliferator-activated receptor-beta/delta inhibits cell proliferation in human HaCaT keratinocytes. Mol Pharmacol. 2008;74:1429–1442. doi: 10.1124/mol.108.050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borland MG, Khozoie C, Albrecht PP, Zhu B, Lee C, Lahoti TS, et al. Stable over-expression of PPARβ/δ and PPARγ to examine receptor signaling in human HaCaT keratinocytes. Cell Signal. 2011;23:2039–2050. doi: 10.1016/j.cellsig.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouillet P, Sapin V, Chazaud C, Messaddeq N, Décimo D, Dollé P, et al. Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech Dev. 1997;63:173–186. doi: 10.1016/s0925-4773(97)00039-7. [DOI] [PubMed] [Google Scholar]
- 35.Brenner RR. Effect of unsaturated acids on membrane structure and enzyme kinetics. Prog Lipid Res. 1984;23:69–96. doi: 10.1016/0163-7827(84)90008-0. [DOI] [PubMed] [Google Scholar]
- 36.Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol. 2002;22:2632–2641. doi: 10.1128/MCB.22.8.2632-2641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burnett KG, Felder MR. Peromyscus alcohol dehydrogenase: lack of cross-reacting material in enzyme-negative animals. Biochem Genet. 1978;16:1093–1105. doi: 10.1007/BF00484530. [DOI] [PubMed] [Google Scholar]
- 38.Careri M, Elviri L, Mangia A, Zagnoni I, Torta F, Cavazzini D, et al. Mass spectrometry techniques for detection of ligand-dependent changes in the conformational flexibility of cellular retinol-binding protein type I localized by hydrogen/deuterium exchange. Rapid Commun Mass Spectrom RCM. 2006;20:1973–1980. doi: 10.1002/rcm.2547. [DOI] [PubMed] [Google Scholar]
- 39.Chai X, Boerman MH, Zhai Y, Napoli JL. Cloning of a cDNA for liver microsomal retinol dehydrogenase. A tissue-specific, short-chain alcohol dehydrogenase. J Biol Chem. 1995;270:3900–3904. doi: 10.1074/jbc.270.8.3900. [DOI] [PubMed] [Google Scholar]
- 40.Chai X, Zhai Y, Popescu G, Napoli JL. Cloning of a cDNA for a second retinol dehydrogenase type II. Expression of its mRNA relative to type I. J Biol Chem. 1995;270:28408–28412. doi: 10.1074/jbc.270.47.28408. [DOI] [PubMed] [Google Scholar]
- 41.Chazaud C, Bouillet P, Oulad-Abdelghani M, Dollé P. Restricted expression of a novel retinoic acid responsive gene during limb bud dorsoventral patterning and endochondral ossification. Dev Genet. 1996;19:66–73. doi: 10.1002/(SICI)1520-6408(1996)19:1<66::AID-DVG7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 42.Chen N, Napoli JL. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J Off Publ Fed Am Soc Exp Biol. 2008;22:236–245. doi: 10.1096/fj.07-8739com. [DOI] [PubMed] [Google Scholar]
- 43.Chen W, Song M-S, Napoli JL. SDR-O: an orphan short-chain dehydrogenase/reductase localized at mouse chromosome 10/human chromosome 12. Gene. 2002;294:141–146. doi: 10.1016/s0378-1119(02)00757-6. [DOI] [PubMed] [Google Scholar]
- 44.Chen AC, Yu K, Lane MA, Gudas LJ. Homozygous deletion of the CRABPI gene in AB1 embryonic stem cells results in increased CRABPII gene expression and decreased intracellular retinoic acid concentration. Arch Biochem Biophys. 2003;411:159–173. doi: 10.1016/s0003-9861(02)00732-4. [DOI] [PubMed] [Google Scholar]
- 45.Chen N, Onisko B, Napoli JL. The nuclear transcription factor RARalpha associates with neuronal RNA granules and suppresses translation. J Biol Chem. 2008;283:20841–20847. doi: 10.1074/jbc.M802314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chytil F, Ong DE. Cellular vitamin A binding proteins. Vitam Horm. 1978;36:1–32. doi: 10.1016/s0083-6729(08)60980-2. [DOI] [PubMed] [Google Scholar]
- 47.Chytil F, Ong DE. Cellular retinol- and retinoic acid-binding proteins in vitamin A action. Fed Proc. 1979;38:2510–2514. [PubMed] [Google Scholar]
- 48.Chytil F, Ong DE. Intracellular vitamin A--binding proteins. Annu Rev Nutr. 1987;7:321–335. doi: 10.1146/annurev.nu.07.070187.001541. [DOI] [PubMed] [Google Scholar]
- 49.Chytil F, Page DL, Ong DE. Presence of cellular retinol and retinoic acid binding proteins in human uterus. Int J Vitam Nutr Res Int Z Für Vitam- Ernährungsforschung J Int Vitaminol Nutr. 1975;45:293–298. [PubMed] [Google Scholar]
- 50.Collins MD, Mao GE. Teratology of retinoids. Annu Rev Pharmacol Toxicol. 1999;39:399–430. doi: 10.1146/annurev.pharmtox.39.1.399. [DOI] [PubMed] [Google Scholar]
- 51.Collins MD, Eckhoff C, Chahoud I, Bochert G, Nau H. 4-Methylpyrazole partially ameliorated the teratogenicity of retinol and reduced the metabolic formation of all-trans-retinoic acid in the mouse. Arch Toxicol. 1992;66:652–659. doi: 10.1007/BF01981505. [DOI] [PubMed] [Google Scholar]
- 52.Cornish-Bowden A. Fundamentals of enzyme kinetics. Portland Press; London: 1995. [Google Scholar]
- 53.Cowan SW, Newcomer ME, Jones TA. Crystallographic studies on a family of cellular lipophilic transport proteins. Refinement of P2 myelin protein and the structure determination and refinement of cellular retinol-binding protein in complex with all-trans-retinol. J Mol Biol. 1993;230:1225–1246. doi: 10.1006/jmbi.1993.1238. [DOI] [PubMed] [Google Scholar]
- 54.Creech Kraft J, Löfberg B, Chahoud I, Bochert G, Nau H. Teratogenicity and placental transfer of all-trans-, 13-cis-, 4-oxo-all-trans-, and 4-oxo-13-cis-retinoic acid after administration of a low oral dose during organogenesis in mice. Toxicol Appl Pharmacol. 1989;100:162–176. doi: 10.1016/0041-008x(89)90099-9. [DOI] [PubMed] [Google Scholar]
- 55.Dela Seña C, Riedl KM, Narayanasamy S, Curley RW, Schwartz SJ, Harrison EH. The human enzyme that converts dietary provitamin A carotenoids to vitamin A is a dioxygenase. J Biol Chem. 2014;289:13661–13666. doi: 10.1074/jbc.M114.557710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deltour L, Foglio MH, Duester G. Metabolic deficiencies in alcohol dehydrogenase Adh1, Adh3, and Adh4 null mutant mice. Overlapping roles of Adh1 and Adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J Biol Chem. 1999;274:16796–16801. doi: 10.1074/jbc.274.24.16796. [DOI] [PubMed] [Google Scholar]
- 57.Delva L, Bastie JN, Rochette-Egly C, Kraïba R, Balitrand N, Despouy G, et al. Physical and functional interactions between cellular retinoic acid binding protein II and the retinoic acid-dependent nuclear complex. Mol Cell Biol. 1999;19:7158–7167. doi: 10.1128/mcb.19.10.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem. 1999;274:23695–23698. doi: 10.1074/jbc.274.34.23695. [DOI] [PubMed] [Google Scholar]
- 59.Drevon CA, Blomhoff R, Rasmussen M, Kindberg GM, Berg T, Norum KR. Retinol esterification in cultured rat liver cells. Biochem J. 1985;230:617–623. doi: 10.1042/bj2300617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.EX, Zhang L, Lu J, Tso P, Blaner WS, Levin MS, et al. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J Biol Chem. 2002;277:36617–36623. doi: 10.1074/jbc.M205519200. [DOI] [PubMed] [Google Scholar]
- 61.Eckhoff C, Nau H. Vitamin A supplementation increases levels of retinoic acid compounds in human plasma: possible implications for teratogenesis. Arch Toxicol. 1990;64:502–503. doi: 10.1007/BF01977634. [DOI] [PubMed] [Google Scholar]
- 62.El Akawi Z, Napoli JL. Rat liver cytosolic retinal dehydrogenase: comparison of 13-cis-, 9-cis-, and all-trans-retinal as substrates and effects of cellular retinoid-binding proteins and retinoic acid on activity. Biochemistry (Mosc) 1994;33:1938–1943. doi: 10.1021/bi00173a042. [DOI] [PubMed] [Google Scholar]
