

Abstract
Purpose of review
Gene-environment (G×E) interactions likely contribute to numerous diseases, but are often difficult to model in the laboratory. Such interactions have been widely hypothesized for Amyotrophic Lateral Sclerosis (ALS); recent controlled laboratory studies are discussed here and hypotheses related to possible mechanisms of action are offered. Using methylmercury exposure and mutated SOD1 to model the impacts of such an interaction, we interpret evidence about their respective mechanisms of toxicity to interrogate the possibility of additive (or synergistic) effects when combined.
Recent work
Recent work has converged on mechanisms of calcium-mediated glutamate-excitotoxicity as a likely contributor in one model of a gene-environment interaction affecting the onset and progression of ALS-like phenotype.
Summary
The current experimental literature on mechanisms of metal-induced neuronal injury, and their relevant interactions with genetic contributions in ALS is sparse, but we describe those studies here and offer several integrative hypotheses about the likely mechanisms involved.
Keywords: Amyotrophic Lateral Sclerosis, gene-environment (G×E) interaction, methylmercury, AMPA receptor, glutamate, calcium homeostasis
Introduction
Many diseases are thought to arise, or hasten in progression, when environmental events interact with genetic predispositions in unfortunate ways. The idea that gene by environment (G×E) interactions cause disease and dysfunction has been applied to many areas of human health, often successfully expanding our understanding of the complexity of factors that influence health and disease states. Epidemiological reports often herald laboratory studies of disease pathogenesis, but for those thought to arise via G×E interactions that path is uniquely complex. Often prototypical environmental events (e.g. exposure to an environmental toxicant and genetic mutations that are known to cause dysfunction) are used to model such an interaction in the laboratory. These studies model a slice of the complexities inherent in the human condition and can yield valuable information regarding the potential mechanisms involved in disease onset and progression, pointing towards meaningful considerations for populations at risk. This review takes a narrow focus on the idea of G×E interactions by confining the discussion to a model of one motor neuron (MN) disease, Amyotrophic Lateral Sclerosis (ALS).
No clear genetic (or environmental) factors are known to cause the vast majority (~90%) of ALS cases, which are referred to collectively as sporadic ALS (sALS), making a G×E interaction exceedingly likely. Because of this, and because there have been several “outbreaks” of ALS or ALS-like syndromes among non-relatives living near one another, much has been written about environmental risk factors for sALS [1, 2]. Among the most commonly described environmental risks are exposure to pesticides, solvents, and the heavy metals lead and mercury; a history of electric shock or physical trauma; military service; and chronic strenuous physical activity [1]. Some hints at possible epigenetic or genetic predispositions to sALS are also beginning to emerge; carriage of some genetic polymorphisms (e.g. coproporphyrinogen oxidase 4 (CPOX4) [3]; Val66Met in brain-derived neurotrophic factor (BDNF) [4]) in combination with mercury exposure was shown to result in heightened motor deficits in humans. Several gene mutations are known to contribute directly to ALS - mutated superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), fused in sarcoma/translocated in sarcoma (FUS), and chromosome 9 open reading frame 72 (C9orf72) genes account for around 65% of all familial ALS (fALS) cases and around 11% of all sALS cases [5–7]. Several reviews have examined the possibility that G×E interactions are indeed relevant for ALS and we direct readers to those publications [8–10], as well as to an alternate perspective (see [11]).
The present review assumes a G×E interaction is relevant to at least some cases of ALS, and instead focuses on the potential neural mechanisms that are affected when genes and the environment converge, manifesting in this disease. While a wealth of circumstantial evidence points toward G×E interactions in ALS, studies (experimental or epidemiological) that directly test this hypothesis are uncommon, underpowered or correlational [12]. The few exceptions to this are well-controlled laboratory animal models that permit cause-effect relations to be identified; those using the environmental neurotoxicant methylmercury (MeHg) and a mutated SOD1 gene as exemplar G×E factors are emphasized below. We do not propose that only one environmental factor will be important in understanding the complex etiology of ALS, nor do we propose that any environmental factor alone will cause ALS to manifest. We also emphasize that many, divergent genetic predispositions could contribute to this interaction. Indeed, we hypothesize that some individuals might possess currently unrecognized single nucleotide polymorphisms (SNPs) that predispose them to developing ALS when confronted with an environmental insult. Finally, we recognize that more than one cell type inevitably participates in the etiology of ALS – certainly including microglia and astrocytes and likely other neurons that synapse on MNs as well. Much in this field simply remains unknown. Here we discuss the progress that has been made towards untangling the complexities inherent in ALS onset and progression, within the context of the G×E interaction model.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a neurodegenerative MN disease in which progressive loss of α and γ MNs of the cerebral cortex, brainstem, and spinal cord occurs [13, 14]. This degeneration first leads to weakness and/or difficulty engaging in voluntary movements. Often symptoms of ALS (e.g. muscle weakness) begin within a single limb or body region [15]. When symptoms begin with speaking or swallowing difficulties it is referred to as bulbar-onset (reflecting the degeneration of MNs in the corticobulbar region of the brainstem). In most cases, partial or total paralysis occurs and respiratory failure or cardiac arrest ultimately results in death. Typically, the onset of ALS is in middle- or late-life, but once it appears its progression is swift; it is generally fatal within 4–5 years following the onset of symptoms [15]. There is no known cure or effective treatment. A number of pharmacotherapies have been tested for the treatment of ALS, however, none to-date have dramatically altered the disease progression. As such, understanding what environmental and/or genetic factors are responsible for the onset and progression of ALS is a goal with significant implications for human health.
