

Abstract
Background:
ETV6/RUNX1 gene fusion is the most frequently seen chromosomal abnormality in childhood acute lymphobastic leukamia (ALL). However, additional genetic changes are known to be required for the development of this type of leukaemia. Therefore, we here aimed to assess the somatic mutational profile of four ALL cases carrying the ETV6/RUNX1 fusion gene using whole-exome sequencing.
Methods:
DNA was isolated from bone marrow samples using a QIAmp DNA Blood Mini kit and subsequently sequenced using the Illumina MiSeq system.
Results:
We identified 12,960 to17,601 mutations in each sample, with a total of 16,466 somatic mutations in total. Some 15,533 variants were single nucleotide polymorphisms (SNPs), 129 were substitutions, 415 were insertions and 389 were deletions. When taking into account the coding region and protein impact, 1,875 variants were synonymous and 1,956 were non-synonymous SNPs. Among non-synonymous SNPs, 1,862 were missense, 13 nonsense, 35 frameshifts, 11 nonstop, 3 misstart, 15 splices disrupt and 17 in-frame indels. A total of 86 variants were located in leukaemia-related genes of which 32 variants were located in the coding regions of GLI2, SP140, GATA2, SMAD5, KMT2C, CDH17, CDX2, FLT3, PML and MOV10L1.
Conclusions:
Detection and identification of secondary genetic alterations are important in identifying new therapeutic targets and developing rationally designed treatment regimens with less toxicity in ALL patients.
Keywords: Whole-exome sequencing, ETV6/RUNX1 fusion gene, acute lymphoblastic leukaemia
Introduction
B-cell precursor acute lymphoblastic leukaemia (ALL) is the most common malignant disorder in children with incidence around 0.9-4.7 per 100,000 children per year worldwide (Awan et al., 2012; Hicks et al., 2013). The t(12;21)(p13;q22) translocation encoding the ETV6/RUNX1- fusion gene is the most common chromosomal abnormality detected in 20-25% of childhood ALL cases (Inaba et al., 2013; Ney-Garcia et al., 2012). The event-free survival (EFS) of childhood ALL patients after first-line therapy are approximately 80% with more favourable prognosis (EFS; 90%) in the presence of the ETV6/RUNX1 fusion gene (Aljamaan et al., 2015; Borst et al., 2012).
Initiation of the ETV6/RUNX1 fusion gene can occur as early as in the prenatal B-cell progenitor cells. However, the fusion gene alone is incapable of causing leukaemia and requires additional genetic changes (Lilljebjorn et al., 2010). Therefore, identification of the genetic changes and genetic evolution that co-exist with ETV6/RUNX1 fusion gene is necessary for both prognosis and disease treatment (Goud et al., 2015; Ilyas et al., 2015). Currently, the tools for identifying genetic changes in childhood leukaemia are array Comparative Genomic Hybridization (array CGH) and Single Nucleotide Polymorphism array (SNP array) (van der Veken and Buijs, 2011; Zakaria et al., 2012). However, the usefulness of these technologies is confined by technology limitations such as genomic probes limitation while array CGH only measures copy number variants (Zhang et al., 2011). With the advancement of sequencing technology such as Next Generation Sequencing (NGS), novel genetic alterations that contribute towards cancer progression have been discovered in neoplastic cells (Kalender Atak et al., 2012; Obata et al., 2015; Valli et al., 2011; Wang et al., 2014a; Wang et al., 2014b).
The whole-exome sequencing technology becomes the major focus and is used extensively in genetic studies because of its robustness, cost-effective and requires less handling of data (Guo et al., 2012; Ku et al., 2012). It can maximize the efficiency of detection by characterizing the majority of protein-coding regions of interest and identifying somatic and germline mutations in the sample within a shorter period of time (Chang et al., 2013; Liu et al., 2012). In this paper, we report whole-exome sequencing of four ALL cases carrying the ETV6/RUNX1 fusion gene and sequence variations in the coding region of the genome as preliminary data on mutational profile in patients in our population.
