

Abstract
Elevated blood glucose and the apolipoprotein (APOE) ε4 allele have both been associated with increased dementia risk; however, the neuropathological mechanisms underlying these associations remain unclear. We examined the impact of APOE genotype and midlife blood glucose on post-mortem vascular and Alzheimer’s disease (AD) neuropathology. Ninety-four participants from the Framingham Heart Study without diagnosed diabetes underwent health examination at midlife and brain autopsy at death. Histopathological measures of vascular and AD neuropathology were obtained and analyzed. Results demonstrated that, among APOE ε4 carriers, elevated blood glucose was associated with more severe AD pathology. There was no such relationship with vascular pathology. In a relatively healthy sample with low vascular risk burden, midlife elevated blood glucose was associated with greater AD pathology among APOE ε4 carriers. A better understanding of interactive effects of APOE genotype and vascular risk on neuropathology has implications for identification of individuals at risk for decline and long-term preventive treatment.
Keywords: Alzheimer’s disease, glucose, diabetes, vascular risk, apolipoprotein E (APOE), neuropathology
INTRODUCTION
Although many factors regarding the underlying medical problems that may potentiate Alzheimer’s disease (AD) are poorly understood, recent research has established links between AD and common cerebrovascular risk factors including diabetes [1]. Several studies have found that, even among nondiabetic individuals, elevated blood glucose is associated with increased risk for dementia [2, 3]. However, despite relatively consistent epidemiological findings linking diabetes and elevated blood glucose to dementia, there are few neuropathological studies examining the underlying neuropathological substrate of this association, and findings have generally been mixed. For example, although diabetes has been linked to cerebral infarcts [4–6], its association with AD neuropathology (amyloid plaques and neurofibrillary tangles [NFT]) is less clear [4, 5, 7–9]. Discrepant findings across studies have been attributed to methodological differences regarding risk factor characterization and assessment, as well as failure to account for potentially confounding variables such as presence of vascular pathologies [1, 9].
To our knowledge, there are no published reports investigating how the apolipoprotein E (APOE) ε4 allele— a well-established risk factor for AD—may modify the relationship between elevated blood glucose and neuropathology in nondiabetic individuals. Furthermore, most investigations of dementia and diabetes or blood glucose have focused on late-life versus midlife exposure despite growing awareness of the importance of a life-long perspective of dementia prevention and, in particular, the need for studies to follow participants beginning in midlife, prior to the typical later-life onset of AD. Thus, in a well-characterized cohort of Framingham Heart Study participants, we examined the combined impact of midlife elevated blood glucose and APOE genotype as related to AD neuropathology, vascular lesions, and clinical diagnosis.
MATERIALS AND METHODS
Participants
The Framingham Heart Study (FHS) began in 1948 to identify risk factors for cardiovascular disease. Participants in the current study were members of the Original cohort and Offspring cohort, which includes biological children of the Original cohort and Offspring spouses. Original and Offspring cohorts have undergone health examinations every two years since 1948 and every four to eight years since 1971, respectively. Participants in the current study are those individuals from these two cohorts that later came to autopsy through the Framingham Brain Donation Program.
Midlife blood glucose and other vascular risk factor data were extracted from the examination closest to midlife (50±5 years). Clinical diagnosis is the diagnosis closest in time to death and is determined at a consensus conference. The diagnostic protocol involves review of medical information from five key sources: (1) FHS health examinations, (2) FHS neurological examinations, (3) neuropsychological evaluations, (4) outside medical records, and (5) a post-mortem family interview inquiring about changes in cognitive and functional status as well as the time course of these changes. Information from these sources is reviewed to determine presence of cognitive impairment and dementia, timing of onset and diagnosis, and subtype of dementia or cognitive impairment. Individuals identified as demented met Diagnostic and Statistical Manual of Mental Disorders, 4th Edition criteria [10]. Those categorized as AD met National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorder Association criteria for possible, probable or definite AD [11]. The diagnosis of vascular dementia was determined based on Alzheimer’s Disease Diagnostic and Treatment Centers and National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) criteria [12]. Clinical diagnostic criteria for other forms of dementia such as Lewy Body disease and frontotemporal dementia were also carefully specified based on published criteria [13, 14]. Mild cognitive impairment (MCI) diagnosis was based on subjective and/or objective cognitive impairment in one or more cognitive domains with essentially normal functional status [15].
Of the 164 participants who came to autopsy between 1997 and 2013, we included those individuals who had midlife blood glucose and APOE genotype data. Individuals were excluded for missing data, APOE ε2/ε4 genotype, and diagnosis of diabetes at midlife. For Original cohort participants (who had not been asked to fast for blood glucose test), diabetes mellitus was defined as blood glucose ≥200 mg/dL or current treatment for diabetes. For Offspring cohort participants (who were asked to fast for blood glucose test), diabetes mellitus was defined as fasting blood glucose ≥126 mg/dL or use of an anti-diabetic therapy. Together these inclusion and exclusion criteria resulted in a final sample of 94 participants for the current study. See Figure 1. The protocol was approved by the Institutional Review Board of Boston University Medical Center. All participants provided written informed consent.
Figure 1.

