

Abstract
Structural models of the complex that regulates the direction of flagellar rotation assume either ~34 or ~25 copies of the protein FliG. Support for ~34 came from crosslinking experiments identifying an intersubunit contact most consistent with that number; support for ~25 came from the observation that flagella can assemble and rotate when FliG is genetically fused to FliF, for which the accepted number is ~25. Here, we have undertaken crosslinking and other experiments to address more fully the question of FliG number. The results indicate a copy number of ~25 for FliG. An interaction between the C-terminal and middle domains, which has been taken to support a model with ~34 copies, is also supported. To reconcile the interaction with a FliG number of ~25, we hypothesize conformational plasticity in an interdomain segment of FliG that allows some subunits to bridge gaps created by the number mismatch. This proposal is supported by mutant phenotypes and other results indicating that the normally helical segment adopts a more extended conformation in some subunits. The FliG amino-terminal domain is organized in a regular array with dimensions matching a ring in the upper part of the complex. The model predicts that FliG copy number should be tied to that of FliF, whereas FliM copy number can increase or decrease according to the number of FliG subunits that adopt the extended conformation. This has implications for the phenomenon of adaptive switch remodeling, in which the FliM copy number varies to adjust the bias of the switch.
Keywords: motility, molecular motors, chemotaxis, protein structure, self-assembly
Introduction
Bacterial flagella are rotating organelles fueled by the membrane gradient of protons or sodium ions [1–3]. Most are capable of both clockwise (CW) and counterclockwise (CCW) rotation; regulated reversals in motor direction are responsible for directed cellular movements such as chemotaxis [4]. Flagella consist of a long, relatively rigid helical filament that functions as propeller, a curved flexible element called the hook that serves as a universal joint, and a structure within the membrane and that extends into the cytosol, termed the basal body [5,6] (Fig. 1). The basal body is surrounded by complexes formed from the integral membrane proteins MotA and MotB (or related orthologs) [7]. These constitute the stator or non-rotating part of the motor [8]. The stator complexes conduct gradient protons (or Na+ ions) [9,10] and couple ion flow to conformational changes that are likely to be the “power-stroke” driving rotation [11]. For reviews of flagellar motility and chemotaxis, see Refs. [12–15].
Fig. 1.
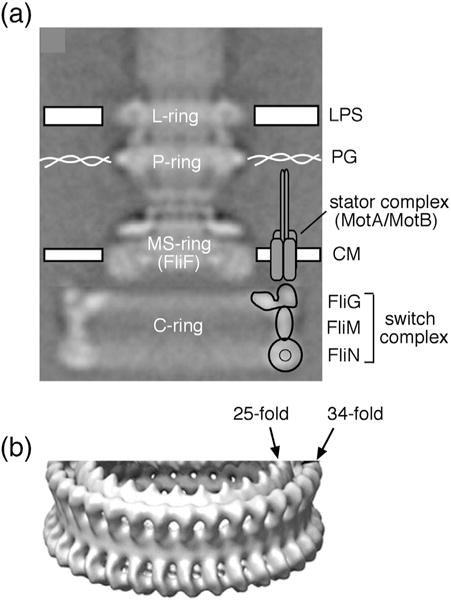
Morphology of the flagellar basal body of Salmonella. (a) EM reconstruction of the flagellar basal body from Thomas et al. [28]. The structure consists of a central rod and a set of rings. The membrane/supra-membrane (MS) ring is in and just above the cytoplasmic membrane (CM). The outer membrane (LPS) and peptidoglycan (PG) are also indicated. One stator complex is shown; motors can contain up to about a dozen independently functioning stator complexes, each attached to peptidoglycan. Approximate positions of the three switch complex proteins are indicated. (b) A tilted view of the switch complex from a higher-resolution EM reconstruction [6]. Density has been rotationally averaged to improve signal to noise. Most of the switch complex has ~34-fold symmetry, but the inner ring in the upper (membrane-proximal) part displays ~25-fold symmetry.
Reversals in motor direction are controlled by a multiprotein assembly termed the switch complex [16–19]. Structurally, the switch complex corresponds to the basal-body C-ring (Fig. 1). In the well-studied model species Escherichia coli and Salmonella typhimurium, the complex is formed from multiple copies of the proteins FliG, FliM, and FliN. FliG functions most directly in the generation of torque and is at the top (i.e., the membrane-proximal part) of the switch complex where it can interact with the stator protein MotA [20,21]. FliM lies just below FliG [22–24], and FliN is at the bottom [25] (Fig. 1). Although there is consensus on this overall organization of the components, certain aspects of switch complex architecture, particularly the arrangement of FliG subunits, remain uncertain.
Models of switch complex architecture must specify the copy number of each protein constituent. Some clues to subunit number can be gleaned from symmetries observed in electron microscopic reconstructions of the basal body of Salmonella. Most parts of the C-ring in Salmonella exhibit ~34-fold symmetry (the ~ symbol denoting the fact that ring size and symmetry can vary from specimen to specimen), whereas an inner (smaller radius) ring near the top of the C-ring, and the membrane/supra-membrane (MS)-ring, displays ~25-fold symmetry [6,26] (Fig. 1b). The MS-ring is constructed from FliF, for which the accepted copy number is ~25. An important difference between published proposals for switch complex architecture concerns FliG copy number: some models postulate ~34 copies of FliG, matching the symmetry of most of the C-ring [24,27], whereas a previous structural proposal from our laboratory assumed a value of ~25 to reflect the symmetry of the MS-ring and the inner ring in the upper part of the complex [23].
Evidence for 25 copies of FliG came from the characterization of a Salmonella mutant in which a small deletion near the end of fliF brought it into frame with fliG to encode a FliF–FliG fusion protein. Cells of this strain make essentially normal-looking basal bodies [28], and the flagellar motors can rotate, although with a CW bias [17]. The normal morphology of these flagellar basal bodies would seem to indicate that FliG and FliF are present in equal numbers, presumably ~25 given the symmetries of the MS-ring [26] and the parts of the C-ring closest to it. However, the available data cannot rule out the possibility that the strain contained a certain amount of native-sized FliG incorporated into the motors along with the FliF–FliG fusion protein, in which case the actual FliG copy number could be ~34.
In the present study, we sought to examine FliG copy number more closely and to develop a model for switch complex architecture that can account for all the data presently available. In the course of the study, a new FliF–FliG fusion was constructed and shown to function essentially like wild type. Native-sized FliG was not detected in the cells; the results thus support a copy number of ~25 for FliG. Additional experiments to examine FliG organization in the switch endorse the suggestion by Lee et al. [27] that in the motor, the C-terminal domain of FliG stacks onto the middle domain of an adjacent subunit, making an interdomain contact similar to one observed in several crystal structures. This stacking interaction has been taken to support a model with ~34 FliG subunits; hence, none of the published models for FliG organization appears consistent with all of the data. Accordingly, we propose a revised model of switch complex architecture that combines the FliG copy number of Paul et al. [23] with the FliGC/FliGM interaction proposed by Lee et al. [27] and Vartanian et al. [24] and that is in keeping with recent solution studies of intermolecular contacts in FliG:FliM complexes [29]. An essential feature of the model is conformational plasticity in FliG that allows the segment joining its middle and C-terminal domains to be helical in most subunits but extended in a subset. The amino-terminal domain of FliG (FliGN) was also examined and found to be present in a closely spaced array, likely corresponding to the inner, 25-fold symmetric ring at the top of the complex.
In the updated model for the switch, the number of FliG subunits is predicted to remain fairly constant and equal to that of FliF, whereas FliM copy number can vary as more or fewer FliG subunits take on the extended conformation. It is thus relevant to the phenomenon of switch remodeling, in which additional copies of FliM can be added to the switch to adjust its intrinsic CW/CCW bias [30–33].
Results
Characterization of a FliF–FliG fusion strain
Support for a FliG copy number of ~25 came primarily from a study by Francis et al. [17] showing that cells with a chromosomally encoded fusion of FliF to FliG produced normal-looking basal bodies and were capable of movement. Although extensive proteolysis of the FliF–FliG fusion protein was shown to not occur, the occurrence of some native-sized FliG in the motors, which might arise from proteolysis or from translation from an internal start site, was not ruled out. We therefore undertook further examination of this FliF–FliG fusion strain. In the originally isolated Salmonella strain [17], a 7-nt deletion in fliF shifted the fliG reading frame into register with fliF and caused five C-terminal residues of FliF to be replaced with Ile. An equivalent strain was engineered in E. coli by making the same 7-nt deletion in a plasmid encoding fliF–fliG (Fig. S1) and by transferring this construct into the chromosome by replacement of a tet cassette. Under the microscope, the FliF–FliG fusion strain displayed highly tumbly motility. Tethered cells rotated almost exclusively CW (Fig. S1), and the strain was nonchemotactic on soft-agar plates (Fig. 2a), presumably owing to the CW switch bias (rates of migration in soft agar for this and other strains are summarized in Table S1.)
Fig. 2.