Despite the seemingly divergent etiology of sALS and fALS, the clinical manifestation and time course of these forms are nearly identical, suggesting a shared pathophysiology. It is possible that with a disease that is associated with some degree of variability (e.g. in age of onset, progression and survival time) that multiple pathways might be differentially responsible. Some of the most common characteristics attributed to ALS include a high sensitivity of MNs to glutamate (Glu)-mediated damage, difficulty buffering changes to internal Ca2+ concentration ([Ca2+]i), dysfunction of mitochondrial processes, the generation of reactive oxygen species (ROS), and various alterations to the cytoskeleton [16, 17]. Because ALS emerges relatively late in life, perhaps following a long silent latency, identifying primary (vs. secondary) pathways is made more difficult. Further complicating the issue, emerging evidence suggests that ALS is not a MN-only disease, as once thought; astrocytes and microglia also seem to contribute to its pathophysiology [18, 19].
Cu2+/Zn2+ Superoxide Dismutase 1 (SOD1) Gene Mutations
Almost all of the mechanisms associated with ALS (mentioned above) have been identified using rodent models, especially mice expressing mutations in the human SOD1, referred to below as SOD1G93A. SOD1G93A has an amino acid substitution of glycine to alanine at residue 93. This mutation leads to a toxic gain of function of the enzyme. The nature of this gain in function is as yet unclear, but is the subject of much investigation [20]. SOD1 is a Cu2+/Zn2+ superoxide dismutase that is ubiquitously expressed in all cells. Over 150 mutations have been identified in the SOD1 sequence (http://alsod.iop.kcl.ac.uk) and unsurprisingly mutant SOD1 appears to elicit neurotoxicity via many mechanisms (see [21] for an excellent review on the mechanisms of mutant SOD1 neurotoxicity). The consequences associated with mutant SOD1 involve MNs, microglia, astrocytes and oligodendrocytes and include endoplasmic reticulum (ER) stress, mitochondrial dysfunction, excitotoxicity, oxidative stress, non-cell autonomous toxicity of neuroglia, axonal transport disruption. SOD1 mutations show a characteristic prion-like propagation. It is even possible that wild-type SOD1 can become misfolded and ultimately contribute to the pathogenesis of sALS [22, 23]. Some of these mechanisms closely resemble those involved in MeHg’s neurotoxicity.
Methylmercury (MeHg)
The scientific community learned of MeHg’s toxicity following a number of catastrophic poisoning events in which discrete populations were exposed to relatively high concentrations of MeHg [24–26]. Perhaps the most infamous were the events in Iraq and Japan occurring in the last century, which have been discussed at length elsewhere [25, 26]. Chronic, low-level exposure to MeHg, however, is of concern for much of the world’s population because the consumption of contaminated seafood serves as a primary route of exposure to this toxicant - meals of tuna and swordfish, for example, both deliver relatively high concentrations of MeHg to the consumer and are common worldwide. Some populations for which seafood constitutes a large portion of the diet, or where MeHg contamination is particularly elevated, may be at even greater risk. These include the Amazon rainforest, the Faroe Islands, artisanal gold mining communities, the Great Lakes region of the U.S., Greenland, and perhaps the Seychelles Islands [27–31].
MeHg consumption has been associated with ALS-like syndromes, which has led to its inclusion in animal models examining a G×E interaction in ALS. The characteristic motor impairments following post-birth MeHg intoxication (e.g. exposure to high concentrations) have largely driven its inclusion in the G×E studies highlighted here. Ataxia, visual disturbances, muscle weakness and sensory abnormalities are all common symptoms of MeHg intoxication [26, 32]. These symptoms are often preceded by lengthy asymptomatic periods, generating the characteristic “silent latency” associated with MeHg exposure [33]. Fetal exposure to high concentrations of MeHg presents differently and is associated with physical deformations and cognitive impairment. Notably, unusually high rates of motor disease and dysfunction were observed following the Iraq poisoning episode, in which a large population consumed very high concentrations of MeHg over a relatively short period of time. An abnormally high percent (14%) of this population presented with “myasthenia gravis-like” syndromes [25], a term used loosely to capture the profound motor disturbances that occurred among those exposed. Similarly, ALS-like diseases have emerged following accidental or occupational exposures to high doses of mercurials [34, 35]. Perhaps lower concentrations, like those found in seafood-rich diets could interact with (currently unrecognized) genetic polymorphisms to hasten (or even trigger) the onset of ALS.