Materials and Methods
Patient samples
Four bone marrow samples of childhood leukaemia patients aspirated in EDTA tubes were obtained at the time of diagnosis from the Kuala Lumpur Paediatric Institute. The patients were confirmed carrying the ETV6/RUNX1 fusion gene by multiplex reverse transcriptase PCR – HemaVision®-28N (DNA Technology A/S, Denmark). This study was reviewed and approved by the Medical Research and Ethics Committee (MREC), Ministry of Health Malaysia.
Exome-sequencing and data analysis
Prior to sequencing, genomic DNA was isolated from individual bone marrow samples using QIAmp DNA Blood Mini kit (Qiagen, Valencia, California) as per manufacturer’s protocol with slight modifications. The quality of DNA was determined using Qubit 2.0 fluorometer and 1% (w/v) agarose gel. Genomic DNA was enriched for coding exons using Truseq® Exome Enrichment Kit (Illumina, San Diego, CA) and subsequently sequenced using Illumina MiSeq system.
Cluster intensities were extracted from the raw image data. Base calling and quality filtering were performed using Illumina pipeline software (RTA). The sequencing was performed using 2x250 paired-end (PE) reads and the raw reads were trimmed at Q-score 20, leaving the high quality reads mapping to the genome. Read files (FASTQ) were generated from the sequencing platform via Illumina’s CASAVA pipeline version 1.8.2. Sequence alignment and variant calling were performed against the human reference genome UCSC NCBI137/ hg19 using CLC Genomic Workbench version 7.5. In this analysis, the parameters were set with strict criteria. The length fraction accepts 80% of the read length with a 95% match to each other. The quality-based variant detection was based on the Neighbourhood Quality Standard (NQS) algorithm. After the filtering process, the FASTQ file was generated into VCF file for further analysis. Variant detection and analysis were subsequently performed using ENLIS Genomics software version 1.7 (Berkeley, CA). The potential somatic mutations were studied if they were non-synonymous, protein-damaging or rare somatic mutations in the leukaemia-related gene. The coding and non-synonymous variants were also predicted if they have the non-benign effect on the protein using Polyphen-2 and SIFT.
Sequencing validation
Primers for PCR amplifying regions with candidate mutations (in FLT3 and CDX2 genes) were designed using Primer3 (http://bioinfo.ut.ee/primer3/). The amplified DNA was sequenced using the BigDye terminator cycle sequencing kit (Life Technologies, Carlsbad, CA, USA) and run on 3730 Capillary DNA Analyzer. The sequences were visualized and analysed using CLC Main Workbench version 7.5.
Results
Exome sequencing performance
The exome sequencing was performed on 4 children with ALL carrying the ETV6/RUNX1 fusion gene. DNA was extracted from individual bone marrow aspirate who were from different ethnicity background (see Table 1). The sequence was aligned to the hg19 genome build (UCSC) using high stringency mapping parameters, which allowed for a maximum of 2 mismatches. The total number of reads generated per sample ranged from 13 to 20 gigabases (Gb). Of the raw reads, 99% could be aligned to the human reference genome with coverage ranged from a minimum of 20 times. Broken paired reads were removed from the raw data, leaving 12 to 18 million unique reads mapping to the genome (see Table 1).
Table 1.
Details of the DNA Extracted from 4 Childhood ALL Cases
| Sample | Age (year) | Sex | Race | Reads mapping to genome | Number of reads after trim | Total mapped % | Reads in pairs |
|---|---|---|---|---|---|---|---|
| Case 1 | 4 | M | Chinese | 14,479,748 | 14,479,657 | 14,350,725 (99.11) | 14,117,286 |
| Case 2 | 6 | M | Chinese | 14,139,404 | 14,139,322 | 14,019,895 (99.16) | 13,722,530 |
| Case 3 | 4 | F | Malay | 19,353,128 | 19,353,012 | 19,213,608 (99.28) | 18,598,962 |
| Case 4 | 3 | F | Malay | 13,102,050 | 13,101,976 | 13,012,367 (99.32) | 12,564,032 |
M, Male; F, Female
Overview of somatic mutations
The data in VCF file format were extensively analyzed using ENLIS Genomic software. This software provides a simple and user-friendly interface which contains different parts in the Analysis Tools; eg. the summary information of input variations, the distribution and details of variation annotation, and genes exploration by disease category (see Figure 1). High stringency parameters for SNP calling were used to allow us to identify variants with lower possibility of false-positive and high confidence results. We identified a range of 12,960-17,601 mutations in each sample (see Table 2).