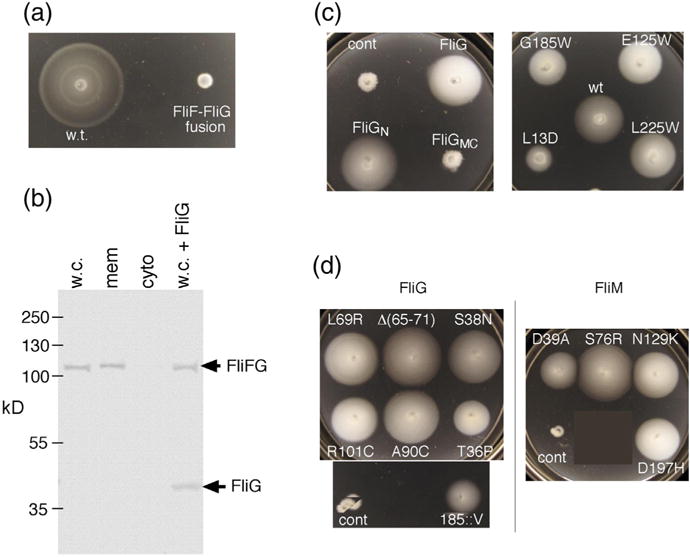
Properties of the original FliF–FliG fusion strain. The strain has a 7-nt deletion near the end of fliF that brings it into frame with fliG while replacing five C-terminal FliF residues with Ile (see Fig. S1 for details). (a) Chemotaxis defect of the strain in a soft-agar migration assay. A tryptone soft-agar plate was spotted with 3 μl of overnight cultures and incubated for 6 h at 32 °C. Under the microscope, cells displayed vigorous tumbling. (b) Anti-FliG immunoblots of cell fractions, and whole cells, showing that native-sized FliG is not present at a significant level. w.c., whole cell; mem, membrane; cyto, cytosol; w.c. + FliG, whole-cell proteins from the fusion strain with native-sized FliG expressed from a plasmid (pDB97, with no induction). (c) Improved motility of the original FliF–FliG fusion strain when FliG or certain FliG variants are expressed from plasmids. The control (cont) is the original fusion strain with nothing else expressed. Plasmids were derivatives of pKP619, induced with 200 μM IPTG. Plates were spotted with 3 μl of overnight cultures and incubated for 12 h at 32 °C. The strain expressing wild-type FliG from the plasmid migrates at ~20% of the wild-type rate. FliGN, FliG amino-terminal domain; FliGMC, FliG middle and C-terminal domains. The right-hand plate shows the effects of expressing point-mutant variants of FliG that have been shown previously to abolish the function of the full-length protein [34,35]. Rescue of the fusion strain by extra FliG is diminished by a mutation in the N-terminal domain (L13D) but not by mutations in the middle or C-terminal domains. (d) Examples of suppressors of the chemotaxis defect in the fusion strain. Strains retained the FliF–FliG fusion and had acquired the indicated mutations in FliG or FliM. The original, unsuppressed FliF–FliG fusion is also shown (labeled “cont”). Plates were incubated for 12 h at 32 °C. Suppressor mutations are listed in Table 1.
Anti-FliG immunoblots of whole cells and of membrane and soluble fractions showed strong bands for the FliF–FliG fusion protein but very little normal-sized FliG, particularly in the membrane fraction (Fig. 2b). The flagella thus contain just the FliF–FliG fusion protein and do not contain significant quantities of FliG. FliM blots showed similar expression and membrane localization of FliM in the fusion strain and wild type (Fig. S2), as expected, given the normal appearance of the basal bodies in Salmonella [17,28]. Together, the results indicate that motors containing equal numbers of FliF and FliG units can be assembled and can rotate vigorously. They are strongly biased toward CW rotation, however, and we next sought to understand this defect.
Treatments that improve the function of the FliF–FliG fusion strain
When wild-type FliG was expressed in the FliF–FliG fusion strain, migration in soft agar was improved to about 20% of wild type (Fig. 2c). Cells in liquid culture showed an increase in smooth swimming, and a larger fraction of tethered cells exhibited CCW rotation (Fig. S1). To determine which parts of FliG are necessary for this effect, we tested deletion and point-mutant variants. A construct consisting of the middle and C-terminal domains (FliGMC) did not improve the migration of the fusion strain, and point mutations in these domains in residues previously found essential for function [34] did not prevent the motility enhancement. The N-terminal domain alone (residues 1–87), which is the part that binds FliF [35], was as effective as the full-length protein (Fig. 2c), and a mutation in the domain (L13D) that weakens the interaction with FliF [35] significantly decreased the motility rescue.
The severe chemotaxis defect in the FliF–FliG fusion strain facilitated the isolation of spontaneous suppressor mutations, which appear as flares of faster-migrating cells arising from streaks inoculated on soft-agar plates. Cells from flares were colony-purified and retested to confirm the improved migration; then, genomic DNA was prepared and used to identify the suppressing mutations by the sequencing of candidate genes (fliG and fliM). All of the faster-migrating derivatives tested retained the original fliF–fliG fusion and had acquired mutations in either fliG or fliM. Examples of mutant phenotypes are shown in Fig. 2d, and the observed mutational changes are listed in Table 1.
Table 1.
Mutations suppressing the CW bias in the FliF–FliG fusion strain
The suppressing mutations do not appear to act by increasing the proteolytic cleavage at the FliF–FliG joint, because immunoblots of several of the suppressor strains (Fig. S3) showed only minimal breakdown of the fusion protein, similar to that seen in the parent strain. FliM levels were likewise unaffected by the suppressing mutations (Fig. S3). Most of the suppressing mutations were single amino acid replacements in FliG (12 mutations) or FliM (4 mutations); a 6-residue deletion and a single-residue insertion in FliG were also isolated. FliM mutations included ones previously shown to give CCW motor bias in a mutational analysis by Macnab and co-workers (Table 1). We conclude that motors with equal numbers of FliF and FliG units (in the form of the fusion protein) can rotate and also switch direction appropriately in response to signals from the chemotaxis pathway, provided that switch bias has been reset by a compensating mutation elsewhere. All but one of the suppressing mutations in FliG were in FliGN (Table 1) and are discussed further below in the context of a model for FliGN organization. The exception was the insertion of a valine after residue 185, which is in the helix joining the FliGM and FliGC domains (Fig. 2d). This mutation was made in plasmid-borne fliG (encoding the normal-sized protein) and was found to cause impaired migration in soft agar (Fig. S4) and smooth swimming, indicative of excessive CCW rotation. This effect prompted us to make a one-residue deletion (of Q186) in the same region. This mutant also showed impaired migration (Fig. S4), in this case associated with highly tumbly motility indicating CW bias.
A better-functioning FliF–FliG fusion strain
The chemotaxis defect in the FliF–FliG fusion strain might be due to a subnormal FliG copy number (if the normal number is really ~34) or might reflect abnormal conformation at the FliF–FliG junction. To determine whether a less-constrained FliF–FliG fusion can function better, we constructed a strain in which the proteins are joined by a nine-residue linker with sequence GSGSGSGSL (Fig. 3a). In soft-agar plates, this strain migrated at around 80% of the wild-type rate (Fig. 3b). Rotation speeds in tethered-cell assays were also close to normal (Fig. 3d). As with the original fusion, very little native-sized FliG was detected on immunoblots (Fig. 3c), and FliM level and localization were similar to wild type (Fig. S2). Thus, motors containing equal numbers of FliF and FliG units can assemble and function with very nearly wild-type efficiency. The bias defect in the original fusion strain is then presumably due to its more constrained FliF–FliG junction. In contrast to the original fusion, migration of the new fusion strain was slowed rather than helped by the expression of native-sized FliG or FliGN in the cells (Fig. S5).
Fig. 3.
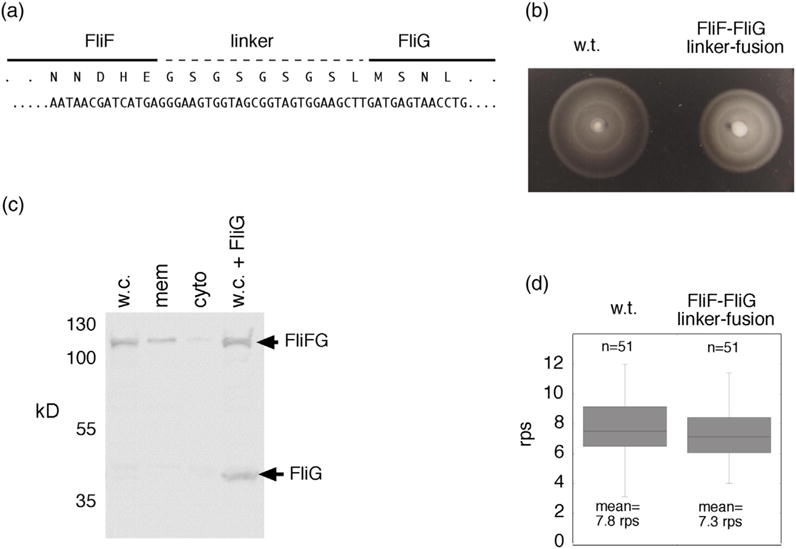
Properties of a different fusion strain that has a linker between FliF and FliG. (a) Nucleotide and amino-acid sequences at the FliF–FliG junction. (b) Motility of the linker-fusion strain in soft agar. A tryptone soft-agar plate was inoculated with 3 μl of overnight cultures and incubated for 8 h at 32 °C. Following a short delay in the onset of motility, the fusion-linker strain migrates at >80% of the wild-type rate. (c) Anti-FliG immunoblots of whole-cell samples and cell fractions showing that native-sized FliG is not present at a significant level in the fusion-linker strain. w.c., whole cell; mem, membrane; cyto, cytosol; w.c. + FliG, whole cells with native-sized FliG expressed from a plasmid (pDB97, with no induction). (d) Rotation speeds of tethered cells of the FliF–FliG fusion-linker strain and of the wild type. Cells were tethered to coverslips by anti-flagellin antibody, and rotation speeds were determined by slow-speed video playback.
