

Abstract
Ischemia-reperfusion brain injury can be iatrogenically-induced secondary to life-saving procedures. Prophylactic treatment of these patients offers a promising prevention for life-long complications. We postulate that a CpG oligodeoxynucleotide (ODN) can provide robust antecedent protection against cerebral ischemic injury with minimal release of pro-inflammatory cytokines, making it an ideal candidate for further clinical development. Mouse and nonhuman primate (NHP) models of cerebral ischemic injury were used to test whether an A-type CpG ODN, which induces minimal systemic inflammatory cytokine responses, can provide prophylactic protection. Extent of injury in the mouse was measured by histological staining of live tissue. In the NHP, injury was assessed 2 and 7 days post-occlusion from T2-weighted magnetic resonance images and neurological and motor deficits were cataloged daily. Plasma cytokine levels were measured using species specific Luminex assays. Prophylactic administration of an A-type CpG ODN provided robust protection against cerebral ischemic injury in the mouse with minimal systemic inflammation. Rhesus macaques treated with D192935, a mixture of human optimized A-type CpG ODNs, had smaller infarcts and demonstrated significantly less neurological and motor deficits following ischemic injury. Our findings demonstrate the translational potential of D19293 5 as a prophylactic treatment for patients at risk of cerebral ischemic injury.
Keywords: CpG ODN, ischemia, preconditioning, neuroprotection, nonhuman primate, stroke
Introduction
Cerebral ischemic injury is the fifth leading cause of death and the leading cause of serious, long-term disability in the United States [1]. Ischemia-reperfusion injury is often iatrogenically-induced secondary to life-saving endovascular or cardiac procedures. Recent advances in neuroimaging indicate that these peri-procedural brain lesions (silent infarcts) are more frequent than originally believed [2-5]. While these procedures may be essential, the long-term implications of these silent infarcts remain unclear [6, 7]. Given that stroke prevention is a primary goal of these procedures, cerebral embolization, whether clinical or subclinical, should be considered a complication worth addressing. For these patients, antecedent therapy could be extremely beneficial.
To combat predictable ischemic injury, the use of “delayed preconditioning” has been proposed, whereby stimulation with low doses of an otherwise harmful insult serves to protect the brain, kidney, or heart from subsequent injurious ischemia [8]. Transient remote ischemic preconditioning using a tourniquet approach in the limbs has emerged as a promising therapeutic modality to protect from ischemia [9-11], although the repeated and daily treatment regimens are quite cumbersome to patients. Pharmacological preconditioning, however, may offer greater translational potential as dosages are easily controlled. We, and others, have demonstrated that pharmacological preconditioning via Toll-like receptor 9 (TLR9) holds great promise for use in prophylactic protection of the brain [12-14] as well as other organs against ischemic injury [15, 16].
TLR9 can be activated with synthetic DNA oligonucleotides that contain unmethylated cytosine-guanine rich regions (CpG ODNs) in a sequence dependent manner. Interestingly, distinct CpG ODN motifs result in divergent immune effects. CpG ODNs with completely phosphorothioate-modified backbones (known as B-class or K-type) are potent inducers of TH1-type cytokines (TNF, IFNγ, IL12) and strongly induce B cell proliferation [17]. On the other hand, CpG ODNs of the A-class (also known as D-type), which contain a central palindromic phosphodiester CpG sequence and a phosphorothioate-modified 3′ poly-G tail, require higher concentrations to induce cytokine secretion and B cell proliferation and they induce higher levels of natural killer cell activation than B-class. Ballas and colleagues also noted that different classes of CpG ODNs had different therapeutic effects on tumor growth, thus the distinct immune modulation of CpG classes alter their therapeutic effect [17].
TLR9 mediated protection against ischemic injury, to date, has only been demonstrated with B-class CpGs [12-16], and although the protection is robust, increased systemic cytokine levels may restrict their use as clinical therapeutics. The A-class CpGs, with their minimal induction of systemic cytokines, may provide a safer alternative for prophylactic protection if, despite their distinct immune modulation, they protect against ischemic injury. Thus, we hypothesize that A-class CpGs can provide robust protection against cerebral ischemic injury with minimal release of pro-inflammatory cytokines, making them ideal for clinical development.
To determine whether A-class CpG ODNs can protect against cerebral ischemic injury, we used the mouse model of middle cerebral artery occlusion (MCAO) to demonstrate efficacy. We show for the first time that A-class CpG ODNs protect against cerebral ischemic injury in the mouse at doses that lack measurable increases in systemic cytokines, contrasting with protection induced by B-class CpG ODNs. Once efficacy was established, we tested an A-class CpG ODN in a nonhuman primate (NHP) model of cerebral ischemic injury, following the recommendations of the Stroke Therapeutics Academic Industry Roundtable (STAIR) [18]. We demonstrate significant protection in the male rhesus macaque with sustained reductions in infarct volume, reduced neurological deficits and improved physical activity with a mixture of three A-class CpGs (D192935) administered prior to surgically induced stroke. These studies demonstrate efficacy in two species for this class of compound, consistent with the STAIR recommendations, and are a valuable precursor to advance these compounds toward human clinical trials [18].
Materials and Methods
Animals
Mice
C57B1/6J mice (males, 8-12w; Jackson Laboratories, West Sacramento, CA) were housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) international-approved facility. The animal protocols met National Institutes of Health guidelines with the approval of Oregon Health and Science University (OHSU) Institutional Animal Care and Use Committee (IACUC).
Nonhuman primates
Adult male rhesus macaques (Macaca mulatta, Chinese origin; n=60), with an average age of 7.49 ± 0.2 years and average body weight of 7.88 ± 0.3 kg, were cared for by the Division of Comparative Medicine at the Oregon National Primate Research Center (ONPRC) in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals. The animal care program is compliant with federal and local regulations regarding the care and use of research animals and is AAALAC accredited. All procedures were approved by the OHSU IACUC.