Figure 1.
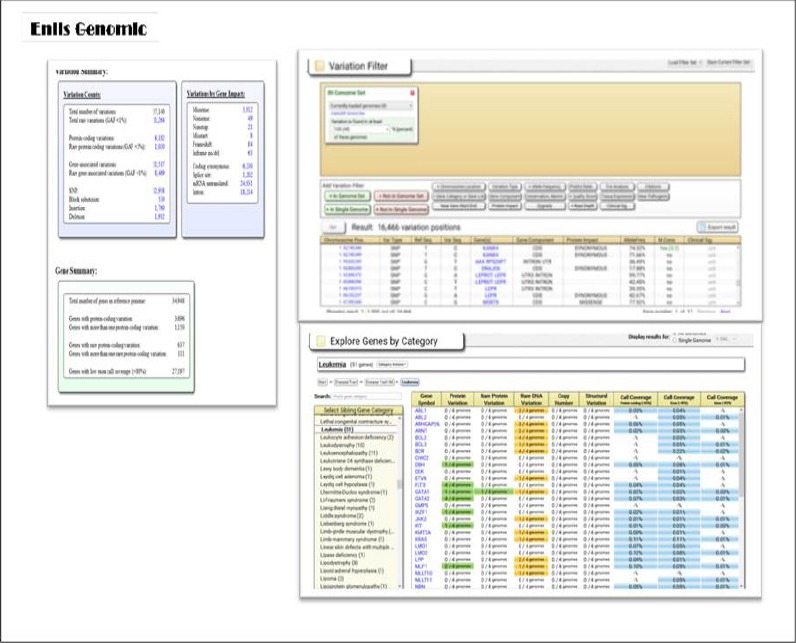
Screenshot of Enlis Genomic Interface
Table 2.
The Distribution of Somatic Mutation Per Cases
| Mutation types | Case 1 | Case 2 | Case 3 | Case 4 |
|---|---|---|---|---|
| Synonymous | 6,412 | 6,230 | 7,993 | 5,902 |
| Missense | 5,985 | 5,912 | 7,595 | 5,734 |
| Splice site | 1,267 | 1,202 | 1,703 | 1,084 |
| Nonsense | 48 | 49 | 65 | 44 |
| Nonstop | 22 | 21 | 26 | 25 |
| Misstart | 9 | 8 | 12 | 9 |
| Frameshift | 94 | 84 | 121 | 88 |
| Inframe ins/del | 62 | 65 | 86 | 74 |
| Total | 13,899 | 13,571 | 17,601 | 12,960 |
In total, an average of 14,508 variants (SNPs and indels) were identified per sample, with 55% of the cases were non-synonymous mutations. An average of 6,307 missense mutations and 72 indels were observed in each sample. The majority of the identified SNPs (97%) were within the coding regions of the genome (see Figure 2).
Figure 2.
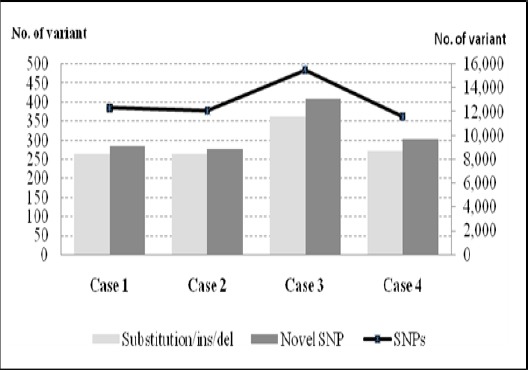
The Distribution of Somatic Mutations in Coding Region of Cases. The bar chart (left scale) represent individual samples carrying substitution/ins/del and novel SNPs. The line chart (right scale) represent single nucleotide polymorphism (SNPs) in individual ETV6/RUNX1 samples.
