

Abstract
Nanoparticles are used to deliver anticancer drugs to solid tumors. However, clinical development of nanoparticles is challenging because of their limitations in physicochemical properties, such as low drug loading efficiency and poor circulation stability. Low drug loading not only causes technical difficulty in administration but also increases the amount of co-delivered carrier materials, imposing biological burdens to patients. Poor circulation stability causes loss of pharmacokinetics benefits of nanoparticles. To overcome these challenges, we developed an albumin-coated nanocrystal (NC) formulation of paclitaxel (PTX) with 90% drug loading and high serum stability. The NC was produced by inducing crystallization of PTX in aqueous medium, coating the surface with albumin, and removing extra non-drug ingredients. Among three types of NC produced with different crystallization conditions, NC crystallized in the medium containing Pluronic F-127 then coated with albumin (“Cim-F-alb”) had the smallest size and the most native albumin, thus showing the most favorable cell interaction profiles (low uptake by J774A.1 macrophages and high uptake by SPARC+ B16F10 melanoma cells). Cim-F-alb remained more stable in undiluted serum than Abraxane, a commercial albumin-based PTX nanoparticle formulation, while maintaining comparable cytotoxicity to those of Abraxane and solvent-dissolved PTX. In a mouse model of B16F10 melanoma, Cim-F-alb showed higher antitumor efficacy than Abraxane at the same dose. This study demonstrates the feasibility and benefits of delivering an anticancer drug using a carrier-free nanoparticle formulation with good circulation stability.
Keywords: Paclitaxel, nanocrystals, albumin, drug delivery, carrier-free, Abraxane, anti-tumor efficacy
Graphical Abstract
1. Introduction
Anticancer drugs are formulated in various types of nanoparticles, based on the premise that small particles will gain a selective access to tumors via hyperpermeable vasculature [1]. However, their clinical development has been tedious because of the limitations in physicochemical properties of nanoparticles [2] and the complexity of human cancer that have been poorly simulated in preclinical models [3–5]. Among physicochemical challenges, low drug loading efficiency and poor circulation stability are critical issues that can overshadow most benefits of nanoparticles. With low drug loading efficiency (typically <20 wt% of total nanoparticle mass [2]), nanoparticles bring a large amount of carrier materials in addition to the drug. Although the carrier materials are judiciously selected and tested for safety, their biological activities and long term effects are not always predicted in the early stage of development, where the outcomes are monitored with a limited number of doses over a short period of time. Accumulated over repeated administrations, slowly degrading (or non-degrading) carrier materials become an increasing biological burden to patients [6, 7]. Moreover, the high fraction of carrier materials increases the total mass to administer, thus increasing the injection volume and/or the concentration of the nanoparticles such that the volume and concentration of a treatment become dose limiting factors [2, 8]. Concentrated nanoparticles can undergo irreversible aggregation and undue clearance by the mononuclear phagocyte system (MPS), negatively affecting the bioavailability of the treatment. Another prevalent but frequently overlooked challenge is the unstable drug retention in nanoparticles during circulation. Many nanoparticles retain drugs stably in buffered saline but release them almost instantaneously when exposed to physiological media containing proteins and lipids [9]. Nanoparticles losing the payload in circulation are unlikely to provide benefits over conventional solution formulations, no matter how stable they are in buffered saline and what they are capable of in cell models.
In recognition of the problems related to low drug loading efficiency, a selected group of drugs have been formulated into small crystalline particles called nanocrystals (NC). Poorly water-soluble drugs such as paclitaxel (PTX) or docetaxel tend to form solid particles in water. With optimal processing methods and surface stabilizers, these poorly-soluble drugs may be made into NC with a size ranging from 100 to 300 nm. The processing methods may be “top-down” (breaking down large particles with high shear stress), “bottom-up” (inducing nucleation of drug molecules by solvent and/or temperature conditions), or combinations of the two [10]. Since NC are produced from the drug itself and a small quantity of surface stabilizers, the drug content in NC is close to 100 wt% [10]. Depending on the lattice energy between drug molecules, the NC can remain quite stable in aqueous media and serve as nanoparticles [11].
As nanoparticles with high drug loading efficiency (i.e., drug content), NC have drawn increasing interest in the field of nanomedicine as a way of delivering poorly water-soluble anticancer drugs. For example, PTX NC were produced by an anti-solvent and reduced temperature and tested in a colon cancer xenograft murine model [12]. Another type of PTX NC were produced by transforming an amorphous mixture of PTX and Pluronic F127 into crystalline particles and tested in two tumor models [13]. As NC were mostly distributed in the MPS organs leaving little drug in circulation [12, 14, 15], the NC were further modified with polyethylene glycol (PEG) by forming NC together with PEGylated PTX [16] or a crosslinkable polymeric amphiphile containing PEG [17]. Recently, iron-tannic acid complex was used to coat PTX nanoassemblies with an intention to reduce their MPS uptake [18]. However, none of these NC or self-assemblies showed better antitumor effects than commercial PTX formulations such as Taxol (surfactant-solubilized PTX) or Abraxane (albumin-bound PTX) at equivalent doses [12, 13, 18, 19], making their therapeutic benefits a moot point.
We hypothesize that the relative inefficiency of NC may be due to the limitations in size control and suboptimal surface properties. With traditional top-down or bottom-up methods, it is difficult to reduce the particle size to below 200 nm, the upper size limit for preferential extravasation through the leaky tumor vasculature [20]. In addition, bare NC with hydrophobic surfaces prone to non-specific protein adsorption are readily subject to MPS uptake [21]. PEG or other hydrophilic ‘stealth’ surface help reduce the MPS uptake of NC, but they can interfere with the retention and intended cellular interactions after NC distribution in tumors [22]. To address these challenges, we use the NC preparation method introduced by Liu et al [13] and further modify the NC surface with serum albumin. By inducing crystallization in the presence of a surfactant intimately associated with the drug, this preparation method creates NC with a size less than 200 nm [13]. Albumin is the most abundant protein in plasma involved in transendothelial transport of nutrients and drugs [23, 24]. As a native protein with a long circulation half-life, albumin bound to hydrophobic nanoparticles can prevent their opsonization and phagocytosis [25, 26]. Moreover, many tumors have a high demand for albumin as a major source of energy and nutrients [27], and, therefore, an increased capacity to take up albumin. Albumin can extravasate not only by paracellular pathway but also via transcellular pathway that involves gp60 (albondin) [23]. Once in tumors, albumin can bind to SPARC (secreted protein acidic and rich in cysteine) expressed in various cancer cells and tumor interstitium [28]. Based on these features, we expect that albumin modification will help NC to avoid MPS uptake, translocate across the tumor endothelium, and stay in tumors.
In this study, we produce the mentioned NC and two additional NC with different sizes and incipient surfaces for albumin binding and compare their cellular interactions to investigate the effect of the size and the status of surface-bound albumin. The best NC form is compared with Abraxane with respect to the dissolution behavior and in vivo antitumor activities. Our results show that small size of NC and native status of albumin lead to favorable cellular interactions and help the NC to outperform Abraxane in a SPARC-positive B16F10 melanoma model.
2. Materials and Methods
2.1. Materials
PTX was a gift of Samyang Biopharm (Seoul, Korea). Pluronic F127 (F127) was a gift from BASF (New York, NY, USA). Abraxane was obtained from Celgene Corporation (Summit, NJ, USA). Human serum albumin (≥96% agarose gel electrophoresis) and hexadecyltrimethylammonium bromide (CTAB) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Reagents for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Bio-Rad (Hercules, CA, USA). Terminal deoxynucleotidyl transferase dUTP nick end labeling kit (DeadEnd Fluorometric TUNEL System) was purchased from Promega (Madison, WI, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2. NC production
NC were prepared in two steps: crystallization in surfactant-containing medium, followed by surface coating with albumin (Fig. 1). The first step followed the method described by Liu et al [13] with modification. Briefly, a mixture of PTX and surfactant (6 mg PTX and 24 mg Pluronic F127 or 2.4 mg of CTAB) was fully dissolved in 3 mL of chloroform in a round-bottom flask. Chloroform was evaporated with a rotary evaporator at 40 °C for 10 min to form a thin film on the wall of the flask. Six milliliters of DI water was added to the film at room temperature. The formed incipient NC were called Cim-F (Crystallized in medium containing F127) and Cim-C (Crystallized in medium containing CTAB), according to the type of surfactants included in the film. The PTX/surfactant film was hydrated for 5 sec to 15 min with bath sonication, and the hydrated suspension was probe-sonicated in an ice-water bath for 15 min with a power level of 40% and a 1:1 duty cycle every 2 sec. In the second step, the NC were stabilized with albumin. Four milligrams of albumin was added to 1 mL of NC suspension (equivalent (eq.) to 1 mg PTX) and incubated at room temperature for 24 h on a rotating rocker. The NC suspension was centrifuged at 135,700 rcf for 10 min at 4 °C to remove excess surfactant and unadsorbed albumin. The pellet was re-suspended in water and collected by centrifugation at 135,700 rcf for 10 min at 4 °C. The formed NC were called Cim-F-alb (Cim-F stabilized with albumin) and Cim-C-alb (Cim-C stabilized with albumin). For confocal microscopy, Cim-F-alb was labeled with Rhodamine B (Cim*-F-alb) by adding 0.1 mg/mL of aqueous rhodamine B solution in the film hydration step. The amount of incorporated rhodamine B was measured based on fluorescence intensity (λex/λem: 540 nm/625 nm) of Cim*-F-alb dissolved in acetonitrile (ACN). Alternatively, Cim-F-alb labeled with Oregon green (Cim#-F-alb) was produced by replacing a small fraction of PTX with Oregon green-conjugated PTX.