NHP Housing
Animals were housed indoors under controlled conditions at a constant temperature of 24 ± 2°C, with 12L:12D photoperiods (∼300 lux) starting at 0700 h and ending at 1900 h daily. Animals were provided enrichments between 1300 h and 1400 h. Animals received fresh drinking water ad libitum and regular meals at 0830 h and 1500 h (Purina High Protein Monkey Chow, Purina Mills, Inc., St. Louis, MO) supplemented with juice, fresh fruit, vegetables and candy treats prior to stroke. Animals received a variety pan of food post-stroke to stimulate appetite and food treats during neurological assessments. Room washing was performed daily between 1000 h and 1100 h, except for the days following surgically-induced stroke.
NHP Selection and Acclimation
Based on availability of equipment and surgical staff, studies were performed on cohorts of 5-8 animals at a time, with 1-2 months separating each cohort. Approximately 3 weeks prior to stroke surgery, rhesus macaques were examined for general health by the attending veterinarian and then moved to study cages for acclimation. At this time an Actiwatch activity monitor (Philips-Respironics, Bend, OR, USA) was attached to each animal's collar, as previously described [19-21], and a blood sample collected to establish prescreen hematology levels. Animals were selected based on normal physical status and clinical laboratory findings. Exclusion criteria included: 1) abnormal hematology and/or clinical chemistry >2 standard deviations from mean historical ranges in adult male Chinese rhesus macaques (data from over 100 healthy individual age/sex-matched controls), 2) recent invasive procedures (e.g., dental cleaning or tooth extraction), 3) presence of serum cytokines or other indicators of inflammation or infection (e.g., tooth abscess, high c-reactive protein levels, visible injury, chronic diarrhea), 4) neurological disorders evident by abnormal motor or cognitive abilities, or 5) demonstration of chronic stress-associated behaviors.
Reagents
ODN1826 (mouse specific B class CpG; TCCATGACGTTCCTGACGTT) and ODN1585 (mouse specific A class CpG; GGggtcaacgttgaGGGGGG) were from InvivoGen (San Diego, CA, USA). D19293 5 CpG ODN, a 1:1:1 mixture of three human optimized phosphorothioate A class (also known as D-type) ODNs (D19: GGtgcatcgatgcaggGGGG; D29:GGtgcaccggtgcaggGGGG; D35:GGtgcatcgatgcaggggGG) was manufactured by Oligos Etc. (Wilsonville, OR, USA). All CpG compounds were diluted in saline. Endotoxin levels were determined to be negligible (<0.125 EU/mg). Nucleotides shown in CAPS are phosphorothioate and those in lowercase are phosphodiester modified.
Drug Treatments
Animals are preconditioned with the appropriate CpG compound 3 days (72 h) prior to surgical occlusion, since this is the time point we have demonstrated to be most efficacious in the mouse for ODN1826 [14] and have shown that a B-type CpG ODN protected the NHP at this time point [12]. Mice: Mice were injected subcutaneously with 100-μL of ODN1826, ODN1585 or saline. For MCAO studies, drug syringes were prepared daily and randomized for order of injection prior to random retrieval of mice from cage. Injections were 3 days prior to surgery.
NHP
A 1-mL fixed dose of D192935 or saline was injected intramuscularly before middle cerebral artery and anterior cerebral artery occlusion (MACAO). For efficacy studies, randomized animals received 0.15 mg/kg (low dose), 0.3 mg/kg (mid dose), 0.6 mg/kg (high dose) or saline 3 days before MACAO. The animals were randomized for drug treatment within a surgical cohort of 5-8 animals by drawing a number from a cup. Each cohort included at least one vehicle treated animal. Surgical staff and researchers were blinded to treatment throughout the study.
Surgical Protocols
Mice
MCAO
Focal cerebral ischemia was induced by MCAO as described previously [14]. In brief, MCAO was performed in anesthetized mice (1.5-2.0% isoflurane) by threading a 7-0 silicone-coated nylon surgical filament (Doccol, Redlands CA) through the external right carotid artery to the internal carotid artery, blocking blood flow at the bifurcation into the MCA and anterior cerebral artery. Following 60 minutes of occlusion, the monofilament was removed and blood flow restored. Cerebral blood flow was monitored throughout the procedure by laser Doppler flowmetry (Transonic System Inc., Ithaca NY). Animals were excluded if blood flow did not maintain a >80% drop from baseline levels during occlusion or if reperfusion levels following occlusion did not return to >50% of baseline. Our lab has historically used these parameters as the hallmark of a successful occlusion and reperfusion. Only one animal was excluded based on these criteria, an animal in the vehicle group, failed to maintain an 80% drop in blood flow for the entire occlusion period. An additional animal was dropped from the ODN1585 group due to an intestinal blockage discovered following euthanasia. MCAO surgery was performed on 35 mice with 27 mice surviving to the 24-hour endpoint (23% mortality rate).
Evaluation of Infarct Size
Twenty-four hours following MCAO, mice were deeply anesthetized with isoflurane and perfused with ice-cold saline containing 2 U/ml sodium heparin. Brains were removed, sectioned and infarct visualized with 2,3,5-triphenyltetrazolium chloride (TTC; Sigma Aldrich, St. Louis MO) as previously described [14]. Sections were imaged and infarct volume measured by an investigator blinded to the experimental conditions using ImageJ software (NIH Image, Bethesda MD). Infarct volume was calculated from slices 2-7 using the indirect method [(contralateral live – ipsilateral live) / contralateral live * 100] to account for edema.
NHP
Two-vessel Occlusion Protocol
The right middle cerebral artery (distal to the orbitofrontal branch) and both anterior cerebral arteries were exposed and occluded with vascular clips for 60 minutes, as previously described [22]. Surgical procedures were conducted by a single surgeon. Briefly, anesthesia was induced with ketamine (∼10 mg/kg, intramuscular injection) and animals were then intubated and maintained under general anesthesia using 0.8% to 1.3% isoflurane vaporized in 100% oxygen. A blood sample was taken and a venous line placed for fluid replacement. An arterial line was established for blood pressure monitoring throughout surgery and to maintain a mean arterial blood pressure of 60-80 mm Hg. End-tidal CO2 and arterial blood gases were continuously monitored to titrate ventilation to achieve a goal PaCO2 of 35- 40 mm Hg. Post-operative analgesia consisted of intramuscular hydromorphone hydrochloride and buprenorphine.