An average of 12,864 SNPs and 291 substitution/ insertion/ deletion (substitution/ ins/ del) were identified per sample, where 2.43% were novel SNPs. By analysing the cases as a group, a total of 16,466 somatic mutations were identified; 15,533 variants were SNPs, 129 substitutions, 415 insertions and 389 deletions. Among these, only a small percentage of the variants (23.26%) were located in the coding region of the genome and had a potential functional impact on the gene expression. The SNPs variants included 1,875 synonymous and 1,956 non-synonymous. For non-synonymous SNPs, it contained 1,862 missense, 13 nonsense, 35 frameshifts, 11 nonstop, 3 misstart, 15 splices disrupt and 17 inframe indels.
Among these, we identified 1,954 variants by removing synonymous or non-frameshift variants (Figure 3). Subsequently, 296 variants were found within genes that are implicated in a disease or trait within the OMIM database, 192 variants in the COSMIC cancer database, and 22 variants were classified as a novel after removing polymorphisms found in dbSNP137.
Figure 3.
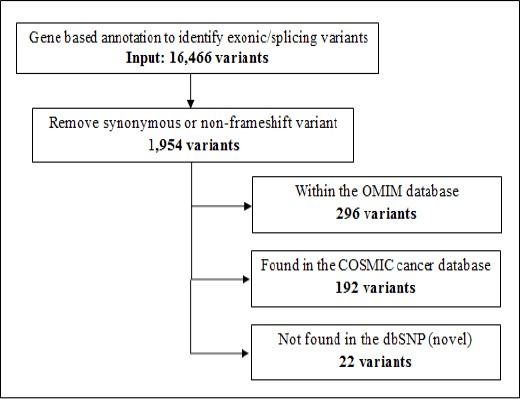
Identification of Novel Variants that Might Be Responsible in the Pathogenesis of ETV6/ RUNX1 Positive ALL. Filtering strategies using ENLIS Genomic software was used to identify somatic mutations in the exomes. Samples sequence was read as group on gene-based annotation to identify exonic or splicing variants. Synonymous or non-frameshift variant was removed from the list. A potential somatic mutations were compared with sites of known mutations in OMIM and COSMIC databases.
Examination of the sequencing variants was dependent on the filtering parameters. To evaluate the relationship of identified variants to the disease, we re-filtered the variants that were only located in the leukaemia-related genes. An average of 335 variants was identified whereby 73 of them were located in the coding region of the genome per sample (Figure 4). Interestingly, when analysing as a group (pool cases), only 86 variants were identified and 32 variants were located in the coding regions. We examined these 32 variants and found that 50% of them were nonsynonymous mutations comprising 15 missense and one frameshift mutation (Table 3).
Figure 4.
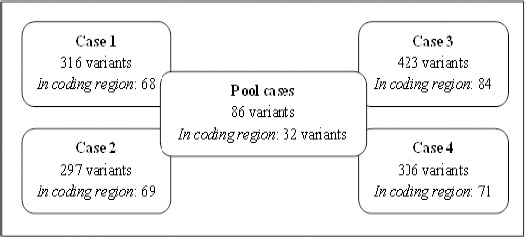
Identification of Variants Located in Leukaemia-Related Genes. Variants filtration was done individually and as a group (pool cases) based on published leukaemia-related genes. Some of these variants were already reported in previous findings elsewhere with different protein impact prediction.
Table 3.