Figure 1.

Schematic description of Cim-F-alb preparation.
For comparison, NC were also prepared by the anti-solvent and temperature-induced crystallization described in the literature [29]. Briefly, 1 mL of 4 mg/mL PTX/ethanol solution was added to 20 mL of DI water and stirred for 10 min in a round-bottom flask immersed in an ice water-filled sonication bath. Ethanol was removed by 10 min rotary evaporation at 40°C. NC were collected on a polycarbonate membrane filter with an average pore size of 100 nm and retrieved in DI water by 1 min bath sonication. The formed NC were called PNC. To form an albumin-coated PNC (PNC-alb), albumin was added to PNC suspension (equivalent to 1 mg/mL PTX) to make 4 mg/mL and incubated at room temperature for 24 h on a rocking shaker. PNC-alb was washed in the same way as above.
2.3. NC characterization
2.3.1. Particle size
The particle size of NC was measured in sodium phosphate buffer (1 mM, pH 7.4) with a Zetasizer Nano-ZS90 (Malvern instruments, Westborough, MA, USA). The particle size was measured at each step of preparation (hydration, albumin coating, and washing).
2.3.2. Morphology
The morphology of NC was examined with a Phillips CM-100 transmission electron microscopy (Hillsboro, OR, USA). Samples were mounted on a 400-mesh Cu grid with Formvar/carbon supporting film and stained with 2% phosphotungstic acid. Images were captured with a SIA L3-C2 megapixel CCD camera (Scientific Instruments and Application, Duluth, GA).
2.3.3. X-ray powder diffraction (XRPD)
Cim-F-alb and its intermediate were analyzed with a Rigaku Smartlab™ diffractometer (Rigaku Americas, Texas, USA) with a Cu-Kα radiation source and a highly sensitive D/tex ultra detector. The powders were placed in glass sample holders, and powder patterns were obtained from 5 to 40° 2θ at a scan speed of 4°/min and a step size of 0.02°. The voltage and current used were 40 kV and 44 mA, respectively
2.3.4. PTX content
The PTX content in NC was determined by HPLC. NC with a premeasured mass were dissolved in a 50:50 mixture of ACN and water at a concentration of 30–60 μg/mL and filtered with 0.45 μm syringe filter prior to analysis. HPLC was performed with an Agilent 1100 HPLC system (Palo Alto, CA), equipped with Ascentis C18 column (25 cm × 4.6 mm, particle size: 5 μm). The mobile phase was a 50:50 mixture of water and ACN and run at 1 mL/min. PTX was detected by a UV detector at a wavelength of 227 nm.
2.3.5. Albumin content and status
The albumin content in NC was determined with SDS-PAGE. NC with a premeasured mass or standard albumin solutions were mixed with 4× Laemmli sample buffer and heated at 95 °C for 5 min. The samples were resolved in a 12% polyacrylamide gel. The gel was stained with QC Colloidal Coomassie Stain (Bio-Rad, Hercules, CA, USA) and imaged with Azure C300 (Dublin, CA, USA). The albumin bands were quantified by densitometry function of the AzureSpot Analysis Software (Dublin, CA, USA). The albumin content was determined by comparing the band intensities of NC samples and standard albumin solutions.
The status of the NC-bound albumin was analyzed by pulse proteolysis [30]. PNC-alb, Cim-C-alb, and Cim-F-alb (eq. to 0.2 mg/mL albumin) were treated with 0.2 mg/mL of thermolysin in HEPES buffer (pH 7.4, 20 mM) containing 100 mM NaCl and 10 mM CaCl2. After 5 min incubation at room temperature, 5 μL of 50 mM EDTA was added to a 15 μL aliquot to quench proteolysis. For the control, NC were treated the same without thermolysin. The treated NC was mixed with 4× Laemmli sample buffer and heated at 95 °C for 5 min. The samples were resolved in 15% polyacrylamide gel and detected with QC Colloidal Coomassie Stain. The protein bands were imaged with Azure C300 to analyze the extent of proteolysis of surface-bound albumin. The percent digestion was calculated as (1-the band intensity of albumin after proteolysis/the band intensity of albumin prior to proteolysis)×100.
The functional status of NC-bound albumin was estimated based on its esterase activity [31]. PNC-alb, Cim-C-alb, and Cim-F-alb (eq. to 0.5 mg/mL albumin) in sodium phosphate buffer (pH 7.4, 0.2 M) was incubated with 100 μM of p-nitrophenyl acetate (pNPA) at room temperature. Immediately and 60 min after the addition of pNPA, a supernatant was separated from the NC suspension by centrifugation and read at 405 nm to quantify p-nitrophenol (product of pNPA hydrolysis). The amount of active albumin was calculated based on the rate of pNPA hydrolysis. The percent active albumin was calculated as the amount of active albumin divided by the amount of total albumin determined with SDS-PAGE.
2.4. Cellular uptake of NC
J774A.1 mouse macrophage cells (ATCC, Manassas, VA, USA) and B16F10 mouse melanoma cells (ATCC, Manassas, VA, USA) were grown in DMEM medium containing 10% fetal bovine serum (FBS) and penicillin (100 IU/mL) and streptomycin (100 μg/mL). Cells were seeded in a 6 well plate at a density of 100,000 cells per well. After overnight incubation, the cell culture medium was replaced with fresh medium, to which 100 μL of PNC-alb, Cim-C-alb, or Cim-F-alb in PBS was added to provide the final concentration equivalent to 30 μg/mL PTX. After incubation with the treatments for 30 min (J774A.1) or 3 h (B16F10), cells were washed with PBS two times to remove unbound NC. The cells in each well were then collected in 1 mL of PBS. A 30 μL fraction of the cell suspension was analyzed with a flow cytometer to determine the cell count. The rest was analyzed to determine PTX taken up by the cells. Briefly, the cell pellet was lysed with 3 cycles of freezing and thawing, followed by probe sonication. The cell lysate was extracted with ethyl acetate, and the content of PTX in the ethyl acetate phase was analyzed with HPLC. Carbamazepine (CBZ) was used as an internal standard during the extraction.
For J774A.1 macrophages, the same experiment was repeated at 4 °C or after pretreatment with polyinosinic acid. When the experiment was performed at 4 °C, the macrophages were equilibrated at 4 °C for 30 min prior to the NC treatment. When polyinosinic acid was used, the macrophages were preincubated with 100 μg/mL of polyinosinic acid in serum-free medium for 30 min, washed once with PBS, and treated with NC in serum-free medium for 30 min. Intracellular PTX content was determined as described above. For B16F10 cells, the same experiment was repeated with and without albumin (5–50 mg/mL) in serum-free medium.
2.5. Cytotoxicity
2.5.1. Cytotoxicity of NC after 3 h treatment
Cytotoxicity of NC was tested with B16F10 cells. B16F10 cells were grown in RPMI-1640 or DMEM medium supplemented with 10% FBS, penicillin (100 IU/mL) and streptomycin (100 μg/mL). B16F10 cells grown in RPMI-1640 were seeded in a 96 well plate at a density of 10,000 cells per well. After overnight incubation, the culture medium was replaced with a fresh cell culture medium, to which PNC-alb, Cim-C-alb, or Cim-F-alb were added to provide the final concentration eq. to 30 μg/mL PTX. After 3 h incubation, cells were washed with PBS twice to remove NC, followed by additional incubation for 21 h in NC-free medium. The cell viability was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Cells were treated with 75 μg of MTT and incubated for 3.5 h. The formazan crystals were dissolved in stop/solubilization solution (50% DMF, 20% SDS, pH 5) and quantified with a SpectraMax M3 microplate reader (Molecular Devices, CA, USA) at the wavelength of 562 nm. The measured absorbance was normalized to the absorbance of control cells that did not receive treatments.
B16F10 cells grown in DMEM medium was tested with propidium iodide (PI) staining due to the production of melanin [32], which interferes with colorimetric assay. B16F10 cells were seeded in a 6 well plate at a density of 100,000 cells per well. After overnight incubation, the medium was replaced with fresh medium, to which 100 μL of PNC-alb, Cim-C-alb, or Cim-F-alb in PBS was added to provide the final concentration eq. to 30 μg/mL PTX. After incubation with NC for 3 h, cells were washed with PBS two times to remove NC, followed by additional incubation for 21 h in NC-free medium. Then, B16F10 cells were trypsinized and stained with PI (1 μg/mL) for 15 min in dark at room temperature. The cells were centrifuged at 300 g for 5 minutes to remove extra PI dye and reconstituted in PBS for flow cytometry (Accuri C6, BD Biosciences, San Jose, CA). At least 15,000 gated events were acquired, and FL-2 channel was monitored to determine the population of PI-stained cells.