- 63.Esteban J, Elabbas LE, Borg D, Herlin M, Akesson A, Barber X, et al. Gestational and lactational exposure to the polychlorinated biphenyl mixture Aroclor 1254 modulates retinoid homeostasis in rat offspring. Toxicol Lett. 2014;229:41–51. doi: 10.1016/j.toxlet.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 64.Everts HB, Sundberg JP, Ong DE. Immunolocalization of retinoic acid biosynthesis systems in selected sites in rat. Exp Cell Res. 2005;308:309–319. doi: 10.1016/j.yexcr.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 65.Farias EF, Ong DE, Ghyselinck NB, Nakajo S, Kuppumbatti YS, Mira Y, Lopez R. Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation, and tumorigenicity. J Natl Cancer Inst. 2005;97:21–29. doi: 10.1093/jnci/dji004. [DOI] [PubMed] [Google Scholar]
- 66.Farjo KM, Moiseyev G, Takahashi Y, Crouch RK, Ma J. The 11-cis-retinol dehydrogenase activity of RDH10 and its interaction with visual cycle proteins. Invest Ophthalmol Vis Sci. 2009;50:5089–5097. doi: 10.1167/iovs.09-3797. [DOI] [PubMed] [Google Scholar]
- 67.Fex G, Johannesson G. Radioimmunological determination of cellular retinol-binding protein in human tissue extracts. Cancer Res. 1984;44:3029–3032. [PubMed] [Google Scholar]
- 68.Fiorella PD, Napoli JL. Expression of cellular retinoic acid binding protein (CRABP) in Escherichia coli. Characterization and evidence that holo-CRABP is a substrate in retinoic acid metabolism. J Biol Chem. 1991;266:16572–16579. [PubMed] [Google Scholar]
- 69.Fiorella PD, Napoli JL. Microsomal retinoic acid metabolism. Effects of cellular retinoic acid-binding protein (type I) and C18-hydroxylation as an initial step. J Biol Chem. 1994;269:10538–10544. [PubMed] [Google Scholar]
- 70.Fiorella PD, Giguère V, Napoli JL. Expression of cellular retinoic acid-binding protein (type II) in Escherichia coli. Characterization and comparison to cellular retinoic acid-binding protein (type I) J Biol Chem. 1993;268:21545–21552. [PubMed] [Google Scholar]
- 71.Folli C, Calderone V, Ottonello S, Bolchi A, Zanotti G, Stoppini M, et al. Identification, retinoid binding, and x-ray analysis of a human retinol-binding protein. Proc Natl Acad Sci U S A. 2001;98:3710–3715. doi: 10.1073/pnas.061455898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Franzoni L, Lücke C, Pérez C, Cavazzini D, Rademacher M, Ludwig C, et al. Structure and backbone dynamics of Apo- and holo-cellular retinol-binding protein in solution. J Biol Chem. 2002;277:21983–21997. doi: 10.1074/jbc.M201994200. [DOI] [PubMed] [Google Scholar]
- 73.Franzoni L, Cavazzini D, Rossi GL, Lücke C. New insights on the protein-ligand interaction differences between the two primary cellular retinol carriers. J Lipid Res. 2010;51:1332–1343. doi: 10.1194/jlr.M002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Futterman S, Andrews JS. The composition of liver Vitamin A Ester and the synthesis of Vitamin A Ester by liver microsomes. J Biol Chem. 1964;239:4077–4080. [PubMed] [Google Scholar]
- 75.Ganfornina MD, Gutiérrez G, Bastiani M, Sánchez D. A phylogenetic analysis of the lipocalin protein family. Mol Biol Evol. 2000;17:114–126. doi: 10.1093/oxfordjournals.molbev.a026224. [DOI] [PubMed] [Google Scholar]
- 76.Gaub MP, Lutz Y, Ghyselinck NB, Scheuer I, Pfister V, Chambon P, et al. Nuclear detection of cellular retinoic acid binding proteins I and II with new antibodies. J Histochem Cytochem Off J Histochem Soc. 1998;46:1103–1111. doi: 10.1177/002215549804601002. [DOI] [PubMed] [Google Scholar]
- 77.Ghyselinck NB, Båvik C, Sapin V, Mark M, Bonnier D, Hindelang C, et al. Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J. 1999;18:4903–4914. doi: 10.1093/emboj/18.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giguère V, Lyn S, Yip P, Siu CH, Amin S. Molecular cloning of cDNA encoding a second cellular retinoic acid-binding protein. Proc Natl Acad Sci U S A. 1990;87:6233–6237. doi: 10.1073/pnas.87.16.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gorry P, Lufkin T, Dierich A, Rochette-Egly C, Décimo D, Dollé P, et al. The cellular retinoic acid binding protein I is dispensable. Proc Natl Acad Sci U S A. 1994;91:9032–9036. doi: 10.1073/pnas.91.19.9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gough WH, VanOoteghem S, Sint T, Kedishvili NY. cDNA cloning and characterization of a new human microsomal NAD + –dependent dehydrogenase that oxidizes all-trans-retinol and 3alpha-hydroxysteroids. J Biol Chem. 1998;273:19778–19785. doi: 10.1074/jbc.273.31.19778. [DOI] [PubMed] [Google Scholar]
- 81.Graham CE, Brocklehurst K, Pickersgill RW, Warren MJ. Characterization of retinal-dehyde dehydrogenase 3. Biochem J. 2006;394:67–75. doi: 10.1042/BJ20050918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grün F, Hirose Y, Kawauchi S, Ogura T, Umesono K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. J Biol Chem. 2000;275:41210–41218. doi: 10.1074/jbc.M007376200. [DOI] [PubMed] [Google Scholar]
- 83.Gustafson AL, Donovan M, Annerwall E, Dencker L, Eriksson U. Nuclear import of cellular retinoic acid-binding protein type I in mouse embryonic cells. Mech Dev. 1996;58:27–38. doi: 10.1016/s0925-4773(96)00554-0. [DOI] [PubMed] [Google Scholar]
- 84.Haeseleer F, Huang J, Lebioda L, Saari JC, Palczewski K. Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. J Biol Chem. 1998;273:21790–21799. doi: 10.1074/jbc.273.34.21790. [DOI] [PubMed] [Google Scholar]
- 85.Harrison EH. Bile salt-dependent, neutral cholesteryl ester hydrolase of rat liver: possible relationship with pancreatic cholesteryl ester hydrolase. Biochim Biophys Acta. 1988;963:28–34. doi: 10.1016/0005-2760(88)90334-7. [DOI] [PubMed] [Google Scholar]
- 86.Harrison EH. Lipases and carboxylesterases: possible roles in the hepatic metabolism of retinol. Annu Rev Nutr. 1998;18:259–276. doi: 10.1146/annurev.nutr.18.1.259. [DOI] [PubMed] [Google Scholar]
- 87.Harrison EH. Lipases and carboxylesterases: possible roles in the hepatic utilization of vitamin A. J Nutr. 2000;130:340S–344S. doi: 10.1093/jn/130.2.340S. [DOI] [PubMed] [Google Scholar]
- 88.Harrison EH. Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim Biophys Acta. 2012;1821:70–77. doi: 10.1016/j.bbalip.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harrison EH, Gad MZ. Hydrolysis of retinyl palmitate by enzymes of rat pancreas and liver. Differentiation of bile salt-dependent and bile salt-independent, neutral retinyl ester hydrolases in rat liver. J Biol Chem. 1989;264:17142–17147. [PubMed] [Google Scholar]
- 90.Harrison EH, Napoli JL. Bile salt-independent retinyl ester hydrolase activities associated with membranes of rat tissues. Methods Enzymol. 1990;189:459–469. doi: 10.1016/0076-6879(90)89323-a. [DOI] [PubMed] [Google Scholar]
- 91.Harrison EH, Blaner WS, Goodman DS, Ross AC. Subcellular localization of retinoids, retinoid-binding proteins, and acyl-CoA:retinol acyltransferase in rat liver. J Lipid Res. 1987;28:973–981. [PubMed] [Google Scholar]
- 92.Heinemann FS, Ozols J. Isolation and structural analysis of microsomal membrane proteins. Front Biosci J Virtual Libr. 1998;3:d483–d493. doi: 10.2741/a295. [DOI] [PubMed] [Google Scholar]
- 93.Helgerud P, Petersen LB, Norum KR. Acyl CoA:retinol acyltransferase in rat small intestine: its activity and some properties of the enzymic reaction. J Lipid Res. 1982;23:609–618. [PubMed] [Google Scholar]