Details of 32 Variants Identified in Four ALL Cases
| No. | Chr: Position | Gene symbol | Ref/ Mut | Protein Impact | Predicted protein change | PolyPhen-2 prediction | SIFT prediction** |
|---|---|---|---|---|---|---|---|
| 1. | 1: 47685455 | TAL1 | T>C | Synonymous | K-311-K | - | - |
| 2. | 2:100218080 | AFF3 | G>A | Synonymous | A-421-A; A-396-A | - | - |
| 3. | 2:121726447 | GLI2 | G>A | Synonymous | S-267-S | - | - |
| 4. | 2:121747406 | G>A | Missense | D-1306-N | Benign | Tolerated | |
| 5. | 2:121747429 | A>G | Synonymous | P-1313-P | - | - | |
| 6. | 2:231149108 | SP140 | G>A | Missense | E-489-K; E-516-K; E-456-K; E-402-K | Benign | Tolerated |
| 7. | 3:128204951 | GATA2 | C>T | Missense | A-164-T | Benign | Tolerated |
| 8. | 3:187447032 | BCL6 | G>A | Synonymous | N-387-N | - | - |
| 9. | 5:135513086 | SMAD5 | InsC | Frameshift | H-439-PS | Unknown | Unknown |
| 10. | 5:176636882 | NSD1 | C>T | Synonymous | C-225-C; C-494-C | - | - |
| 11. | 5:176637149 | G>A | Synonymous | E-314-E; E-583-E | - | - | |
| 12. | 5:176721198 | T>C | Synonymous | L-2008-L; L-2277-L | - | - | |
| 13. | 7:151927021 | KMT2C | C>A | Missense | C-988-F | Unknown | Damaging |
| 14. | 7:151932908 | T>C | Synonymous | L-921-L | - | - | |
| 15. | 7:151945007 | C>T | Missense | G-838-S | Unknown | Damaging | |
| 16. | 7:151945204 | G>A | Missense | S-772-L | Unknown | Damaging | |
| 17. | 7:151970856 | T>A | Missense | T-316-S | Possibly damaging | Tolerated | |
| 18. | 7:151970931 | G>A | Missense | L-291-F | Probably damaging | Unknown | |
| 19. | 8:90967711 | NBN | A>G | Synonymous | D-399-D | - | - |
| 20. | 8:95143172 | CDH17 | T>G | Missense | E-739-A | Benign | Tolerated |
| 21. | 8:95143186 | C>G | Missense | E-734-D | Benign | Tolerated | |
| 22. | 9:139391636 | NOTCH1 | G>A | Synonymous | D-2185-D | - | - |
| 23. | 10:104160434 | NFKB2 | A>G | Synonymous | A-607-A | - | - |
| 24. | 13:28537317 | CDX2* | G>A | Missense | P-293-S | Benign | Tolerated |
| 25. | 13:28624294 | FLT3* | G>A | Missense | T-227-M | Probably damaging | Tolerated |
| 26. | 15:74328116 | PML | A>G | Missense | S-772-G; S-724-G | Unknown | Tolerated |
| 27. | 19:15271771 | NOTCH3 | G>A | Missense | A-2223-V | Benign | Tolerated |
| 28. | 19:15285052 | T>C | Synonymous | P-1521-P | - | - | |
| 29. | 19:15295134 | G>A | Synonymous | C-846-C | - | - | |
| 30. | 19:15302844 | T>C | Synonymous | A-202-A | - | - | |
| 31. | 22:50555619 | MOV10L1 | C>T | Synonymous | L-431-L; L-411-L | - | - |
| 32. | 22:50582626 | A>G | Missense | Q-820-R; Q-800-R | Benign | Tolerated |
Chr, chromosome; Ref, reference; Mut, mutant;
Validated with PCR and cycle sequencing (data not shown);
SIFT prediction score ≤0.05
Sixteen mutations occurred in genes previously identified to be related to leukaemia. The mutated genes include GLI2, SP140, GATA2, SMAD5, KMT2C, CDH17, CDX2, FLT3, PML and MOV10L1. These variants were heterozygous and were predicted to alter the amino acid composition of the resulting proteins. Six variants (38%) were protein-damaging, according to either Polyphen or SIFT whereas others had either unknown /benign/tolerated significance.