Study Groups, Timeline and Analysis strategy
Three days prior to stroke surgery, animals were randomized by number drawing from a cup and intramuscularly injected in the lateral aspect of the thigh with D192935 (0.15 mg/kg, 0.3 mg/kg, or 0.6 mg/kg) or vehicle (saline). Thirteen animals received 0.15 mg/kg, seventeen received 0.3 mg/kg, and seven received 0.6 mg/kg. The vehicle control animals numbered twenty-three in total. After stroke surgery, all animals were recovered in their home cages (48 h) prior to processing for magnetic resonance imaging (MRI) to assess the severity of damage. This is our primary readout of induced damage. Animals were scanned again 7 days after stroke surgery to access the progression of the damage (Fig. 1). Data were analyzed using both an intention-to-treat (ITT) and a modified ITT (mITT) model. In the ITT analyses all NHPs are included, with the exception of one animal that died from an accidental overdose during surgery, unrelated to study treatments. In animals that did not survive to the primary MRI (48 h), a max infarct volume was assigned equivalent to the largest infarct measured at 48 h in the study (11.21%). The mITT analysis excluded two animals that did not survive to the 48-h time point as well as two additional animals described below. Of the 60 animals, 59 survived the surgery, 57 survived to the 48-h time point (5% mortality) and 47 survived to complete the study at 7 days (22% mortality).
Figure 1. Nonhuman primate study design.
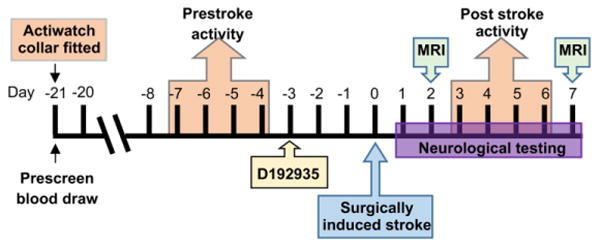
NHPs were anesthetized, fitted with an Actiwatch activity monitor and baseline blood drawn 21 days prior to surgically induced stroke. For activity analysis, measurements from day -7 to day -4 were used as the pre-stroke activity value and values from day 3 to day 6 were used as post stroke activity. NHPs were injected with D192935 3 days prior to surgery induced stroke. On the day of surgery (day 0) animals were anesthetized and blood drawn immediately prior to surgery. Following surgery neurological testing was performed on day 1 to day 7 and animals were anesthetized for MRIs on day 2 and day 7.
mITT Study Exclusions
A total of 59 NHPs survived the MACAO surgery and are included in the ITT analysis. One animal from the 0.15 mg/kg group died during surgery prior to occlusion from an accidental overdose of anesthesia, this animal is not included in either analysis. For the mITT analysis, a total of four animals were excluded. Two animals were euthanized within 24 h of surgery due to severe pathologies, one from the 0.3-mg/kg group and one from the vehicle-treated group. Two additional animals were excluded from the vehicle group, one due to a suspected pituitary tumor identified at necropsy and one which had a white blood cell count 6 standard deviations above the group mean at the time of surgery (data not shown).
Infarct measurements
Infarct volume was measured from T2-weighted MRI performed after 2 and 7 days of reperfusion [12]. We have previously published that histologic analyses performed 2 days post-stroke correlates with T2 infarct measurements [12]. All scans were performed on a Siemen's 3T Trio system, housed on-campus near the surgical suite at ONPRC. The protocol included a turbo spin-echo T2-weighted scan, with TR=5280 ms, TE=57 ms, number of averages=4, an echo train length of 5, and a refocusing pulse flip angle of 120°. The entire brain was imaged with a 0.5 × 0.5 mm in-plane resolution and a slice thickness of 1 mm. Infarct volume was determined via a semi-automated workflow that incorporated the magnetization-prepared rapid acquisition gradient echo (MPRAGE), fluid attenuation inversion recovery (FLAIR), spin echo multi contrast (SE-MC) and T2-weighted turbo spin echo (T2TSE) sequences. In brief, the FMRIB Software Library (FSL) Brain Extraction Tool (BET) was used to separate the brain in the MPRAGE, FLAIR, SE-MC and T2TSE images from surrounding skull. Extracted brain images were cleaned up using a “hand-drawn” region of interest (ROI) mask and co-registering the extracted brain and ROI masks to the brain extracted MPRAGE using the FSL Linear Registration Tool (FLIRT). Finally, “approximate” lesion ROIs were calculated by thresholding the FLAIR brain images. Visual inspection of automated ROIs was performed by a researcher blinded to treatment. Infarct volume was calculated as the percent of total brain by taking the ROI lesion volume divided by the extracted brain volume. The infarct measurement for one animal in the 0.6mg/kg group was not determined due to a technical problem in the MRI processing. All other data for this animal was available and included in the study.
Neurological assessment
Neurological assessments were performed daily between 0700 h and 0830 h by a single observer as previously described [12]. In brief, our scale evaluates motor function and behavior (mental status) with higher scores representing better functional outcomes (100=normal). Motor function is scored from 1 to 70, according to severity of hemiparesis in the left extremities. A score of 10=severe hemiparesis, 25=moderate hemiparesis, 40= slight hemiparesis, 55=favors normal side, or 70=normal ability. Behavior and alertness are scored ranging from 1 to 20, with l=unresponsive, 5=aware but inactive, 15=aware but less active, and 20=normal. Facial deficit was scored as l=one-sided paralysis or 5=normal facial movement. Visual deficit was scored as l=present and 5=absent. As analgesics were found to have minimal effect on our provoked assessments, scores are reported for days 1–7 post-stroke. For animals euthanized prior to study end, the last daily value was carried forward. For the ITT analysis, a value of 8, equivalent to the lowest measured value of an animal with severe pathologies, was used for a 0.3 mg/kg treated animal euthanized prior to neurological testing on day 1.