2.5.2. IC50 determination
Cytotoxicity of Cim-F-alb was tested with B16F10 cells and SKOV-3 human ovarian cancer cells (ATCC, Manassas, VA, USA) over a range of PTX concentrations. SKOV-3 cells were grown in RPMI-1640 medium containing 10% FBS and penicillin (100 IU/mL) and streptomycin (100 μg/mL). Both cells were treated with Cim-F-alb for 24 h, followed by post-treatment incubation in NC-free medium for 12–48 h. Free PTX and Abraxane were tested in the same manner for comparison. Cim-F-alb and Abraxane were directly reconstituted in PBS and serially diluted by a factor of 10. Free PTX solution was prepared as PTX stock solution in 50% DMSO at a concentration of 1 mg/mL and diluted in PBS. The maximum possible concentration of DMSO in the medium was 5% v/v. SKOV-3 cells and B16F10 cells grown in RPMI-1640 were tested with the MTT assay, and B16F10 cells grown in DMEM were tested with PI staining and flow cytometry. IC50 was calculated with GraphPad Prism 6 (La Jolla, CA, USA).
2.6. Intracellular localization of Cim-F-alb
Cim*-F-alb uptake by B16F10 cells was observed with confocal microscopy. B16F10 cells were seeded in a 35 mm dish with a glass window (MatTek) at a density of 50,000 cells per dish. After 24 h, the medium was replaced with fresh medium, and the cells were incubated with LysoTracker Green DND-26 (25 nM) for 30 min. After washing, cells were incubated with Cim*-F-alb or Cim#-F-alb (30 μg/mL PTX eq.) or free rhodamine B solution (0.3 μg/mL) for 30 min in complete medium. Following two washes with PBS, the cells were fixed with 4% paraformaldehyde in PBS for 10 min. After 5 min nucleus staining with Hoechst 33342 (2 μg/mL), the fixed cells were imaged with a Nikon A1R confocal microscope. Hoechst and LysoTracker Greeen DND-26 were detected with λex/λem of 407 nm/425–475 nm and 488 nm/500–550 nm, respectively. Cim*-F-alb or free Rhodamine B was detected with λex/λem of 561 nm/570–620 nm. Cim#-F-alb was detected with λex/λem of 488 nm/500–550 nm. The same experiment was repeated at 4 °C. Prior to Cim*-F-alb treatment, the cells were equilibrated at 4 °C for 30 min following LysoTracker labeling.
2.7. Dissolution of Cim-F-alb in PBS and FBS
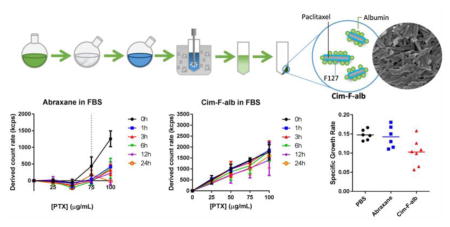
Based on the linear relationship between the number of particles and light scattering intensity, the derived count rate (i.e., absolute light scattering) was monitored as a measure of particle dissolution in PBS or undiluted FBS. Cim-F-alb and Abraxane equivalent to PTX 25 100 μg/mL were incubated in PBS or FBS at 37 °C for 24 h with periodical measurements of their derived count rates using a Zetasizer Nano-ZS90. The FBS suspension was sampled at 6 h to quantify dissolved PTX by that time. A supernatant was separated from the sample by 10 min centrifugation at 135,700 rcf and analyzed as described in 2.4.
Dissolution of Cim-F-alb and Abraxane was monitored by measuring changes in light scattering intensities over time. Briefly, 10 μL of particle suspensions equivalent to 2 μg or 30 μg of PTX was added to 990 μL of FBS or PBS containing 0.2% Tween80 (PBST) to make the final volume 1 mL. After gentle mixing, a series of measurements were performed at 25 °C using a Zetasizer Nano-ZS90. Each measurement was done for 2 runs at 2 sec, at the measurement position of 4.65 mm and with an attenuator setting of 10.
2.8. In vivo activity of Cim-F-alb
2.8.1. Anti-tumor activity
All animal procedures were approved by Purdue Animal Care and Use Committee, in conformity with the NIH guidelines for the care and use of laboratory animals. Eight to ten week old male C57BL/6 mice were obtained from Envigo (Indianapolis, IN, USA) and acclimatized for 1 week prior to the procedure. Each mouse received a subcutaneous injection of 106 B16F10 melanoma cells in the upper flank of the right hind leg. When the tumor was palpable (12 days after tumor cell inoculation), the animals were randomly assigned to 3 groups (n=6 for PBS and Abraxane-treated group, n=7 for Cim-F-alb group) and treated with 4 tail vein injections of PBS, Abraxane, or Cim-F-alb (equivalent to 15 mg/kg of PTX in 100 μL per injection) at 3-day interval. Abraxane and Cim-F-alb were prepared in sterile PBS freshly on the day of treatment. Tumor volume and body weights were monitored every day. The length (L) and width (W) of each tumor were measured with a digital caliper, and the volume (V) was calculated according to the modified ellipsoid formula: V = (L×W2)/2. Specific growth rate of a tumor was calculated as ΔlogV/Δt (t: time in days) [33]. One day after the last injection, mice were sacrificed for the tumor tissue analysis. The excised tumors were fixed in 10% neutral buffered formalin solution or frozen at -80 °C until analysis. Animals losing weight in excess of 20% body weight or with tumors reaching >10% body weight were humanely euthanized prior to the end of the study.
2.8.2. PTX content in tumor
The frozen tumors were homogenized in PBS using a Tissue Master 125 homogenizer (Omni International). One milliliter of the homogenate was mixed with 3 mL of ethyl acetate with 35 μg of CBZ as an internal standard and rotated for 1 h for PTX extraction. After centrifugation at 3,724 rcf for 10 min, 2.8 mL of ethyl acetate layer was evaporated and dissolved in a 1:1 mixture of ACN/water for HPLC analysis.
2.8.3. TUNEL assay of tumor sections
The fixed tumor tissues were embedded in paraffin, sectioned, and mounted on a glass slide for TUNEL assay (DeadEnd Fluorometric TUNEL system; Promega). Images were taken with a Nikon A1R confocal microscope. Two randomly selected fields per tissue were analyzed with ImageJ (National Institutes of Health, Bethesda, MD) to count apoptotic cells and nuclei. Percent apoptotic cells were calculated as the ratio of the number of apoptotic cells to the number of the nuclei.
2.9. Statistical analysis
All statistical analysis was performed with GraphPad Prism 6. All in vitro data were analyzed with one-way or two-way ANOVA test to determine the difference of means among groups, followed by Tukey’s or Sidak’s multiple comparisons test. In vivo data were analyzed with one-way ANOVA, followed by Tukey’s multiple comparison test unless specified otherwise. A value of p< 0.05 was considered statistically significant.
3. Results and Discussion
3.1. Preparation of PNC-alb, Cim-C-alb, and Cim-F-alb
Albumin-coated PTX NC were prepared in two steps: First, incipient NC were prepared by inducing PTX crystallization with an anti-solvent and low temperature [29] or hydrating a dry film of PTX/surfactant mixture [13]. Subsequently, albumin was added to the NC suspension and incubated with the NC to let the protein adsorb on the NC surface.
The incipient NC prepared by the anti-solvent and low-temperature induced crystallization (PNC) showed a z-average of 321.7 ± 34.1 nm. The NC formed from a dry film of PTX-surfactant mixture (Cim-F and Cim-C) were substantially smaller: 154.8 ± 33.1 nm (Cim-F) and 237.8 ± 18.7 nm (Cim-C). The relatively small size is attributable to the surfactant present in the crystallization medium. In a dry film, PTX and F127 form an amorphous mixture making contacts at a molecular level as shown by the XRPD pattern (Fig. 2). PTX transforms to crystalline particles through the hydration and sonication step (Fig. 2). During hydration, F127, a triblock copolymer of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) (PEO-PPO-PEO), starts to dissolve and adsorb to the NC surface via the hydrophobic PPO block, exposing the hydrophilic PEO domains to water. Present at the interface between the incipient NC and the medium, F127 effectively suppresses the crystal growth and their agglomeration. Consistently, Liu et al observed the formation of large crystals in the absence of F127, ascertaining the critical role of F127 in forming small NC [13]. As a surfactant, CTAB is considered to have a similar function.
Figure 2.