Studies of the pathology of MeHg toxicity that use rodent models have demonstrated that 1) MeHg accumulates in, and degenerates, α MNs in the spinal cord, 2) when MNs become saturated with MeHg, ataxia is observed and 3) neurophagia, atrophy and degeneration occur in the anterior horn of the lumbar region of the spinal cord [36–38]. More recent studies have demonstrated that MeHg exposure leads to a concentration-dependent incidence of cell death in MNs that could occur as a result of alterations in intracellular Ca2+ ([Ca2+]i) homeostasis [39, 40]. Acute in vitro exposure to low concentrations (0.1 – 1.5 μM) of MeHg in primary spinal cord MNs leads to alterations in [Ca2+]i. Disruption of [Ca2+]i is mediated in part by the ionotropic Glu receptors: N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) [40]. Taken together, these studies demonstrate that MNs degenerate after exposure to MeHg, and that this can occur as a result of the observed alterations in [Ca2+]i that are mediated by ionotropic Glu receptors. This observation is similar to one mechanism of pathogenesis observed in ALS. Studies in other neuronal cells, including cerebellar granule cells, have shown that MeHg exposure causes alterations in neurotransmitter release resulting in an increase of Glu release [41] and [Ca2+]i [40, 42], mitochondrial damage [43], increases in ROS [44, 45], and impaired function of the excitatory astrocytic Glu transporter (excitatory amino acid transporter 2 (EAAT2)) [46]. Again, these are among the mechanisms of toxicity observed in sALS and fALS [13].
Clearly, MeHg exposure is associated with a number of deleterious effects on cells, but the ability for MeHg to disrupt divalent cation regulation (and the related excitotoxicity that occurs as a result) is of particular interest with respect to ALS [40]. Even very low concentrations of MeHg (e.g. sub- and low-μM) can increase levels of [Ca2+]i in single cells or synapses [47–49] and precipitate cell death following a delay. The spontaneous release of Glu is stimulated by MeHg, which can itself disrupt the homeostasis of intracellular divalent cations [41]. MeHg’s effects on mitochondrial Ca2+ have been shown to contribute to elevated levels of [Ca2+]i and cell death [43, 50]. Behavioral deficits, including both motoric and cognitive dysfunction, that result from MeHg exposure have been linked to disrupted [Ca2+]i homeostasis [51]. Further, the oral administration of a Ca2+ channel antagonist concomitant with MeHg exposure ameliorated the behavioral toxicity associated with MeHg [51] in rodents. Thus, [Ca2+]i homeostasis disruption is also a valid mechanism at the level of the whole organism.
Evidence of a G×E Interaction
Recently, the explicit focus of our own work has been to determine if a G×E interaction of metal-induced neuronal injury can contribute to the development (or progression) of ALS. We have successfully demonstrated that the time course of mutant SOD1G93A-induced motor dysfunction is hastened by chronic MeHg exposure [52]. Mice expressing the human SOD1G93A mutation were given chronic, low concentrations of MeHg as they aged. These “dual-hit” animals displayed profound motor dysfunction (e.g. rotorod failure) weeks before the unexposed SOD1 mutants; wild type animals receiving MeHg at the same level never displayed this motor impairment. At the time of impairment, these animals had elevated [Ca2+]i in MNs of the brainstem. Death and dysfunction of brainstem MNs models “bulbar onset” ALS – this occurs when symptoms begin in the craniofacial regions. This excess [Ca2+]i was associated with Ca2+-permeable AMPA receptors: the AMPA/Kainate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) delayed the MeHg-induced increase in [Ca2+]i in all SOD1G93A mice (irrespective of MeHg exposure) but the Ca2+ permeable AMPA receptor antagonist 1-naphthyl acetyl spermine (NAS) further reduced [Ca2+]i in the MeHg exposed SOD1 mutants compared to those untreated with MeHg [52]. These results support a hypothesis that a MeHg-induced increase in Ca2+ and cell death of MNs are mediated in part by Ca2+ permeable AMPA receptors, which enhances the dysfunction associated with the SOD1 mutation.
Increases in [Ca2+]i among these “dual hit” animals was replicated in spinal cord MNs [53], under conditions of acute MeHg exposure (in spinal cord tissue). The spinal cord is a target of particular interest in understanding ALS etiology, as it models the limb onset (or, non-bulbar) forms of this disease. Intracellular Ca2+ regulation was tracked via fluo-4 epifluorescence following acute 20μM MeHg exposure in spinal cord slice from SOD1G93A mutants and their appropriate controls. As a function of exposure time, acute treatment with MeHg caused a greater increase in fluo-4 fluorescence in the spinal cord tissue of SOD1G93A mice compared to control mice. These data further suggest that MeHg interacts with the SOD1G93A mutation to enhance neuronal dysfunction (mediated via intracellular Ca2+ dysregulation among neurons of the spinal cord). This, again, likely demonstrates evidence of a G×E interaction relevant to the etiology of ALS. In both cases, an environmental event was shown to speed the onset and progression of the ALS phenotype caused by the SOD1 mutation (i.e. the genetic contribution) in a way that was not seen with either the gene or environmental event alone. Ongoing and future projects include other genetic models of ALS and as additional genetic targets become known this paradigm can be expanded and tested within numerous (genetic) contexts. These data are of conceptual importance as they demonstrate, unequivocally, that the possibility of a G×E interaction in the onset and progression of this disease exists.