- 94.Heller J. Interactions of plasma retinol-binding protein with its receptor. Specific binding of bovine and human retinol-binding protein to pigment epithelium cells from bovine eyes. J Biol Chem. 1975;250:3613–3619. [PubMed] [Google Scholar]
- 95.Heller M, Bok D. A specific receptor for retinol binding protein as detected by the binding of human and bovine retinol binding protein to pigment epithelial cells. Am J Ophthalmol. 1976;81:93–97. doi: 10.1016/0002-9394(76)90198-7. [DOI] [PubMed] [Google Scholar]
- 96.Herr FM, Ong DE. Differential interaction of lecithin-retinol acyltransferase with cellular retinol binding proteins. Biochemistry (Mosc) 1992;31:6748–6755. doi: 10.1021/bi00144a014. [DOI] [PubMed] [Google Scholar]
- 97.Hoegberg P, Schmidt CK, Fletcher N, Nilsson CB, Trossvik C, Gerlienke Schuur A, et al. Retinoid status and responsiveness to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking retinoid binding protein or retinoid receptor forms. Chem Biol Interact. 2005;156:25–39. doi: 10.1016/j.cbi.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 98.Imaoka S, Wan J, Chow T, Hiroi T, Eyanagi R, Shigematsu H, et al. Cloning and characterization of the CYP2D1-binding protein, retinol dehydrogenase. Arch Biochem Biophys. 1998;353:331–336. doi: 10.1006/abbi.1998.0644. [DOI] [PubMed] [Google Scholar]
- 99.Isken A, Golczak M, Oberhauser V, Hunzelmann S, Driever W, Imanishi Y, et al. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008;7:258–268. doi: 10.1016/j.cmet.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jamison RS, Newcomer ME, Ong DE. Cellular retinoid-binding proteins: limited proteolysis reveals a conformational change upon ligand binding. Biochemistry (Mosc) 1994;33:2873–2879. doi: 10.1021/bi00176a017. [DOI] [PubMed] [Google Scholar]
- 101.Jette C, Peterson PW, Sandoval IT, Manos EJ, Hadley E, Ireland CM, et al. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. J Biol Chem. 2004;279:34397–34405. doi: 10.1074/jbc.M314021200. [DOI] [PubMed] [Google Scholar]
- 102.Jiang W, Napoli JL. Reorganization of cellular retinol-binding protein type 1 and lecithin:retinol acyltransferase during retinyl ester biosynthesis. Biochim Biophys Acta. 2012;1820:859–869. doi: 10.1016/j.bbagen.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jiang W, Napoli JL. The retinol dehydrogenase Rdh10 localizes to lipid droplets during acyl ester biosynthesis. J Biol Chem. 2013;288:589–597. doi: 10.1074/jbc.M112.402883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jing Y, Waxman S, Mira-y-Lopez R. The cellular retinoic acid binding protein II is a positive regulator of retinoic acid signaling in breast cancer cells. Cancer Res. 1997;57:1668–1672. [PubMed] [Google Scholar]
- 105.Jurukovski V, Markova NG, Karaman-Jurukovska N, Randolph RK, Su J, Napoli JL, et al. Cloning and characterization of retinol dehydrogenase transcripts expressed in human epidermal keratinocytes. Mol Genet Metab. 1999;67:62–73. doi: 10.1006/mgme.1999.2840. [DOI] [PubMed] [Google Scholar]
- 106.Kakkad BP, Ong DE. Reduction of retinaldehyde bound to cellular retinol-binding protein (type II) by microsomes from rat small intestine. J Biol Chem. 1988;263:12916–12919. [PubMed] [Google Scholar]
- 107.Kane MA, Chen N, Sparks S, Napoli JL. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem J. 2005;388:363–369. doi: 10.1042/BJ20041867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kane MA, Folias AE, Napoli JL. HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Anal Biochem. 2008;378:71–79. doi: 10.1016/j.ab.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kane MA, Folias AE, Wang C, Napoli JL. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal Chem. 2008;80:1702–1708. doi: 10.1021/ac702030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kane MA, Folias AE, Pingitore A, Perri M, Obrochta KM, Krois CR, et al. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A. 2010;107:21884–21889. doi: 10.1073/pnas.1008859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kane MA, Folias AE, Wang C, Napoli JL. Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: a potential mechanism of ethanol toxicity. FASEB J Off Publ Fed Am Soc Exp Biol. 2010;24:823–832. doi: 10.1096/fj.09-141572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kane MA, Bright FV, Napoli JL. Binding affinities of CRBPI and CRBPII for 9-cis-retinoids. Biochim Biophys Acta. 2011;1810:514–518. doi: 10.1016/j.bbagen.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kane MA, Folias AE, Pingitore A, Perri M, Krois CR, Ryu JY, et al. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol Cell Biol. 2011;31:3277–3285. doi: 10.1128/MCB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kasus-Jacobi A, Ou J, Bashmakov YK, Shelton JM, Richardson JA, Goldstein JL, et al. Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2003;278:32380–32389. doi: 10.1074/jbc.M304969200. [DOI] [PubMed] [Google Scholar]
- 115.Kato M, Blaner WS, Mertz JR, Das K, Kato K, Goodman DS. Influence of retinoid nutritional status on cellular retinol- and cellular retinoic acid-binding protein concentrations in various rat tissues. J Biol Chem. 1985;260:4832–4838. [PubMed] [Google Scholar]
- 116.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 117.Kawaguchi R, Zhong M, Kassai M, Ter-Stepanian M, Sun H. STRA6-catalyzed vitamin A influx, efflux, and exchange. J Membr Biol. 2012;245:731–745. doi: 10.1007/s00232-012-9463-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kawaguchi R, Zhong M, Sun H. Real-time analyses of retinol transport by the membrane receptor of plasma retinol binding protein. J Vis Exp JoVE. 2013;17:e50169. doi: 10.3791/50169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kedishvili NY. Enzymology of retinoic acid biosynthesis and degradation. J Lipid Res. 2013;54:1744–1760. doi: 10.1194/jlr.R037028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kedishvili NY, Chumakova OV, Chetyrkin SV, Belyaeva OV, Lapshina EA, Lin DW, et al. Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1) J Biol Chem. 2002;277:28909–28915. doi: 10.1074/jbc.M202588200. [DOI] [PubMed] [Google Scholar]
- 121.Kim Y-K, Wassef L, Hamberger L, Piantedosi R, Palczewski K, Blaner WS, et al. Retinyl ester formation by lecithin: retinol acyltransferase is a key regulator of retinoid homeostasis in mouse embryogenesis. J Biol Chem. 2008;283:5611–5621. doi: 10.1074/jbc.M708885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kohlstedt K, Gershome C, Trouvain C, Hofmann W-K, Fichtlscherer S, Fleming I. Angiotensin-converting enzyme (ACE) inhibitors modulate cellular retinol-binding protein 1 and adiponectin expression in adipocytes via the ACE-dependent signaling cascade. Mol Pharmacol. 2009;75:685–692. doi: 10.1124/mol.108.051631. [DOI] [PubMed] [Google Scholar]
- 123.Kuppumbatti YS, Bleiweiss IJ, Mandeli JP, Waxman S, Mira-Y-Lopez R. Cellular retinol-binding protein expression and breast cancer. J Natl Cancer Inst. 2000;92:475–480. doi: 10.1093/jnci/92.6.475. [DOI] [PubMed] [Google Scholar]
- 124.Kuppumbatti YS, Rexer B, Nakajo S, Nakaya K, Mira-y-Lopez R. CRBP suppresses breast cancer cell survival and anchorage-independent growth. Oncogene. 2001;20:7413–7419. doi: 10.1038/sj.onc.1204749. [DOI] [PubMed] [Google Scholar]
- 125.Labrecque J, Bhat PV, Lacroix A. Purification and partial characterization of a rat kidney aldehyde dehydrogenase that oxidizes retinal to retinoic acid. Biochem Cell Biol Biochim Biol Cell. 1993;71:85–89. doi: 10.1139/o93-013. [DOI] [PubMed] [Google Scholar]