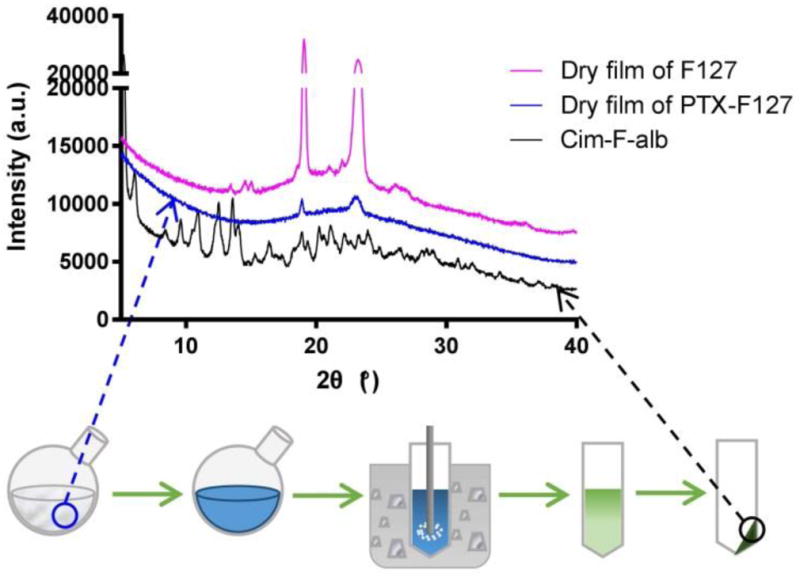
X-ray powder diffraction patterns of a dry film of F127, a dry film of PTX and F127 mixture, and Cim-F-alb.
The second step involved albumin coating of the NC surface and the removal of excess albumin and surfactants (for Cim-F-alb and Cim-C-alb) by repeated centrifugation and resuspension in water. Excess surfactants were removed for potential safety issues. Although F127 is generally considered a nontoxic excipient and has been used as an emulsifying agent in parenteral products [34], studies have reported that F127 could produce hypercholesterolemia in mice after single intraperitoneal (IP) injection at a dose of 500 mg/kg [35] and induce reversible changes in the renal filtration capacity in rats after IP administration at 1000 mg/kg [36]. CTAB is also considered nontoxic, but free CTAB was reported to be cytotoxic in HT-29 colon cancer cells [37], HeLa cervical cancer cells [38], and K562 chronic myelogenous leukemia cells [39]. In the absence of excess surfactants, the surface-bound albumin helped to prevent agglomeration of NC during the purification step. Without albumin, the incipient particles (PNC, Cim-F, and Cim-C) formed micrometer-scale agglomerates upon centrifugation, which were not redispersed to the original size (Fig. 3a). On the other hand, NC coated with albumin withstood centrifugation better and maintained their submicron sizes. After the final step, Cim-F-alb had the smallest z-average (235.6 ± 36.5 nm), and Cim-C-alb and PNC-alb were similar (351.9 ± 75.3 nm and 358.9 ± 79.6 nm) (Fig. 3a).
Figure 3.

(a) Particle size of NC with and without albumin coating as a function of the number of centrifugation, measured by DLS. (b) TEM images and size analysis. TEM images of PNC-alb, Cim-C-alb, and Cim-F-alb were analyzed for size distribution. Scale bars: 500 nm.
The size of Cim-F-alb could be further reduced by adjusting the duration of hydration. Compared to 15 min, 5 sec hydration produced smaller particles with a z-average of 196.7 ± 34.6 nm (Supporting Fig. 1). Reflecting the smaller size, Cim-F-alb formed by 5 sec hydration showed accelerated dissolution in PBS containing 0.2% Tween 80 (PBST) as compared to those formed by 15 min hydration (Supporting Fig. 1). The dependence of particle size on the hydration time indicates that NC grew during the hydration step whereas the subsequent sonication step helped perturb the NC growth. Cim-F-alb could be lyophilized and reconstituted to the original size with trehalose as a lyoprotectant (Supporting Fig. 2).
3.2. Morphology and composition of PNC-alb, Cim-C-alb, and Cim-F-alb
All NC showed a long rod shape, but their size and aspect ratio were slightly different according to the media in which the incipient NC were prepared (Fig. 3b). PNC-alb showed a rod shape with a length of 254.7 ± 119 nm and a width of 76.5 ± 21 nm (n = 172 with ImageJ). Cim-C-alb and Cim-F-alb showed a higher aspect ratio with smaller widths. Cim-C-alb had an average length of 342.5 nm ± 132 nm and a width of 33.5 ± 7 nm (n = 195), while Cim-F-alb had a length of 198.4 ± 80 nm and a width of 35.8 ± 6 nm (n = 188). The rod shape is expected to be beneficial for systemic drug delivery as it helps avoid the particle uptake by the MPS [40]. The prismatic morphology suggests the crystalline nature of NC, which was consistent with the result of XRPD (Fig. 2). Cim-F-alb displayed sharp peaks typical of crystalline solids, similar to PNC [29].
The PTX contents in PNC-alb, Cim-C-alb, and Cim-F-alb were 88.5 ± 4.2wt% (n=4), 79.2 ± 5.1wt% (n=4), 88.7 ± 2.5wt% (n=4), respectively. The albumin contents were 7.6 ± 1.4wt% (PNC-alb), 14.8 ± 0.7wt% (Cim-C-alb), and 9.7 ± 0.9wt% (Cim-F-alb) (Supporting Fig. 3). The amount of remaining F127 and CTAB were not directly measured, but according to the mass balance their contents were estimated to be 1–6wt%. While albumin is an acidic protein with an isoelectric point of pH 4.7, the amount of surface-bound albumin did not directly correlate with the zeta potential of the albumin-bound NC (Supporting Fig. 3). PNC-alb and Cim-C-alb had similar zeta potentials despite the significant difference in the albumin contents. Cim-F-alb was not as negatively charged as PNC-alb, even though the albumin contents were comparable. Based on this observation, we hypothesized that the conformation and arrangement of surface-bound albumin might be different depending on the platform to which albumin was binding (i.e., hydrophobic surface of PNC-alb, cationic surface of Cim-C, and hydrophilic surface of Cim-F). To test this, we investigated the status of albumin bound to NC.
3.3. Evaluation of the status of albumin bound on PNC-alb, Cim-C-alb, and Cim-F-alb
The NC were produced with commercially available albumin, but SDS-PAGE analysis found additional proteins to be associated with the NC (Supporting Fig. 4). The non-albumin proteins, identified by LC-MS/MS, are attributable to impurities incompletely removed from human serum plasma during the production. Interestingly, Cim-C-alb showed more intense non-albumin bands than PNC-alb and Cim-F-alb. The preferential enrichment of non-albumin proteins is attributed to the cationic surface of Cim-C.
We then analyzed the conformation of albumin bound to NC. Typical methods to assess the protein conformation, such as circular dichroism and synchronous fluorescence spectroscopy, were not suitable for analyzing the surface-bound albumin because they require the protein solution contain no substances to interfere with optical measurements. Therefore, we resorted to pulse proteolysis for evaluating the status of albumin bound to NC [30]. The principle of pulse proteolysis is that folded and unfolded proteins have different susceptibilities to proteolysis: i.e., protease exposed to a mixture of unfolded and folded proteins for a short time will preferentially digest unfolded proteins. Accordingly, the extent of protein unfolding can be estimated based on the band intensity of intact protein in the gel electrophoresis. When NC were subjected to pulse proteolysis by thermolysin, Cim-C-alb showed the highest degree of digestion (67.8 ± 13.4%) (Fig. 4a, b). PNC-alb and Cim-F-alb showed similar fractions of digestion of 18.4 ± 7.2% and 14.5 ± 2.6%, respectively. This indicates that albumin on Cim-C-alb was more unfolded than those of PNC-alb and Cim-F-alb.
Figure 4.