Physical activity measurement
As previously described [19-21], physical activity was monitored using an Actiwatch 64 monitor (Philips-Respironics), which comprises a piezoelectric accelerometer and 64 kilobytes of storage memory. The Actiwatch records the integration of intensity, amount, and duration of movement in all directions with a force sensitivity of 0.05 g and a maximum sampling frequency of 32 Hz. Each animal is fitted with an Actiwatch placed inside a protective case (Philips-Respironics) and then attached to a lightweight loose-fitting aluminum collar (Primate Products, Inc., Immokalee, FL) approximately 3 weeks prior to surgery, as described previously [20]. Continuous recordings were made prior to surgery and up to 7 days after surgery. Devices were programmed to collect data in 60-second epochs and data were downloaded using a dedicated reader (Philips-Respironics). Data were interpreted and actigrams drawn using Actiware-Sleep version 3.4 software (Cambridge Neurotechnology Ltd, Cambridge, United Kingdom). The mean daytime activity (defined as activity during the period between 0700 h and 1859 h) was calculated. The mean daily baseline activity for each individual animal was calculated as the 4 consecutive days just prior to drug administration on Day-3 (Fig. 1; Individual mean baseline activity = (ActivityDay-7 + ActivityDay-6 + ActivityDay-5 + ActivityDay-4)/4). Following stroke, we found that analgesics administered post-surgery affected activity on Days 1 and 2. Therefore, data analysis of activity post-stroke included activity measurements beginning on Day 3 post-surgery continuing to Day 6. The percent change from mean baseline value was calculated for each of the four days using the following equation: % change = (post-stroke activity (Day x) / mean baseline activity)* 100. For animals euthanized prior to study end, the last measured daily value was carried forward. Animals euthanized prior to a full day of post-stroke data were assigned a value of 97.4%, a value equivalent to the most severe pathology in the study. There was no data collected for one animal in the 0.3 mg/kg group because of a device malfunction, this animal was not included in the activity analyses.
Systemic cytokine response
Mouse plasma cytokine levels
Blood was collected via cardiac puncture under isoflurane anesthesia. Plasma cytokine levels were evaluated using a mouse specific cytokine 20-plex Luminex assay (Life Technologies) and a mouse ProcartaPlex IFNα/β panel (eBioscience).
NHP plasma cytokine levels
Blood was collected from anesthetized animals just prior to and 0.5, 1, 3, and 5 h post-intramuscular injection of D192935 (0.3 mg/kg). Plasma cytokine levels were evaluated using a primate specific cytokine 29-plex Luminex assay (Life Technologies).
Statistical analyses
All statistical analyses were performed using Prism (Graphpad Software, La Jolla, CA). Group means were compared using a one-way ANOVA with Dunnett's multiple comparison post-hoc test unless otherwise noted in the figure legend. Longitudinal group means were compared using a two-way repeated measures ANOVA for treatment and time with Dunnett's post-hoc tests. Data represent mean ± standard error of the mean (SEM). Differences were considered statistically significant when p<0.05. Group sizes for all experiments are based on power analysis of previous experiments using these ischemia models. Sample size was selected to control type I error at 0.05 (alpha error) and achieve a power ∼80% (treatment effect size of >25%). In the NHP studies larger numbers of vehicle treated animals are included to take into account the complexity of performing these studies over an extended period of time, since only a small number of animals can be processed in a single cohort. We included a minimum of one vehicle-treated animal in each cohort, to monitor for any drift in surgical performance. In addition, the small number of animals in the 0.6 mg/kg group reflects a cessation of the study group when no improvement in outcome measures was evident. Study performance was monitored by a non-blinded researcher not involved in the study.
Results
Prophylactic administration of an A-type CpG protects mice against cerebral ischemic injury
We have previously shown that B-type CpGs provide significant protection in both a mouse and NHP model of stroke when administered 3 days prior to experimentally induced transient cerebral ischemic injury [12, 14]. To determine whether an A-type CpG could also provide protection we compared CpG ODN1585, an A-type CpG, to the B-type CpG, ODN1826 that we had previously shown to be protective at a dose of 0.8mg/kg [14]. Mice were preconditioned with ODN1826 or ODN1585 (0.8mg/kg) 3 days prior to MCAO. As expected, preconditioning with ODN1826 resulted in a significantly smaller infarct volume (23.50 +/- 5.4%) compared to vehicle treated animals (37.54 +/- 9.4%), a 37% decrease (Fig. 2). ODN1585, when given at an equivalent dose, demonstrated a similar reduction in injury with a nearly 30% decrease in infarct size (26.43+/- 3.4%). These data indicate that ODN1585, an A-class CpG, provides robust protection against cerebral ischemic injury in the mouse. We next assessed the differences in systemic inflammatory cytokine induction. Mice were given either ODN1826 (0.8 mg/kg) or ODN1585 at two different doses (0.8 or 1.6 mg/kg) and plasma cytokine levels measured at 3 h post-dose. We examined the efficacious dose (0.8mg/kg) of ODN1585 and a dose that was 2-fold higher (1.6mg/kg) to test our hypothesis that ODN1585 induces a significantly lower inflammatory response than ODN1826. As expected, ODN1826 significantly increased plasma levels of TNF as well as IL1β, IL10, IL6, IL12, MCP1, IL5, IL2, IP10, MIG and KC when compared to saline controls (Fig. 3). In contrast, ODN1585 treatment induced no increase in any of the cytokines tested even at a dose twice that of ODN1826 (Fig. 3). In addition, no increase in cytokine levels were seen for ODN1585 (0.8 mg/kg) at later times (6 or 24 h) post injection (data not shown) and no increase in IFNα or β was detected with either type of CpG ODN at the concentrations or times tested (data not shown). These data suggest that a substantial increase in inflammatory cytokines, which can be detrimental in certain situations, is not required for protection and that A-class CpGs may provide a safer route to clinical translation for protection of patients against ischemic injury.
Figure 2. Prophylactic treatment of mice with ODN1585 provides significant protection against MCAO.
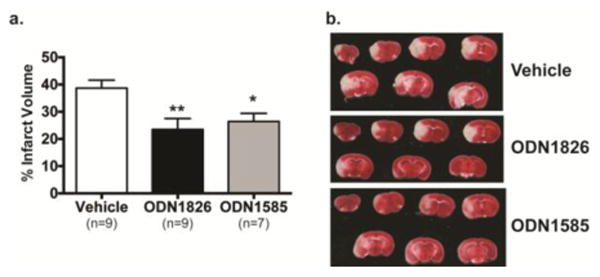
Mice were administered vehicle, ODN1826 or ODN1585 (0.8 mg/kg) 3 days prior to MCAO (60 min). Infarct volume was determined 24 h following MCAO by TTC staining, a) Values graphed are group means ± SEM; *p<0.05, **p<0.01 versus vehicle by one-way ANOVA followed by Dunnett's multiple comparison test, b) Representative images of TTC stained slices 2-8.