- 126.Lagace TA, Ridgway ND. The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833:2499–2510. doi: 10.1016/j.bbamcr.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 127.Lee M, Lucia SP. Effect of ethanol on the mobilization of vitamin A in the dog and in the rat. Q J Stud Alcohol. 1965;26:1–9. [PubMed] [Google Scholar]
- 128.Lee MO, Manthey CL, Sladek NE. Identification of mouse liver aldehyde dehydrogenases that catalyze the oxidation of retinaldehyde to retinoic acid. Biochem Pharmacol. 1991;42:1279–1285. doi: 10.1016/0006-2952(91)90266-8. [DOI] [PubMed] [Google Scholar]
- 129.Lee S-A, Belyaeva OV, Kedishvili NY. Biochemical characterization of human epidermal retinol dehydrogenase 2. Chem Biol Interact. 2009;178:182–187. doi: 10.1016/j.cbi.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lei Z, Chen W, Zhang M, Napoli JL. Reduction of all-trans-retinal in the mouse liver peroxisome fraction by the short-chain dehydrogenase/reductase RRD: induction by the PPAR alpha ligand clofibrate. Biochemistry (Mosc) 2003;42:4190–4196. doi: 10.1021/bi026948i. [DOI] [PubMed] [Google Scholar]
- 131.Leo MA, Lieber CS. Normal testicular structure and reproductive function in deermice lacking retinol and alcohol dehydrogenase activity. J Clin Invest. 1984;73:593–596. doi: 10.1172/JCI111248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Levi L, Lobo G, Doud MK, von Lintig J, Seachrist D, Tochtrop GP, et al. Genetic ablation of the fatty acid-binding protein FABP5 suppresses HER2-induced mammary tumori-genesis. Cancer Res. 2013;73:4770–4780. doi: 10.1158/0008-5472.CAN-13-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Levin MS, Locke B, Yang NC, Li E, Gordon JI. Comparison of the ligand binding properties of two homologous rat apocellular retinol-binding proteins expressed in Escherichia coli. J Biol Chem. 1988;263:17715–17723. [PubMed] [Google Scholar]
- 134.Li E, Norris AW. Structure/function of cytoplasmic vitamin A-binding proteins. Annu Rev Nutr. 1996;16:205–234. doi: 10.1146/annurev.nu.16.070196.001225. [DOI] [PubMed] [Google Scholar]
- 135.Li E, Qian SJ, Winter NS, d’ Avignon A, Levin MS. Fluorine nuclear magnetic resonance analysis of the ligand binding properties of two homologous rat cellular retinol-binding proteins expressed in Escherichia coli. J Biol Chem. 1991;266:3622–3629. [PubMed] [Google Scholar]
- 136.Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 137.Lin M, Napoli JL. cDNA cloning and expression of a human aldehyde dehydrogenase (ALDH) active with 9-cis-retinal and identification of a rat ortholog, ALDH12. J Biol Chem. 2000;275:40106–40112. doi: 10.1074/jbc.M008027200. [DOI] [PubMed] [Google Scholar]
- 138.Lin B, White JT, Ferguson C, Wang S, Vessella R, Bumgarner R, et al. Prostate short-chain dehydrogenase reductase 1 (PSDR1): a new member of the short-chain steroid dehydrogenase/reductase family highly expressed in normal and neoplastic prostate epithelium. Cancer Res. 2001;61:1611–1618. [PubMed] [Google Scholar]
- 139.Lin M, Zhang M, Abraham M, Smith SM, Napoli JL. Mouse retinal dehydrogenase 4 (RALDH4), molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells. J Biol Chem. 2003;278:9856–9861. doi: 10.1074/jbc.M211417200. [DOI] [PubMed] [Google Scholar]
- 140.Linke T, Dawson H, Harrison EH. Isolation and characterization of a microsomal acid retinyl ester hydrolase. J Biol Chem. 2005;280:23287–23294. doi: 10.1074/jbc.M413585200. [DOI] [PubMed] [Google Scholar]
- 141.Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J Biol Chem. 2005;280:40226–40234. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- 142.Liu C, Russell RM, Seitz HK, Wang XD. Ethanol enhances retinoic acid metabolism into polar metabolites in rat liver via induction of cytochrome P4502E1. Gastroenterology. 2001;120:179–189. doi: 10.1053/gast.2001.20877. [DOI] [PubMed] [Google Scholar]
- 143.Liu L, Tang X-H, Gudas LJ. Homeostasis of retinol in lecithin: retinol acyltransferase gene knockout mice fed a high retinol diet. Biochem Pharmacol. 2008;75:2316–2324. doi: 10.1016/j.bcp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu J, Cistola DP, Li E. Two homologous rat cellular retinol-binding proteins differ in local conformational flexibility. J Mol Biol. 2003;330:799–812. doi: 10.1016/s0022-2836(03)00629-6. [DOI] [PubMed] [Google Scholar]
- 145.MacDonald PN, Ong DE. Binding specificities of cellular retinol-binding protein and cellular retinol-binding protein, type II. J Biol Chem. 1987;262:10550–10556. [PubMed] [Google Scholar]
- 146.MacDonald PN, Ong DE. A lecithin:retinol acyltransferase activity in human and rat liver. Biochem Biophys Res Commun. 1988;156:157–163. doi: 10.1016/s0006-291x(88)80818-0. [DOI] [PubMed] [Google Scholar]
- 147.MacDonald PN, Ong DE. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J Biol Chem. 1988;263:12478–12482. [PubMed] [Google Scholar]
- 148.Mahadevan S, Murthy SK, Krishnamurthy S, Ganguly J. Studies on vitamin A esterase. 4. The hydrolysis and synthesis of vitamin A esters by rat intestinal mucosae. Biochem J. 1961;79:416–424. doi: 10.1042/bj0790416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Malpeli G, Stoppini M, Zapponi MC, Folli C, Berni R. Interactions with retinol and retinoids of bovine cellular retinol-binding protein. Eur J Biochem FEBS. 1995;229:486–493. [PubMed] [Google Scholar]
- 150.Markova NG, Pinkas-Sarafova A, Karaman-Jurukovska N, Jurukovski V, Simon M. Expression pattern and biochemical characteristics of a major epidermal retinol dehydrogenase. Mol Genet Metab. 2003;78:119–135. doi: 10.1016/s1096-7192(02)00226-3. [DOI] [PubMed] [Google Scholar]
- 151.Markova NG, Pinkas-Sarafova A, Simon M. A metabolic enzyme of the short-chain dehydrogenase/reductase superfamily may moonlight in the nucleus as a repressor of promoter activity. J Invest Dermatol. 2006;126:2019–2031. doi: 10.1038/sj.jid.5700347. [DOI] [PubMed] [Google Scholar]
- 152.Marwarha G, Berry DC, Croniger CM, Noy N. The retinol esterifying enzyme LRAT supports cell signaling by retinol-binding protein and its receptor STRA6. FASEB J Off Publ Fed Am Soc Exp Biol. 2014;28:26–34. doi: 10.1096/fj.13-234310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matsuzaka Y, Okamoto K, Tsuji H, Mabuchi T, Ozawa A, Tamiya G, et al. Identification of the hRDH-E2 gene, a novel member of the SDR family, and its increased expression in psoriatic lesion. Biochem Biophys Res Commun. 2002;297:1171–1180. doi: 10.1016/s0006-291x(02)02344-6. [DOI] [PubMed] [Google Scholar]
- 154.McCaffery P, Lee MO, Wagner MA, Sladek NE, Dräger UC. Asymmetrical retinoic acid synthesis in the dorsoventral axis of the retina. Dev Camb Engl. 1992;115:371–382. doi: 10.1242/dev.115.2.371. [DOI] [PubMed] [Google Scholar]
- 155.McCaffery P, Koul O, Smith D, Napoli JL, Chen N, Ullman MD. Ethanol increases retinoic acid production in cerebellar astrocytes and in cerebellum. Brain Res Dev Brain Res. 2004;153:233–241. doi: 10.1016/j.devbrainres.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 156.Miller WH. The emerging role of retinoids and retinoic acid metabolism blocking agents in the treatment of cancer. Cancer. 1998;83:1471–1482. doi: 10.1002/(sici)1097-0142(19981015)83:8<1471::aid-cncr1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 157.Mira-Y-Lopez R, Zheng WL, Kuppumbatti YS, Rexer B, Jing Y, Ong DE. Retinol conversion to retinoic acid is impaired in breast cancer cell lines relative to normal cells. J Cell Physiol. 2000;185:302–309. doi: 10.1002/1097-4652(200011)185:2<302::AID-JCP15>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 158.Mittag T, Franzoni L, Cavazzini D, Schaffhausen B, Rossi GL, Günther UL. Retinol modulates site-specific mobility of apo-cellular retinol-binding protein to promote ligand binding. J Am Chem Soc. 2006;128:9844–9848. doi: 10.1021/ja0616128. [DOI] [PubMed] [Google Scholar]
