

Abstract
Organophosphate (OP) and organophosphate ester (OPE) adducts of albumin are valuable biomarkers for retrospective verification of exposure. In the present study, our goal was to determine whether OPE flame retardants (OPE FRs) and OPE plasticizers can covalently bind to human serum albumin (HSA), which would allow the resulting adducts to be used to evaluate exposure. Eleven OPE FRs and plasticizers were examined in a HSA-adduct in vitro assay. Pure HSA was incubated with the target OPEs, as well as with an OP insecticide (profenofos) positive control. After enzymatic cleavage with pepsin or Glu-C, the digested albumin was analyzed by matrix-assisted laser desorption ionization tandem time-of-flight mass spectrometry (MALDI-TOF/TOF-MS) and liquid chromatography-quadrupole time-of-flight mass spectrometry (LC-Q-ToF-MS). Under optimized HSA assay conditions, tyrosine adducts were formed at Y411 and Y148/Y150 with a characteristic mass shift for phosphorylation (Δm/z 166) for the profenofos positive control. However, no such phosphorylated peptides were detected for the 11 target OPEs. This negative result suggests that these OPEs have very different affinities from the OP insecticide. They are less reactive or they may specifically interact with other proteins.
Keywords: Human serum albumin, Protein adduct, Organophosphate ester, Flame retardant, Plasticizer, Biomarker
Graphical abstract
1. Introduction
Organophosphates (OPs) are a large class of chemicals, and organophosphate esters (OPEs) are widely used as insecticides, herbicides, flame retardants, plasticizers, and even as toxic nerve agents (Reemtsma et al. 2008; Thompson et al. 2010; van der Veen and de Boer 2012; Morgan and de Boer 2013;). OPE flame retardants (OPE FRs) and plasticizers are high production-volume chemicals used in a variety of industries, including plastics, furniture, textile, electronics, construction, vehicle, and petroleum industries (Marklund et al. 2005; Reemtsma et al. 2008; Wei et al. 2015; van der Veen and de Boer 2012; Wang et al. 2010). In 2001, worldwide usage of OPE FRs was estimated at 186,000 tons (Hartmann et al. 2004). OPE FRs used in Western Europe accounted for about 91,000 tons per year in 2006, and there was an increase of 2.5% in 2001–2005 and 7.1% in 2005–2006 (Reemtsma et al. 2008). In China, annual production of OPE FRs reached approximately 70,000 tons in 2007 and is predicted to increase by 15% annually (Wang et al. 2010). The EPA reported that up to 50 million lbs of chlorinated OPE FRs were produced or imported into the United States in 2012 (EPA, 2015). North American annual production of tris(2-chloroisopropyl) phosphate (TCIPP), tris(1,3-dichloro-2-propyl) phosphate (TDCIPP), and tris(2-chloroethyl) phosphate (TCEP) increased from less than 14,000 tons per year in 1986 to 38,000 tons per year in 2012 (Schreder et al. 2016). Production and usage of OPE FRs may even increase due to restrictions and regulations on the use of polybrominated diphenyl ether (PBDE) flame retardants.
The wide array of OP structural types, functional groups and substitutions results in dramatic differences in their chemical, physical and biochemical properties. Usage of OPEs as flame retardants and plasticizers are of increasing concern because of their potential adverse effects on the environment and human health (van der Veen and de Boer 2012; Wei et al. 2015). Most OPEs are used as additives instead of being chemically bonded to host materials and constitute 1% to 30% of the total composition by weight (Morgan and Gilman 2013). Therefore, they are constantly released into the environment via volatilization, abrasion and leaching from host materials during their lifetime. A growing number of reports has shown the detection and measurement of OPE FRs and OPE plasticizers in various environmental samples including indoor and outdoor air, indoor dust, water, sediments, soils and landfill leachates, biota samples and human plasma and breast milk (Marklund et al. 2003 and 2005; Hartmann et al. 2004; Reemtsma et al. 2008; Mercier et al. 2011; van der Veen and de Boer 2012; McGoldrick et al. 2014; Brommer and Harrad 2015; Hallanger et al. 2015; Wei et al. 2015; He et al. 2016; Greaves et al. 2016a).