Figure 3. ODN1585 induces a minimal systemic inflammatory response as compared to ODN1826.

Mice (n=4/treatment group) were subcutaneously administered either saline, ODN1826 (0.8 mg/kg) or ODN1585 (0.8 or 1.6 mg/kg) and blood collected 3 h after injection. Plasma levels of cytokines were measured with a mouse specific cytokine 20-plex Luminex assay. Values are group means ± SEM; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way ANOVA followed by Tukey's multiple comparison test.
Prophylactic administration of a mixture of human specific A-type CpGs, significantly reduces cortical damage in NHPs
To determine whether A-type CpGs would induce neuroprotection in a NHP model of stroke injury, D192935 (a mixture of three human optimized A-type CpGs) or vehicle was administered to male NHPs 3 days before experimental stroke (Fig. 1). Three different doses (0.15, 0.3 and 0.6 mg/kg) were examined and all three doses were well tolerated by the rhesus macaque with no apparent toxicities. Vital physiological measures were record just prior to, during and immediately following occlusion (Table 1). A significant drop in systolic blood pressure was detected just prior to occlusion in the 0.15 mg/kg group compared to vehicle treated animals. The difference was not sustained, with readings during and following occlusion comparable. No other significant difference between groups was detected. Extent of injury was assessed by T2-weighted MR imaging 48 h after reperfusion (Fig. 4a). We found that systemic administration of D192935 significantly reduced cortical ischemic volumetric damage by ∼50% at both 0.15 mg/kg (3.76 +/- 0.9% total brain) and 0.3 mg/kg (3.94 +/-0.7%) compared to vehicle treated animals in our mITT analysis (6.71 +/- 0.7%; Fig. 4b). The higher dose (0.6 mg/kg) did not provide protection (8.18 +/- 0.9%; Fig. 4b), however, this is not unexpected because mechanisms of preconditioning-induced protection are generally associated with low dose treatments and protection is lost at higher doses [8]. When all data was included in our ITT analysis, there was a significant overall treatment effect by one-way ANOVA (p=0.0175), however no significant difference in individual treatments vs vehicle by Dunnet's multiple comparison test was evident (Fig. 4c). We also examined the progression of the infarct injury between groups from day 2 to day 7. Only animals that had data at both time points were included. Importantly a significant reduction in infarct was still evident out to 7 days post stroke with both the 0.15 and 0.3 mg/kg doses compared to vehicle treated animals (Fig. 4d), demonstrating that protection was long-lasting and not just a delay in injury progression.
Table 1. Vital Physiological measurements during surgical induction of ischemia.
| Heart Rate (beats/min) | Temperature (°C) | SpO2 | EtCO2 (mm Hg) | Systolic Blood Pressure (mm Hg) | Diastolic Blood Pressure (mm Hg) | |
|---|---|---|---|---|---|---|
| PRIOR TO OCCLUSION | ||||||
| Vehicle | 116.9 + /- 2.8 | 37.4 + /-0.1 | 97.7 + /- 0.5 | 37.6 + /- 0.6 | 73.2 + /-3.6 | 37.7 + /-2.0 |
| 0.15mg/kg D192935 | 123.8 + /- 6.3 | 37.2 + /-0.1 | 98.3 + /- 0.4 | 35.9 + /-1.1 | 57.5 + /- 3.4** | 31.5 + /-1.7 |
| 0.3mg/kg D192935 | 117.3 + /- 3.3 | 37.3 + /-0.1 | 98.1 + /-0.5 | 35.7 + /-0.9 | 70.2 + /- 2.0 | 35.4 + /-1.4 |
| 0.6mg/kg D192935 | 116.5 + /- 4.3 | 37.5 +/- 0.2 | 97.8 + /- 0.5 | 36.7 + /- 1.1 | 69.5 + /- 5.5 | 33.2 + /- 4.0 |
| DURING OCCLUSION | ||||||
| Vehicle | 120.4 + /- 2.5 | 37.9 + /- 0.1 | 98.6 + /- 0.4 | 38.0 +/- 0.6 | 79.0 + /- 2.6 | 40.6 + /- 1.2 |
| 0.15mg/kg D192935 | 125.4 + /- 3.9 | 37.9 + /- 0.07 | 98.6 +/- 0.3 | 37.0 +/- 0.8 | 72.0 + /- 2.8 | 37.5 + /- 1.8 |
| 0.3mg/kg D192935 | 122.2 + /- 3.1 | 37.7 + /- 0.1 | 98.4 +/- 0.5 | 36.1 +/- 0.8 | 80.4 +/- 2.4 | 42.5 + /- 2.0 |
| 0.6mg/kg D192935 | 124.2 + /- 4.4 | 38.0 + /- 0.1 | 97.8 +/- 0.7 | 38.7 +/- 0.9 | 77.0 +/- 3.8 | 35.7 +/- 2.4 |
| POST OCCLUSION | ||||||
| Vehicle | 119.9 + /- 2.7 | 38.0 + /-0 .1 | 98.7 + /- 0.3 | 41.8 + /- 1.5 | 85.7 + /- 3.6 | 43.4 + /- 1.9 |
| 0.15mg/kg D192935 | 125.5 + /- 4.0 | 38.0 + /- 0.07 | 97.8 + /- 0.4 | 41.9 + /- 1.6 | 74.4 + /- 2.5 | 38.1 + /- 1.9 |
| 0.3mg/kg D192935 | 118.3 +/- 3.4 | 37.8 + /- 0.1 | 98.4 + /- 0.4 | 39.8 + /- 1.4 | 81.1 + /- 3.0 | 42.3 + /- 2.0 |
| 0.6mg/kg D192935 | 122.4 +/- 5.7 | 37.9 + /- 0.1 | 98.0 + /- 0.6 | 41.8 + /- 2.4 | 78.8 + /- 5.2 | 37.0 + /- 3.5 |
Group means ± SEM. Repeated measures two-way analysis of variance (time, treatment);
p<0.01 vs Vehicle by Tukey's post-hoc analysis.