Flow diagram of participant selection process
Blood Glucose and Vascular Risk Factors
Elevated blood glucose was defined as fasting glucose ≥100 mg/dL (offspring cohort) or random glucose ≥140 mg/dL (original cohort) [16]. No participants were taking anti-diabetic medication. To assess overall vascular risk burden, additional vascular risk factors were assessed at midlife. Obesity was defined as body mass index ≥30 kg/m2; hypertension was defined as systolic blood pressure ≥140 mm Hg, diastolic blood pressure ≥90 mm Hg, or use of antihypertensive medications; history of cardiovascular disease was based on history of coronary heart disease (myocardial infarction, angina pectoris, coronary insufficiency), cardiac failure, and intermittent claudication [17]; serum total cholesterol was measured; and cigarette smokers were identified based on current smoking at midlife.
Neuropathological Assessment
Brain autopsies were performed by a single neuropathologist (ACM), who was blind to all clinical information, using established Framingham Brain Donation Program protocols. Details of the neuropathological assessment have been previously published [15]. Briefly, brains were received fresh and examined grossly. The frontal, temporal and occipital poles were removed from one hemisphere and snap frozen at −80 C. The remaining tissue was fixed in 4% periodate-lysine-paraformaldehyde at 4 degrees Celsius for a minimum of two weeks. Tissue blocks were taken from 30 regions including two levels of the midbrain; two levels of pons; medulla; two levels of the cerebellum; the olfactory bulb; subcortical regions of the brain including the caudate, putamen, nucleus accumbens, amygdala, entorhinal cortex, globus pallidus, substantia innominata, anterior hippocampus, hippocampus at the level of the lateral geniculate, and two levels of the thalamus; the pre- and post-central gyrus; the frontal lobe including inferior frontal, superior frontal, and dorsolateral middle frontal regions, and anterior cingulate; the temporal lobe including anterior temporal, superior temporal, superior temporal posterior regions; posterior cortex including the posterior cingulate, calcarine, inferior parietal, superior parietal and visual association regions.
Tissue blocks were embedded in paraffin, cut into 10-μm thick sections, and stained with luxol fast blue, hematoxylin and eosin, Bielschowsky silver method, and immunocytochemistry for phosphorylated tau protein (AT8) and Aβ protein.
Alzheimer’s Disease Lesions
Neurofibrillary tangles (NFT) were counted using AT8 immunostained sections and rated on a semi-quantitative scale for three medial temporal structures (amygdala, entorhinal cortex, hippocampus) and five neocortical regions (inferior parietal, middle frontal, superior temporal, calcarine, visual association cortices). For analytical purposes, we focused on MTL and cortical NFT densities given that they are areas thought to be affected early and later in the course of AD, respectively. The four point semiquantitative scale ranges from 1+ (indicating 1–10 NFT/microscopic field for the MTL structures and a maximum density of 1 NFT per 200 × microscopic field for the cortical regions) to 4+ (corresponding to ≥31 NFT/field for MTL structures and ≥10 NFT/field for cortical regions). Density of diffuse and neuritic plaques was determined by Aβ immunostaining and Bielschowsky silver stain in these same regions [15]. Brains were staged for the degree of NFT pathology using a modification of Braak and Braak’s scheme [15]. Neuropathological diagnosis of AD was performed based on recommendations of the National Institute on Aging (NIA)-Reagan criteria [19] and Braak and Braak Stage.
Vascular Lesions
Participant brains were assessed for forms of vascular pathology including: 1) chronic macroscopic infarcts (>10 mm), 2) macroscopic lacunes (small [<10 mm] infarcts) 3) microinfarcts (encephalomalacic lesions ≤2 mm in diameter not identifiable on gross inspection of the brain), 4) degree of cerebral arteriosclerosis (hyaline thickening of arteriolar wall evaluated in four regions of deep white matter and basal ganglia), and 5) degree of atherosclerosis in the circle of Willis. A semi-quantitative measure of vascular pathology severity was calculated based on the presence versus absence and severity of the above forms of pathology (Atherosclerotic Injury Score). The presence and severity of cerebral amyloid angiopathy (CAA) was assessed semi-quantitatively in middle frontal, inferior parietal, superior temporal, and calcarine cortices.
Statistical Analyses
Multivariate linear and logistic regression analyses were performed to examine associations among blood glucose, APOE genotype, and neuropathology. APOE genotype was coded as ε4 carrier (i.e., individuals with at least one copy of the APOE ε4 gene) versus non-carrier (i.e., participants with no copies of the APOE ε4 gene). Main effects and interactions were examined in separate models. Models with interaction terms were performed in order to examine the modifying effect of APOE genotype on the relationship between midlife blood glucose level and neuropathological burden. All models were adjusted for age at blood glucose assessment, sex, time from midlife vascular risk factor assessment to death, and obesity, smoking status, systolic blood pressure, and total cholesterol at midlife. Logistic regression analyses were conducted to examine the associations among blood glucose, APOE genotype, and clinical diagnosis (i.e., normal cognition, MCI, or dementia).
Three sets of secondary analyses were performed. First, analyses were performed adjusting for late life diabetes to determine whether findings were due to late life diabetes onset in people with elevated blood glucose at midlife. Second, analyses were performed adjusting for late life elevated blood glucose to determine whether results were due to late life elevated blood glucose regardless of the presence or absence of diabetes. Third, analyses additionally adjusting for cohort given that our definition of elevated blood glucose was not uniform across the original and offspring cohorts (i.e., data from the original cohort consisted of casual glucose samples whereas the offspring cohort consisted of fasting glucose samples). Significance levels of 0.05 were used for all tests. Analyses were performed using Statistical Analyses System software version 9.3 (SAS Institute, Cary, NC).