Discussion
In the present study, we have sequenced four cases of ETV6/RUNX1 positive childhood ALLs using Next Generation Sequencing (NGS), in particular, whole-exome sequencing. This has allowed us, for the first time, to obtain an overview of the mutational profile of this subtype in our local population. After sequencing, each sample generated an average of 14.75 million paired reads with 99% matched to the reference genome (Table 1). Characterization of the matched sequences identified variants in different protein impact (Table 2, Figure 2). Analysis of the SNPs/Indels, based on information from public databases such as OMIM, COSMIC cancer, and dbSNPs, gave a general view of the genetic variations in disease-related and variant novelty (Figure 3).
To explore the potential genetic variations involved in leukaemia, we further re-filtered the variants located only in previously reported leukaemia genes, adapted from leukaemia databases and published journals (Borst, et al, 2012; Lilljebjorn et al., 2012; Zakaria, et al, 2012). Identification of variants by single case analysis and as a group is essential to reduce large numbers of variation into small subsets (Figure 4). We identified 32 somatic exome mutations located in the coding region of leukaemia-related genes (Table 3). These mutations may or may not have an association with disease development; hence, functional evaluation of the outcome of each mutation would be specifically beneficial to determine their contribution to leukemogenesis. These mutations were randomly selected for validation using Sanger sequencing (data not shown).
The outcome of the mutations which described the functions of the affected genes, either amino acid change was predicted to be ‘damaging’ or ‘probably damaging’ was modelled using the prediction tools PolyPhen-2 and SIFT (Table 3). Six mutations were identified as a potential driver mutation that was likely to affect the function of the translated protein. Interestingly, one of these ‘damaging’ mutations occurred in FLT3 gene and the rest were found in KMT2C gene.
Previous study by Lilljebjorn et al (2012) also found a mutation in the FLT3 gene with SIFT prediction of damaging effect (p.D835H) and can lead to leukemogenesis in both lymphoid and myeloid leukaemias (Lilljebjorn, et al, 2012). FLT3 encodes a class III receptor tyrosine-protein kinase that plays a role as a cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and dendritic cells (Gilliland and Griffin, 2002; Levis and Small, 2003). It also promotes phosphorylation of SHC1, AKT1, FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, MAPK1/ERK2 and /or MAPK3/ERK1 and STAT5A and/or STAT5B and activation of the downstream effectors mTOR and RAS signalling (Takahashi, 2011). Mutation in this gene may result in an active proliferation of the cell and resistance to apoptosis via activation of multiple signalling pathways. The presence of a mutation in FLT3 gene can also cause disruption in their function and contribute to oncogenic events (Armstrong et al., 2004; Chang, et al, 2013; Lilljebjorn, et al, 2012).
KMT2C is the central component of the MLL2/3 complex that encodes histone methyltransferase, which represents a specific tag for epigenetic transcriptional activation (Li et al., 2013). The expression of this gene may be involved in leukemogenesis and developmental disorder. Mutations in KMT2C were reported by Abel Gonzalez-Perez et al (2013). They analysed the relative importance of mutations in chromatin regulatory factors group for the development of tumorigenesis and found 17 mutational frequency of KMT2C in a hematopoietic cell. They concluded that mutations in certain genes correlate with broad expression changes across cancer cell lines. These mutations could contribute to tumorigenesis in cells of the corresponding tissues by at least one mechanism (Gonzalez-Perez et al., 2013).
Single nucleotide mutations are known for their contribution to leukaemia progression from previous studies, but none of them was found to be recurrent in our four ETV6/RUNX1-positive ALLs. However, the protein changes and their impact due to the mutations can simply predicted using appropriate tools. Lilljebjorn et al., (2012) also reported that sequencing of two ETV6/RUNX1-positive ALLs results in 12 somatic mutations that had no recurring event in extended ALL samples. Three genes; SERPINB1, PPL, and ZNF546 with synonymous mutations are known from COSMIC to be mutated in cancer. They concluded that, although most of the identified somatic mutations were synonymous and comprehensively described as gene mutations in other types of cancers in COSMIC and NCBI database, it may become a potential candidate-driver mutation (Lilljebjorn, et al, 2012). These findings showed that high mutation rates in somatic cells, or that the silent mutations are important, either, by affecting splicing or translation of the proteins. Thus, their potential roles as secondary genetic changes required for the development of ETV6/RUNX1-positive ALLs are plausible (Borst, et al, 2012). Apart from that, these findings are important for cancer mutation cataloguing such as for the COSMIC and the Mitelman database of chromosome aberrations in cancer (Rabbani et al., 2013).