Figure 4. Reduction in injury following MCA and ACA occlusion in NHPs preconditioned with D192935.

D192935 or vehicle was administered via intramuscular injection to male NHPs 3 days prior to the onset of surgical occlusion. T2-weighted MR imaging was performed on 2 and 7 days following reperfusion. a) Representative images of day 2 T2-MRI scans, b) mITT subpopulation and c) ITT analysis of infarct volumes measured as percent of whole brain on day 2. Values denote group means ± SEM; *p<0.05, **p<0.01 versus vehicle by one-way ANOVA followed by Dunnett's multiple comparison test, d) Infarct progression between day 2 and 7 of treatment groups excluding animals that were dropped from study prior to day 7 MRI. **p<0.01, ***p<0.001 versus vehicle at day 7 by two-way repeated measures ANOVA followed by Dunnett's multiple comparison test.
D192935 significantly reduces stroke induced neurological deficits in NHPs
Direct and indirect interactions were performed daily in order to elicit specific motor functions for evaluation of neurological function post stroke as previously described [12]. In our mITT analysis, daily neurological scores showed groups receiving 0.15 or 0.3 mg/kg of D192935 had significantly higher scores than vehicle on days 1-5 and on days 1-7, respectively (Fig. 5a). In addition, the scores within treatment groups did not vary significantly by day (Fig. 5a). When all data was included in the ITT analysis, groups receiving 0.3 mg/kg were still higher than vehicle at days 4, 5 and 7 but the 0.15 mg/kg group differences did not persist (Fig. 5b). These data demonstrate that preconditioning with D19293 5 protects against neurological deficits induced by stroke.
Figure 5. Improvement in post-occlusion neurological and activity measurements in NHPs preconditioned with D192935.

Neurological deficits were evaluated for the 7 days following surgical occlusion, a) mITT subpopulation and b) ITT data are graphed as the group mean ± SEM for individual days with 100 being the equivalent of no deficit. *veh vs 0.15 mg/kg p<0.05, **veh vs 0.15mg/kg p<0.01, +veh vs 0.3 mg/kg p<0.05, ++veh vs. 0.3 mg/kg p<0.01, +++veh vs 0.3 mg/kg p<0.0001 by two-way repeated measures ANOVA followed by Dunnett's multiple comparison test. c,d) Physical activity was monitored daily both prior to and following surgical occlusion using accelerometry collars. Pre-stroke activity levels from d-7 to d-4 were used to derive a mean baseline activity level for each animal. Following stroke, activity levels from d3 to d6 of the study were used to derive individual post-stroke mean activity levels. mITT subpopulation (c) and ITT (d) data are presented as group means ± SEM of the percent in reduction in daytime activity following stroke compared to pre-stroke values for individual animals. *veh vs 0.15 mg/kg p<0.05, **veh vs 0.15 mg/kg p<0.01, +veh vs 0.3 mg/kg p<0.05, ++veh vs. 0.3 mg/kg p<0.01 by two-way repeated measures ANOVA followed by Dunnett's multiple comparison test.
D192935 results in significant improvement of physical activity deficits associated with stroke in NHPs
We have previously shown that monitoring of physical activity using the Actiwatch device provides an objective measure to quantify changes in spontaneous motor activity in the rhesus macaque stroke model, with changes in post stroke activity correlating with infarct size and neurological assessment [21]. Animals were acclimatized to the Actiwatch collars and baseline measurements recorded prior to surgery. Following stroke, the mITT analysis of daily activity scores showed groups receiving 0.15 or 0.3 mg/kg had significant reductions in scores on days 5 and 6 (49.36+/-9.00%, 50.45+/-7.93% vs. 77.46+/-5.26% vehicle on day 5, and 42.63+/-12.38%, 45.30+/-9.10% vs. 78.05+/-4.15% vehicle on day 6, respectively), in addition the group receiving 0.15 mg/kg had significant reductions in scores on day 3 (55.55+/-10.41% vs. 80.25+/-3.50%), while the other differences were smaller and not significant on day 3 and 4 (Fig. 5c). When all data was included in the ITT analysis, only the differences on day 6 remained (Fig. 5d). These results further demonstrate the efficacy of D192935 to significantly reduce brain injury and physical deficits associated with stroke.
Systemic plasma cytokine induction in D192935 treated NHPs
To assess the systemic inflammatory response to D192935, three primates were assessed pre- and post-injection with 0.3 mg/kg D192935. Blood samples taken just prior to injection and then 0.5, 1, 3 and 5 h post drug administration were analyzed. This subset of animals was necessary to measure cytokines at relevant time points since the study constraints on the surgically induced stroke group did not allow for bleeding of animals at these early time points. Examination of cytokines assessed in the mouse studies revealed that the primate response was much more heterogeneous, with baseline values differing between animals. One NHP showed little to no induction of inflammatory cytokines (NHP#3; Fig. 6), consistent with our mouse responses to the A-type CpG, in which no systemic cytokine induction was observed. A minor inflammatory response was seen in NHP#2 in which D192935 induced IL6 with a slight increase in MCP1 (Fig. 6). In contrast NHP #1 had measurable induction of IL6, IL12, TNF, IL2, and MIG (Fig. 6). We were unable to perform correlation analysis on systemic cytokine responses and D192935 induction of neuroprotection in these animals because they were not included in our stroke group. Although the small n makes it impossible to draw conclusions from this data set, the disparity of responses between the three animals does suggest that systemic cytokines may not be essential for the induction of protection in the NHP. Further studies would be needed to confirm that protection is not dependent on a systemic inflammatory response in the NHP.
Figure 6. Heterogeneity in the systemic cytokine response to D192935 in individual NHPs.

Blood was collected from three individual NHPs immediately prior to intramuscular injection of D192935 (0.3 mg/kg) and following injection at 0.5, 1, 3 and 5 h. Plasma levels of cytokines were measured with a primate specific cytokine 29-plex Luminex assay. Data represents the individual animal responses to D192935.