RESULTS
Demographic variables and participant characteristics
Midlife blood glucose and vascular risk factors were assessed at a mean age of 50 years. The mean age at death was 84 years. During the period of time between blood glucose assessment and death, 20 participants sustained a stroke and 41 developed dementia. Of those who sustained a stroke, five had elevated blood glucose at midlife (two APOE ε4 carriers and three noncarriers) and 15 did not have elevated blood glucose at midlife (four APOE ε4 carriers and 11 noncarriers). At the midlife assessment approximately 20% of the sample was classified as having elevated blood glucose. Nearly half of the participants had no vascular risk factors (elevated blood glucose, hypertension, cardiovascular disease, current smoking) and only 20% had two or more vascular risk factors. Approximately 25% of participants were APOE ε4 carriers (Table 1). APOE ε4 carriers and non-carriers did not significantly differ in terms of the proportion of individuals who had elevated blood glucose at midlife (30.4% of APOE ε4 carriers and 16.9% of non-carriers; Fisher’s exact p = 0.08) and at late life (38.1% of APOE ε4 carriers and 35.7% of non-carriers; χ2 = 0.04, p = 0.85). It should be noted that late life blood glucose data was missing for 18% of the sample.
Table 1.
Participant Demographics and Risk Factor Characteristics
| Variable | |
|---|---|
| N | 94 |
| Demographics | |
| Age at vascular risk factor assessment (years; mean ± SD) | 50±2 |
| Time between vascular risk factor assessment and death (years; mean ± SD) | 34±11 |
| Cohort, n (%) Original (first generation) Offspring (second generation) |
48 (51.1) 46 (48.9) |
| Women, n (%) | 47 (50.0) |
| Education, n (%) No HS Degree HS Degree Some College College Degree |
8 (8.8) 24 (26.4) 25 (27.5) 34 (37.3) |
| APOE genotype, n (%) 22 23 33 34 44 ε4 carrier, n (%) |
1(1.0) 12 (12.8) 58 (61.7) 23 (24.5) 0 23 (24.5) |
| Vascular Risk Factors | |
| Systolic Blood Pressure (mmHg; mean ± SD) Total sample APOE ε4 carriers APOE ε4 non-carriers |
123±17 119±15 125±17 |
| Elevated blood glucose, n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
19 (20.2) 7 (30.4) 12 (16.9) |
| Hypertension, n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
24 (25.5) 7 (30.4) 17 (23.9) |
| History of CVD, n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
1 (1.1) 0 1 (1.4) |
| Atrial fibrillation, n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
1 (1.1) 1 (4.4) 0 |
| Obesity (body mass index ≥30), n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
12 (12.8) 2 (8.7) 10 (14.1) |
| Smoking, n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
27 (28.7) 7 (30.4) 20 (28.2) |
| Aggregate Vascular Risk (based on sum of above risk factors [i.e., obesity, HTN, CVD, elevated blood glucose, Smoking], %) No vascular risk factors 1 vascular risk factor ≥2 vascular risk factors |
44 (46.8) 31 (33.0) 19 (20.2) |
| Use of anti-hypertensive medication, n (%) Total sample APOE ε4 carriers APOE ε4 non-carriers |
6 (6.5) 1 (4.4) 5 (7.3) |
| Cognitive Functioning | |
| Clinical Diagnosis, n (%) Normal MCI Dementia AD |
37 (39.4) 16 (17.05) 41 (43.6) 29 (30.9) |
| Neuropathology | |
| Braak Stage, n (%) I–III, VII (Not AD) IV–VI (AD) |
65 (69.2) 29 (30.9) |
| NIA Reagan, n (%) 1–2 (AD) 3–4 (Not AD) |
36 (38.3) 58 (61.7) |
| Cortical neuritic plaques, n (%) Absent Present |
29 (30.9) 65 (69.1) |
| Medial temporal neuritic plaques, n (%) Absent Present |
38 (40.4) 56 (59.6) |
Abbreviations: SD = standard deviation; HS = high school; APOE = apolipoprotein E; CVD = cardiovascular disease; HTN = hypertension; AD = Alzheimer’s disease; NIA = National Institute on Aging
Effects of midlife elevated blood glucose and APOE genotype on neuropathology
Across all participants, elevated midlife blood glucose was significantly associated with greater density of medial temporal NFT (β±SE: 1.97±0.88, p<.05). There were no other significant main effects of elevated blood glucose (p-values>.05; Table 2). Across all participants, the presence of the APOE ε4 allele was significantly associated with AD pathology, including greater density of medial temporal NFT (β±SE: 2.13±0.77, p≤ .01) and cortical NFT (β±SE: 5.73±1.90, p≤.01), presence of cortical diffuse plaques (OR[95% CI]: 6.86[1.26–37.50], p<.05), medial temporal neuritic plaques (OR[95% CI]: 5.77[1.52–21.89], p≤.01), cortical neuritic plaques (OR[95% CI]: 10.23[1.75–59.94], p≤.01), and higher Braak staging (β±SE: 1.24±0.44, p≤.01). The presence of the APOE ε4 allele was also significantly associated with the presence of CAA (OR[95% CI]: 16.76[1.85–152.13], p<.05). APOE genotype was not associated with the presence of vascular pathology (p-values >.05). All models were adjusted for age, sex, time between midlife vascular risk assessment and death, and presence of additional vascular risk factors at midlife.