Interaction between FliGM and FliGC domains
In a previous communication [23], we were critical of a switch complex model proposed by Lee et al. [27] largely on the grounds that it is based on ~34 rather than ~25 copies of FliG. The present results reinforce the view that the motor contains only about 25 copies of FliG. However, recent crosslinking results of Baker et al. [36], and spectroscopic studies of Sircar et al. [29], provide support for an interaction between FliGC and FliGM that is a central feature of the Lee et al. model. In similar crosslinking experiments, we had failed to observe this interaction. Previous experiments used HA-tagged proteins and fairly high-level expression from plasmids [23]. The recent findings [36] prompted us to revisit this question using additional Cys pairs, untagged protein, and other experimental improvements.
Cysteine replacements were made at a number of positions around the hypothesized FliGC–FliGM interface (Table S2). Function was tested in soft-agar motility assays, and Cys replacements that permitted nearly normal function were combined in pairs. Function of the double mutants was tested, and those that supported substantial motility were used in cross-linking experiments. Initial experiments used intact cells and FliG variants expressed from a plasmid, with I2, hydrogen peroxide, or Cu-phenanthroline as oxidizing agents. Products of crosslinking were characterized on FliG immunoblots.
On treatment with oxidant, several of the Cys pairs gave significant yield of FliG dimer and, in some cases, larger multimers (Table S2). Greatest yields were with I2; representative results with this oxidant are shown in Fig. 4a. Crosslinking was especially efficient for the 158C/214C mutant. These residues are near each other at the FliGC/FliGM interface in FliG crystal structures, having Cβ–Cβ distances of 5.1 Å, 5.2 Å, and 6.3 Å, respectively, in the structures from Thermotoga maritima (1LKV [37]), Aquifex aeolicus (3HJL [27]), and Helicobacter pylori (3USY [38]). The 158C/214C mutation pair and another one that showed significant crosslinking (159C/217C) were next transferred into the chromosome to verify that the crosslinking was not an artifact of expression from plasmids. The chromosomally expressed Cys-mutant proteins supported migration at rates comparable to (158C/214C) or about half (159C/217C) of wild type (Fig. S6). On treatment with I2, the 158C/214C protein again crosslinked efficiently to form a ladder of products. The 159/217 pair, which is somewhat more exposed in the crystal structures, showed relatively little crosslinking with I2 but formed a ladder on treatment with the bifunctional reagent bis-maleimidohexane (BMH) (Fig. 4a). The three Cys pairs that were studied previously by Baker et al. (159/218, 165/195, and 166/194) [36] using plasmid-based constructs were also examined here using chromosomal expression; all formed ladders upon oxidation (Fig. S7). In soft-agar motility assays, two of the mutants showed fairly severe impairments, but the 166/194 mutant retained ~65% of wild-type function (Fig. S7). We conclude, in agreement with Lee and co-workers [27,36] and Vartanian et al. [24], that FliGC does interact with FliGM in the motor, in a manner similar to the contact seen in crystal structures and in the pulsed dipolar electron spin resonance (ESR) studies of Sircar et al. [29].
Fig. 4.
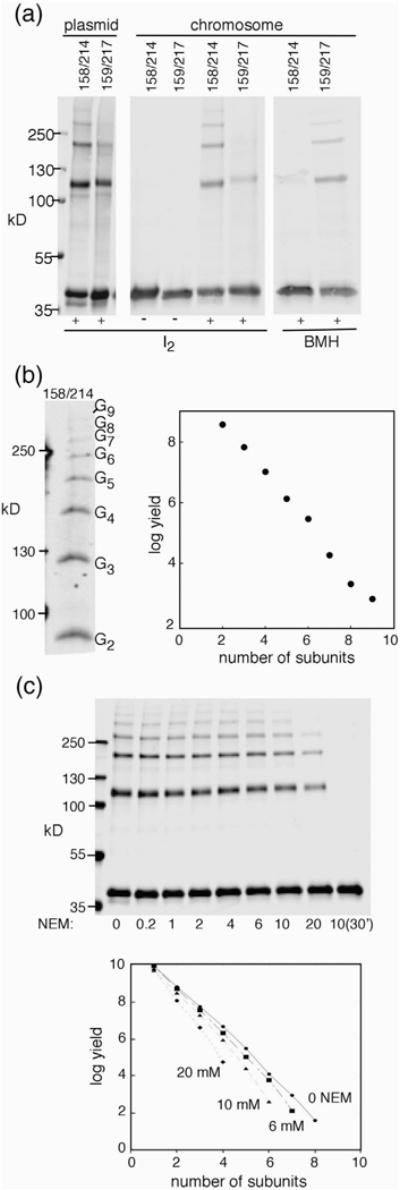
Crosslinking between positions in the middle and C-terminal domains of FliG. (a) Examples of the crosslinking of double-Cys mutant proteins, using either plasmid-based expression (derivatives of pKP619 induced with 40 μM IPTG) or expression from the normal chromosomal locus. (b) Left: ladder of products observed with the chromosomally expressed 158/214 pair, resolved on a gel using less than the usual amount of bis-acrylamide (acrylamide:bis-acrylamide ratio 80:1). Right: Yield versus product size for the ladders produced by the 158/214 Cys pair. Bands were quantified by densitometry using Image-J [53]. (c) Blocking of the 158–214 crosslinking by pretreatment with NEM. Blocking was for 3 min prior to treatment with iodine for all except for the rightmost sample, which was blocked for 30 min. Proteins were resolved on a 4–20% gradient gel.
In crosslinking experiments with the chromosomally expressed 158C/214C mutant, product ladders extended to nine FliG subunits or more. Yield showed an approximately exponential decrease with increasing size (Fig. 4b), as expected if each crosslink forms with equal, less-than-100% probability. In our previous model, FliGC domains were proposed to occur in closely spaced groups of only three or four separated by gaps due to FliG absences. The crosslinked ladders observed with the 158C/214C mutant appear inconsistent with groupings this small and suggest instead that the FliG subunits are continuously connected within a larger array. Conceivably, FliG subunits might normally occur in groups of just three or four but undergo rapid rearrangements that allow gaps to migrate along the array during a crosslinking experiment to allow the trapping of FliG chains larger than those present in the functioning motor. This was tested in an experiment using pretreatment with N-ethyl maleimide (NEM). The side chains of Cys residues at gap positions should be relatively exposed and susceptible to blocking, and pretreatment with NEM should then yield shortened ladders of a characteristic (presumably three- or four-subunit) size. This was not observed: treatment with increasing concentrations of NEM decreased the average product size, but the simple negative exponential dependence was retained, with no indication of a particular favored product size (Fig. 4c).
Although it is unlikely, given the nearly normal function of the new FliF–FliG fusion, it remains formally possible that the wild-type motor contains 34 copies of FliG. In this case, the long ladders would just reflect the presence of a continuous array of all 34 FliG subunits. Then, in a FliF–FliG fusion strain that is limited to only ~25 copies of FliG, about 9 gaps should be present and should limit the size of crosslinked products to 3 or 4 subunits. To test this possibility, the 158C/214C Cys pair was introduced into the new fusion strain, and crosslinking was carried out as before. Although ladders were slightly shorter than with the native-sized FliG, products with as many as six subunits were still observed, and yields again followed a negative exponential dependence with no indication of being limited to groups of three or four (Fig. 5a and b). An NEM-blocking experiment showed a general decrease in crosslink yield but again with no indication of clusters of a particular size (Fig. 5b).
Fig. 5.
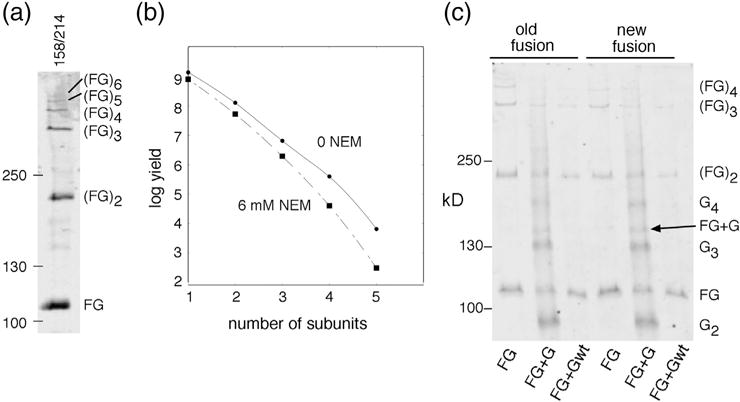
(a) Iodine-induced disulfide crosslinking of positions 158 and 214 in the contest of the new (i.e., linker-containing) FliF–FliG fusion protein, expressed from the chromosome. (b) Product yield versus size (number of crosslinked subunits), and effect of pretreatment with NEM (6 mM for 3 min). (c) Crosslinking of native-sized, plasmid-expressed FliG to the FliF–FliG fusion proteins. Results with both the old and new fusion constructs are shown. The FliF–FliG fusions contained the 158C/214C replacements. The plasmid-expressed FliG either carried the same Cys replacements or was wild type (with no Cys residues), as indicated; plasmids were derivatives of pKP619, induced with 40 μM IPTG.