- 159.Moise AR, Golczak M, Imanishi Y, Palczewski K. Topology and membrane association of lecithin: retinol acyltransferase. J Biol Chem. 2007;282:2081–2090. doi: 10.1074/jbc.M608315200. [DOI] [PubMed] [Google Scholar]
- 160.Molotkov A, Fan X, Duester G. Excessive vitamin A toxicity in mice genetically deficient in either alcohol dehydrogenase Adh1 or Adh3. Eur J Biochem FEBS. 2002;269:2607–2612. doi: 10.1046/j.1432-1033.2002.02935.x. [DOI] [PubMed] [Google Scholar]
- 161.Nadauld LD, Shelton DN, Chidester S, Yost HJ, Jones DA. The zebrafish retinol dehydrogenase, rdh1l, is essential for intestinal development and is regulated by the tumor suppressor adenomatous polyposis coli. J Biol Chem. 2005;280:30490–30495. doi: 10.1074/jbc.M504973200. [DOI] [PubMed] [Google Scholar]
- 162.Napoli JL. Retinol metabolism in LLC-PK1 Cells. Characterization of retinoic acid synthesis by an established mammalian cell line. J Biol Chem. 1986;261:13592–13597. [PubMed] [Google Scholar]
- 163.Napoli JL. Chem Biol Synth Retin. Boca Raton: CRC Press; 1990. The biogenesis of retinoic acid: a physiologically significant promoter of differentiation; pp. 229–49. [Google Scholar]
- 164.Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim Biophys Acta. 1999;1440:139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 165.Napoli JL. Effects of ethanol on physiological retinoic acid levels. IUBMB Life. 2011;63:701–706. doi: 10.1002/iub.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Napoli JL. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim Biophys Acta. 2012;1821:152–167. doi: 10.1016/j.bbalip.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Napoli JL, McCormick AM. Tissue dependence of retinoic acid metabolism in vivo. Biochim Biophys Acta. 1981;666:165–175. doi: 10.1016/0005-2760(81)90102-8. [DOI] [PubMed] [Google Scholar]
- 168.Napoli JL, Race KR. The biosynthesis of retinoic acid from retinol by rat tissues in vitro. Arch Biochem Biophys. 1987;255:95–101. doi: 10.1016/0003-9861(87)90298-0. [DOI] [PubMed] [Google Scholar]
- 169.Napoli JL, Pacia EB, Salerno GJ. Cholate effects on all-trans-retinyl palmitate hydrolysis in tissue homogenates: solubilization of multiple kidney membrane hydrolases. Arch Biochem Biophys. 1989;274:192–199. doi: 10.1016/0003-9861(89)90430-x. [DOI] [PubMed] [Google Scholar]
- 170.Napoli JL, Posch KP, Fiorella PD, Boerman MH. Physiological occurrence, biosynthesis and metabolism of retinoic acid: evidence for roles of cellular retinol-binding protein (CRBP) and cellular retinoic acid-binding protein (CRABP) in the pathway of retinoic acid homeostasis. Biomed Pharmacother Bioméd Pharmacothérapie. 1991;45:131–143. doi: 10.1016/0753-3322(91)90101-x. [DOI] [PubMed] [Google Scholar]
- 171.Napoli JL, Posch KC, Burns RD. Microsomal retinal synthesis: retinol vs. holo-CRBP as substrate and evaluation of NADP, NAD and NADPH as cofactors. Biochim Biophys Acta. 1992;1120:183–186. doi: 10.1016/0167-4838(92)90267-h. [DOI] [PubMed] [Google Scholar]
- 172.Napoli JL, Boerman MH, Chai X, Zhai Y, Fiorella PD. Enzymes and binding proteins affecting retinoic acid concentrations. J Steroid Biochem Mol Biol. 1995;53:497–502. doi: 10.1016/0960-0760(95)00096-i. [DOI] [PubMed] [Google Scholar]
- 173.Nau H. Teratogenicity of isotretinoin revisited: species variation and the role of all-trans-retinoic acid. J Am Acad Dermatol. 2001;45:S183–S187. doi: 10.1067/mjd.2001.113720. [DOI] [PubMed] [Google Scholar]
- 174.Newcomer ME. Retinoid-binding proteins: structural determinants important for function. FASEB J Off Publ Fed Am Soc Exp Biol. 1995;9:229–239. doi: 10.1096/fasebj.9.2.7781925. [DOI] [PubMed] [Google Scholar]
- 175.Newcomer ME, Jamison RS, Ong DE. Structure and function of retinoid-binding proteins. Subcell Biochem. 1998;30:53–80. doi: 10.1007/978-1-4899-1789-8_3. [DOI] [PubMed] [Google Scholar]
- 176.Nishiwaki S, Kato M, Okuno M, Kanai M, Muto Y. Purification and partial characterization of a novel cellular retinol-binding protein, type three, from the piscine eyes. Biochim Biophys Acta. 1990;1037:192–199. doi: 10.1016/0167-4838(90)90167-e. [DOI] [PubMed] [Google Scholar]
- 177.Norris AW, Cheng L, Giguère V, Rosenberger M, Li E. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim Biophys Acta. 1994;1209:10–18. doi: 10.1016/0167-4838(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 178.Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000;348(Pt 3):481–495. [PMC free article] [PubMed] [Google Scholar]
- 179.Noy N, Blaner WS. Interactions of retinol with binding proteins: studies with rat cellular retinol-binding protein and with rat retinol-binding protein. Biochemistry (Mosc) 1991;30:6380–6386. doi: 10.1021/bi00240a005. [DOI] [PubMed] [Google Scholar]
- 180.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, et al. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT) J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Obrochta KM, Kane MA, Napoli JL. Effects of diet and strain on mouse serum and tissue retinoid concentrations. PLoS One. 2014;9:e99435. doi: 10.1371/journal.pone.0099435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ong DE. Purification and partial characterization of cellular retinol-binding protein from human liver. Cancer Res. 1982;42:1033–1037. [PubMed] [Google Scholar]
- 183.Ong DE. A novel retinol-binding protein from rat. Purification and partial characterization. J Biol Chem. 1984;259:1476–1482. [PubMed] [Google Scholar]
- 184.Ong DE. Cellular transport and metabolism of vitamin A: roles of the cellular retinoid-binding proteins. Nutr Rev. 1994;52:S24–S31. doi: 10.1111/j.1753-4887.1994.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 185.Ong DE, Chytil F. Retinoic acid-binding protein in rat tissue. Partial purification and comparison to rat tissue retinol-binding protein. J Biol Chem. 1975;250:6113–6117. [PubMed] [Google Scholar]
- 186.Ong DE, Chytil F. Cellular retinoic acid-binding protein from rat testis. Purification and characterization. J Biol Chem. 1978;253:4551–4554. [PubMed] [Google Scholar]
- 187.Ong DE, Chytil F. Cellular retinol-binding protein from rat liver. Purification and characterization. J Biol Chem. 1978;253:828–832. [PubMed] [Google Scholar]
- 188.Ong DE, Chytil F. Immunochemical comparison of vitamin A binding proteins of rat. J Biol Chem. 1979;254:8733–8735. [PubMed] [Google Scholar]
- 189.Ong DE, Page DL. Quantitation of cellular retinol-binding protein in human organs. Am J Clin Nutr. 1986;44:425–430. doi: 10.1093/ajcn/44.3.425. [DOI] [PubMed] [Google Scholar]
- 190.Ong DE, Crow JA, Chytil F. Radioimmunochemical determination of cellular retinol- and cellular retinoic acid-binding proteins in cytosols of rat tissues. J Biol Chem. 1982;257:13385–13389. [PubMed] [Google Scholar]