Table 2.
Main effect of APOE genotype, main effect of elevated blood glucose (EBG), and interaction between APOE genotype and EBG on neuropathology
| APOE ε4 β ± SE or OR [95% CI] |
Elevated Blood Glucose (EBG) β ± SE or OR [95% CI] |
Interaction between APOE genotype and EBG p value |
|
|---|---|---|---|
| AD pathology | |||
| Medial temporal AT8 NFT density | 2.13 ± 0.77** | 1.97 ± 0.88* | 0.017 |
| Cortical AT8 NFT density | 5.73 ± 1.90** | 1.13 ± 2.23 | 0.179 |
| Medial temporal diffuse plaques (>0) | 3.56 [0.95–13.38] | 0.93 [0.27–3.23] | 0.965 |
| Cortical diffuse plaques (>0) | 6.86 [1.26–37.50]* | 1.25 [0.34–4.60] | 0.973 |
| Medial temporal neuritic plaques (>0) | 5.77 [1.52–21.89]** | 1.04 [0.33–3.29] | 0.964 |
| Cortical neuritic plaques (>0) | 10.23 [1.75–59.94]** | 1.00 [0.28–3.56] | 0.972 |
| Braak and Braak Stage | 1.24 ± 0.44** | −0.03 ± 0.52 | 0.064 |
| Vascular pathology | |||
| Atherosclerotic Injury Score | −0.17 ± 0.76 | −0.08 ± 0.86 | 0.119 |
| Atherosclerosis of the circle of Willis (>0) | 1.14 [0.37–3.54] | 2.87 [0.72–11.42] | 0.741 |
| Arteriosclerosis (>0) | 1.23 [0.29–5.19] | 2.48 [0.45–13.50] | 0.973 |
| Cerebral amyloid angiopathy | |||
| CAA (>0) | 16.76 [1.85–152.13]* | 0.60 [0.18–2.00] | 0.971 |
p<0.05,
p≤0.01
β represents difference in adjusted means; SE = standard error; AD = Alzheimer’s disease; NFT = neurofibrillary tangles; OR = odds ratio; CI = confidence interval; APOE = apolipoprotein E; CAA = cerebral amyloid angiopathy
Main effects and interactions were examined in separate models. All models adjusted for age; sex; time between midlife vascular risk factor assessment and death; and obesity, systolic blood pressure, total cholesterol, and smoking status at midlife.
There were significant interactions whereby APOE genotype modified the relationship between midlife elevated blood glucose and neuropathology (Table 2). Specifically, among APOE ε4 carriers, elevated blood glucose was associated with significantly greater density of medial temporal NFT (β±SE: 7.41±2.26, p=0.006) and higher Braak staging (β±SE: 3.54±1.12, p =0.007). See Table 3 and Figure 2.
Table 3.
Association between midlife elevated blood glucose and neuropathology stratified by APOE ε4 status (for those interactions where p < 0.10)
| Elevated Blood Glucose β ± SE (p) |
|
|---|---|
| AD pathology | |
| Medial temporal AT8 NFT APOE ε4− APOE ε4+ |
0.30±1.04 (0.771) 7.41±2.26 (0.006) |
| Braak and Braak Stage APOE ε4− APOE ε4+ |
−0.91±0.61 (0.140) 3.54±1.12 (0.007) |
β represents difference in adjusted means; SE = standard error; AD = Alzheimer’s disease; NFT = neurofibrillary tangles; APOE = apolipoprotein E
All models are adjusted for age; sex; time between midlife vascular risk assessment and death; and obesity, systolic blood pressure, total cholesterol, and smoking status at midlife
Figure 2. Mean medial temporal neurofibrillary tangle density by APOE genotype and elevated blood glucose.
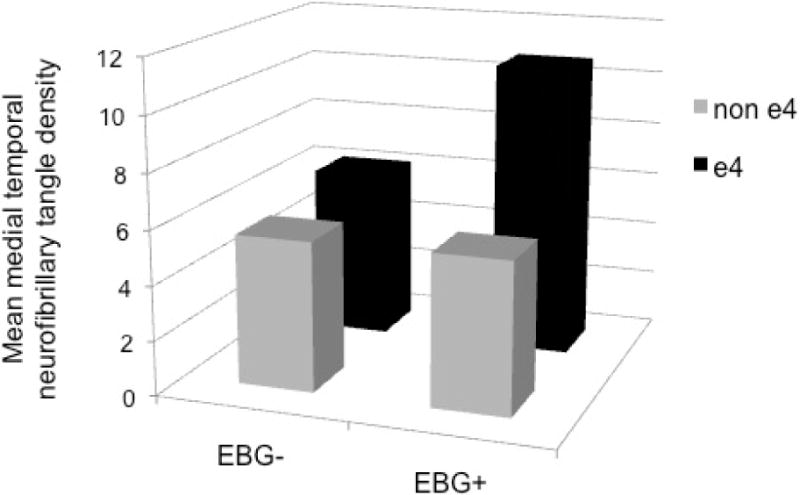
Mean medial temporal neurofibrillary tangle (NFT) density adjusted for age at blood glucose assessment, sex, time from midlife blood glucose assessment to death, and obesity, smoking status, systolic blood pressure, and total cholesterol at midlife. APOE genotype significantly modified the relationship between elevated blood glucose and medial temporal NFT density (p = 0.017). EBG = elevated blood glucose.