Compared to OP insecticides, knowledge is limited on the toxicity and biological effects of OPE FRs and OPE plasticizers, as was recently reviewed by Wei et al. (2015) and Greaves and Letcher (2016b). Among these OPEs, some chlorinated OPE FRs, such as TCEP and TDCIPP are suspected carcinogens (EPA 2015). Animal studies have shown that OPE elicited effects on kidneys, liver and neurological systems, while thyroid effects and developmental and reproductive toxicity were more variable across studies (EPA 2015; Wei et al. 2015). Neurotoxic effects have been observed for TCEP, tri-n-butyl phosphate (TNBP) and triphenyl phosphate (TPHP) (Meeker and Stapleton 2010). Some compounds, such as TPHP, are suspected of being sensitizers for allergies (Camarasa and Serrabaldrich 1992) and are potent human blood monocyte carboxylesterase inhibitors (Wei et al. 2015). However, paradoxical conclusions were reported on toxicity of OPE FRs and OPE plasticizers, and these mostly resulted from a problem in quantitative risk assessment and a number of data gaps (de Ree et al. 2014).
Various enzymatic studies show that OP insecticides inhibit acetylcholinesterase (AChE) (McDonough and Shih 1997). However, there is still a debate on the toxic effects and current thought suggests that OP toxicity cannot be attributed entirely to inhibition of AChE (Casida and Quistad 2004; EPA 2015). Recently, investigators have begun to investigate covalent OP adduction of non-AChE protein targets as a possible causative step in other toxic responses (Thompson et al. 2010; Schopfer et al. 2010; Marsillach et al. 2013; Crow et al. 2014). Serum albumin was found to form a covalent bond with OPs (Casida and Quistad 2004; Peeples et al. 2005; Schopfer et al. 2010; Crow et al. 2014) and these covalent adducts with blood proteins might provide a new biomarker of OP exposure (Li et al. 2007; Williams et al. 2007; John et al. 2008 and 2010; Crow et al. 2014). In contrast to the original OPs that often have quite short half-lives in vivo, OP adducts to albumin are relatively stable and more resistant to oxime therapy (Peeples et al. 2005).
Human serum albumin (HSA) adducts can be used as a biomarker for OP insecticides and chemical warfare agent exposure (Li et al. 2007; Schopfer et al. 2010; John et al. 2010; Crow et al. 2014). However, to our knowledge, there is no report in the literature on phosphorylation of HSA by OPE FRs and OPE plasticizers. The objective of the present study was to determine whether common commercial OPE FRs and OPE plasticizers can covalently form adducts with HSA via nucleophilic sites similar to that reported for OP insecticides and chemical warfare agents. In order to achieve sensitive and reproducible results in the present study, the conditions for the specific analysis of stable OPE-HSA adducts formed in vitro were optimized with an OP insecticide as the positive control assay. The results, both positive and negative, were confirmed by matrix-assisted laser desorption ionization tandem time-of-flight mass spectrometry (MALDI-TOF/TOF-MS) and liquid chromatography-quadrupole time-of-flight mass spectrometry (LC-Q-TOF-MS).