If the FliG copy number is normally ~25 but is not absolutely linked to FliF number, it might be possible to force additional copies of native-sized FliG into the fusion strains. Results above (Fig. 2c) suggest that extra copies of at least the FliGN domain might insert. This was tested in a crosslinking experiment in the new FliF–FliG fusion carrying the 158C/214C Cys replacements in FliG, with native-sized FliG carrying the same Cys replacements expressed from a plasmid. Treatment with iodine produced a ladder of products similar to that seen in the fusion strain, but with small amounts of an additional product at the position expected for a FliFG + FliG heterodimer (Fig. 5c). The low yield indicates that the native FliG does not incorporate readily, and the low level of this mixed (FG + G) product in experiments in the simple fusion strains (“FG” lanes in Fig. 5) constitutes further evidence that those motors do not contain native-sized FliG.
Organization of the FliGC and FliGM domains
On the basis of the contact observed in crystal structures, Lee et al. [27] developed a model in which each FliGC domain rests on FliGM of the adjacent subunit. Subunit relationships in this model (or any similar model that involves FliGC/FliGM stacking) will thus depend on the secondary structure and orientation of the segment connecting the FliGM and FliGC domains (comprising residues 170–193 in the E. coli protein). This segment forms a continuous helix in some crystal structures (1LKV, 3HJL) but not others (e.g., 4FHR, 3AJC, 3SOH, 3USY). In most crystal structures, the first half of the segment is packed closely against the FliGM domain. Exceptions are a structure (3AJC) where the helix is engaged in the same packing interactions except with the FliGM domain of a different subunit, and another (1LKV) where the helix swivels far away from the FliGM domain and is stabilized in a four-helix bundle involving four FliG subunits in an evidently nonnative arrangement (Fig. S8). We previously reported intramolecular FliG crosslinks (179/121 and 183/121) that support the close packing of the helix against FliGM [39]. This experiment was repeated here using chromosomally expressed and untagged proteins, including one additional Cys pair (113/183) that should report on the disposition of the linking helix. Intramolecular crosslinking was observed for all three pairs (Fig. S9). Accordingly, a model was constructed that assumes the inward, FliGM-contacting position for the helix (Fig. S10).
The present results, and those of Baker et al. [36], indicate that our previous contention of a direct contact between FliGC and FliMM is not correct or at least is not the primary way in which FliGC is supported. The present results also indicate, however, that the copy number of FliG is ~25 rather than ~34. The middle domain of FliG interacts with the middle domain of FliM, through interfaces that have been seen in multiple crystal structures (4QRM, 4FHR, 4FQ0) and that are well conserved (involving an “EHPQR” motif on FliGM and a “GGXG” motif in FliMM). The question that arises is how to place an array of ~25 FliG subunits onto a ring of ~34 FliMM domains: the number mismatch should give rise to gaps where FliGC is without a neighboring FliGM to rest upon (Fig. S10B). While such “orphan” FliGC domains might in principle be held in place by different contacts (for example, a FliGC–FliMM interaction), the crosslinking results (Fig. 4) indicate that the FliGC–FliGM interaction connects arrays of at least nine contiguous subunits. Alternatively, the FliGC domain at a gap position might find a way of reaching across the gap to interact with FliGM in the next position over. This hypothesis was tested in crosslinking and mutational experiments.
Evidence that the segment connecting FliGM to FliGC can extend
To reach FliGM of a non-adjacent subunit, a FliGC domain would need to cross a gap of about 3.5 nm (the spacing between FliMM domains in the underlying ring). In certain of the crystal structures, and in our working model (Fig. S10), the segment between FliGM and FliGC is a helix of about 20 aa (commencing at residue 169 in the E. coli protein). The needed extension could be obtained if most of this helix melted into an extended conformation. Such melting appears plausible: the helix showed a tendency to melt in MD simulations [40], and it is predicted to have only marginally helical character for much of its length (using PsiPred [41] with FliG sequences from E. coli and a variety of other species) and is helical for only half its length in crystal structures of the protein from H. pylori [38].
In the melted-helix model, an extended segment that bridges the gap caused by a FliG absence will pass over a FliMM domain whose top is exposed because it is not supporting a FliGM. Residues near the middle of the segment, which are packed against the FliGM domain when the segment is helical, would shift to a position near the top of this FliMM if the segment takes on extended conformation (Fig. S10C). This was tested in a crosslinking experiment with Cys residues introduced at the top of FliMM (residues 73 or 74) and at positions in FliG near the middle of the connecting segment (residues 179 and 183). The Cys-mutant FliG and FliM proteins were expressed from their chromosomal loci. Crosslinking was induced with I2, H2O2, or Cu-phenthroline; results with H2O2 are shown in Fig. 6a. A crosslinked product was observed on both FliM and FliG immunoblots when Cys was present at position 183 in FliG and at position 73 or 74 in FliM. The 183/73 mutant retained nearly wild-type motility in soft agar while the 183/74 mutant was significantly impaired (rate ~25% of wild type). Residue 183 is packed against the FliGM domain in the majority of the crystal structures (the exception being 1LKV, where the helix is stabilized in the evidently non-physiological four-subunit bundle illustrated in Fig. S8). The observed crosslinking of FliG position 183 to FliM positions 73 and 74 would therefore require a significant displacement, in the direction hypothesized in the melted-helix model. With Cys at position 179 in FliG instead, a crosslinked product was seen on FliG blots at the size of a FliG dimer, but not on FliM blots, and was still observed, although more weakly, in the absence of any Cys replacement in FliM (Fig. 6a). This suggests that position 179 in FliG can crosslink to itself, particularly when FliM is mutant. The FliG-179C replacement appears to decrease the stability of the complex; although tolerated by itself, this substitution completely eliminated motility when combined with FliM-74C.
Fig. 6.
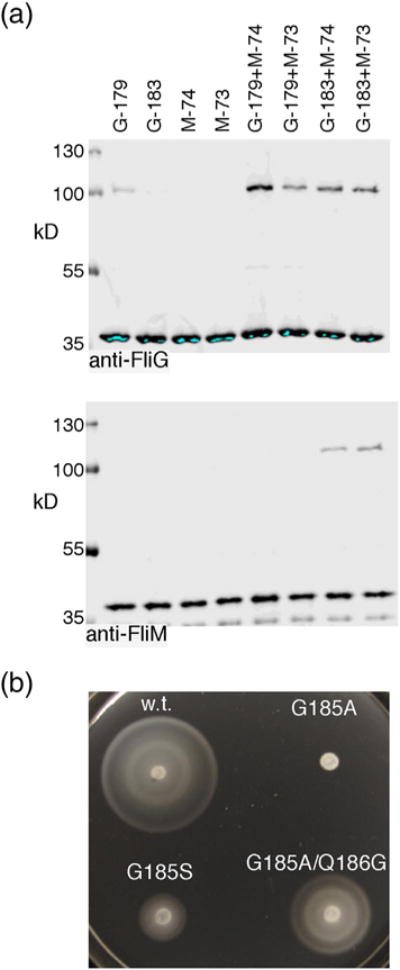
(a) Crosslinking of position 183 in the FliGM–FliGC connecting segment to positions 73 and 74 at the top of FliMM. Mutant FliG and FliM proteins were expressed from the chromosome. Crosslinking was induced with 50 mM H2O2 in all lanes. (b) Effects of mutations in the FliGM–FliGC connecting helix.
A further test of the melted-helix hypothesis was made using a mutational approach. In E. coli FliG, a glycine residue at position 185 should be a significant factor in the only-marginal helical propensity of the segment (because helical conformation is disfavored by glycine [42]). Crystal structures of the T. maritima protein indicate that position 185 is exposed, and the backbone geometry is typical for an α-helix. Nevertheless, migration in soft agar was prevented when G185 was mutated to the strongly helix-stabilizing residue alanine (Fig. 6b). The G185A mutant exhibited tumbly motility, indicative of CW motor bias. This functional disruption does not appear to be due to the restriction of backbone angles or other steric factors involving the alanine side chain, because replacement with serine—larger but less helix-stabilizing than alanine—caused only a mild reduction in chemotaxis (Fig. 6b). Moreover, the defect in the G185A mutant was largely rescued when the adjacent residue Q186 was replaced with glycine (making the G185A/Q186G double mutant; Fig. 6b). These results suggest that the marginally helical character of the segment is important for function and can be provided in more than one way (in the present case, by glycine at either position 185 or 186).