Effects of midlife elevated blood glucose and APOE genotype on neuropathological and clinical diagnosis
Although there were no main effects of midlife elevated blood glucose on neuropathological or clinical diagnosis (p-values >.05), there were significant main effects of APOE genotype such that APOE ε4 carriers were significantly more likely to be diagnosed with AD based upon Braak staging and NIA-Reagan neuropathologic criteria. In addition, APOE ε4 carriers were significantly more likely to be clinically diagnosed with dementia (all forms) and AD specifically (Table 4). APOE genotype did not significantly modify the relationship between midlife elevated blood glucose and neuropathological or clinical diagnosis (p-values >.05).
Table 4.
Main effect of APOE genotype, main effect of elevated blood glucose (EBG), and interaction between APOE genotype and EBG on neuropathological and clinical diagnosis
| Neuropathological Diagnosis | APOE ε4 OR [95% CI] |
Elevated Blood Glucose (EBG) OR [95% CI] |
Interaction between APOE genotype and EBG p value |
|---|---|---|---|
| Alzheimer’s disease (Braak and Braak Stage) | 4.46 [1.50–13.29]** | 1.62 [0.48–5.47] | 0.200 |
| Alzheimer’s disease (NIA/Reagan) | 5.77 [1.88–17.70]** | 1.32 [0.41–4.30] | 0.560 |
| Clinical Diagnosis | |||
| Cognitively normal | Ref | Ref | |
| Mild cognitive impairment | 2.17 [0.40–11.94] | 2.59 [0.41–16.33] | 0.999 |
| Dementia (all forms) | 5.63 [1.50–21.20]** | 2.73 [0.71–10.55] | 0.963 |
| Alzheimer’s disease | 8.66 [1.68–44.77]** | 2.18 [0.45–10.62] | 0.952 |
p<0.05,
p≤0.01
Abbreviations: OR = odds ratio; CI = confidence interval; APOE = apolipoprotein E; NIA = National Institute on Aging
All models are adjusted for age; sex; time between midlife vascular risk assessment and death; and obesity, systolic blood pressure, total cholesterol, and smoking status at midlife
Secondary analyses adjusting for late life diabetes, late life elevated blood glucose, and cohort effect
When analyses were performed a second time adjusting for late life diabetes, all findings remained statistically and qualitatively similar. When analyses were performed a third time additionally adjusting for late life elevated blood glucose, the majority of the findings remained similar. The association between cortical diffuse plaques and APOE genotype, association between elevated blood glucose and medial temporal NFT density, interaction between elevated blood glucose and APOE genotype on Braak and Braak stage, and associations between APOE genotype and clinical diagnosis no longer met statistical significance. When analyses were performed a fourth time additionally adjusting for cohort/type of blood glucose test (i.e., random versus fasting), findings were generally not changed. The one exception was related to the association between midlife elevated blood glucose and Braak Stage among APOE ε4 carriers (see Table 3). After controlling for cohort effect/type of glucose test, there was no longer a statistically significant association and instead a trend toward an association between elevated blood glucose and higher Braak stage among APOE ε4 carriers was revealed (β±SE: 2.90±1.62, p=.098).
DISCUSSION
In this prospective, community-based sample, we found that midlife elevated blood glucose predicted the later development of AD tangle pathology in individuals with the APOE ε4 genotype. Presence of the APOE ε4 allele significantly modified the association between elevated blood glucose and AD tangle pathology independently of other known risk factors for AD, including age, history of smoking, and midlife obesity, hypertension, and high cholesterol. Importantly, findings did not change when adjusting for presence of late life diabetes, suggesting that results were not due to late life diabetes in people with elevated glucose during midlife. These results suggest that higher levels of blood glucose—even decades before diagnosis and death—and even among nondiabetics—may exert deleterious effects on the aging brain among those at genetic risk for AD.
Most previous studies that have examined associations between glucose metabolism and neuropathological outcome have focused on diabetes itself, and far fewer have specifically examined the impact of APOE genotype on this association. Our findings are in line with autopsy studies wherein individuals with both diabetes and the APOE ε4 allele demonstrated an elevated risk of AD-associated neuropathology [5, 7]. Our findings also parallel those from various cohorts that demonstrate a significant interaction of APOE ε4 genotype and diabetes that increases the risk of cognitive decline [20–23], and they extend these findings to a relatively healthy sample of middle-aged adults with elevated blood glucose versus diagnosed diabetes.
Suggested mechanisms by which diabetes may affect the brain and negatively impact cognition include changes to brain vasculature, alterations in cerebral insulin signaling, insulin resistance, glucose toxicity, oxidative stress, accumulation of advanced glycation end products (AGEs), increased inflammation, and/or ischemia [24, 25]. The precise mechanism by which elevated blood glucose and APOE genotype interact to increase AD neuropathology is unclear. It has been suggested that AGEs and APOE may play a role in neurodegeneration given co-localization of AGEs in NFTs, senile plaques, and CAA in AD and other neurodegenerative diseases [26]. Indeed, APOE ε4 is associated with a 3-fold greater AGE-binding activity compared to the APOE ε3 isoform indicating that age by APOE ε4 interactions may contribute to the formation of dense amyloid deposits as well as NFTs [27]. Interestingly, although APOE ε4 carriers with elevated blood glucose at midlife demonstrated greater density of medial temporal NFT and higher Braak staging, there were no significant interactions on density of neuritic or diffuse plaques. These findings dovetail with evidence showing that hyperinsulinemia and impaired cortical insulin resistance may be linked to tau hyperphosphorylation [28, 29]. Collectively, these findings indicate that altered glucose metabolism may also be linked to processes promoting tau accumulation.