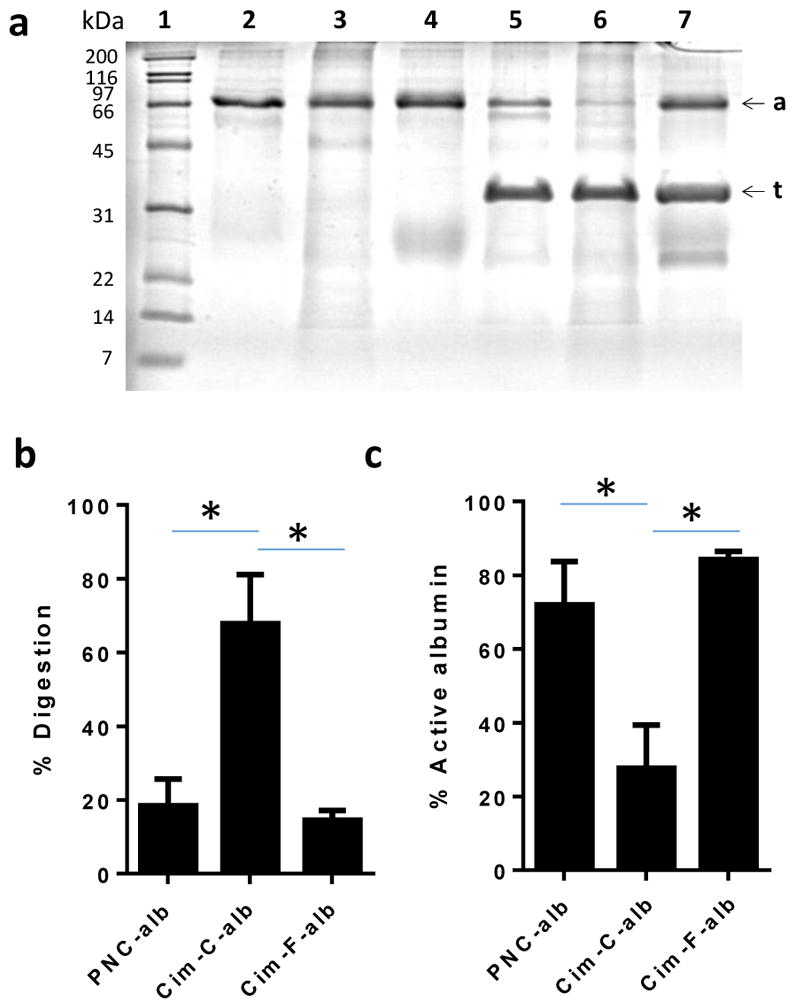
(a) Representative SDS-PAGE gel image of pulse proteolysis. PNC-alb, Cim-C-alb, and Cim-F-alb were incubated with thermolysin for 5 min. Lanes 1: size markers; Lane 2: PNC-alb; Lane 3: Cim-C-alb, Lane 4: Cim-F-alb; Lane 5: PNC-alb + thermolysin; Lane 6: Cim-C-alb + thermolysin; and Lane 7: Cim-F-alb + thermolysin. ‘a’ denotes albumin (66 kDa), and ‘t’ denotes thermolysin (35 kDa). (b) % Digestion of albumin. % digested albumin was determined by albumin band intensity after proteolysis / albumin band intensity prior to proteolysis. n = 4 independently and identically performed experiments. (c) Active albumin content estimated by esterase assay. % active albumin was calculated by dividing esterase active albumin / the amount of albumin determined by SDS-PAGE. n = 3 independently and identically performed experiments. *: p<0.01 by Tukey’s multiple comparisons test.
Albumin has esterase-like activity [41], which is affected by its conformation change [42]. Therefore, the extent of intact (i.e., active) albumin can be determined by measuring its ability to hydrolyze pNPA to p-nitrophenol. PNC-alb and Cim-F-alb showed similar % active albumin, but Cim-C-alb showed much lower % active albumin (Fig. 4c). This indicates that albumin on Cim-C-alb underwent greater conformation change than PNC-alb and Cim-F-alb, consistent with the proteolysis assay.
3.4. Cellular uptake of PNC-alb, Cim-C-alb, and Cim-F-alb by macrophages and cancer cells
We expected that NC-cell interactions would be affected by the surface-bound albumin. Cellular uptake of NC was tested with J774A.1 murine macrophages and B16F10 melanoma cells, which represent the MPS and cancer cells overexpressing SPARC [43], respectively. The extent of cellular uptake of NC was measured by analyzing the PTX content in the collected cell pellets after a shot term exposure of NC to the cells. The NC concentration was kept at 30 μg/mL, much higher than 9.6 μg/mL, the solubility of amorphous PTX in 10% FBS measured in 6 h, so that the majority of NC remain intact during the incubation period (30 min or 3 h).
In J774A.1 macrophages, Cim-C-alb was taken up significantly more than PNC-alb and Cim-F-alb (Fig. 5a). The difference between PNC-alb and Cim-F-alb was insignificant. Cellular uptake of Cim-C-alb at 4 °C was significantly lower than that at 37 °C and not different from those of PNC-alb and Cim-F-alb, indicating that the relatively high Cim-C-alb uptake was energy-dependent endocytosis. The macrophage uptake of Cim-C-alb is attributable to the conformation change of the surface bound albumin, which may have been recognized by scavenger receptors of macrophages. To test this, macrophages were pretreated with polyinosinic acid, a known inhibitor of scavenger receptor-mediated endocytosis [44]. After polyinosinic acid treatment, macrophage uptake of Cim-C-alb was reduced to the same level as other NC (Fig. 5b). Neither PNC-alb nor Cim-F-alb uptake was affected by polyinosinic acid treatment. This result confirms that Cim-C-alb with denatured albumin was preferentially taken up by macrophages via scavenger receptors. In contrast, PNC-alb and Cim-F-alb with more native albumin avoided the scavenger receptor-mediated endocytosis by macrophages, resulting in relatively small macrophage uptake. The albumin on Cim-F-alb rather helps reduce macrophage uptake of the NC (Supporting Fig. 5), consistent with its dysopsonin function reported in the literature [25, 26].
Figure 5.

(a) PTX uptake by J774A.1 macrophages after 30 min incubation with PNC-alb, Cim-C-alb, and Cim-F-alb (equivalent to 30 μg/mL PTX) at 4°C or 37°C. n = 3 independently and identically performed experiments. (b) PTX uptake by J774A.1 macrophages after 30 min incubation with PNC-alb, Cim-C-alb, and Cim-F-alb (equivalent to 30 μg/mL PTX) with and without pre-treatment of polyinosinic acid. n = 4 independently and identically performed experiments. *: p<0.05; **: p < 0.01; #: p<0.001 by Sidak's multiple comparisons test.
NC uptake by B16F10 cells showed a different trend. Cim-F-alb was taken up most by B16F10 cells, and Cim-C-alb least (Fig. 6a). Consistent with the cellular uptake, Cim-F-alb treatment induced greater cell death (Fig. 6b) and lower metabolic activity (Fig. 6c) in B16F10 cells than PNC-alb and Cim-C-alb. We confirmed that the relatively high cytotoxicity of Cim-F-alb was not due to F127, which constitutes 1.6wt% of the total NC mass. F127 itself did not have any toxicity in B16F10 cells at a concentration up to 1 mg/mL (Supporting Fig. 6). PNC-alb mixed with 10 μg/mL F127 (18 times higher than the F127 content in Cim-F-alb at an equivalent PTX dose) showed the same level of cytotoxicity as PNC-alb and lower toxicity than Cim-F-alb (Supporting Fig. 6). This confirms that there was no additional cytotoxicity attributable to F127. In other words, the relatively high cytotoxicity of Cim-F-alb is a consequence of the enhanced cellular uptake, not the effect of residual F127. The preferential uptake of Cim-F-alb by B16F10 cells is attributed to the native conformation and arrangement of the surface-bound albumin, which facilitates its interaction with SPARC expressed on B16F10 cells. PNC-alb and Cim-F-alb had comparable albumin contents (Supporting Fig. 3, 4). Pulse proteolysis and esterase activity indicate that the conformations of albumin bound to the two NC were also similar (Fig. 4). However, the significant difference in zeta potential (Supporting Fig. 3) suggests that the orientation of albumin on the NC surface have been affected by the nature of the exposed surface. Between hydrophobic PNC surface and hydrophilic Cim-F surface, the latter may have provided a favorable platform for albumin to position itself to interact with SPARC. It is also possible that the small size of Cim-F-alb has contributed to the greater uptake by B16F10 cells than PNC-alb uptake. Cim-C-alb did not have advantages in either size or albumin conformation, which accounts for their relatively low cellular uptake and activity in B16F10 cells.
Figure 6.
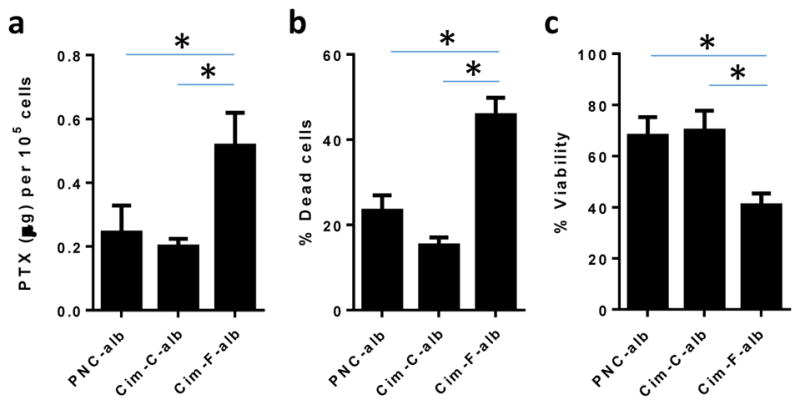
(a) PTX uptake by B16F10 melanoma cells after 3h incubation with PNC-alb, Cim-C-alb, and Cim-F-alb (equivalent to 30 μg/mL PTX). (b–c) Cytotoxicity of NC in B16F10 cells measured after 3h incubation with NC and 21h additional incubation in treatment-free medium, measured by propiodium iodide staining and flow cytometry (b) and MTT assay (c). n = 3 independently and identically performed experiments (a, b). n = 5 measurements (c). *: p <0.05 by Tukey’s multiple comparisons test.