- 191.Ong DE, Kakkad B, MacDonald PN. Acyl-CoA-independent esterification of retinol bound to cellular retinol-binding protein (type II) by microsomes from rat small intestine. J Biol Chem. 1987;262:2729–2736. [PubMed] [Google Scholar]
- 192.Ong DE, MacDonald PN, Gubitosi AM. Esterification of retinol in rat liver. Possible participation by cellular retinol-binding protein and cellular retinol-binding protein II. J Biol Chem. 1988;263:5789–5796. [PubMed] [Google Scholar]
- 193.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, et al. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 194.Orland MD, Anwar K, Cromley D, Chu C-H, Chen L, Billheimer JT, et al. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: diacylglycerol acyltransferase 1. Biochim Biophys Acta. 2005;1737:76–82. doi: 10.1016/j.bbalip.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 195.Ottonello S, Petrucco S, Maraini G. Vitamin A uptake from retinol-binding protein in a cell-free system from pigment epithelial cells of bovine retina. Retinol transfer from plasma retinol-binding protein to cytoplasmic retinol-binding protein with retinyl-ester formation as the intermediate step. J Biol Chem. 1987;262:3975–3981. [PubMed] [Google Scholar]
- 196.Ottonello S, Scita G, Mantovani G, Cavazzini D, Rossi GL. Retinol bound to cellular retinol-binding protein is a substrate for cytosolic retinoic acid synthesis. J Biol Chem. 1993;268:27133–27142. [PubMed] [Google Scholar]
- 197.Parker RO, Crouch RK. Retinol dehydrogenases (RDHs) in the visual cycle. Exp Eye Res. 2010;91:788–792. doi: 10.1016/j.exer.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Penzes P, Napoli JL. Holo-cellular retinol-binding protein: distinction of ligand-binding affinity from efficiency as substrate in retinal biosynthesis. Biochemistry (Mosc) 1999;38:2088–2093. doi: 10.1021/bi982228t. [DOI] [PubMed] [Google Scholar]
- 199.Penzes P, Wang X, Napoli JL. Enzymatic characteristics of retinal dehydrogenase type I expressed in Escherichia coli. Biochim Biophys Acta. 1997;1342:175–181. doi: 10.1016/s0167-4838(97)00102-7. [DOI] [PubMed] [Google Scholar]
- 200.Penzes P, Wang X, Sperkova Z, Napoli JL. Cloning of a rat cDNA encoding retinal dehydrogenase isozyme type I and its expression in E. coli. Gene. 1997;191:167–172. doi: 10.1016/s0378-1119(97)00054-1. [DOI] [PubMed] [Google Scholar]
- 201.Persaud SD, Lin Y-W, Wu C-Y, Kagechika H, Wei L-N. Cellular retinoic acid binding protein I mediates rapid non-canonical activation of ERK1/2 by all-trans retinoic acid. Cell Signal. 2013;25:19–25. doi: 10.1016/j.cellsig.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Persson B, Kallberg Y. Classification and nomenclature of the superfamily of short-chain dehydrogenases/reductases (SDRs) Chem Biol Interact. 2013;202:111–115. doi: 10.1016/j.cbi.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 203.Persson B, Kallberg Y, Bray JE, Bruford E, Dellaporta SL, Favia AD, et al. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem Biol Interact. 2009;178:94–98. doi: 10.1016/j.cbi.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ. Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci U S A. 1996;93:13090–13095. doi: 10.1073/pnas.93.23.13090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Piantedosi R, Ghyselinck N, Blaner WS, Vogel S. Cellular retinol-binding protein type III is needed for retinoid incorporation into milk. J Biol Chem. 2005;280:24286–24292. doi: 10.1074/jbc.M503906200. [DOI] [PubMed] [Google Scholar]
- 206.Picozzi P, Marozzi A, Fornasari D, Benfante R, Barisani D, Meneveri R, et al. Genomic organization and transcription of the human retinol dehydrogenase 10 (RDH10) gene. FEBS Lett. 2003;554:59–66. doi: 10.1016/s0014-5793(03)01089-5. [DOI] [PubMed] [Google Scholar]
- 207.Pierzchalski K, Yu J, Norman V, Kane MA. CrbpI regulates mammary retinoic acid homeostasis and the mammary microenvironment. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27:1904–1916. doi: 10.1096/fj.12-219410. [DOI] [PubMed] [Google Scholar]
- 208.Porter SB, Fraker LD, Chytil F, Ong DE. Localization of cellular retinol-binding protein in several rat tissues. Proc Natl Acad Sci U S A. 1983;80:6586–6590. doi: 10.1073/pnas.80.21.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Posch KC, Napoli JL. Multiple retinoid dehydrogenases in testes cytosol from alcohol dehydrogenase negative or positive deermice. Biochem Pharmacol. 1992;43:2296–2298. doi: 10.1016/0006-2952(92)90191-k. [DOI] [PubMed] [Google Scholar]
- 210.Posch KC, Enright WJ, Napoli JL. Retinoic acid synthesis by cytosol from the alcohol dehydrogenase negative deermouse. Arch Biochem Biophys. 1989;274:171–178. doi: 10.1016/0003-9861(89)90428-1. [DOI] [PubMed] [Google Scholar]
- 211.Posch KC, Boerman MH, Burns RD, Napoli JL. Holocellular retinol binding protein as a substrate for microsomal retinal synthesis. Biochemistry. 1991;30:6224–6230. doi: 10.1021/bi00239a021. [DOI] [PubMed] [Google Scholar]
- 212.Posch KC, Burns RD, Napoli JL. Biosynthesis of all-trans-retinoic acid from retinal. Recognition of retinal bound to cellular retinol binding protein (type I) as substrate by a purified cytosolic dehydrogenase. J Biol Chem. 1992;267:19676–19682. [PubMed] [Google Scholar]
- 213.Quadro L, Hamberger L, Gottesman ME, Wang F, Colantuoni V, Blaner WS, et al. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology. 2005;146:4479–4490. doi: 10.1210/en.2005-0158. [DOI] [PubMed] [Google Scholar]
- 214.Quick TC, Ong DE. Vitamin A metabolism in the human intestinal Caco-2 cell line. Biochemistry (Mosc) 1990;29:11116–11123. doi: 10.1021/bi00502a015. [DOI] [PubMed] [Google Scholar]
- 215.Randolph RK, Simon M. Metabolism of all-trans-retinoic acid by cultured human epidermal keratinocytes. J Lipid Res. 1997;38:1374–1383. [PubMed] [Google Scholar]
- 216.Randolph RK, Winkler KE, Ross AC. Fatty acyl CoA-dependent and -independent retinol esterification by rat liver and lactating mammary gland microsomes. Arch Biochem Biophys. 1991;288:500–508. doi: 10.1016/0003-9861(91)90227-a. [DOI] [PubMed] [Google Scholar]
- 217.Rask L, Peterson PA. In vitro uptake of vitamin A from the retinol-binding plasma protein to mucosal epithelial cells from the monkey’s small intestine. J Biol Chem. 1976;251:6360–6366. [PubMed] [Google Scholar]
- 218.Ratnam KK, Feng X, Chuang PY, Verma V, Lu T-C, Wang J, et al. Role of the retinoic acid receptor-α in HIV-associated nephropathy. Kidney Int. 2011;79:624–634. doi: 10.1038/ki.2010.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Reeves PG. Components of the AIN-93 diets as improvements in the AIN-76A diet. J Nutr. 1997;127:838S–841S. doi: 10.1093/jn/127.5.838S. [DOI] [PubMed] [Google Scholar]
- 220.Reeves PG, Nielsen FH, Fahey GC., Jr AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 221.Rexer BN, Ong DE. A novel short-chain alcohol dehydrogenase from rats with retinol dehydrogenase activity, cyclically expressed in uterine epithelium. Biol Reprod. 2002;67:1555–1564. doi: 10.1095/biolreprod.102.007021. [DOI] [PubMed] [Google Scholar]
- 222.Rexer BN, Zheng WL, Ong DE. Retinoic acid biosynthesis by normal human breast epithelium is via aldehyde dehydrogenase 6, absent in MCF-7 cells. Cancer Res. 2001;61:7065–7070. [PubMed] [Google Scholar]
- 223.Reynier M. Pyrazole inhibition and kinetic studies of ethanol and retinol oxidation catalyzed by rat liver alcohol dehydrogenase. Acta Chem Scand. 1969;23:1119–1129. doi: 10.3891/acta.chem.scand.23-1119. [DOI] [PubMed] [Google Scholar]