One proposed model of neurodegeneration suggests that amyloid-β (Aβ) protein deposition drives AD pathogenesis and secondarily induces the formation of abnormal tau protein, followed by tau-mediated neuronal injury, metabolic and structure brain changes, and culminating with cognitive and functional impairments [30]. However, other evidence suggests that Aβ is not required to develop neurodegeneration within AD-affected regions[31]. Furthermore, recent studies suggest that the various biomarkers of neurodegeneration (FDG-PET, CSF tau, structural MRI) are not interchangeable [32]. That is, different ‘neuronal injury’ biomarkers showed important disagreements in classifying subjects and in predicting progression [32]. Our findings suggest that the mandatory initial appearance of amyloidosis prior to tau alterations may be altered in the presence of APOE ε4 and elevated blood glucose. That is, genetic and neural injury risks may trump amyloidosis in the AD pathophysiological cascade.
Although diabetes has been shown to be associated with increased risk for cerebral infarcts [5, 8, 33] and vascular dementia [34], we did not observe such an association with elevated blood glucose in our sample. However, a recent study that modeled diabetic status in midlife and cognitive functioning and MR measures of brain volume, infarcts, and white matter hyperintensities in late life found that global brain volume (rather than markers of vascular pathology) was in the causal pathway for the association between diabetes and cognitive decline [35]. Furthermore, we previously found that, in a sample of patients with autopsy-confirmed AD who had mild CVD, diabetes predicted the presence of CVD only when considered in combination with additional vascular risk factors [36]. Such findings highlight the importance of investigating aggregate risk in the vascular contribution to dementia. However, it may be the case that some individuals in our sample, with an average age of 50, may not have yet expressed abnormalities in glucose metabolism.
After additionally adjusting statistical models for late life elevated blood glucose, the majority of findings remained similar including the significant interaction between midlife elevated blood glucose and APOE genotype on medial temporal neurofibrillary tangles; however, some findings were attenuated and no longer statistically significant. Both midlife and late life diabetes have been associated with increased risk of Alzheimer’s disease [37] and findings from a meta-analysis suggested that the magnitude of association is similar in both age groups [38]. Although both midlife and late life elevated blood glucose may represent important risk factors, we focused on midlife elevated blood glucose given that many previous studies of dementia risk have not included midlife data as well as the hope that midlife may present opportunities for earlier intervention aimed at preventing cognitive decline. It is possible that including midlife and late life elevated blood glucose as an independent variable and covariate, respectively, in our models may have led to overadjustment in our secondary analyses.
Strengths of the current study include its well-characterized, community-based sample; prospective study design; intensive, ongoing follow-up for several decades; and pathologic diagnoses made blind to clinical or demographic information [39]. However, despite these strengths, there are some weaknesses that are important to note. First, we did not find any significant interactive effects of midlife elevated blood glucose and APOE genotype on diagnosis, and findings may have differed if we had examined diagnosis at an earlier age. In addition, the number of participants in some subgroups was relatively small and the sample was generally healthy with relatively few vascular risk factors, which may have limited our findings. Moreover, we measured blood glucose levels, and results may have differed if we examined a different glucose measure (e.g., glycated hemoglobin level). In addition, due to different collection procedures (i.e., random/casual versus fasting) and therefore different thresholds for elevated blood glucose and diabetes across participants, we did not investigate blood glucose as a continuous variable. Given power considerations, we were unable to examine gene-dose effects or the specific influence of the APOE ε2 allele. Finally, as is typical among neuropathology studies, we assessed segments of brain tissue in only one hemisphere. We may have therefore underestimated existing pathology—particularly evidence of CVD that may be patchy and irregularly distributed throughout the brain—and, as such, a more exhaustive autopsy approach may be more useful to quantify this type of neuropathology.
Despite these limitations, our finding of an interaction between elevated midlife glucose levels and APOE ε4 genotype is especially important when taking into consideration the public health impacts of diabetes and AD coupled with epidemiological data showing that rates of both disease states are expected to dramatically increase. In the search for reliable biomarkers of preclinical dementia, examination of the combination or interaction of risk factors in evaluating dementia risk may have potential use. Among older patients with diabetes, the effects of tighter glycemic control on cognition have been inconclusive [40–42]. However, it is possible that earlier intervention in individuals who do not yet have diabetes may be more effective, particularly in light of recent findings demonstrating that diabetes medications may reduce AD neuropathology [43]. The present findings, along with other studies, may inform future trials of glucose lowering in prediabetes during midlife, particularly among those who are already at risk for dementia by virtue of possession of the APOE ε4 allele.