To confirm the involvement of albumin in Cim-F-alb uptake process, B16F10 cells were incubated with Cim-F (NC identically prepared omitting albumin coating) and Cim-F-alb. PTX uptake was greater with Cim-F-alb (Fig. 7a), confirming the contribution of albumin to cellular uptake of the NC. Cim-F-alb uptake by B16F10 cells was inhibited in the presence of 20 mg/mL of albumin (Fig. 7b). This result indicates that Cim-F-alb and albumin share the same receptor, consistent with the native conformation. It is also worth noting that the competition for the common receptor occurs at 20 mg/mL, higher than typical albumin concentration in tissue interstitium [45]. This suggests that cellular uptake of Cim-F-alb may not be much affected by interstitial albumin. However, we cannot exclude the possibility that some tumors have higher level of interstitial albumin than normal tissues and may negatively impact cellular uptake of Cim-F-alb.
Figure 7.

(a) PTX uptake by B16F10 cells after 3h incubation with Cim-F or Cim-F-alb (equivalent to 30 μg/mL PTX). n = 3 independently and identically performed experiments; *: p<0.01 by unpaired two-tailed t-test. (b) PTX uptake by B16F10 cells after 3 h incubation with Cim-F-alb (equivalent to 30 μg/mL PTX) in the presence of extra albumin. n = 3 independently and identically performed experiments; *: p<0.01 by Tukey’s multiple comparison test vs. no albumin control. (c) Intracellular localization of rhodamine B doped Cim*-F-alb (left) or free Rhodamine B (right) in B16F10 cells after 30 min incubation. (d) Overlay image of all fluorescence channels of B16F10 cells incubated with rhodamine B doped Cim*-F-alb.
Cim-F-alb taken up by B16F10 cells was located with confocal microscopy. Cim-F-alb was fluorescently labeled by doping with a small quantity of rhodamine B. We chose rhodamine B as a fluorescence marker for tracing Cim-F-alb uptake, because the dye maintained constant fluorescence intensity at a pH range of 5 to 7 (Supporting Fig. 7), suitable for intracellular imaging. It is well established that an organic dye such as rhodamine B can be stably incorporated in organic crystal lattice with minimal change in crystallization behavior [29], and NC similarly doped with fluorescent dyes have previously been used to visualize their cellular uptake [46]. To ensure that the incorporated rhodamine B represents Cim-F-alb, free rhodamine B was tested in parallel. In addition, another set of labeled Cim-F-alb was prepared by replacing a small fraction of PTX with Oregon green-conjugated PTX (Cim#-F-alb) and compared with Cim*-F-alb. The rhodamine B-doped NC (Cim*-F-alb) showed the same size and shape as Cim-F-alb and thus qualified as its representative (Supporting Fig. 8). B16F10 cells incubated with Cim*-F-alb for 30 min showed punctate signals of rhodamine B (Fig. 7c). Those incubated with an equivalent dose of free rhodamine B only showed dim diffuse fluorescence, indicating that the punctate signals shown with Cim*-F-alb represent NC, not the dye leached out of the NC. B16F10 cells treated with Cim#-F-alb (covalently labeled Cim-F-alb) also showed punctate signals similar to those of Cim*-F-alb, confirming the cellular uptake of Cim-F-alb (Supporting Fig. 9). The NC uptake was completely abolished at 4 °C (Supporting Fig. 10). The fluorescence of Cim*-F-alb overlapped with that of LysoTracker (Fig. 7d) with a Pearson’s correlation coefficient (R) of 0.71 (Supporting Fig. 11), which indicates a high degree of colocalization (perfect colocalization: R=1; no colocalization: R=0; and perfect exclusion: R=-1). These results show that Cim*-F-alb were taken up by the energy-dependent endocytosis pathway and partly trafficked to late-endo/lysosomes.
The in vitro cellular uptake studies identify Cim-F-alb as the most desirable NC among the three types of NC. Cim-F-alb shows less macrophage uptake than Cim-C-alb and greater uptake by B16F10 melanoma cells than PNC-alb and Cim-C-alb, most likely due to the small size and favorable albumin conformation and arrangement on the surface. Therefore, Cim-F-alb was used in the subsequent studies.
3.5. Dissolution of Cim-F-alb
To predict the circulation stability of Cim-F-alb, we evaluated the dissolution of Cim-F-alb in PBS and undiluted FBS, comparing with Abraxane, a commercial albumin-bound PTX formulation. The as-received PTX, which was amorphous (Supporting Fig. 12), dissolved in 10% FBS, 50% FBS, and undiluted FBS up to 9.6 ± 2.6 μg/mL, 47.5 ± 11.4 μg/mL, 71.9 ± 8.2 μg/mL, respectively, in 6 h at 37 °C (Supporting Fig. 13). Due to the crystallinity, we expected that Cim-F-alb would dissolve in each medium no more than the measured values at comparable conditions.
Particle dissolution was monitored by light scattering according to the method reported by Anhalt et al. [47], which depends on the linear relationship between the number of particles and absolute scattering intensities (i.e., derived count rate). The scattering intensity of particle suspension decreases in proportion to the solid content in the suspension. When the particles completely dissolve, the scattering intensity matches that of blank medium. Therefore, if the particles dissolve in a medium, the scattering intensity vs. concentration plot shows two linear segments with distinct slopes: a flat segment followed by the second segment with a slope corresponding to the increasing solid fraction [47]. The solubility is calculated from the intersection point of the two segments.
Abraxane in PBS and FBS followed this pattern, with solubility of 25 μg/mL and 75 μg/mL (as PTX equivalent), respectively (Fig. 8a). Twenty five μg/mL is higher than the solubility of amorphous as-received PTX in PBS we reported previously (~0.2 μg/mL [9]). This may be explained by the solubilizing effect of albumin present in the Abraxane formulation (225 μg/mL albumin in Abraxane eq. to 25 μg/mL PTX) in addition to its amorphous status (Supporting Fig. 14). The solubility of Abraxane in FBS (75 μg/mL) was comparable to amorphous PTX solubility in FBS (71.9. μg/mL). The linearity of scattering intensity vs. concentration plot started to disappear as soon as in 1 h, indicating that the dissolution occurred quickly, which is consistent with the literature [48]. The PTX dissolved in FBS over 6 h was proportional to the concentration of Abraxane, which further confirms complete dissolution of Abraxane up to 75 μg/mL (Supporting Fig. 13). In contrast, Cim-F-alb in PBS showed a linear relationship over the entire concentration range without evident change in slope, which indicates that Cim-F-alb remained as particles at all concentrations tested. The linear pattern was persistent in FBS, with slight decrease in the slope attributable to partial dissolution of PTX (no higher than 8.3 ± 3.0 μg/mL when measured after 6h incubation, Supporting Fig. 13). The contrast between Abraxane and Cim-F-alb in FBS is worthwhile to note. This suggests that Abraxane in blood will dissociate into albumin-bound PTX molecules at <75 μg/mL, whereas Cim-F-alb in blood will mostly remain as nanoparticles at least for 24 h.
Figure 8.

(a) Derived count rate of Cim-F-alb and Abraxane in PBS (top) and undiluted FBS (bottom) at 37 °C. For samples in FBS, the values were obtained by subtracting the derived count rate of FBS from those of particle suspensions in FBS at each time point. n = 3 independently and identically performed experiments; (b) Dissolution rate of Cim-F-alb, PNC, and Abraxane in FBS at PTX concentration of 2 μg/mL and 30 μg/mL.
The dissolution of Abraxane and Cim-F-alb in FBS was monitored over time to confirm this difference in the stability in serum. Abraxane in FBS was immediately indistinguishable from FBS at 2 μg/mL and 30 μg/mL, indicating its rapid dissolution (Fig. 8b). On the other hand, Cim-F-alb in FBS showed higher count rates than that of FBS over 15 min (duration of observation), indicating that Cim-F-alb maintained the particle status at both concentrations. We may predict the fates of systemically administered Abraxane and Cim-F-alb based on these results. Considering that the typical PTX dose in mice is 15–40 mg/kg in a blood volume of 1.5–2.5 mL [49], the initial PTX concentration is expected to be 6–26.7 μg/mL. At this concentration range, Cim-F-alb is likely to circulate as nanoparticles, whereas Abraxane will dissociate instantaneously into individual albumin molecules bound to PTX. In other words, during the initial circulation and biodistribution phase, Cim-F-alb and Abraxane will circulate in different forms – nanoparticles and PTX-bound albumin molecules, respectively.
We then compared Cim-F-alb, Abraxane, and solvent-dissolved PTX with respect to the cytotoxicity. All three groups showed similar dose response curves with comparable IC50 values irrespective of the cell lines, treatment conditions, or assay methods (Supporting Table 1, Supporting Fig. 15). One-way ANOVA analysis of IC50 values indicated no statistically significant difference between groups. This result indicates that Cim-F-alb, dissolved in the medium and/or taken up as NC (at the higher end of the concentration range: 50,000 and 100,000 nM, in particular), provided similar effects as free drug or Abraxane.
3.6. In vivo evaluation of Cim-F-alb
We tested if PTX circulating as nanoparticles would have any advantages over Abraxane in antitumor effects. According to the prevalent premise of nanomedicine, nanoparticles can provide greater tumor accumulation than free drug by reducing renal clearance and allowing for selective access to tumors [50, 51]. Cim-F-alb is uniquely suited for testing this hypothesis because it contains the same components as Abraxane (albumin and PTX) but differs from Abraxane in the circulation stability. This allows us to exclude the potential contribution of the carrier materials, which often account for the majority of nanoparticle mass and add confounding effects to the therapeutic outcomes [52].