Understanding the mechanism(s) underlying this interaction should prove useful for identifying the potential targets of other environmental and genetic factors relevant to ALS. In brief, we have hypothesized ([based partly on [52]) that exposure to MeHg disrupts MN divalent cation homeostasis, thereby stimulating MN Glu release. MeHg subsequently inhibits astrocyte Glu uptake by excitatory amino acid transporters (EAATs) [54]. In so doing, MeHg affects the functional relationship between MNs and their attendant astrocytes and generates ROS [54]. However, the proximal events in this process could be MN- or astrocyte-directed. So by using MeHg-treated SOD1 mice with distinct SOD1 mutations we have begun to address the temporal relationship that exists between astrocyte and MN-directed effects. The interdependence of this pair may ultimately contribute to an enhanced development of ALS phenotype. The concomitant effects of increased ROS generation and Ca2+-dependent excitotoxicity are likely accelerating the course of ALS development.
Convergence on a Mechanism: Glutamate-Induced Excitotoxicity
Ca2+-mediated, Glu-induced excitotoxicity is thought to be an important cellular-level mechanism that underlies MN loss in ALS and dysfunction in MeHg toxicity [41] – and by extension, in the G×E interaction discussed here. Increased Glu release results in increased [Ca2+]i and activating proteins, which can cause mitochondrial toxicity and the generation of ROS. Moreover, because both MeHg and ALS are associated with increased ROS generation, the MeHg-induced increase of oxidative stress likely contributes to an early onset of ALS. An enhancement in the sensitivity of MNs to Glu-induced excitotoxicity [55] is also hypothesized to contribute to the development of ALS, and is the process that might be especially relevant to the increases in Glu release that are associated with exposure to MeHg [41]. Elevated concentrations of Glu and aspartate have been found in the cerebrospinal fluid of patients with ALS [56], which supports this emphasis on Glu-induced excitotoxicity. In this way, excitotoxicity ultimately results in cell damage and death via Ca2+-mediated pathways. Increased [Ca2+]i is integral in this pathogenesis, and recent reports from our laboratory have shown that MeHg exposure can accelerate increases in [Ca2+]i in MNs from the brainstem [52] and spinal cord [53] of SOD1G93A mutants.
The importance of AMPA receptors
The extension of these findings has led to subsequent, ongoing projects by our group, designed to identify the role of Glu receptors in MeHg-induced toxicity. Examining AMPA receptor function has been central to this aim. AMPA receptors are the ligand-gated Glu receptors in the central nervous system that mediate fast excitatory neurotransmission. When one of the four AMPA receptor subunits, GluA2, is not present (or is in an RNA unedited state), AMPA receptors become Ca2+ permeable. In ALS, increased levels of unedited GluA2 have been observed. Adenosine deaminase acting on RNA 2 (ADAR2) edits GluA2, and in vitro studies have shown that Glu-induced excitotoxicity can cause dysfunction of ADAR2, leading to increased expression of Ca2+-permeable GluA2 in neuronal cells [57, 58]. MeHg exposure causes increased Glu release [41], impaired function of excitatory amino acid transporter EAAT2 [46], increased expression of GluA2 in the brainstem [59] and increases in [Ca2+]i in MNs mediated through AMPA receptors [40, 52]. One hypothesis here is that chronic MeHg exposure impairs ADAR2 function, leading to an increase in Ca2+ permeable AMPA receptors and as a result increased [Ca2+]i. Preliminary evidence has indicated that MeHg exposure increases the expression of AMPA receptor subunits, especially GluA2, and decreases the RNA editing enzyme ADAR2 in brainstem hypoglossal tissue from rats, a region rich in MNs, to a greater extent in SOD1G93A MNs compared to control [59]. See Fig.1 for a schematic representation of the mechanisms thought to be relevant to both ALS and MeHg exposure.
Figure 1.
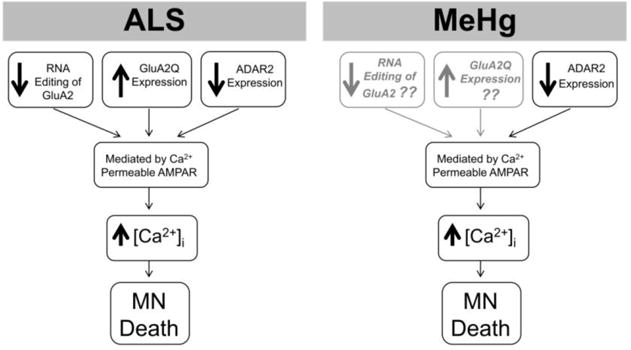
Role of AMPA receptors in ALS and MeHg toxicity on MNs. In ALS, AMPA receptors contribute to the alterations in intracellular Ca2+. This effect is in part due to the decrease in RNA editing of the GluA2, which results from a decrease in ADAR2. That contributes the increase in the unedited form of GluA2 (GluA2Q) containing receptors in MNs. This is one of the mechanisms that contribute to MN cell death in ALS. MeHg-induced Ca2+ dysregulation in MNs is also mediated in part by Ca2+ permeable AMPA receptors. Our preliminary studies have shown that there is a decrease of ADAR2. However, we do not know how and if the GluA2 subunit is affected by MeHg exposure. This should be the focus of future studies.