Detection of variable and rare somatic mutations in our study of the four samples of ETV6/RUNX1 subtypes ALL in our population supported the notion that rearrangement of this fusion gene alone is insufficient for leukemogenesis. Although the sample size is relatively small, the overall genetic variation in the sample showed some similarity and could be potentially used for further reference. One or more secondary genetic alterations are indeed required for progression of the disease. The number and type of mutations detected may differ from what was previously described in leukaemia genomes of similar or different subtypes. Identification and characterization of mutational profiles are certainly important in identifying new therapeutic targets and developing rationally designed treatment regimen with less toxicity.
Statement conflict of Interest
The authors declare that they have no competing interests.
Acknowledgements
The authors would like to thank the Director General of Health, Ministry of Health Malaysia for approval to publish this scientific paper. We would also like to thank the Deputy Director General of Health (Research and Technical Support), and the Director of the Institute for Medical Research (IMR) for their support. This research was funded by the Ministry of Health Malaysia (JPP-IMR 07-042 and JPP-IMR 14-030).
References
- Aljamaan K, Aljumah TK, Aloraibi S, et al. Low frequency of ETV6-RUNX1 (t 12;21) in Saudi Arabian pediatric acute lymphoblastic leukemia patients:Association with clinical parameters and early remission. Asian Pac J Cancer Prev. 2015;16:7523–7. doi: 10.7314/apjcp.2015.16.17.7523. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Mabon ME, Silverman LB, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004;103:3544–6. doi: 10.1182/blood-2003-07-2441. [DOI] [PubMed] [Google Scholar]
- Awan T, Iqbal Z, Aleem A, et al. Five most common prognostically important fusion oncogenes are detected in the majority of Pakistani pediatric acute lymphoblastic leukemia patients and are strongly associated with disease biology and treatment outcome. Asian Pac J Cancer Prev. 2012;13:5469–75. doi: 10.7314/apjcp.2012.13.11.5469. [DOI] [PubMed] [Google Scholar]
- Borst L, Wesolowska A, Joshi T, et al. Genome-wide analysis of cytogenetic aberrations in ETV6/RUNX1- positive childhood acute lymphoblastic leukaemia. Br J Haematol. 2012;157:476–82. doi: 10.1111/j.1365-2141.2012.09083.x. [DOI] [PubMed] [Google Scholar]
- Chang VY, Basso G, Sakamoto KM, et al. Identification of somatic and germline mutations using whole exome sequencing of congenital acute lymphoblastic leukemia. BMC Cancer. 2013;13:1–6. doi: 10.1186/1471-2407-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Jene-Sanz A, Lopez-Bigas N. The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 2013;14:1–15. doi: 10.1186/gb-2013-14-9-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goud TM, Al Salmani KK, Al Harasi SM, et al. Importance of FISH combined with Morphology, Immunophenotype and cytogenetic analysis of childhood/ adult acute lymphoblastic leukemia in Omani patients. Asian Pac J Cancer Prev. 2015;16:7343–50. doi: 10.7314/apjcp.2015.16.16.7343. [DOI] [PubMed] [Google Scholar]