Cim-F-alb or Abraxane equivalent to 15 mg/kg PTX was administered to C57BL/6 mice bearing subcutaneous B16F10 tumors by tail vein injection on days 0, 3, 6, and 9 (total PTX dose = 60 mg/kg). Cim-F-alb was well tolerated at this dose and caused no significant loss of body weight (Fig. 9a) or abnormal blood chemistry (data not shown). This dose was also below the reported maximum tolerated dose of Abraxane [8]. Therefore, 15 mg/kg q3d×4 was considered a sufficiently safe condition to test the anti-tumor activities of two formulations. At this dose, Abraxane did not attenuate the growth of B16F10 tumors as compared to PBS control (Fig. 9b). On the other hand, Cim-F-alb treatment resulted in significant delay in tumor growth (p<0.01 vs PBS or Abraxane, Tukey’s multiple comparisons test). One mouse in each of the PBS and Abraxane-treated groups had to be sacrificed on day 7 according to the humane endpoint, and all animals in these groups had tumors with ≥1000 mm3 in size by day 10. In contrast, there was only one animal with a tumor >1000 mm3 in the Cim-F-alb treated group on day 10. Tumor growth was also expressed as the specific growth rate, which is appropriate in assessing the exponential growth of tumor [33] (Fig. 9c). The Cim-F-alb group showed a significantly lower specific growth rate than the PBS- or Abraxane-treated groups (p<0.05 vs. both groups, Tukey’s multiple comparisons test). To compare the effect of treatment on the tissue level, tumors collected one day after the last dose were analyzed with respect to the number of apoptotic cells and the PTX content. Tumor sections from the Cim-F-alb group showed a significantly higher number of TUNEL+ apoptotic cells than those of PBS- or Abraxane-treated groups (Fig. 10a, b, Supporting Fig. 16, 17). Consistently, the PTX content in tumor was significantly higher in the Cim-F-alb group (27.4 ± 22 μg/g) than in the Abraxane treatment group (13.8 ± 6 μg/g) (Fig. 10c).
Figure 9.

In vivo activity of Cim-F-alb and Abraxane in C57BL/6 mice. (a) Body weight change of animals receiving PBS, Abraxane or Cim-F-alb, expressed in box-and-whisker plot. The box extends from the 25th to 75th percentiles, the line in the middle of the box indicates the median, and the whiskers go down to the smallest value and up to the largest. (b) Box-and-whisker plot of B16F10 tumor volumes: Mice were treated with PBS (black; n=6), Abraxane (blue; n=6), or Cim-F-alb (red; n=7) at 15 mg/kg q3d×4. Arrows indicate treatment times. One mouse in each of the Abraxane and PBS group was sacrificed on day 7 due to the large size of the tumor. (c) Specific growth rate of B16F10 tumor = ΔlogV/Δt (V: tumor volumes; t: time in days). Horizontal bars = means. *: p<0.05 by Tukey’s multiple comparisons test.
Figure 10.

(a) Representative photographs of deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end (TUNEL)-stained B16F10 tumor sections. (b) Quantitative analysis of TUNEL-stained sections. % apoptotic cells = number of apoptotic cells / total number of nuclei measured by ImageJ. Data are represented as averages and standard deviations of 12 images for PBS- and Abraxane groups and 14 images for Cim-F-alb group (2 images from each tumor section). *: p<0.01 by Tukey’s multiple comparisons test. (c) PTX content in B16F10 tumor tissues. n = 6 for Abraxane and n = 7 for Cim-F-alb group. Horizontal bars = means. *: p<0.05 by Mann-Whitney test.
We examined if the superior performance of Cim-F-alb relative to Abraxane was due to preferable cellular uptake of the NC. Macrophages exposed to Cim-F-alb and Abraxane for 30 min showed no difference in PTX uptake (Supporting Fig. 18), which may be explained by the fact that both are small enough to avoid phagocytic uptake. Cim-F-alb uptake by B16F10 cells in 3 h were rather less than Abraxane uptake (Supporting Fig. 18). This suggests that, with both Cim-F-alb and Abraxane containing albumin that helps interact with SPARC-positive B16F10 cells, the molecularly dispersed Abraxane be more efficient than nanopaticulate Cim-F-alb in entering the cancer cells. Therefore, the advantage of Cim-F-alb over Abraxane is not at the cellular level.
Taken together, these results support that Cim-F-alb circulating as nanoparticles have reached tumors better than Abraxane which dissociates into PTX-bound albumin molecules upon intravenous injection. Although pharmacokinetics and biodistribution of the two formulations remain to be compared, the in vitro cell studies and dissolution tests let us predict that the surface-bound albumin and particle stability in serum may contribute to the preferential tumor accumulation and retention of Cim-F-alb.
4. Conclusion
To develop a carrier-free nanoparticle formulation of PTX with good circulation stability, PTX NC (Cim-F-alb) were produced by crystallizing PTX in a medium containing Pluronic F127 and stabilizing the NC surface with native albumin. Cim-F-alb had a smaller size and native albumin than other NC produced by a different crystallization method or surfactant. The small size and native conformation of surface-bound albumin resulted in reduced uptake by J774A.1 macrophages and increased uptake by SPARC-positive B16F10 melanoma cells. Cim-F-alb outperformed Abraxane in a mouse model of B16-F10 melanoma. The in vitro dissolution profile in undiluted serum suggests that the superior performance of Cim-F-alb is likely due to the circulation stability of the nanoparticles, which leads to their preferential tumor accumulation and retention.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health NIBIB R01EB017791 and the Ronald W. Dollens Graduate Scholarship support for J.P. and B.S. This work was also supported by the Indiana Clinical and Translational Sciences Institute, funded in part by grant #UL1 TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. We thank Samyang Biopharm (Seoul, Korea) for the kind donation of paclitaxel and the NAL Pharmaceuticals Ltd. (Monmouth Junction, NJ, USA) for the gift support. We also thank Prof. Chiwook Park for the help with pulse proteolysis, Victoria Hedrick at the Purdue Proteomics Core for the LC-MS/MS analysis, and Andrew Wakefield for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 2.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials. 2016;1:16014. [Google Scholar]
- 3.Nichols JW, Bae YH. EPR: Evidence and fallacy. J Control Release. 2014;190:451–464. doi: 10.1016/j.jconrel.2014.03.057. [DOI] [PubMed] [Google Scholar]
- 4.Park K. Drug delivery of the future: Chasing the invisible gorilla. J Control Release. 2015 doi: 10.1016/j.jconrel.2015.10.048.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality and possibility. J Control Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mc CW, Jackson G, Grieble HG. Treatment of chronic pyelonephritis: II. Short-term intravenous administration of single and multiple antibacterial agents; acidosis and toxic nephropathy from a preparation of intravenous nitrofurantoin. AMA Arch Intern Med. 1959;104:710–719. [Google Scholar]
- 7.Laine GA, Hossain SM, Solis RT, Adams SC. Polyethylene glycol nephrotoxicity secondary to prolonged high-dose intravenous lorazepam. Ann Pharmacother. 1995;29:1110–1114. doi: 10.1177/106002809502901107. [DOI] [PubMed] [Google Scholar]
- 8.Ernsting MJ, Murakami M, Undzys E, Aman A, Press B, Li SD. A docetaxel-carboxymethylcellulose nanoparticle outperforms the approved taxane nanoformulation, abraxane, in mouse tumor models with significant control of metastases. J Control Release. 2012;162:575–581. doi: 10.1016/j.jconrel.2012.07.043. [DOI] [PubMed] [Google Scholar]
- 9.Abouelmagd SA, Sun B, Chang AC, Ku YJ, Yeo Y. Release kinetics study of poorly water-soluble drugs from nanoparticles: Are we doing it right? Mol Pharm. 2015;12:997–1003. doi: 10.1021/mp500817h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller RH, Gohla S, Keck CM. State of the art of nanocrystals–special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78:1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Fuhrmann K, Gauthier MA, Leroux JC. Targeting of injectable drug nanocrystals. Mol Pharm. 2014;11:1762–1771. doi: 10.1021/mp5001247. [DOI] [PubMed] [Google Scholar]