- 224.Rhinn M, Schuhbaur B, Niederreither K, Dollé P. Involvement of retinol dehydroge-nase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc Natl Acad Sci U S A. 2011;108:16687–16692. doi: 10.1073/pnas.1103877108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Rieck M, Meissner W, Ries S, Müller-Brüsselbach S, Müller R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) beta/delta: a comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol Pharmacol. 2008;74:1269–1277. doi: 10.1124/mol.108.050625. [DOI] [PubMed] [Google Scholar]
- 226.Rigtrup KM, Ong DE. A retinyl ester hydrolase activity intrinsic to the brush border membrane of rat small intestine. Biochemistry (Mosc) 1992;31:2920–2926. doi: 10.1021/bi00126a011. [DOI] [PubMed] [Google Scholar]
- 227.Rigtrup KM, Kakkad B, Ong DE. Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: probable identity with brush border phospholipase B. Biochemistry (Mosc) 1994;33:2661–2666. doi: 10.1021/bi00175a039. [DOI] [PubMed] [Google Scholar]
- 228.Rigtrup KM, McEwen LR, Said HM, Ong DE. Retinyl ester hydrolytic activity associated with human intestinal brush border membranes. Am J Clin Nutr. 1994;60:111–116. doi: 10.1093/ajcn/60.1.111. [DOI] [PubMed] [Google Scholar]
- 229.Ross AC. Retinol esterification by mammary gland microsomes from the lactating rat. J Lipid Res. 1982;23:133–144. [PubMed] [Google Scholar]
- 230.Ross AC. Retinol esterification by rat liver microsomes. Evidence for a fatty acyl coenzyme A: retinol acyltransferase. J Biol Chem. 1982;257:2453–2459. [PubMed] [Google Scholar]
- 231.Ross AC. Cellular metabolism and activation of retinoids: roles of cellular retinoid-binding proteins. FASEB J Off Publ Fed Am Soc Exp Biol. 1993;7:317–327. doi: 10.1096/fasebj.7.2.8440409. [DOI] [PubMed] [Google Scholar]
- 232.Ross AC, Goodman DS. Intracellular binding proteins for retinol and retinoic acid: comparison with each other and with serum retinol-binding protein. Fed Proc. 1979;38:2515–2518. [PubMed] [Google Scholar]
- 233.Ross AC, Takahashi YI, Goodman DS. The binding protein for retinol from rat testis cytosol. Isolation and partial characterization. J Biol Chem. 1978;253:6591–6598. [PubMed] [Google Scholar]
- 234.Ross AC, Adachi N, Goodman DS. The binding protein for retinoic acid from rat testis cytosol: isolation and partial characterization. J Lipid Res. 1980;21:100–109. [PubMed] [Google Scholar]
- 235.Ruff SJ, Ong DE. Cellular retinoic acid binding protein is associated with mitochon-dria. FEBS Lett. 2000;487:282–286. doi: 10.1016/s0014-5793(00)02366-8. [DOI] [PubMed] [Google Scholar]
- 236.Rühl R, Plum C, Elmazar MM, Nau H. Embryonic subcellular distribution of 13-cis- and all-trans-retinoic acid indicates differential cytosolic/nuclear localization. Toxicol Sci Off J Soc Toxicol. 2001;63:82–89. doi: 10.1093/toxsci/63.1.82. [DOI] [PubMed] [Google Scholar]
- 237.Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem. 1999;274:3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- 238.Ruiz A, Ghyselinck NB, Mata N, Nusinowitz S, Lloyd M, Dennefeld C, et al. Somatic ablation of the Lrat gene in the mouse retinal pigment epithelium drastically reduces its retinoid storage. Invest Ophthalmol Vis Sci. 2007;48:5377–5387. doi: 10.1167/iovs.07-0673. [DOI] [PubMed] [Google Scholar]
- 239.Ruiz A, Mark M, Jacobs H, Klopfenstein M, Hu J, Lloyd M, et al. Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Invest Ophthalmol Vis Sci. 2012;53:3027–3039. doi: 10.1167/iovs.11-8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Saari JC, Crabb JW. Focus on molecules: cellular retinaldehyde-binding protein (CRALBP) Exp Eye Res. 2005;81:245–246. doi: 10.1016/j.exer.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 241.Saari JC, Futterman S. Separable binding proteins for retinoic acid and retinol in bovine retina. Biochim Biophys Acta. 1976;444:789–793. doi: 10.1016/0304-4165(76)90325-1. [DOI] [PubMed] [Google Scholar]
- 242.Saari JC, Futterman S, Bredberg L. Cellular retinol- and retinoic acid-binding proteins of bovine retina. Purification and properties. J Biol Chem. 1978;253:6432–6436. [PubMed] [Google Scholar]
- 243.Sani BP. Localization of retinoic acid-binding protein in nuclei. Biochem Biophys Res Commun. 1977;75:7–12. doi: 10.1016/0006-291x(77)91281-5. [DOI] [PubMed] [Google Scholar]
- 244.Sani BP, Donovan MK. Localization of retinoic acid-binding protein in nuclei and the nuclear uptake of retinoic acid. Cancer Res. 1979;39:2492–2496. [PubMed] [Google Scholar]
- 245.Sani BP, Hill DL. Retinoic acid: a binding protein in chick embryo metatarsal skin. Biochem Biophys Res Commun. 1974;61:1276–1282. doi: 10.1016/s0006-291x(74)80422-5. [DOI] [PubMed] [Google Scholar]
- 246.Sato M, Lieber CS. Hepatic vitamin A depletion after chronic ethanol consumption in baboons and rats. J Nutr. 1981;111:2015–2023. doi: 10.1093/jn/111.11.2015. [DOI] [PubMed] [Google Scholar]
- 247.Schmidt CK, Brouwer A, Nau H. Chromatographic analysis of endogenous retinoids in tissues and serum. Anal Biochem. 2003;315:36–48. doi: 10.1016/s0003-2697(02)00662-0. [DOI] [PubMed] [Google Scholar]
- 248.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc Natl Acad Sci U S A. 2008;105:7546–7551. doi: 10.1073/pnas.0709981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Shaw N, Elholm M, Noy N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor beta/delta. J Biol Chem. 2003;278:41589–41592. doi: 10.1074/jbc.C300368200. [DOI] [PubMed] [Google Scholar]
- 251.Shih MYS, Kane MA, Zhou P, Yen CLE, Streeper RS, Napoli JL, et al. Retinol esterification by DGAT1 is essential for retinoid homeostasis in murine skin. J Biol Chem. 2009;284:4292–4299. doi: 10.1074/jbc.M807503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shingleton JL, Skinner MK, Ong DE. Characteristics of retinol accumulation from serum retinol-binding protein by cultured Sertoli cells. Biochemistry (Mosc) 1989;28:9641–9647. doi: 10.1021/bi00451a015. [DOI] [PubMed] [Google Scholar]
- 253.Shingleton JL, Skinner MK, Ong DE. Retinol esterification in Sertoli cells by lecithin-retinol acyltransferase. Biochemistry (Mosc) 1989;28:9647–9653. doi: 10.1021/bi00451a016. [DOI] [PubMed] [Google Scholar]
- 254.Siegenthaler G, Saurat JH, Ponec M. Retinol and retinal metabolism. Relationship to the state of differentiation of cultured human keratinocytes. Biochem J. 1990;268:371–378. doi: 10.1042/bj2680371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Sima A, Parisotto M, Mader S, Bhat PV. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim Biophys Acta. 2009;1790:1660–1664. doi: 10.1016/j.bbagen.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 256.Song M-S, Chen W, Zhang M, Napoli JL. Identification of a mouse short-chain dehydrogenase/reductase gene, retinol dehydrogenase-similar. Function of non-catalytic amino acid residues in enzyme activity. J Biol Chem. 2003;278:40079–40087. doi: 10.1074/jbc.M304910200. [DOI] [PubMed] [Google Scholar]
- 257.Ström K, Gundersen TE, Hansson O, Lucas S, Fernandez C, Blomhoff R, et al. Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL. FASEB J Off Publ Fed Am Soc Exp Biol. 2009;23:2307–2316. doi: 10.1096/fj.08-120923. [DOI] [PubMed] [Google Scholar]