- Guo Y, Long J, He J, et al. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13:1–10. doi: 10.1186/1471-2164-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks C, Miele L, Koganti T, et al. Analysis of patterns of gene expression variation within and between ethnic populations in pediatric B-ALL. Cancer Inform. 2013;12:155–73. doi: 10.4137/CIN.S11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas AM, Ahmad S, Faheem M, et al. Next generation sequencing of acute myeloid leukemia:influencing prognosis. BMC Genomics. 2015;16:5. doi: 10.1186/1471-2164-16-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381:1943–55. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalender Atak Z, De Keersmaecker K, Gianfelici V, et al. High accuracy mutation detection in leukemia on a selected panel of cancer genes. PLoS One. 2012;7:1–11. doi: 10.1371/journal.pone.0038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CS, Cooper DN, Ziogas DE, et al. Research and clinical applications of cancer genome sequencing. Curr Opin Obstet Gynecol. 2012;25:3–10. doi: 10.1097/GCO.0b013e32835af17c. [DOI] [PubMed] [Google Scholar]
- Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17:1738–52. doi: 10.1038/sj.leu.2403099. [DOI] [PubMed] [Google Scholar]
- Li WD, Li QR, Xu SN, et al. Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood. 2013;121:1478–9. doi: 10.1182/blood-2012-12-470559. [DOI] [PubMed] [Google Scholar]
- Lilljebjorn H, Rissler M, Lassen C, et al. Whole-exome sequencing of pediatric acute lymphoblastic leukemia. Leukemia. 2012;26:1602–7. doi: 10.1038/leu.2011.333. [DOI] [PubMed] [Google Scholar]
- Lilljebjorn H, Soneson C, Andersson A, et al. The correlation pattern of acquired copy number changes in 164 ETV6/RUNX1-positive childhood acute lymphoblastic leukemias. Hum Mol Genet. 2010;19:3150–8. doi: 10.1093/hmg/ddq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Shen E, Min Q, et al. Exome-assistant:a rapid and easy detection of disease-related genes and genetic variations from exome sequencing. BMC Genomics. 2012;13:1–7. doi: 10.1186/1471-2164-13-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney-Garcia DR, Liehr T, Bhatt S, et al. Childhood B-cell progenitor acute lymphoblastic leukemia presenting a three-way t(11;12;21)(q14;p13;q22) with a RUNX1 gene signal on chromosome 11. Int J Hematol. 2012;95:112–4. doi: 10.1007/s12185-011-0981-x. [DOI] [PubMed] [Google Scholar]
- Obata M, Tsutsumi S, Makino S, et al. Whole-exome sequencing confirmation of a novel heterozygous mutation in RUNX1 in a pregnant woman with platelet disorder. Platelets. 2015;26:364–9. doi: 10.3109/09537104.2014.912750. [DOI] [PubMed] [Google Scholar]
- Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2013;59:5–15. doi: 10.1038/jhg.2013.114. [DOI] [PubMed] [Google Scholar]
- Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia:biology and therapeutic implications. J Hematol Oncol. 2011;4:13. doi: 10.1186/1756-8722-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli R, Marletta C, Pressato B, et al. Comparative genomic hybridization on microarray (a-CGH) in constitutional and acquired mosaicism may detect as low as 8% abnormal cells. Mol Cytogenet. 2011;4:1–6. doi: 10.1186/1755-8166-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veken LT, Buijs A. Array CGH in human leukemia:from somatics to genetics. Cytogenet Genome Res. 2011;135:260–70. doi: 10.1159/000330629. [DOI] [PubMed] [Google Scholar]
- Wang H, Li W, Guo R, et al. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int J Cancer. 2014a;135:2783–94. doi: 10.1002/ijc.28922. [DOI] [PubMed] [Google Scholar]
- Wang L, Swierczek SI, Drummond J, et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Leukemia. 2014b;28:935–8. doi: 10.1038/leu.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakaria Z, Ahid MF, Ismail A, et al. Chromosomal aberrations in ETV6/RUNX1-positive childhood acute lymphoblastic leukemia using 244K oligonucleotide array comparative genomic hybridization. Mol Cytogenet. 2012;5:1–6. doi: 10.1186/1755-8166-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Kim YM, Yang X, et al. A possible 5’-NRIP1/UHRF1-3’ fusion gene detected by array CGH analysis in a Ph+ALL patient. Cancer Genet. 2011;204:687–91. doi: 10.1016/j.cancergen.2011.11.006. [DOI] [PubMed] [Google Scholar]