Discussion
The neuroprotective potential of B-type CpG ODNs and other TLR ligands as prophylactic treatments against cerebral ischemic injury has been well documented in rodents [23]. Furthermore, B-type CpGs have been shown by our group to provide robust protection in a NHP model of stroke [12]. Here we assessed whether A-type CpGs, which induce a low inflammatory systemic response, also induce protection. Our studies demonstrated that a mouse specific A-type CpG administered prior to MCAO significantly reduced ischemic injury at a dose that resulted in no induction of inflammatory cytokines. The protection was similar to that observed with a B-type CpG, which, however, induced significant increases in plasma levels of multiple proinflammatory cytokines. We furthered the translational importance of this promising prophylactic neuroprotective strategy by demonstrating that an A-type CpG also provides significant protection against cerebral ischemic injury in the NHP, which is a more relevant model of ischemic brain injury for therapeutic development.
Although immune activators have demonstrated success as prophylactic treatments for reduction of brain injury in experimental models of cerebral ischemia [13, 14, 24-28], their modulation of inflammation has dampened enthusiasm for clinical use. Systemic infection is often associated with poorer outcomes in stroke patients [29, 30]. Although most of the clinical studies refer to post-stroke infection, some studies report that infection in the month preceding ischemic stroke is a significant risk factor for cerebral infarction [29]. Retrospective studies of ischemic stroke patients indicate the prevalence of infection in the month preceding stroke ranges from 18-40%, with the week preceding stroke ranging from 10-35% [29]. In addition, patients undergoing cardiovascular procedures often times have comorbidities such as diabetes that can be aggravated by activation of inflammation [31]. Therefore, it is important to understand the systemic responses engaged following prophylactic treatment of patients with immune activators, so that potential negative consequences can be mitigated. Our finding that A-type CpGs provide protection against cerebral ischemia without an increase in systemic inflammation not only identifies a potentially safer therapeutic agent, but demonstrates that increases in systemic inflammatory cytokines are not required for prophylactic neuroprotection.
The demonstration that a mixture of A-type CpGs provides robust protection in a relevant NHP model of stroke indicates that these compounds are efficacious not only in rodents, but in higher order species as well. Prophylactic treatment with D192935 not only provided acute neuroprotection at 48 h by MRI, but infarct reduction was also observed through 7 days, consistent with significant attenuation of behavioral deficits in D192935-treated animals. As the neurological assessment is subjective, we also used accelerometry technology to monitor movement, a technique we used previously in monkeys [21] and that is also used in stroke patients to assess movement disorders [32]. This quantitative measure indicated that doses of D192935 that reduced infarct size also significantly improved post-stroke activity. These studies indicate that the accelerometer represents an important non-invasive means to quantitatively measure the effects of stroke and provide unbiased behavioral information for drug development.
Due to study constraints and the scarcity of NHPs as a resource, examination of inflammatory cytokine responses to D192935 was not possible in the stroke study group, and a separate small cohort of NHPs was used. As expected in a heterogeneous population such as NHPs, cytokine levels, even at baseline, were variable between individual animals. Responses to D192935 were also variable, with one animal mounting an inflammatory response, while the other two showed little to no induction of inflammatory cytokines. Unfortunately, it is hard to draw conclusions from such a small n, however, given the fact that we saw a significant reduction in injury in the primates as a group, the heterogeneity of the response in these animals suggests that detection of systemic inflammatory cytokines may not correlate with efficacy, because two of the three animals had minimal to no increases in any cytokines. Further pharmacokinetic studies of D192935 in a larger cohort of NHPs would greatly enhance these studies, however, due to the cost and limited access to these animals these studies were not possible. However, our findings are consistent with the mouse data for A-type CpGs and further supports the premise that prophylactic protection against cerebral ischemic injury can be induced without induction of potentially harmful systemic inflammation.
The demonstration that A-type CpG ODNs can provide prophylactic protection against cerebral ischemic injury in both mouse and NHP experimental models provides important insights for the development of these compounds as promising therapeutics with consideration of the STAIR criteria [18]. This is the second independent study in male NHPs identifying CpG ODNs as a class of drug with potential antecedent therapy for ischemic injury. We are developing a female NHP stroke model for testing a range of neuroprotective compounds, which may provide important information regarding the effects of sex on neuroprotective efficacy.
In summary, our positive results showing A-type CpG ODNs provide robust neuroprotection against ischemic damage adds to the previous data showing that CpG ODNs are potent neuroprotective agents when used as prophylactic treatments against ischemia. Moreover, the finding that A-type CpG ODNs provide such protection without the co-induction of a strong inflammatory response, increases their potential to be used safely in a clinical setting. Taken together, these results continue to support the idea that preconditioning with CpG ODNs offers a very promising approach for neuroprotection to patients at risk of cerebral ischemia.
Acknowledgments
We thank Laurie Renner for support with behavioral assessments and animal handling and Dr. Jessica Minnier for statistical support. The authors would like to thank Sara Christensen and Valerie Conrad for excellent technical support in the mouse studies and Dr. Mingyue Liu for the mouse middle cerebral artery occlusion (MCAO) surgeries. This work was supported by National Institutes of Health grants NS064953 (HFU, MSP, SGK), NS062381 (MSP) and OD011092 (HFU, SGK).
Funding: This work was supported by National Institutes of Health grants NS064953 (HFU, MSP, SGK), NS062381 (MSP) and OD011092 (HFU, SGK).
Footnotes
Compliance with Ethical Standards: Conflict of Interest: Frances Rena Bahjat declares that she has no conflict of interest. G. Alexander West declares that he has no conflict of interest. Steven Kohama declares that he has no conflict of interest. Christine Glynn declares that she has no conflict of interest. Henryk Urbanski declares that he has no conflict of interest. Theodore Hobbs declares that he has no conflict of interest. Eric Earl declares that he has no conflict of interest. Susan Stevens declares that she has no conflict of interest. Mary Stenzel-Poore declares that she has no conflict of interest.