- 12.Hollis CP, Weiss HL, Leggas M, Evers BM, Gemeinhart RA, Li T. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: Lessons learned of the EPR effect and image-guided drug delivery. J Control Release. 2013;172:12–21. doi: 10.1016/j.jconrel.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, Park JY, Zhang Y, Conwell C, Liu Y, Bathula SR, Huang L. Targeted cancer therapy with novel high drug-loading nanocrystals. J Pharm Sci. 2010;99:3542–3551. doi: 10.1002/jps.22112. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Li X, Wang L, Xu Y, Cheng X, Wei P. Formulation and pharmacokinetic evaluation of a paclitaxel nanosuspension for intravenous delivery. Int J Nanomedicine. 2011;6:1497–1507. doi: 10.2147/IJN.S21097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei L, Ji Y, Gong W, Kang Z, Meng M, Zheng A, Zhang X, Sun J. Preparation, physical characterization and pharmacokinetic study of paclitaxel nanocrystals. Drug Dev Ind Pharm. 2015;41:1343–1352. doi: 10.3109/03639045.2014.950272. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Hu H, Zhang H, Dai W, Wang X, Wang X, Zhang Q. Effects of PEGylated paclitaxel nanocrystals on breast cancer and its lung metastasis. Nanoscale. 2015;7:10790–10800. doi: 10.1039/c4nr07450e. [DOI] [PubMed] [Google Scholar]
- 17.Fuhrmann K, Schulz JD, Gauthier MA, Leroux JC. PEG nanocages as non-sheddable stabilizers for drug nanocrystals. ACS Nano. 2012;6:1667–1676. doi: 10.1021/nn2046554. [DOI] [PubMed] [Google Scholar]
- 18.Shen G, Xing R, Zhang N, Chen C, Ma G, Yan X. Interfacial cohesion and assembly of bioadhesive molecules for design of long-term stable hydrophobic nanodrugs toward effective anticancer therapy. ACS Nano. 2016;10:5720–5729. doi: 10.1021/acsnano.5b07276. [DOI] [PubMed] [Google Scholar]
- 19.Lu Y, Wang ZH, Li T, McNally H, Park K, Sturek M. Development and evaluation of transferrin-stabilized paclitaxel nanocrystal formulation. J Control Release. 2014;176:76–85. doi: 10.1016/j.jconrel.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotech. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134:2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 22.Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: Advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36:892–899. doi: 10.1248/bpb.b13-00059. [DOI] [PubMed] [Google Scholar]
- 23.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 24.Minshall RD, Malik AB. Transport across the endothelium: Regulation of endothelial permeability. Handb Exp Pharmacol. 2006:107–144. doi: 10.1007/3-540-32967-6_4. [DOI] [PubMed] [Google Scholar]
- 25.Beukers H, Deierkauf FA, Blom CP, Deierkauf M, Riemersma JC. Effects of albumin on the phagocytosis of polystyrene spherules by rabbit polymorphonuclear leucocytes. J Cell Physiol. 1978;97:29–36. doi: 10.1002/jcp.1040970105. [DOI] [PubMed] [Google Scholar]
- 26.Peng Q, Zhang S, Yang Q, Zhang T, Wei XQ, Jiang L, Zhang CL, Chen QM, Zhang ZR, Lin YF. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34:8521–8530. doi: 10.1016/j.biomaterials.2013.07.102. [DOI] [PubMed] [Google Scholar]
- 27.Stehle G, Sinn H, Wunder A, Schrenk HH, Stewart JCM, Hartung G, Maier-Borst W, Heene DL. Plasma protein (albumin) catabolism by the tumor itself—implications for tumor metabolism and the genesis of cachexia. Crit Rev Oncol Hematol. 1997;26:77–100. doi: 10.1016/s1040-8428(97)00015-2. [DOI] [PubMed] [Google Scholar]
- 28.Elsadek B, Kratz F. Impact of albumin on drug delivery—new applications on the horizon. J Control Release. 2012;157:4–28. doi: 10.1016/j.jconrel.2011.09.069. [DOI] [PubMed] [Google Scholar]
- 29.Zhao R, Hollis CP, Zhang H, Sun L, Gemeinhart RA, Li T. Hybrid nanocrystals: Achieving concurrent therapeutic and bioimaging functionalities toward solid tumors. Mol Pharm. 2011;8:1985–1991. doi: 10.1021/mp200154k. [DOI] [PubMed] [Google Scholar]
- 30.Park C, Marqusee S. Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2:207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 31.John JM, Sivakami S, Dongre PM. Elucidation of structural and functional properties of albumin bound to gold nanoparticles. J Biomol Struct Dyn. 2016:1–38. doi: 10.1080/07391102.2016.1144223. [DOI] [PubMed] [Google Scholar]
- 32.Diawpanich P. DMEM enhances tyrosinase activity in B16 mouse melanoma cells and human melanocytes. Warasan Songkhla Nakharin. 2008;30:603–610. [Google Scholar]
- 33.Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 2007;67:3970–3975. doi: 10.1158/0008-5472.CAN-06-3822. [DOI] [PubMed] [Google Scholar]
- 34.Rowe RC, Sheskey PJ, Owen SC AP Association. Handbook of Pharmaceutical Excipients. Pharmaceutical Press; 2006. [Google Scholar]
- 35.Palmer WK, Emeson EE, Johnston TP. Poloxamer 407–induced atherogenesis in the C57BL/6 mouse. Atherosclerosis. 1998;136:115–123. doi: 10.1016/s0021-9150(97)00193-7. [DOI] [PubMed] [Google Scholar]
- 36.Li C, Palmer WK, Johnston TP. Disposition of poloxamer 407 in rats following a single intraperitoneal injection assessed using a simplified colorimetric assay. J Pharm Biomed Anal. 1996;14:659–665. doi: 10.1016/0731-7085(95)01621-x. [DOI] [PubMed] [Google Scholar]
- 37.Alkilany AM, Nagaria PK, Hexel CR, Shaw TJ, Murphy CJ, Wyatt MD. Cellular uptake and cytotoxicity of gold nanorods: Molecular origin of cytotoxicity and surface effects. Small. 2009;5:701–708. doi: 10.1002/smll.200801546. [DOI] [PubMed] [Google Scholar]
- 38.Niidome T, Yamagata M, Okamoto Y, Akiyama Y, Takahashi H, Kawano T, Katayama Y, Niidome Y. PEG-modified gold nanorods with a stealth character for in vivo applications. J Control Release. 2006;114:343–347. doi: 10.1016/j.jconrel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 39.Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1:325–327. doi: 10.1002/smll.200400093. [DOI] [PubMed] [Google Scholar]
- 40.Chu KS, Hasan W, Rawal S, Walsh MD, Enlow EM, Luft JC, Bridges AS, Kuijer JL, Napier ME, Zamboni WC, DeSimone JM. Plasma, tumor and tissue pharmacokinetics of docetaxel delivered via nanoparticles of different sizes and shapes in mice bearing SKOV-3 human ovarian carcinoma xenograft. Nanomedicine. 2013;9:686–693. doi: 10.1016/j.nano.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurono Y, Kushida I, Tanaka H, Ikeda K. Esterase-like activity of human serum albumin. Viii. Reaction with amino acid p-nitrophenyl esters. Chem Pharm Bull. 1992;40:2169–2172. doi: 10.1248/cpb.40.2169. [DOI] [PubMed] [Google Scholar]
- 42.Watanabe H, Tanase S, Nakajou K, Maruyama T, Kragh-Hansen U, Otagiri M. Role of Arg-410 and Tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem J. 2000;349:813–819. doi: 10.1042/bj3490813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoang B, Ernsting MJ, Roy A, Murakami M, Undzys E, Li SD. Docetaxel-carboxymethylcellulose nanoparticles target cells via a sparc and albumin dependent mechanism. Biomaterials. 2015;59:66–76. doi: 10.1016/j.biomaterials.2015.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang HY, Wu LX, Reinhard BM. Scavenger receptor mediated endocytosis of silver nanoparticles into J774A.1 macrophages is heterogeneous. ACS Nano. 2012;6:7122–7132. doi: 10.1021/nn302186n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellmerer M, Schaupp L, Brunner GA, Sendlhofer G, Wutte A, Wach P, Pieber TR. Measurement of interstitial albumin in human skeletal muscle and adipose tissue by open-flow microperfusion. Am J Physiol Endocrinol Metab. 2000;278:E352–E356. doi: 10.1152/ajpendo.2000.278.2.E352. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Li T. Cellular uptake mechanism of paclitaxel nanocrystals determined by confocal imaging and kinetic measurement. AAPS J. 2015;17:1126–1134. doi: 10.1208/s12248-015-9774-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anhalt K, Geissler S, Harms M, Weigandt M, Fricker G. Development of a new method to assess nanocrystal dissolution based on light scattering. Pharm Res. 2012;29:2887–2901. doi: 10.1007/s11095-012-0795-4. [DOI] [PubMed] [Google Scholar]
- 48.Desai N. Nab technology: A drug delivery platform utilising endothelial gp60 receptor-based transport and tumour-derived sparc for targeting. Drug Delivery Report Winter 2007/2008. 2008:37–41. [Google Scholar]
- 49.Riches AC, Sharp JG, Thomas DB, Smith SV. Blood volume determination in the mouse. J Physiol. 1973;228:279–284. doi: 10.1113/jphysiol.1973.sp010086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 51.van Vlerken LE, Duan Z, Little SR, Seiden MV, Amiji MM. Biodistribution and pharmacokinetic analysis of paclitaxel and ceramide administered in multifunctional polymer–blend nanoparticles in drug resistant breast cancer model. Mol Pharm. 2008;5:516–526. doi: 10.1021/mp800030k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeo Y, Kim BK. Drug carriers: Not an innocent delivery man. AAPS J. 2015;17:1096–1104. doi: 10.1208/s12248-015-9789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.