While helix extension at the gap positions allows for a continuous chain of FliGC/FliGM interactions in the present model, the FliGM domains themselves are hypothesized to remain in groups of three or four separated by gaps. A Cys pair in FliGM (117/166) was previously shown to crosslink efficiently into ladders in experiments using plasmid-expressed proteins [43]. The chromosomally expressed 117C/166C protein was tested here and was found to be crosslinked even prior to treatment with oxidant, exhibiting dimer, trimer, and trace amounts of tetramer (Fig. S11). Oxidation with iodine gave ladders with as many as eight subunits, but in contrast to the situation with the 158/214 pair, this was effectively blocked by pretreatment with NEM (10 μM), so that oxidation gave, besides the pre-existing products, a significant increase of only tetramer (Fig. S11). Both the size distribution prior to oxidation (trimer and smaller) and the NEM-induced restriction to tetramer and smaller appear consistent with the occurrence of clusters of three or four closely spaced FliGM domains. In this hypothesis, the longer ladders observed in unblocked samples would result from the trapping of non-native states (juxtapositions of more than four FliGM domains) that occur as FliGM domains that move within the array in the course of the crosslinking.
Organization of FliGN domains
A recent crystal structure [35] revealed the details of the FliF/FliG binding interaction that attaches the switch complex to the MS-ring. The interaction is quite strong and involves the insertion of two helices in the C-terminal part of FliF (FliFC) into deep hydrophobic grooves on FliGN. The relationship between adjacent FliGN domains remains uncertain. We sought to address this question through additional crosslinking experiments, using the FliFC/FliGN structure to guide the placement of Cys residues.
In the structure of the FliGC/FliGN complex, FliGN exhibits a surface hydrophobic patch formed (in E. coli) from the side chains of M15, F26, V34, and M41. Hydrophobic character is strongly conserved at these positions, suggesting their involvement in a functionally important contact. Cys replacements made for the crosslinking study therefore included a number in this patch. Other Cys replacements were made in the helices near the C terminus of the domain (H5 and H6), at positions chosen on the basis of conserved hydrophobic character and the results of initial experiments. Mutant FliG proteins were expressed from the chromosomal locus. Cross-linking was induced using I2 or BMH, and products were characterized on FliG immunoblots. Examples of I2-induced crosslinking are shown in Fig. 7, and results for all FliGN Cys pairs for both I2 and BMH are summarized in Table S3. Non-crosslinked and single-Cys controls are shown in Fig. S12.
Fig. 7.
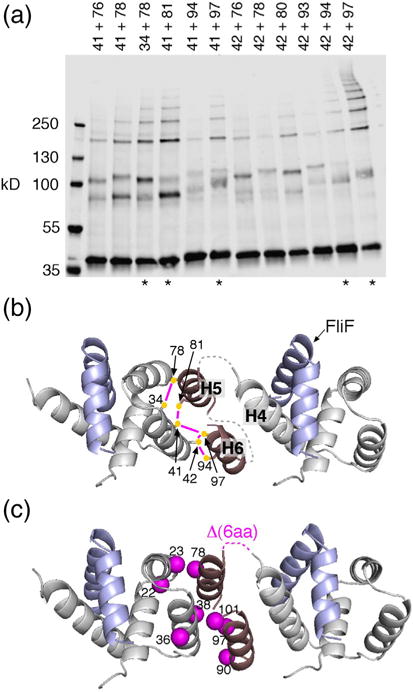
Organization of the FliG N-terminal domain. (a) I2-induced crosslinking through pairs of Cys residues in FliGN. Mutant FliG proteins were expressed from the chromosome. Asterisks at the bottom indicate the five highest-yield pairs. (b) Model for the arrangement of FliGN domains based on the crosslinking. The five highest-yield Cys pairs are shown, connected by magenta lines. Dashed lines indicate segments with predicted non-regular secondary structure, 4–5 residues in length, whose conformation is uncertain. Numbers are for the protein of E. coli. (c) Mapping the suppressors of the original FliF–FliG fusion (Fig. 2d and Table 1) onto the model. Mutations cluster at the FliGN–FliGN interface. One of the suppressors is a 6-aa deletion that corresponds with the loop connecting H4 and H5.
On treatment with I2, several Cys pairs in FliGN gave rise to ladders of crosslinked products. Crosslinking was most efficient for the 42/97, 42/94, and 41/81 pairs, which formed ladders of 6–9 subunits. The formation of extensive ladders indicates that FliGN, like the other domains of FliG, is present in a regular, closely spaced array. To develop a model of FliGN arrangement, we assumed that H5 and H6 are distinct helices, as observed in A. aeolicus FliG crystal structure [27] and as indicated by secondary-structure prediction tools (which place helices H5 and H6 at around residues 74–83 and 87–98 in the E. coli protein) [41,44]. Residues that crosslinked most efficiently were positioned near each other, and the N terminus of H5 was placed near the C terminus of H4. Helices H5 and H6 were positioned side by side, because residues near the middles of both (80 and 81 in H5, 97 in H6) formed strong crosslinks to adjacent residues (41 and 42) in the hydrophobic patch. An antiparallel orientation of H5 and H6 was assumed because the segment linking them is short. In the resulting model (Fig. 7b), the spacing between FliGN units is approximately 4 nm. The interface between adjacent domains is formed from the conserved hydrophobic patch at one end of the domain and hydrophobic groups in H5 and H6 at the other end. Notably, all of the FliGN point mutations that suppressed the chemotactic defect of the original fusion strain (Table 1) map to this interface (Fig. 7c). One suppressor was a six-residue deletion in the loop connecting H4 and H5; although not included explicitly in the structural model (due to its uncertain conformation), this loop would also be in proximity to the FliGN–FliGN interface.
Binding of wild-type FliG to the FliF part of the fusion
The bias-normalizing effect of FliG expressed in the original fusion strain was shown to involve the N-terminal domain and was prevented by mutations in the domain that disrupt its binding to FliFC (Fig. 2c) [35]. This suggests that the “extra” FliG can form normal contacts with the FliF part of the fusion protein, in spite of the presence of the covalently attached FliG moiety. Interaction between the FliF–FliG fusion protein and native-sized FliG was examined in a crosslinking experiment with Cys introduced at position 528 in the FliF part of the fusion protein and at three positions in native-sized, plasmid-expressed FliG (residues 17, 56, and 59) that are near FliF-528 in the crystal structure and that were previously shown to crosslink to it [35]. Results of an experiment using H2O2 as oxidant are shown in Fig. S13. In both the old and new fusion strains, plasmid-expressed FliG was observed to crosslink to the FliF part of the fusion protein. Yields were low but followed the same pattern of relative intensities (56 > 59 > 17) observed in the previous crosslinking study with unfused proteins [35]. Thus, the FliGN domain of the wild-type protein can bind to the FliF part of the fusion protein to displace the covalently linked FliGN moiety to at least a small extent.
Discussion
The previous characterization of a FliF–FliG fusion strain [17] left open the question of whether a significant amount of native-sized FliG might be incorporated into the motor alongside the fusion protein. While native-sized FliG was not detected in basal bodies from the fusion strain, it was also not detected in basal-body preparations from wild-type cells, because the relatively harsh purification methods used at the time caused FliG to be lost from the basal-body preparations except when fused covalently to FliF. The present results with both the original fusion strain and the fusion with a nine-residue linker indicate that FliG of normal size is present at only a very low level both in whole cells and in the membrane fraction. The assembly and function of the flagella in the fusion strains must therefore be due to the FliF–FliG fusion protein, and the nearly normal function of the new fusion-linker strain implies that flagella containing equal numbers of FliF and FliG can assemble, produce normal torque, and switch direction properly in response to signals from the chemotaxis pathway. The accepted value for FliF copy number is ~25, varying somewhat from specimen to specimen [6,26,45]. Accordingly, we suggest that FliG is present in ~25 copies in the motor, as had been supposed on the basis of the original study [17]. A corollary is that the number of FliG subunits does not equal that of FliM, for which the consensus value is ~34 as determined from the symmetry of the lower and middle parts of the C-ring [6,46]. Models based on equal numbers of FliG and FliM subunits thus appear unlikely.
An important, and debated, feature of FliG organization concerns the interaction between FliGC and FliGM. Previously, we suggested that this interaction does not occur in the motor, except possibly at an intermediate stage of assembly [23]. On the basis of the present results and the recent findings of Sircar et al. [29] and Baker et al. [36], we believe that the interaction does occur as Lee et al. proposed [27] and has a central role in the organization of FliG subunits in the motor. The failure to detect the interaction in previous experiments [23] may have been due to excessive expression of the plasmid-expressed protein, to the use of the HA tag, which caused a partial motility defect, or to both.