Finally, preliminary studies from our group have demonstrated that low-dose MeHg exposure leads to a concentration-dependent cell death following increases in [Ca2+]i that are mediated by AMPA receptors and alterations in AMPA receptor expression in human MNs [60]. Thus, if this is the case in normal human MNs, in a population that has a predisposition to ALS due to genetic mutations, degeneration of these cells may occur faster when exposed to MeHg. Based off these preliminary results and supporting literature it is hypothesized that MeHg action in MNs can have an additive (or perhaps synergistic) effect in ALS-predisposed populations – and, this could have been the case in the mouse study [52] that used the SOD1G93A mouse model.
Conclusions
The result of recent work performed in our lab suggests that Glu-mediated, Ca2+-dependent MN damage can result from a G×E interaction. This was among the first definitive demonstrations of a specific contribution of a persistent environmental contaminant to the development of the ALS phenotype in animal models. MeHg induces multiple effects on neurons that could heighten their sensitivity to subsequent Glu-mediated excitotoxicity due to disease process, SNPs or additional exposure to environmental agents. It also disrupts Glu uptake by astrocytes, which would exacerbate such effects.
The underlying hypothesis here, on which the work reviewed above is based, is that some individuals might possess currently unrecognized SNPs that predispose them to developing ALS when confronted with an environmental insult. In this way, an environmental insult might act to tip the balance of cell damage in the favor of this disease (and possibly others). Sufficient evidence already exists linking exposure to many environmental chemicals including metals and pesticides, which are ubiquitous in the environment, to MN disease. Considering the vast prevalence of diseases that lack any clear genetic cause and the known impact of one’s environment or lifestyle on health outcomes, it is prudent to examine seriously the role of environmental contributions to such diseases. The scope of the present review was narrowed to include an examination of one model of the G×E phenomenon: the impact of MeHg exposure on the onset and progression of a “humanized” mouse expressing an SOD1 mutation model of ALS. It is worth noting that it is not the intention of the present review to propose that MeHg alone causes ALS and it is acknowledged that some of the genetic predispositions to ALS are at present unknown. What is apparent is that a further understanding of these genetic and environmental risk factors, and how they interact, will be integral to a prevention of disease via the reduction of exposures to environmental contributors, particularly for potentially susceptible individuals.
One important implication of the glutamate-induced excitotoxicity that we discuss here, within the context of MeHg exposure, is that other environmental factors that similarly disrupt calcium homeostasis or otherwise elicit a similar trajectory of neuronal dysfunction should be carefully considered as relevant to ALS disease progression. We would also like to acknowledge that, although limited to-date, evidence is continuing to mount that demonstrates the relevance of G×E interactions in ALS (beyond MeHg exposure). For instance, the administration of statins (HMG-CoA reductase inhibitors) has been shown to hasten the ALS-like phenotype of SOD1 mutant rodents [61]; exposure to the toxin Beta-methylamino-L-alanine (BMAA) hastens motor dysfunction in zebrafish SOD1 mutants [62]; and, finally, there is emerging interest in examining G×E interactions that are protective against the ALS-like phenotype seen in mouse models of ALS (e.g. dietary restriction [63], vitamin D3 supplementation [64]), which, in a different way also addresses the interactions among environment and genetics in the onset and progression of ALS.
Acknowledgments
Supported by grants NIEHS T32 ES00725527 (Jordan M. Bailey, Alexandra Colón-Rodríguez) and NIH Grant R01 ES024064 (Jordan M. Bailey, Alexandra Colón-Rodríguez, William D. Atchison).
Abbreviations
- G×E
Gene, environment interaction
- ALS
Amyotrophic Lateral Sclerosis
- sALS
sporadic Amyotrophic Lateral Sclerosis
- fALS
familial Amyotrophic Lateral Sclerosis
- SOD1
superoxide dismutase 1
- FUS
fused in sarcoma/translocated in sarcoma
- TDP-43
TAR DNA-binding protein 43
- C9orf72
chromosome 9 open reading frame 72
- MN
motor neuron
- CPOX4
coproporphyrinogen oxidase 4
- BDNF
brain-derived neurotrophic factor
- MeHg
Methylmercury
- SNP
single nucleotide polymorphisms
- Glu
glutamate
- [Ca2+]i
internal calcium concentration
- NMDA
N-methyl-D-aspartate
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ROS
reactive oxygen species
- EAAT
excitatory amino acid transporter
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- NAS
1-naphthyl acetyl spermine
- GluA2
AMPA receptor subunit 2
- ADAR2
adenosine deaminase acting on RNA
Footnotes
Conflict of Interest
Jordan M. Bailey, Alexandra Colón-Rodríguez, and William D. Atchison declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
References
Papers of particular interest, published recently, have been highlighted as:
•Of importance
••Of outstanding importance
- 1•.Wang MD, Little J, Gomes J, Cashman NR, Krewski D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology. 2016 doi: 10.1016/j.neuro.2016.06.015. This study provides a thorough description of all known risk factors associated with ALS. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell JD. Amyotrophic lateral sclerosis: toxins and environment. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2000;1(4):235–50. doi: 10.1080/14660820050515061. [DOI] [PubMed] [Google Scholar]
- 3.Echeverria D, Woods JS, Heyer NJ, Rohlman D, Farin FM, Li T, et al. The association between a genetic polymorphism of coproporphyrinogen oxidase, dental mercury exposure and neurobehavioral response in humans. Neurotoxicology and Teratology. 2006;28(1):39–48. doi: 10.1016/j.ntt.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Echeverria D, Woods JS, Heyer NJ, Rohlman DS, Farin FM, Bittner AC, Jr, et al. Chronic low-level mercury exposure, BDNF polymorphism, and associations with cognitive and motor function. Neurotoxicology and Teratology. 2005;27(6):781–96. doi: 10.1016/j.ntt.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Current Neurology and Neuroscience Reports. 2006;6(1):37–46. doi: 10.1007/s11910-996-0008-9. [DOI] [PubMed] [Google Scholar]
- 6.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. The Lancet Neurology. 2012;11(4):323–30. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Human mutation. 2013;34(6):812–26. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- 8•.Oskarsson B, Horton DK, Mitsumoto H. Potential environmental factors in Amyotrophic Lateral Sclerosis. Neurologic clinics. 2015;33(4):877–88. doi: 10.1016/j.ncl.2015.07.009. This study describes the known environmental factors associated with ALS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nature Reviews Neurology. 2013;9(11):617–28. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 10.Trojsi F, Monsurro MR, Tedeschi G. Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: state of the art and research perspectives. International Journal of Molecular Sciences. 2013;14(8):15286–311. doi: 10.3390/ijms140815286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. The New England Journal of Medicine. 2001;344(22):1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 12.Factor-Litvak P, Al-Chalabi A, Ascherio A, Bradley W, Chio A, Garruto R, et al. Current pathways for epidemiological research in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration. 2013;14(Suppl 1):33–43. doi: 10.3109/21678421.2013.778565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nature Reviews Neuroscience. 2001;2(11):806–19. doi: 10.1038/35097565. This study describes the machanics of motor neuron dysfunction in ALS. [DOI] [PubMed] [Google Scholar]
- 14.Tandan R, Bradley WG. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Annals of Neurology. 1985;18(3):271–80. doi: 10.1002/ana.410180302. [DOI] [PubMed] [Google Scholar]
- 15.Bryant PR, Geis CC, Moroz A, O’Neill BJ, Bogey RA. Stroke and neurodegenerative disorders. 4. Neurodegenerative disorders. Archives of Physical Medicine and Rehabilitation. 2004;85(3 Suppl 1):S21–33. doi: 10.1053/j.apmr.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–5. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature Reviews Neuroscience. 2006;7(9):710–23. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 18•.Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. This study, importantly, describes the role of glial cells in the dysfunction associated with ALS. [DOI] [PubMed] [Google Scholar]
- 19.Hedlund E, Isacson O. ALS model glia can mediate toxicity to motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3(6):575–6. doi: 10.1016/j.stem.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science (New York, NY) 1994;264(5166):1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y, Homma K, Ichijo H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Advances in Biological Regulation. 2016;60:95–104. doi: 10.1016/j.jbior.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(13):5074–9. doi: 10.1073/pnas.1115402109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PloS One. 2010;5(7):e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barber TE. Inorganic mercury intoxication reminiscent of amyotrophic lateral sclerosis. Journal of Occupational Medicine: Official publication of the Industrial Medical Association. 1978;20(10):667–9. [PubMed] [Google Scholar]
- 25.Rustam H, Von Burg R, Amin-Zaki L, El Hassani S. Evidence for a neuromuscular disorder in methylmercury poisoning. Archives of Environmental Health. 1975;30(4):190–5. doi: 10.1080/00039896.1975.10666674. [DOI] [PubMed] [Google Scholar]
- 26.Eto K. Pathology of Minamata disease. Toxicologic Pathology. 1997;25(6):614–23. doi: 10.1177/019262339702500612. [DOI] [PubMed] [Google Scholar]
- 27.Goncalves A, Goncalves NN. Human exposure to mercury in the Brazilian Amazon: a historical perspective. Pan American Journal of Public Health. 2004;16(6):415–9. doi: 10.1590/s1020-49892004001200008. [DOI] [PubMed] [Google Scholar]