Acknowledgments
Funding Sources
The authors thank the dedicated participants of the Framingham Heart Study. This research was made possible by the Framingham Heart Study’s National Heart, Lung, and Blood Institute contract (NIH/NHLBI Contract #N01-HC-25195), grants from the National Institutes of Health (National Institute on Aging K24 AG26431, AG033040, AG16495, AG08122, and National Institute of Neurological Disorders and Stroke NS17950) and VA Clinical Science Research & Development (Career Development Award-2 1IK2CX000938 to KJB).
Footnotes
Conflicts
There are no conflicts to report.
References
- 1.Vagelatos NT, Eslick GD. Type 2 Diabetes as a Risk Factor for Alzheimer’s Disease: The Confounders, Interactions, and Neuropathology Associated With This Relationship. Epidemiol Rev. 2013 doi: 10.1093/epirev/mxs012. [DOI] [PubMed] [Google Scholar]
- 2.Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, Haneuse S, Craft S, Montine TJ, Kahn SE, McCormick W, McCurry SM, Bowen JD, Larson EB. Glucose levels and risk of dementia. N Engl J Med. 2013;369:540–548. doi: 10.1056/NEJMoa1215740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerti L, Witte AV, Winkler A, Grittner U, Rujescu D, Floel A. Higher glucose levels associated with lower memory and reduced hippocampal microstructure. Neurology. 2013;81:1746–1752. doi: 10.1212/01.wnl.0000435561.00234.ee. [DOI] [PubMed] [Google Scholar]
- 4.Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, Bennett DA. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67:1960–1965. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- 5.Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 6.Sarwar N, Aspelund T, Eiriksdottir G, Gobin R, Seshasai SR, Forouhi NG, Sigurdsson G, Danesh J, Gudnason V. Markers of dysglycaemia and risk of coronary heart disease in people without diabetes: Reykjavik prospective study and systematic review. PLoS Med. 2010;7:e1000278. doi: 10.1371/journal.pmed.1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malek-Ahmadi M, Beach T, Obradov A, Sue L, Belden C, Davis K, Walker DG, Lue L, Adem A, Sabbagh MN. Increased Alzheimer’s disease neuropathology is associated with type 2 diabetes and ApoE epsilon.4 carrier status. Curr Alzheimer Res. 2013;10:654–659. doi: 10.2174/15672050113109990006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. 2010;75:1195–1202. doi: 10.1212/WNL.0b013e3181f4d7f8. [DOI] [PubMed] [Google Scholar]
- 9.Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, Purohit DP, Perl DP, Davidson M, Mohs RC, Haroutunian V. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci. 2005;60:471–475. doi: 10.1093/gerona/60.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Association AP. Diagnostic and statistical manual of mental disorders (DSM-IV) America Psychiatric Association; Washington, D.C.: 1994. [Google Scholar]
- 11.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 12.Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 13.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9:417–423. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 14.Miller BL, Ikonte C, Ponton M, Levy M, Boone K, Darby A, Berman N, Mena I, Cummings JL. A study of the Lund-Manchester research criteria for frontotemporal dementia: clinical and single-photon emission CT correlations. Neurology. 1997;48:937–942. doi: 10.1212/wnl.48.4.937. [DOI] [PubMed] [Google Scholar]
- 15.Au R, Seshadri S, Knox K, Beiser A, Himali JJ, Cabral HJ, Auerbach S, Green RC, Wolf PA, McKee AC. The Framingham Brain Donation Program: neuropathology along the cognitive continuum. Curr Alzheimer Res. 2012;9:673–686. doi: 10.2174/156720512801322609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genuth S, Alberti KG, Bennett P, Buse J, Defronzo R, Kahn R, Kitzmiller J, Knowler WC, Lebovitz H, Lernmark A, Nathan D, Palmer J, Rizza R, Saudek C, Shaw J, Steffes M, Stern M, Tuomilehto J, Zimmet P. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003;26:3160–3167. doi: 10.2337/diacare.26.11.3160. [DOI] [PubMed] [Google Scholar]
- 17.Wolf PA, D’Agostino RB, Belanger AJ, Kannel WB. Probability of stroke: a risk profile from the Framingham Study. Stroke. 1991;22:312–318. doi: 10.1161/01.str.22.3.312. [DOI] [PubMed] [Google Scholar]
- 18.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 19.Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 20.Haan MN, Shemanski L, Jagust WJ, Manolio TA, Kuller L. The role of APOE epsilon4 in modulating effects of other risk factors for cognitive decline in elderly persons. JAMA. 1999;282:40–46. doi: 10.1001/jama.282.1.40. [DOI] [PubMed] [Google Scholar]
- 21.Bangen KJ, Beiser A, Delano-Wood L, Nation DA, Lamar M, Libon DJ, Bondi MW, Seshadri S, Wolf PA, Au R. APOE Genotype Modifies the Relationship between Midlife Vascular Risk Factors and Later Cognitive Decline. J Stroke Cerebrovasc Dis. 2013 doi: 10.1016/j.jstrokecerebrovasdis.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalmijn S, Feskens EJ, Launer LJ, Kromhout D. Cerebrovascular disease, the apolipoprotein e4 allele, and cognitive decline in a community-based study of elderly men. Stroke. 1996;27:2230–2235. doi: 10.1161/01.str.27.12.2230. [DOI] [PubMed] [Google Scholar]