A model that accounts for all of the data must therefore combine the FliGC–FliGM interaction from the model of Lee and co-workers [27] with the FliG copy number (~25) of the Paul et al. model [23]. A working hypothesis is shown in Fig. 8. An important element of the model is the variable conformation of the segment linking FliGM to FliGC: the majority of segments are assumed helical, as observed in most crystal structures, but a subset (about nine, equaling the difference between FliM and FliG copy numbers) adopts an extended conformation that allows them to bridge the gaps in the array resulting from the number mismatch. Both FliG and FliM subunits then occur in multiple environments within the switch complex, as suggested previously [23], but in an arrangement different from that proposed before. In particular, although the segments in extended conformation are likely to contact the top of FliMM, the direct FliGC–FliMM interaction proposed before [23], which would interrupt the array of FliGC–FliGM interactions and shorten the ladders of 158/214 crosslinked products, appears unlikely. Evidence for that interaction came from crosslinking and glutathione S-transferase-pull-down experiments. Crosslinks in that study were weaker than those reported here and may have reflected the effects of overexpression from plasmids. Subsequent work has shown that FliG is prone to non-specific interactions in glutathione S-transferase-pull-down experiments: an initially reported interaction with H-NS [39] was subsequently shown to be unlikely [47]. In experiments with FliMM as well, the present investigators have found that washes stringent enough to give reliably clean negative controls do not preserve a robust interaction with FliG.
Fig. 8.
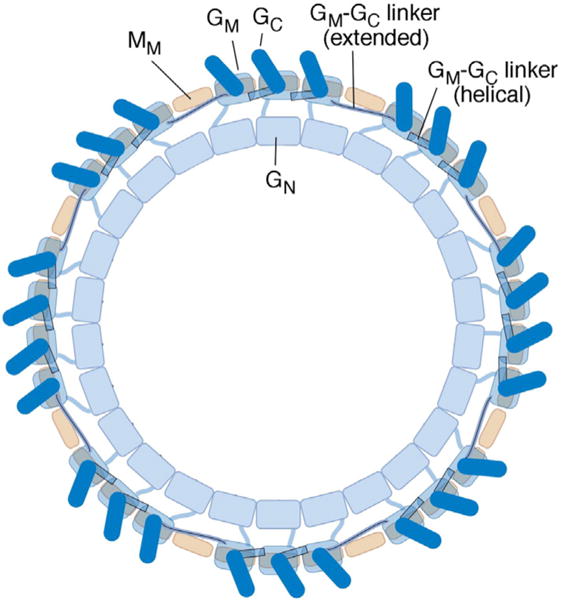
Model for the organization of FliG subunits in the flagellar switch complex. The view is from the top (the membrane-proximal side). The three domains of FliG, and the middle domain of FliM, are shown. Copy numbers of FliG and FliM vary somewhat from specimen to specimen [46]; the complex pictured has 26 FliG and 34 FliM subunits. Each FliGM domain (present in 26 copies) rests on a FliMM domain (present in a 34-fold array). To accommodate the copy-number mismatch while allowing every FliGC to stack onto the FliGM domain of an adjacent subunit, the segment linking FliGM to FliGC takes on an extended rather than helical conformation at the gap positions.
The recent crystal structure of a FliGN–FliFC complex [35] provided guidance for targeted crosslinking experiments to examine the organization of FliGN. The results also shed light on the dispositions of FliGN helices H5 and H6, which are positioned differently in different crystal structures [27,35]. In the resulting model (Fig. 7), adjacent FliGN domains contact each other directly in an array with an inter-subunit spacing of about 4 nm. Twenty-five such FliGN units would form a ring with a diameter of ~32 nm, a satisfactory match to the inner-ring feature seen in electron microscopic (EM) reconstructions (which exhibits ~25-fold symmetry [6]). We note that helices H4 and H5 are assumed to be distinct helices in the model, even though they form a single continuous helix in one of the molecules in the asymmetric unit of the FliFC/FliGN crystal structure, and small-angle x-ray scattering data suggest an extended conformation in the isolated domain in solution [35]. They are distinct helices in the other molecule in the asymmetric unit of that structure and also in the crystal structure of A. aeolicus FliG [27]. Secondary structure predictions using diverse FliG sequences [44] strongly favor distinct helices, and certain FliG sequences (e.g., those from Rhodobacter sphaeroides or Fulvimarina pelagi) contain Gly-or Pro-rich segments in the predicted intervening linker.
In both the present model and our previous proposal for switch complex architecture [23], there is a symmetry mismatch between the inner part of the C-ring (25-fold) and the outer parts (34-fold). In our previous model, FliGM was assumed to be a part of the inner ring, and the segment linking FliGM to FliGC was required to take on different conformations to accommodate the symmetry mismatch. In the present model, the symmetry mismatch occurs between the FliGN and FliGM domains (Fig. 8), which, in E. coli, are joined by the segment comprising approximately residues 99–106. This segment is helical in the Aquifex crystal structure [27] but has strong coil character in a Jpred [44] pre diction based on diverse FliG sequences. Certain FliG sequences give strong indications of flexibility, such as the stretch GGGG in Thiobacillus denitrificans, and GGGS in Idiomarina loihiensis. We suggest that segment 99–106 is a flexible linker that has the major role in reconciling the different symmetries in the inner (FliGN) and outer (FliGM/FliGC) parts of the complex.
The presence of a continuous inner ring of interacting FliGN domains provides a framework for rationalizing the CW bias of the original fusion strain. We suggest that the strong CW bias in this strain results from force exerted by this inner ring on the outer parts of the switch. The suppressing mutations in FliGN (Table 1 and Fig. 7c) would then act by weakening the interaction between adjacent FliGN domains, effectively fragmenting the ring and relieving the strain. Supplementation with additional FliGN domains (Fig. 2c) could alleviate the strain either by enlarging the inner ring or by binding to the FliF part of the resident fusion protein (Fig. 7d) to disrupt the ring. The newer FliF–FliG fusion (Fig. 3) appears to not induce such stresses, presumably because its linker allows a more normal conformation. Given the nearly normal motility of this strain, it is not surprising that its function was harmed rather than helped by extra FliG (Fig. S5).
If the copy number of FliG is tied to that of FliF, a typical motor will contain about 25 copies of FliG, and that number should remain fairly fixed (or change only slowly as the MS-ring itself loses or gains subunits). The FliM number, treated as ~34 in the discussion above, is not tied directly to that of other components and should be free to vary over some range, with additional FliM units being accommodated by the conversion of more FliG subunits to the extended conformation. These considerations are relevant to the process of adaptive remodeling, in which overly CCW-biased motors tend to incorporate additional FliM (and also FliN) subunits and become more CW-biased in the process [30–33]. Motors that turn exclusively CCW have been found to incorporate as many as 44 copies of FliM [31], which would require a substantial increase in the diameter of the C-ring. Such enlargement would be accommodated in the present model by allowing ~19, rather than ~9, FliG subunits to adopt the extended conformation. This would increase the diameter of the outer (FliGC/FliGM) but not the inner (FliGN) parts of the switch; the resulting strains might contribute to the changes in intrinsic motor bias (in this case, increased CW probability) that accompany remodeling.
Most discussions of motor physiology and proton-utilization stoichiometry have assumed ~25 copies of FliG, as was indicated by the original fusion study [17]. The present results validate this number but suggest that there might be gaps in the array of FliGC domains. Physiological studies of the motor have shown that rotation occurs in steps of about 14° [48,49], corresponding to ~25 steps per revolution. The present model predicts that the steps might actually be of two kinds, most slightly smaller than the average but some—those at the gap position—significantly larger. Additional physiological studies are likely to shed additional light on this question and might provide a crucial test of the model.
Previous studies indicated that switching might involve a rocking motion in FliMM, leading to a fairly large rotation of the FliGC domain [50]. While previous conclusions regarding FliM will likely hold, movements in the upper parts of the switch may be altered, owing to the new model for FliG organization. Further experiments are thus needed to examine the movements responsible for direction switching.
Materials and Methods
Strains and media
E. coli strains were derivatives of RP437, a gift from J.S. Parkinson. A full listing of strains and plasmids is provided in Table S4. Site-directed mutations were made using the QuikChange method (Stratagene). DNA sequencing and oligonucleotide synthesis were carried out by core facilities at the University of Utah and by Genewiz, LLC (South Plainfield, NJ). Mutations were transferred into the chromosome using the lambda-Red recombinase method [51] and confirmed by sequencing. Tryptone Broth (TB) medium contained 10 g tryptone and 5 g NaCl per liter. LB medium contained the same plus 5 g yeast extract. Soft-agar motility plates contained TB media and Bacto-agar at 2.6 g/l. Ampicillin was used at 100 μg/ml in liquid media and selective plates and at 50 μg/ml in soft-agar motility plates. Chloramphenicol was used at 50 μg/ml in liquid media and selective plates and at 25 μg/ml in motility plates. IPTG and sodium salicylate were prepared as aqueous 0.1-M stocks and used at the concentrations indicated in the figures.
Motility assay
Motility in soft (0.26%) agar was measured as described previously [52]. Rates are reported relative to wild-type controls measured in the same experiment. In experiments that measure the effects of expressing FliG or its mutant variants in the FliF–FliG fuision strains, the strains (EKS17 or EKS102) were transformed with variants of plasmid pKP619 and cultured in the presence of 200 μM IPTG. Controls carried the parent vector pRR48. To assay the effects of fliG mutations on motility, we transformed mutant variants of pKP619 into the fliG null strain DB225 and cultured it in the presence of 100 μM IPTG.
Rates are reported relative to controls carrying the fliG+ plasmid pKP619.