- 28.Grandjean P, Budtz-Jorgensen E, White RF, Jorgensen PJ, Weihe P, Debes F, et al. Methylmercury exposure biomarkers as indicators of neurotoxicity in children aged 7 years. American Journal of Epidemiology. 1999;150(3):301–5. doi: 10.1093/oxfordjournals.aje.a010002. [DOI] [PubMed] [Google Scholar]
- 29.Madsen ER, DeWeese AD, Kmiecik NE, Foran JA, Chiriboga ED. Methods to develop consumption advice for methylmercury-contaminated walleye harvested by Ojibwe tribes in the 1837 and 1842 ceded territories of Michigan, Minnesota, and Wisconsin, USA. Integrated Environmental Assessment and Management. 2008;4(1):118–24. doi: 10.1897/ieam_2007-026.1. [DOI] [PubMed] [Google Scholar]
- 30.Hansen JC, Tarp U, Bohm J. Prenatal exposure to methyl mercury among Greenlandic polar Inuits. Archives of Environmental Health. 1990;45(6):355–8. doi: 10.1080/00039896.1990.10118754. [DOI] [PubMed] [Google Scholar]
- 31.Myers GJ, Davidson PW. Prenatal methylmercury exposure and children: neurologic, developmental, and behavioral research. Environmental Health Perspectives. 1998;106(Suppl 3):841–7. doi: 10.1289/ehp.98106841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang LW. Neurotoxic effects of mercury–a review. Environmental research. 1977;14(3):329–73. doi: 10.1016/0013-9351(77)90044-5. [DOI] [PubMed] [Google Scholar]
- 33•.Weiss B, Clarkson TW, Simon W. Silent latency periods in methylmercury poisoning and in neurodegenerative disease. Environmental Health Perspectives. 2002;110(Suppl 5):851–4. doi: 10.1289/ehp.02110s5851. This study highlights an important aspect of MeHg exposure (long latency periods) and how that can contribute to the presentation of neurodegenerative diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Praline J, Guennoc AM, Limousin N, Hallak H, de Toffol B, Corcia P. ALS and mercury intoxication: a relationship? Clinical Neurology and Neurosurgery. 2007;109(10):880–3. doi: 10.1016/j.clineuro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz S, Husstedt I, Bertram HP, Kuchelmeister K. Amyotrophic lateral sclerosis after accidental injection of mercury. Journal of Neurology, Neurosurgery, and Psychiatry. 1996;60(6):698. doi: 10.1136/jnnp.60.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moller-Madsen B. Localization of mercury in CNS of the rat. III. Oral administration of methylmercuric chloride (CH3HgCl) Fundamental and Applied Toxicology: official journal of the Society of Toxicology. 1991;16(1):172–87. [PubMed] [Google Scholar]
- 37.Moller-Madsen B. Localization of mercury in CNS of the rat. An autometallographic study. Pharmacology & Toxicology. 1994;75(Suppl 1):1–41. doi: 10.1111/j.1600-0773.1994.tb01927.x. [DOI] [PubMed] [Google Scholar]
- 38.Su M, Wakabayashi K, Kakita A, Ikuta F, Takahashi H. Selective involvement of large motor neurons in the spinal cord of rats treated with methylmercury. Journal of the Neurological Sciences. 1998;156(1):12–7. doi: 10.1016/s0022-510x(98)00030-6. [DOI] [PubMed] [Google Scholar]
- 39.Chapman LA, Chan HM. Inorganic mercury pre-exposures protect against methyl mercury toxicity in NSC-34 (neuron × spinal cord hybrid) cells. Toxicology. 1999;132(2–3):167–78. doi: 10.1016/s0300-483x(98)00151-6. [DOI] [PubMed] [Google Scholar]
- 40•.Ramanathan G, Atchison WD. Ca2+ entry pathways in mouse spinal motor neurons in culture following in vitro exposure to methylmercury. Neurotoxicology. 2011;32(6):742–50. doi: 10.1016/j.neuro.2011.07.007. This study demonstrates a key mechanism by which MeHg results in neuronal dysfunction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan Y, Atchison WD. Methylmercury-induced increase of intracellular Ca2+ increases spontaneous synaptic current frequency in rat cerebellar slices. Molecular Pharmacology. 2007;71(4):1109–21. doi: 10.1124/mol.106.031286. [DOI] [PubMed] [Google Scholar]
- 42.Limke TL, Heidemann SR, Atchison WD. Disruption of intraneuronal divalent cation regulation by methylmercury: are specific targets involved in altered neuronal development and cytotoxicity in methylmercury poisoning? Neurotoxicology. 2004;25(5):741–60. doi: 10.1016/j.neuro.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 43.Limke TL, Atchison WD. Acute exposure to methylmercury opens the mitochondrial permeability transition pore in rat cerebellar granule cells. Toxicology and Applied Pharmacology. 2002;178(1):52–61. doi: 10.1006/taap.2001.9327. [DOI] [PubMed] [Google Scholar]
- 44.Aschner M, Syversen T, Souza DO, Rocha JB, Farina M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Brazilian Journal of Medical and Biological Research. 2007;40(3):285–91. doi: 10.1590/s0100-879x2007000300001. [DOI] [PubMed] [Google Scholar]
- 45•.Dreiem A, Seegal RF. Methylmercury-induced changes in mitochondrial function in striatal synaptosomes are calcium-dependent and ROS-independent. Neurotoxicology. 2007;28(4):720–6. doi: 10.1016/j.neuro.2007.03.004. This study demonstrates the role of calcium dysregulation in MeHg exposure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aschner M, Yao CP, Allen JW, Tan KH. Methylmercury alters glutamate transport in astrocytes. Neurochemistry International. 2000;37(2–3):199–206. doi: 10.1016/s0197-0186(00)00023-1. [DOI] [PubMed] [Google Scholar]