2. Experimental
2.1. Materials
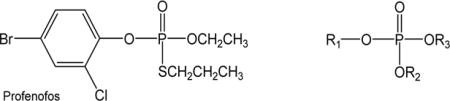
Table 1 shows the chemical structures, names and abbreviations of OPE FRs and OPE plasticizers that were examined in the present study. TCEP, TNBP, TPHP, tripropyl phosphate (TPP), tris(2-butoxyethyl) phosphate (TBOEP), tris(2-ethylhexyl) phosphate (TEHP), triethyl phosphate (TEP) and 2-ethylhexyl-diphenyl phosphate (EHDPP) were purchased from Sigma-Aldrich (St. Louis, MO, USA). TDCIPP and tri(methylphenyl) phosphate (TMPP) were purchased from TCI America (Portland, OR, USA). Tris(2-chloroisopropyl) phosphate (TCIPP) was purchased from AK Scientific (Union City, CA, USA). The organophosphate insecticide profenofos, which was used as a positive control and to optimize the methods, was purchased from Sigma-Aldrich and its molecular structure was shown in Table 1. HSA (fatty acid free, purity > 99%), endoproteinase Glu-C from Staphylococcus aureus V8 (Glu-C, sequencing grade) and pepsin from porcine gastric mucosa were purchased from Sigma–Aldrich. α-Cyano-4-hydroxycinnamic acid (HCCA) and peptide calibration standard II were from Bruker (Billerica, MA, USA). DL-dithiothreitol (DTT) was from Acros Organics (New Jersey, USA) and iodoacetamide (IAA) was from BioRad (Hercules, CA, USA). Millipore ziptips C18 (10 μL pipette tip) and Vivaspin™ 500 centrifugal concentrators VS0102 (cut-off 10 kDa) were obtained from Sigma-Aldrich and Sartorius Stedim North America Inc. (New York, USA), respectively. HPLC grade acetonitrile, ammonium hydroxide (NH4OH), ammonium bicarbonate (NH4HCO3) and trifluoroacetic acid (TFA, >99% pure) were from Fisher Scientific (Waltham, MA, USA). Water was purified on a Milli-Q Advantage A10 system (Millipore, Billerica, MA, USA).
Table 1.
Profenofos and 11 organophosphate ester (OPE) flame retardants and plasticizers
| ||||
|---|---|---|---|---|
|
| ||||
| No. | Chemical names | Abbreviation | Substituents | CAS |
| 1 | Tris(2-chloroethyl) phosphate | TCEP | R1=R2=R3= −CH2CH2Cl | 115-96-8 |
| 2 | Tripropyl phosphate | TPP | R1=R2=R3= −CH2CH2CH3 | 513-08-6 |
| 3 | Tris(2-chloroisopropyl) phosphate | TCIPP | R1=R2=R3= −CH(CH3)CH2Cl | 13674-84-5 |
| 4 | Tris(1,3-dichloro-2-propyl) phosphate | TDCIPP | R1=R2=R3= −CH(CH2Cl)2 | 13674-87-8 |
| 5 | Triphenyl phosphate | TPHP | R1=R2=R3= −C6H5 | 115-86-6 |
| 6 | Tri-n-butyl phosphate | TnBP | R1=R2=R3= −CH2(CH2)2CH3 | 126-73-8 |
| 7 | Tris(methylphenyl) phosphate* | TMPP | R1=R2=R3= −C6H4CH3 | 1330-78-5 |
| 8 | Tris(2-butoxyethyl) phosphate | TBOEP | R1=R2=R3= −CH2CH2OCH2(CH2)2CH3 | 78-51-3 |
| 9 | Tris(2-ethylhexyl) phosphate | TEHP | R1=R2=R3= −CH2CH(C2H5)CH2(CH2)2CH3 | 78-42-2 |
| 10 | Triethyl phosphate | TEP | R1=R2=R3= −CH2CH3 | 78-40-0 |
| 11 | 2-Ethylhexyl-diphenyl phosphate | EHDPP | R1= −CH2CH(C2H5)CH2(CH2)2CH3 R2=R3= −C6H5 | 1241-94-7 |
mixture of o, p, m isomers
2.2. Sample preparation and digestion
Individual standard solutions of profenofos and the target OPEs were diluted to 2 mg/mL with acetonitrile and kept in a refrigerator at 4 °C until further use. Fatty acid-free HSA solution was prepared at a concentration of 0.5 mg/ml in water. An aliquot of 10 μL of the OPE acetonitrile solution (2 mg/mL) was added to 100 μL of fresh HSA water solution (0.5 mg/ml). This mixture was incubated at 37 °C under gentle shaking for 24 h. The incubation mixtures were then subjected to ultrafiltration using a Vivaspin 500 centrifugal concentrator, which was pre-washed three times with 400 μL of water, for a period of 5 min at 14000 × g to concentrate the protein and separate unreacted OPE from the mixture solution. After the filtrate was discarded, the HSA residue was washed seven times with 400 μL of water. Concentrated HSA fractions (about 25 μL) were transferred into a vial. The concentrator was rinsed with 25 μL of water and the rinsate was combined with the HSA fractions. This adduct assay HSA solution (about 50 μL) was stored at −20 °C until further processing.