Crosslinking
Disulfide crosslinking was carried out using intact cells, essentially as described previously [43]. For most crosslinking experiments, single- and double-Cys mutant proteins were expressed from the normal chromosomal loci. Exceptions that used plasmid-expressed proteins are noted in the figures. Cells were cultured overnight to saturation in LB, then diluted 100-fold into TB plus antibiotic and inducer, and recultured at 32 °C to mid-log (typically 5 h). Cells were collected by centrifugation and washed into XL buffer [200 mM Na-phosphate and 150 mM NaCl (pH 7.5)]. Oxidation with I2 (1 mM) iodine or Cu-phenanthroline (0.2 mM) was carried out as described previously [43]. Reaction was quenched by the addition of 2.2 μl 1 M NEM stock. For oxidation by hydrogen peroxide, 100 μl samples of cells were mixed with 5.1 μl 0.98 M peroxide (final peroxide concentration 48 mM), cells were incubated at room temperature for 5 min, and peroxide was removed by the addition of catalase (1 μl of an 85,000 units/ml stock) followed by incubation for 5 min at room temperature. Remaining sulfhydryls were quenched by the addition of 2 μl 0.5 M NEM. Samples were mixed with an equal volume of 2X non-reducing loading buffer and heated at 95 °C for 10 min before loading on SDS-PAGE gels. For the crosslinking of native-sized FliG to the FliF–FliG fusion protein, the FliF I528C mutations were made in the chromosomally encoded fliF–fliG fusion, and Cys-containing FliG variants were expressed from derivatives of plasmid pKP619, induced with 200 μM IPTG. Oxidation was by iodine, Cu-phenanthroline, or hydrogen peroxide, as described above. For crosslinking experiments using the bifunctional reagent BMH, the reagent was prepared as a 50-mM stock in DMSO and used at a final concentration of 1 mM. Following a 30-min incubation on ice, reaction was quenched with 2.5 μl 2-mercaptoethanol.
SDS page and immunoblotting
Proteins were resolved in 7.5% or 10% SDS-PAGE gels and transferred onto nitrocellulose using a Transblot turbo apparatus (BioRad). Gels for most experiments used a 40:1 acrylamide:bis-acrylamide ratio, but larger pore size (80:1 acrylamide: bis-acrylamide) was found to be more effective for resolving ladders of high-MW products observed with the 158C/214C mutant, particularly with the FliF–FliG fusion protein. Then, 4–20% gradient gels (BioRad) were also used for some experiments involving large product ladders, as indicated in the figures. Rabbit polyclonal antibodies against FliG and FliM were used at 1:1000 dilution in a solution containing phosphate-buffered saline (pH 7.4), 0.1% gelatin, and 0.01% Na-azide. Immunoblots were visualized and analyzed using the LiCor Odyssey infrared-imaging system.
Membrane fractionation
Cells of the fusion strains (EKS17 having the 7-nt deletion or EKS102 containing a linker) were cultured overnight at 32 °C, diluted 100-fold into TB, and then grown at 32 °C for 4.5 h (to OD600 of about 0.6). For some experiments, cells were transformed with plasmid pDB97 to allow the overexpression of FliG (as indicated in the appropriate lanes in the figures). OD600 was measured to adjust the cell density; then, equal numbers of cells were pelleted and resuspended in 200 μl of lysis buffer [50 mM Tris (pH 8.0), 0.5 M sucrose, 10 mM EDTA, and 0.2 mg/ml lysozyme]. Cells were incubated on ice for 1 h then subjected to osmotic shock by the addition of 1.8 ml of ice-cold water. Samples were sonicated briefly (Branson model 450 or model 250; power 3, duty cycle 50% for 10 s) and then centrifuged (16,000g for 30 min at 4 °C) to separate membrane and cytosolic fractions. Pellets (membrane) were resuspended in 1× reducing dye. The supernatant (cytosol) was transferred to a new tube, mixed with 10% trichloroacetic acid solution and incubated on ice for 10 min, then centrifuged (16,000g for 10 min at 4 °C), washed with acetone, and centrifuged once more. Precipitated protein was resuspended in 1× reducing dye and immunoblotted with anti-FliG and anti FliM antibodies.
Tethering
Cells were grown overnight at 32 °C in TB, then diluted 100-fold in TB containing antibiotic and inducer, and grown at 32 °C for 3.5 h. Flagella were shorn by blending (15-ml cells; 15-s blending, 10-s rest, 15-s blending, 10-s rest). After shearing, cells were viewed microscopically to ensure the complete loss of motility. A 3-ml aliquot was removed, and cells were pelleted by centrifugation at 4 °C, resuspended in 3 ml of 1:3 motility buffer [0.1 mM methionine, 10 mM Na-EDTA, 70 mM NaCl, and .075% lactic acid (pH adjusted to 7.0)], repelleted, and resuspended in 3 ml of 1:3 motility buffer. A 100-μl aliquot was removed to a micro-centrifuge tube, anti-FliC antibody (20 μl of a 1:5 dilution in PBS) was added, and cells were pipetted onto a glass cover slide that was placed inside a humidified chamber and incubated for 30 min. During the incubation period, a tunnel slide was prepared. At the end of incubation, the tunnel slide was inverted and placed on top of the cover slide containing the incubated cells and antibody. Any cells not tethered to the cover slide were washed away using 1:3 motility buffer. Tethered cells were viewed microscopically. Rotational rates were measured by slow-speed video playback.
Supplementary Material
Acknowledgments
We thank Shahid Khan for helpful discussions and comments on the manuscript. This work is supported by NIH grant GM-46683 to D.F.B. and B.R.C. M.L. was supported by NIH Training grant T32GM008500.
Abbreviations
- CW
clockwise
- CCW
counterclockwise
- MS
membrane/supra-membrane
- BMH
bis-maleimidohexane
- NEM
N-ethyl maleimide
- EM
electron microscopic
- TB
Tryptone broth
Appendix A. Supplementary Data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2017.02.014.
Footnotes
Edited by Charalampo Kalodimos
Note
Recent electron miscopic reconstructions at higher resolution indicate that the MS-ring actually has 34-fold symmetry rather than the ~25-fold symmetry that had been accepted (Keiichi Namba, private communication). This new finding alters the interpretation of the present results, as follows. In the purified basal body, no mismatch between FliG and FliM copy numbers is expected; thus, the extended conformation of FliG hypothesized to span the gaps at mismatch positions should not occur. Flagellar motors in cells have been found to undergo remodeling that results in the presence of substantially larger numbers of FliM subunits, however. Thus, the subunit mismatch is still expected to occur in cells under most circumstances, and a means of accommodating it, such as the hypothesized FliG extension, is still necessary. The presence of additional copies of FliF would result in a tighter packing of FliG N-terminal domains but would not otherwise affect the present conclusions regarding FliG domain organization.