Ethical Approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Bendszus M, Stoll G. Silent cerebral ischaemia: hidden fingerprints of invasive medical procedures. Lancet Neurol. 2006;5:364–372. doi: 10.1016/S1474-4422(06)70412-4. [DOI] [PubMed] [Google Scholar]
- 3.Bonati LH, Jongen LM, Haller S, Flach HZ, Dobson J, Nederkoorn PJ, et al. New ischaemic brain lesions on MRI after stenting or endarterectomy for symptomatic carotid stenosis: a substudy of the International Carotid Stenting Study (ICSS) Lancet Neurol. 2010;9:353–362. doi: 10.1016/S1474-4422(10)70057-0. [DOI] [PubMed] [Google Scholar]
- 4.Shibazaki K, Iguchi Y, Kimura K, Ueno Y, Inoue T. New asymptomatic ischemic lesions on diffusion-weighted imaging after cerebral angiography. J Neurol Sci. 2008;266:150–155. doi: 10.1016/j.jns.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Rosenkranz M, Gerloff C. New ischemic brain lesions after carotid artery stenting. J Cardiovasc Surg (Torino) 2013;54:93–99. [PubMed] [Google Scholar]
- 6.Barber PA, Hach S, Tippett LJ, Ross L, Merry AF, Milsom P. Cerebral ischemic lesions on diffusion-weighted imaging are associated with neurocognitive decline after cardiac surgery. Stroke. 2008;39:1427–1433. doi: 10.1161/STROKEAHA.107.502989. [DOI] [PubMed] [Google Scholar]
- 7.Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–1222. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 8.Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends in Neuroscience. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 9.Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology. 2012;79:1853–1861. doi: 10.1212/WNL.0b013e318271f76a. [DOI] [PubMed] [Google Scholar]
- 10.Wagner R, Piler P, Bedanova H, Adamek P, Grodecka L, Freiberger T. Myocardial injury is decreased by late remote ischaemic preconditioning and aggravated by tramadol in patients undergoing cardiac surgery: a randomised controlled trial. Interact Cardiovasc Thorac Surg. 2010;11:758–762. doi: 10.1510/icvts.2010.243600. [DOI] [PubMed] [Google Scholar]
- 11.Walsh SR, Nouraei SA, Tang TY, Sadat U, Carpenter RH, Gaunt ME. Remote ischemic preconditioning for cerebral and cardiac protection during carotid endarterectomy: results from a pilot randomized clinical trial. Vasc Endovascular Surg. 2010;44:434–439. doi: 10.1177/1538574410369709. [DOI] [PubMed] [Google Scholar]
- 12.Bahjat FR, Williams-Karnesky RL, Kohama SG, West GA, Doyle KP, Spector MD, et al. Proof of concept: pharmacological preconditioning with a Toll-like receptor agonist protects against cerebrovascular injury in a primate model of stroke. J Cereb Blood Flow Metab. 2011;31:1229–1242. doi: 10.1038/jcbfm.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu C, Ha T, Wang X, Liu L, Zhang X, Kimbrough EO, et al. The TLR9 ligand, CpG-ODN, induces protection against cerebral ischemia/reperfusion injury via activation of PI3K/Akt signaling. J Am Heart Assoc. 2014;3:e000629. doi: 10.1161/JAHA.113.000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YI, Park JE, Martinez-Hernandez A, Yi AK. CpG DNA prevents liver injury and shock-mediated death by modulating expression of interleukin-1 receptor-associated kinases. J Biol Chem. 2008;283:15258–15270. doi: 10.1074/jbc.M709549200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markowski P, Boehm O, Goelz L, Haesner AL, Ehrentraut H, Bauerfeld K, et al. Pre-conditioning with synthetic CpG-oligonucleotides attenuates myocardial ischemia/reperfusion injury via IL-10 up- regulation. Basic Res Cardiol. 2013;108:376. doi: 10.1007/s00395-013-0376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballas ZK, Krieg AM, Warren T, Rasmussen W, Davis HL, Waldschmidt M, et al. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J Immunol. 2001;167:4878–4886. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]
- 18.Fisher M, Feuerstein G, Howells DW, Hum PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haley GE, Landauer N, Renner L, Weiss A, Hooper K, Urbanski HF, et al. Circadian activity associated with spatial learning and memory in aging rhesus monkeys. Exp Neurol. 2009;217:55–62. doi: 10.1016/j.expneurol.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urbanski HF. Circadian Variation in the Physiology and Behavior of Humans and Nonhuman Primates. In: Raber J, editor. Animal Models of Behavioral Analysis Neuromethods. Vol. 50. Springer; New York: 2011. pp. 217–235. Chapter 9. [Google Scholar]
- 21.Urbanski HF, Kohama SG, West GA, Glynn C, Williams-Karnesky RL, Earl E, et al. Changes in spontaneous activity assessed by accelerometry correlate with extent of cerebral ischemia-reperfusion injury in the nonhuman primate. Transl Stroke Res. 2012;3:442–451. doi: 10.1007/s12975-012-0191-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West GA, Golshani KJ, Doyle K, Lessov NS, Hobbs TR, Kohama SG, et al. A new model of cortical stroke in the rhesus macaque. Journal of Cereberal Blood Flow and Metabolism. 2009;29:1175–1186. doi: 10.1038/jcbfm.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. 2009;158:1007–1020. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents the cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- 25.Packard AE, Hedges JC, Bahjat FR, Stevens SL, Conlin MJ, Salazar AM, et al. Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2012;32:242–247. doi: 10.1038/jcbfm.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, et al. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 28.Hickey EJ, You X, Kaimaktchiev V, Stenzel-Poore M, Ungerleider RM. Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 2007;133:1588–1596. doi: 10.1016/j.jtcvs.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 29.Emsley HC, Hopkins SJ. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol. 2008;7:341–353. doi: 10.1016/S1474-4422(08)70061-9. [DOI] [PubMed] [Google Scholar]
- 30.Emsley HC, Smith CJ, Tyrrell PJ, Hopkins SJ. Inflammation in acute ischemic stroke and its relevance to stroke critical care. Neurocrit Care. 2008;9:125–138. doi: 10.1007/s12028-007-9035-x. [DOI] [PubMed] [Google Scholar]
- 31.Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13:465–476. doi: 10.1038/nrd4275. [DOI] [PubMed] [Google Scholar]
- 32.Strømmen AM, Christensen T, Jensen K. Quantitative measurement of physical activity in acute ischemic stroke and transient ischemic attack. Stroke. 2014;45:3649–3655. doi: 10.1161/STROKEAHA.114.006496. [DOI] [PubMed] [Google Scholar]