- 258.Sun H. Membrane receptors and transporters involved in the function and transport of vitamin A and its derivatives. Biochim Biophys Acta. 2012;1821:99–112. doi: 10.1016/j.bbalip.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sun G, Alexson SE, Harrison EH. Purification and characterization of a neutral, bile salt-independent retinyl ester hydrolase from rat liver microsomes. Relationship To rat carboxylesterase ES-2. J Biol Chem. 1997;272:24488–24493. doi: 10.1074/jbc.272.39.24488. [DOI] [PubMed] [Google Scholar]
- 260.Szuts EZ, Harosi FI. Solubility of retinoids in water. Arch Biochem Biophys. 1991;287:297–304. doi: 10.1016/0003-9861(91)90482-x. [DOI] [PubMed] [Google Scholar]
- 261.Takahashi Y, Moiseyev G, Farjo K, Ma J-X. Characterization of key residues and membrane association domains in retinol dehydrogenase 10. Biochem J. 2009;419:113–122. 1–122. doi: 10.1042/BJ20080812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Takase S, Ong DE, Chytil F. Transfer of retinoic acid from its complex with cellular retinoic acid-binding protein to the nucleus. Arch Biochem Biophys. 1986;247:328–334. doi: 10.1016/0003-9861(86)90591-6. [DOI] [PubMed] [Google Scholar]
- 263.Thumser AE, Moore JB, Plant NJ. Fatty acid binding proteins: tissue-specific functions in health and disease. Curr Opin Clin Nutr Metab Care. 2014;17:124–129. doi: 10.1097/MCO.0000000000000031. [DOI] [PubMed] [Google Scholar]
- 264.Tzimas G, Nau H. The role of metabolism and toxicokinetics in retinoid teratogenesis. Curr Pharm Des. 2001;7:803–831. doi: 10.2174/1381612013397708. [DOI] [PubMed] [Google Scholar]
- 265.Van Thiel DH, Gavaler J, Lester R. Ethanol inhibition of vitamin A metabolism in the testes: possible mechanism for sterility in alcoholics. Science. 1974;186:941–942. doi: 10.1126/science.186.4167.941. [DOI] [PubMed] [Google Scholar]
- 266.Venepally P, Reddy LG, Sani BP. Analysis of the effects of CRABP I expression on the RA-induced transcription mediated by retinoid receptors. Biochemistry (Mosc) 1996;35:9974–9982. doi: 10.1021/bi9603276. [DOI] [PubMed] [Google Scholar]
- 267.Vogel S, Mendelsohn CL, Mertz JR, Piantedosi R, Waldburger C, Gottesman ME, et al. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J Biol Chem. 2001;276:1353–1360. doi: 10.1074/jbc.M005118200. [DOI] [PubMed] [Google Scholar]
- 268.Vonesch JL, Nakshatri H, Philippe M, Chambon P, Dollé P. Stage and tissue-specific expression of the alcohol dehydrogenase 1 (Adh-1) gene during mouse development. Dev Dyn Off Publ Am Assoc Anat. 1994;199:199–213. doi: 10.1002/aja.1001990305. [DOI] [PubMed] [Google Scholar]
- 269.Vreeland AC, Yu S, Levi L, de Barros RD, Noy N. Transcript Stabilization by the RNA-Binding Protein HuR Is Regulated by Cellular Retinoic Acid-Binding Protein 2. Mol Cell Biol. 2014;34:2135–2146. doi: 10.1128/MCB.00281-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Wang X-D. Alcohol, vitamin A, and cancer. Alcohol Fayettev N. 2005;35:251–258. doi: 10.1016/j.alcohol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 271.Wang X, Penzes P, Napoli JL. Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. J Biol Chem. 1996;271:16288–16293. doi: 10.1074/jbc.271.27.16288. [DOI] [PubMed] [Google Scholar]
- 272.Wang J, Bongianni JK, Napoli JL. The N-terminus of retinol dehydrogenase type 1 signals cytosolic orientation in the microsomal membrane. Biochemistry (Mosc) 2001;40:12533–12540. doi: 10.1021/bi011396+. [DOI] [PubMed] [Google Scholar]
- 273.Wang C, Kane MA, Napoli JL. Multiple retinol and retinal dehydrogenases catalyze all-trans-retinoic acid biosynthesis in astrocytes. J Biol Chem. 2011;286:6542–6553. doi: 10.1074/jbc.M110.198382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Williams JB, Napoli JL. Metabolism of retinoic acid and retinol during differentiation of F9 embryonal carcinoma cells. Proc Natl Acad Sci U S A. 1985;82:4658–4662. doi: 10.1073/pnas.82.14.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Williams JB, Napoli JL. Inhibition of retinoic acid metabolism by imidazole antimycotics in F9 embryonal carcinoma cells. Biochem Pharmacol. 1987;36:1386–1388. doi: 10.1016/0006-2952(87)90102-x. [DOI] [PubMed] [Google Scholar]
- 276.Wolf G. Identification of a membrane receptor for retinol-binding protein functioning in the cellular uptake of retinol. Nutr Rev. 2007;65:385–388. doi: 10.1301/nr.2007.aug.385-388. [DOI] [PubMed] [Google Scholar]
- 277.Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, et al. The molecular basis of retinoid absorption: a genetic dissection. J Biol Chem. 2008;283:13510–13519. doi: 10.1074/jbc.M800777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Wongsiriroj N, Jiang H, Piantedosi R, Yang KJZ, Kluwe J, Schwabe RF, et al. Genetic dissection of retinoid esterification and accumulation in the liver and adipose tissue. J Lipid Res. 2014;55:104–114. doi: 10.1194/jlr.M043844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Wu BX, Chen Y, Chen Y, Fan J, Rohrer B, Crouch RK, et al. Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE. Invest Ophthalmol Vis Sci. 2002;43:3365–3372. [PubMed] [Google Scholar]
- 280.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 281.Yen C-LE, Monetti M, Burri BJ, Farese RV. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J Lipid Res. 2005;46:1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- 282.Yost RW, Harrison EH, Ross AC. Esterification by rat liver microsomes of retinol bound to cellular retinol-binding protein. J Biol Chem. 1988;263:18693–18701. [PubMed] [Google Scholar]
- 283.Yu S, Levi L, Siegel R, Noy N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ) J Biol Chem. 2012;287:42195–42205. doi: 10.1074/jbc.M112.410381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zachman RD, Grummer MA. The interaction of ethanol and vitamin A as a potential mechanism for the pathogenesis of Fetal Alcohol syndrome. Alcohol Clin Exp Res. 1998;22:1544–1556. [PubMed] [Google Scholar]
- 285.Zhai Y, Higgins D, Napoli JL. Coexpression of the mRNAs encoding retinol dehydrogenase isozymes and cellular retinol-binding protein. J Cell Physiol. 1997;173:36–43. doi: 10.1002/(SICI)1097-4652(199710)173:1<36::AID-JCP5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 286.Zhai Y, Sperkova Z, Napoli JL. Cellular expression of retinal dehydrogenase types 1 and 2: effects of vitamin A status on testis mRNA. J Cell Physiol. 2001;186:220–232. doi: 10.1002/1097-4652(200102)186:2<220::AID-JCP1018>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 287.Zhang M, Chen W, Smith SM, Napoli JL. Molecular characterization of a mouse short chain dehydrogenase/reductase active with all-trans-retinol in intact cells, mRDH1. J Biol Chem. 2001;276:44083–44090. doi: 10.1074/jbc.M105748200. [DOI] [PubMed] [Google Scholar]
- 288.Zhang M, Hu P, Napoli JL. Elements in the N-terminal signaling sequence that determine cytosolic topology of short-chain dehydrogenases/reductases. Studies with retinol dehydrogenase type 1 and cis-retinol/androgen dehydrogenase type 1. J Biol Chem. 2004;279:51482–51489. doi: 10.1074/jbc.M409051200. [DOI] [PubMed] [Google Scholar]
- 289.Zhang M, Hu P, Krois CR, Kane MA, Napoli JL. Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse. FASEB J Off Publ Fed Am Soc Exp Biol. 2007;21:2886–2896. doi: 10.1096/fj.06-7964com. [DOI] [PubMed] [Google Scholar]
- 290.Zizola CF, Frey SK, Jitngarmkusol S, Kadereit B, Yan N, Vogel S. Cellular retinol-binding protein type I (CRBP-I) regulates adipogenesis. Mol Cell Biol. 2010;30:3412–3420. doi: 10.1128/MCB.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]