- 47.Edwards JR, Marty MS, Atchison WD. Comparative sensitivity of rat cerebellar neurons to dysregulation of divalent cation homeostasis and cytotoxicity caused by methylmercury. Toxicology and Applied Pharmacology. 2005;208(3):222–32. doi: 10.1016/j.taap.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 48.Limke TL, Otero-Montanez JK, Atchison WD. Evidence for interactions between intracellular calcium stores during methylmercury-induced intracellular calcium dysregulation in rat cerebellar granule neurons. The Journal of Pharmacology and Experimental Therapeutics. 2003;304(3):949–58. doi: 10.1124/jpet.102.042457. [DOI] [PubMed] [Google Scholar]
- 49.Marty MS, Atchison WD. Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicology and Applied Pharmacology. 1997;147(2):319–30. doi: 10.1006/taap.1997.8262. [DOI] [PubMed] [Google Scholar]
- 50.Levesque PC, Atchison WD. Disruption of brain mitochondrial calcium sequestration by methylmercury. The Journal of Pharmacology and Experimental Therapeutics. 1991;256(1):236–42. [PubMed] [Google Scholar]
- 51•.Bailey JM, Hutsell BA, Newland MC. Dietary nimodipine delays the onset of methylmercury neurotoxicity in mice. Neurotoxicology. 2013;37:108–17. doi: 10.1016/j.neuro.2013.03.011. This study demonstrates that MeHg-induced calicum dysreguation can be observed at the level of behavior, and that drugs acting on calcium can influence the deleterious effects of MeHg exposure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52••.Johnson FO, Yuan Y, Hajela RK, Chitrakar A, Parsell DM, Atchison WD. Exposure to an environmental neurotoxicant hastens the onset of amyotrophic lateral sclerosis-like phenotype in human Cu2+/Zn2+ superoxide dismutase 1 G93A mice: glutamate-mediated excitotoxicity. The Journal of Pharmacology and Experimental Therapeutics. 2011;338(2):518–27. doi: 10.1124/jpet.110.174466. This study directly demonstrates a G×E interaction in ALS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bailey JM, Yuan Y, Atchison WD. Methylmercury exposure alters fluo-4 fluorescence in spinal cord slices of mice expressing the human Cu2+/Zn2+ superoxide dismutase 1 (hSOD1) gene mutation. Society for Neuroscience. 2016 46th Annual Meeting(ALS) [Google Scholar]
- 54.Atchison WD. Is chemical neurotransmission altered specifically during methylmercury-induced cerebellar dysfunction? Trends in Pharmacological Sciences. 2005;26(11):549–57. doi: 10.1016/j.tips.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 55.Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacology & Therapeutics. 1999;81(3):163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 56.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, et al. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Annals of Neurology. 1990;28(1):18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 57.Mahajan SS, Ziff EB. Novel toxicity of the unedited GluR2 AMPA receptor subunit dependent on surface trafficking and increased Ca2+-permeability. Molecular and Cellular Neurosciences. 2007;35(3):470–81. doi: 10.1016/j.mcn.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58•.Mahajan SS, Thai KH, Chen K, Ziff E. Exposure of neurons to excitotoxic levels of glutamate induces cleavage of the RNA editing enzyme, adenosine deaminase acting on RNA 2, and loss of GLUR2 editing. Neuroscience. 2011;189:305–15. doi: 10.1016/j.neuroscience.2011.05.027. This study demonstrates the mechanics of glutamate excitotoxicity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colón-Rodríguez A, Hajela RK, Atchison WD. Brain region-dependent effects of methylmercury on expression of ligand and voltage-gated calcium channels in rat. The Toxicologist, Supplement to Toxicological Sciences. 2014;138(1) Abstract #1384. [Google Scholar]
- 60.Colón-Rodríguez A, Hajela RK, Atchison WD. Methylmercury alters intracellular calcium concentrations in human-induced pluripotent stem cell motor neurons in a concentration-dependent manner. The Toxicologist, Supplement to Toxicological Sciences. 2017 Abstract #1143. [Google Scholar]
- 61.Su XW, Nandar W, Neely EB, Simmons Z, Connor JR. Statins accelerate disease progression and shorten survival in SOD1(G93A) mice. Muscle Nerve. 2016;54(2):284–91. doi: 10.1002/mus.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powers S, Kwok S, Lovejoy E, Lavin T, Sher R. Embryonic Exposure to the Environmental Neurotoxin BMAA Negatively Impacts Early Neuronal Development and Progression of Neurodegeneration in the Sod1-G93R Zebrafish Model of Amyotrophic Lateral Sclerosis. Toxicol Sci. 2017 Jan 25; doi: 10.1093/toxsci/kfx020. pii: kfx020. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 63.Bhattacharya A, Bokov A, Muller FL, Jernigan AL, Maslin K, Diaz V, Richardson A, Van Remmen H. Dietary restriction but not rapamycin extends disease onset and survival of the H46R/H48Q mouse model of ALS. Neurobiol Aging. 2012;33(8):1829–32. doi: 10.1016/j.neurobiolaging.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 64.Gianforcaro A1, Hamadeh MJ. Dietary vitamin D3 supplementation at 10× the adequate intake improves functional capacity in the G93A transgenic mouse model of ALS, a pilot study. CNS Neurosci Ther. 2012;18(7):547–57. doi: 10.1111/j.1755-5949.2012.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]