- 23.Dore GA, Elias MF, Robbins MA, Elias PK, Nagy Z. Presence of the APOE epsilon4 allele modifies the relationship between type 2 diabetes and cognitive performance: the Maine-Syracuse Study. Diabetologia. 2009;52:2551–2560. doi: 10.1007/s00125-009-1497-2. [DOI] [PubMed] [Google Scholar]
- 24.Biessels GJ. Cerebral complications of diabetes: clinical findings and pathogenetic mechanisms. Neth J Med. 1999;54:35–45. doi: 10.1016/s0300-2977(98)00134-x. [DOI] [PubMed] [Google Scholar]
- 25.Bornstein NM, Brainin M, Guekht A, Skoog I, Korczyn AD. Diabetes and the brain: issues and unmet needs. Neurol Sci. 2014 doi: 10.1007/s10072-014-1797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, Koike T, Wakayama I, Yanagihara R, Garruto R, Amano N, Makita Z. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am J Pathol. 1998;153:1149–1155. doi: 10.1016/S0002-9440(10)65659-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li YM, Dickson DW. Enhanced binding of advanced glycation endproducts (AGE) by the ApoE4 isoform links the mechanism of plaque deposition in Alzheimer’s disease. Neurosci Lett. 1997;226:155–158. doi: 10.1016/s0304-3940(97)00266-8. [DOI] [PubMed] [Google Scholar]
- 28.Freude S, Plum L, Schnitker J, Leeser U, Udelhoven M, Krone W, Bruning JC, Schubert M. Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes. 2005;54:3343–3348. doi: 10.2337/diabetes.54.12.3343. [DOI] [PubMed] [Google Scholar]
- 29.Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, Kondo T, Alber J, Galldiks N, Kustermann E, Arndt S, Jacobs AH, Krone W, Kahn CR, Bruning JC. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. 2004;101:3100–3105. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer’s disease neurodegenerative biomarkers are associated with decreased cognitive function but not beta-amyloid in cognitively normal older individuals. J Neurosci. 2013;33:5553–5563. doi: 10.1523/JNEUROSCI.4409-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toledo JB, Weiner MW, Wolk DA, Da X, Chen K, Arnold SE, Jagust W, Jack C, Reiman EM, Davatzikos C, Shaw LM, Trojanowski JQ. Neuronal injury biomarkers and prognosis in ADNI subjects with normal cognition. Acta Neuropathol Commun. 2014;2:26. doi: 10.1186/2051-5960-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksson M, Carlberg B, Eliasson M. The disparity in long-term survival after a first stroke in patients with and without diabetes persists: the Northern Sweden MONICA study. Cerebrovasc Dis. 2012;34:153–160. doi: 10.1159/000339763. [DOI] [PubMed] [Google Scholar]
- 34.Cheng PY, Sy HN, Wu SL, Wang WF, Chen YY. Newly diagnosed type 2 diabetes and risk of dementia: a population-based 7-year follow-up study in Taiwan. J Diabetes Complications. 2012;26:382–387. doi: 10.1016/j.jdiacomp.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Roberts RO, Knopman DS, Przybelski SA, Mielke MM, Kantarci K, Preboske GM, Senjem ML, Pankratz VS, Geda YE, Boeve BF, Ivnik RJ, Rocca WA, Petersen RC, Jack CR., Jr Association of type 2 diabetes with brain atrophy and cognitive impairment. Neurology. 2014;82:1132–1141. doi: 10.1212/WNL.0000000000000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bangen KJ, Nation DA, Delano-Wood L, Weissberger GH, Hansen LA, Galasko DR, Salmon DP, Bondi MW. Aggregate effects of vascular risk factors on cerebrovascular changes in autopsy-confirmed Alzheimer’s disease. Alzheimers Dement. 2015;11:394–403. e391. doi: 10.1016/j.jalz.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010;67:505–512. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Kimm H, Lee PH, Shin YJ, Park KS, Jo J, Lee Y, Kang HC, Jee SH. Mid-life and late-life vascular risk factors and dementia in Korean men and women. Arch Gerontol Geriatr. 2011;52:e117–122. doi: 10.1016/j.archger.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Chui HC, Zheng L, Reed BR, Vinters HV, Mack WJ. Vascular risk factors and Alzheimer’s disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res Ther. 2012;4:1. doi: 10.1186/alzrt98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koekkoek PS, Ruis C, van den Donk M, Biessels GJ, Gorter KJ, Kappelle LJ, Rutten GE. Intensive multifactorial treatment and cognitive functioning in screen-detected type 2 diabetes–the ADDITION-Netherlands study: a cluster-randomized trial. J Neurol Sci. 2012;314:71–77. doi: 10.1016/j.jns.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 41.Luchsinger JA, Palmas W, Teresi JA, Silver S, Kong J, Eimicke JP, Weinstock RS, Shea S. Improved diabetes control in the elderly delays global cognitive decline. J Nutr Health Aging. 2011;15:445–449. doi: 10.1007/s12603-011-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan CM, Freed MI, Rood JA, Cobitz AR, Waterhouse BR, Strachan MW. Improving metabolic control leads to better working memory in adults with type 2 diabetes. Diabetes Care. 2006;29:345–351. doi: 10.2337/diacare.29.02.06.dc05-1626. [DOI] [PubMed] [Google Scholar]
- 43.Beeri MS, Schmeidler J, Silverman JM, Gandy S, Wysocki M, Hannigan CM, Purohit DP, Lesser G, Grossman HT, Haroutunian V. Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology. 2008;71:750–757. doi: 10.1212/01.wnl.0000324925.95210.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]