References
- 1.Berg HC, Anderson RA. Bacteria swim by rotating their flagellar filaments. Nature. 1973;245:380–382. doi: 10.1038/245380a0. [DOI] [PubMed] [Google Scholar]
- 2.Larsen SH, Adler J, Gargus JJ, Hogg RW. Chemomechanical coupling without ATP: the source of energy for motility and chemotaxis in bacteria, Proc. Natl Acad Sci U S A. 1974;71:1239–1243. doi: 10.1073/pnas.71.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirota N, Kitada M, Imae Y. Flagellar motors of alkalophilic Bacillus are powered by an electrochemical potential gradient of Na+ FEBS Lett. 1981;132:278–280. [Google Scholar]
- 4.Larsen SH, Reader RW, Kort EN, Tso WW, Adler J. Change in direction of flagellar rotation is the basis of the chemotactic response in E. coli. Nature. 1974;249:74–77. doi: 10.1038/249074a0. [DOI] [PubMed] [Google Scholar]
- 5.DePamphilis ML, Adler J. Fine structure and isolation of the hook-basal body complex of flagella from Escherichia coli and Bacillus subtilis. J Bacteriol. 1971;105:384–395. doi: 10.1128/jb.105.1.384-395.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas DR, Francis NR, Xu C, DeRosier DJ. The three-dimensional structure of the flagellar rotor from a clockwise-locked mutant of Salmonella enterica Serovar typhimurium. J Bacteriol. 2008;188:7039–7048. doi: 10.1128/JB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan S, Dapice M, Reese TS. Effects of mot gene expression on the structure of the flagellar motor. J Mol Biol. 1988;202:575–584. doi: 10.1016/0022-2836(88)90287-2. [DOI] [PubMed] [Google Scholar]
- 8.Chun SY, Parkinson JS. Bacterial motility: membrane topology of the Escherichia coli MotB protein. Science. 1988;239:276–278. doi: 10.1126/science.2447650. [DOI] [PubMed] [Google Scholar]
- 9.Blair DF, Berg HC. The MotA protein of E. coli is a proton-conducting component of the flagellar motor. Cell. 1990;60:439–449. doi: 10.1016/0092-8674(90)90595-6. [DOI] [PubMed] [Google Scholar]
- 10.Sato K, Homma M. Functional reconstitution of the Na(+)-driven polar flagellar motor component of Vibrio alginolyticus. J Biol Chem. 2000;275:5718–5722. doi: 10.1074/jbc.275.8.5718. [DOI] [PubMed] [Google Scholar]
- 11.Kojima S, Blair DF. Conformational change in the stator of the bacterial flagellar motor. Biochemistry. 2001;40:13,041–13,050. doi: 10.1021/bi011263o. [DOI] [PubMed] [Google Scholar]
- 12.Kojima S, Blair DF. The bacterial flagellar motor: structure and function of a complex molecular machine. Int Rev Cytol. 2004;233:93–134. doi: 10.1016/S0074-7696(04)33003-2. [DOI] [PubMed] [Google Scholar]
- 13.Wadhams GH, Armitage JP. Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol. 2004;5:1024–1037. doi: 10.1038/nrm1524. [DOI] [PubMed] [Google Scholar]
- 14.Sowa Y, Berry RM. Bacterial flagellar motor. Q Rev Biophys. 2008;41:103–132. doi: 10.1017/S0033583508004691. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto YV, Minamino T. Structure and function of the bidirectional bacterial flagellar motor. Biomol Ther. 2014;18:217–234. [Google Scholar]
- 16.Yamaguchi S, Aizawa SI, Kihara M, Isomura M, Jones CJ, Macnab RM. Genetic evidence for a switching and energy-transducing complex in the flagellar motor of Salmonella typhimurium. J Bacteriol. 1986;168:1172–1179. doi: 10.1128/jb.168.3.1172-1179.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francis NR, Irikura VM, Yamaguchi S, DeRosier DJ, Macnab RM. Localization of the Salmonella typhimurium flagellar switch protein FliG to the cytoplasmic M-ring face of the basal body. Proc Natl Acad Sci U S A. 1992;89:6304–6308. doi: 10.1073/pnas.89.14.6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis NR, Sosinsky GE, Thomas D, DeRosier DJ. Isolation, characterization and structure of bacterial flagellar motors containing the switch complex. J Mol Biol. 1994;235:1261–1270. doi: 10.1006/jmbi.1994.1079. [DOI] [PubMed] [Google Scholar]
- 19.Khan S, Khan IH, Reese TS. New structural features of the flagellar base in S. typhimurium revealed by rapid-freeze electron microscopy. J Bacteriol. 1991;173:2888–2896. doi: 10.1128/jb.173.9.2888-2896.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou J, Lloyd SA, Blair DF. Electrostatic interactions between rotor and stator in the bacterial flagellar motor. Proc Natl Acad Sci U S A. 1998;95:6436–6441. doi: 10.1073/pnas.95.11.6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takekawa N, Kojima S, Homma M. Contribution of many charged residues at the stator-rotor interface of the Na+-driven flagellar motor to torque generation in Vibrio alginolyticus. J Bacteriol. 2014;196:1377–1385. doi: 10.1128/JB.01392-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SY, Lowder B, Bilwes AM, Blair DF, Crane BR. Structure of FliM provides insight into the assembly of the switch compex in the bacterial flagella motor. Proc Natl Acad Sci U S A. 2006;103:11,886–11,891. doi: 10.1073/pnas.0602811103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paul K, Gonzalez-Bonet G, Bilwes AM, Crane BR, Blair DF. Architecture of the flagellar rotor. EMBO J. 2011;30:2962–2971. doi: 10.1038/emboj.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vartanian AS, Paz A, Fortgang EA, Abramson J, Dahlquist FW. Structure of flagellar motor proteins in complex allows for insights into motor structure and switching. J Biol Chem. 2012;287:35,779–35,783. doi: 10.1074/jbc.C112.378380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paul K, Blair DF. Organization of FliN subunits in the flagellar motor of Escherichia coli. J Bacteriol. 2006;188:2502–2511. doi: 10.1128/JB.188.7.2502-2511.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H, Yonekura K, Namba K. Structure of the rotor of the bacterial flagellar motor revealed by electron cryomicroscopy and single-particle image analysis. J Mol Biol. 2004;337:105–113. doi: 10.1016/j.jmb.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 27.Lee LK, Ginsburg MA, Crovace C, Donohoe M, Stock D. Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature. 2010;466:996–1000. doi: 10.1038/nature09300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas D, Morgan DG, DeRosier DJ. Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. J Bacteriol. 2001;183:6404–6412. doi: 10.1128/JB.183.21.6404-6412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sircar R, Peter P, Borbat PR, Bhatnager J, Beyersdorf MS, Halkides CJ, et al. Assembly states of FliM and FliG within the flagellar switch complex. J Mol Biol. 2015;427:867–886. doi: 10.1016/j.jmb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan J, Branch RW, Hosu BG, Berg HC. Adaptation at the output of the chemotaxis signalling pathway. Nature. 2012;484:233–236. doi: 10.1038/nature10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lele PP, Branch RW, Nathan VS, Berg HC. Mechanism for adaptive remodeling of the bacterial flagellar switch. Proc Natl Acad Sci U S A. 2012;109:20,018–20,022. doi: 10.1073/pnas.1212327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delalez NJ, Berry RM, Armitage JP. Stoichiometry and turnover of the bacterial flagellar switch protein FliN. MBio. 2014;5:e01216–14. doi: 10.1128/mBio.01216-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Branch RW, Sayegh MN, Shen C, Nathan VSJ, Berg HC. Adaptive remodelling by FliN in the bacterial rotary motor. J Mol Biol. 2014;426:3314–3324. doi: 10.1016/j.jmb.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown PN, Terrazas M, Paul K, Blair DF. Mutational analysis of the flagellar protein FliG: sites of interaction with FliM and implications for organization of the switch complex. J Bacteriol. 2007;189:305–312. doi: 10.1128/JB.01281-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynch MJ, Levenson R, Kim EA, Sircar R, Blair DF, Dahlquist FW, et al. Cofolding of a FliF-FliG split domain forms the basis of the MS:C ring interface within the bacterial flagellar motor. Structure. 2017;25:317–328. doi: 10.1016/j.str.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker MA, Hynson RM, Ganuelas LA, Mohammadi NS, Liew CW, Rey AA, et al. Domain-swap polymerization drives the self-assembly of the bacterial flagellar motor. Nat Struct Mol Biol. 2016;23:197–203. doi: 10.1038/nsmb.3172. [DOI] [PubMed] [Google Scholar]
- 37.Brown PN, Hill CP, Blair DF. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J. 2002;21:3225–3234. doi: 10.1093/emboj/cdf332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lam KH, Ip WS, Lam YW, Chan SO, Ling TKW, Au SWN. Multiple conformations of the FliG C-terminal domain provide insight into flagellar motor switching. Structure. 2012;20:315–325. doi: 10.1016/j.str.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 39.Paul K, Carlquist WC, Blair DF. Adjusting the spokes of the flagellar motor with the DNA-binding protein H-NS. J Bacteriol. 2011;193:5914–5922. doi: 10.1128/JB.05458-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandini A, Morcos F, Khan S. The gearbox of the flagellar motor switch. Structure. 2016;24:1209–1220. doi: 10.1016/j.str.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 42.Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowder BJ, Duyvesteyn MD, Blair DF. FliG subunit arrangement in the flagellar rotor probed by targeted cross-linking. J Bacteriol. 2005;187:5640–5647. doi: 10.1128/JB.187.16.5640-5647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones CJ, Macnab RM, Okino H, Aizawa SI. Stoichiometric analysis of the flagellar hook-(basal body) complex of Salmonella typhimurium. J Mol Biol. 1990;212:377–387. doi: 10.1016/0022-2836(90)90132-6. [DOI] [PubMed] [Google Scholar]
- 46.Young HS, Dang H, Lai Y, DeRosier DJ, Khan S. Variable symmetry in Salmonella typhimurium flagellar motors. Biophys J. 2003;84:571–577. doi: 10.1016/S0006-3495(03)74877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim EA, Blair DF. Function of the histone-like protein H-NS in motility in Escherichia coli: multiple regulatory roles rather than direct action at the flagellar motor. J Bacteriol. 2015;197:3110–3120. doi: 10.1128/JB.00309-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sowa Y, Rowe AD, Leake MC, Yakushi T, Homma M, Ishijima A, et al. Direct observation of steps in rotation of the bacterial flagellar motor. Nature. 2005;437:916–919. doi: 10.1038/nature04003. [DOI] [PubMed] [Google Scholar]
- 49.Nakamura S, Kami-Ike N, Yokota JP, Minamino T, Namba K. Evidence for symmetry in the elementary process of bidirectional torque generation by the bacterial flagellar motor. Proc Natl Acad Sci U S A. 2010;107:17,616–17,620. doi: 10.1073/pnas.1007448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paul K, Brunstetter D, Titen S, Blair DF. A molecular mechanism of direction switching in the flagellar motor of Escherichia coli. Proc Natl Acad Sci U S A. 2011;108:17,171–17,176. doi: 10.1073/pnas.1110111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang H, Blair DF. Regulated underexpression of the FliM protein of Escherichia coli and evidence for a location in the flagellar motor distinct from the MotA/MotB torque generators. J Bacteriol. 1995;177:3485–3495. doi: 10.1128/jb.177.12.3485-3495.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, Eliceiri KW. NIH image to image J: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sockett H, Yamaguchi S, Kihara M, Irikura VM, Macnab RM. Molecular analysis of the flagellar switch protein FliM of Salmonella typhimurium. J Bacteriol. 1992;174:793–806. doi: 10.1128/jb.174.3.793-806.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.