Prior to digestion, 25 μL of 4 mM DTT in 50 mM ammonium bicarbonate buffer (pH 8.2) was added to 25 μL of the adduct assay solution and incubated at 55 °C for 40 min to rupture the disulfide bonds in HSA. After cooling, 12.5 μL of 55 mM IAA in 25 mM ammonium bicarbonate buffer was added and the solution was incubated in the dark for 1 h at room temperature to alkylate the free cysteine groups generated in HSA. A 2.5 μL of 150 mM DTT in 50 mM ammonium bicarbonate buffer was then added into the solution to remove any remaining IAA, followed by digestion of the modified HSA by endoproteinase Glu-C or pepsin.
Endoproteinase Glu-C digestion was done by mixing 7.5 μL of 250 μg/mL Glu-C solution in 25 mM ammonium bicarbonate buffer with the modified HSA solution and then incubated at 37 °C for 17 h.
Prior to pepsin digestion, the pH of the modified HSA solution was adjusted to 2–3 by addition of 20 μL of 100 mM HCl. HSA was digested with 3 μL of 250 μg/mL pepsin in 10 mM HCl for 17 h at 37 °C.
After digestion (with Glu-C or pepsin), the sample was concentrated to about 20 μL using a centrifugal vacuum concentrator (Eppendorf Vacufuge plus). The sample was then desalted with a ZipTip-C18 pipette tip, according to the manufacturer’s instructions.
A blank control sample of HSA was prepared in the same manner as described above using aqueous acetonitrile solution as a test solution for each batch of samples. In this study all assays were performed in triplicate.
2.3. MALDI-TOF/TOF-MS analysis
The desalted HSA digest samples were analyzed with MALDI-TOF/TOF-MS and HCCA was used as the MALDI matrix. HCCA was dissolved at a concentration of 10 mg/mL in a solvent mixture of 50% acetonitrile, 47.5% water and 2.5%TFA. A 1 μL aliquot of the HSA digest sample was applied onto a stainless-steel target plate, air-dried, and overlaid with 1 μL HCCA solution and allowed to air-dry at room temperature. MALDI-TOF-MS experiments were carried out with an UltraflexIII MALDI-TOF/TOF mass spectrometer (Bruker, Billerica, MA) equipped with Smartbeam laser system. Mass spectra of the peptides were acquired in reflector positive mode. The following parameters were set for peptide detection: ion source 1, 25.08 kV; ion source 2, 21.72 kV; lens, 9.28 kV; reflector 1, 26.42 kV; reflector 2, 13.867 kV. For MS/MS experiments, LIFT mode was used under the following parameters: ion source 1, 8.01 kV; ion source 2, 7.21 kV; lens, 3.61 kV; reflector 1, 29.50 kV; reflector 2, 13.91 kV; LIFT 1, 19.08 kV; and LIFT 2, 2.71 kV. The measurements were performed in a mass range from m/z 300–4000 Da. Four thousand laser shots were accumulated in 200 shot increments and the sum of all shots represented the final mass spectrum. External mass calibration was carried out with the peptide calibration standard II from Bruker. FlexControl software (v.3.4) was used for controlling the system and data were processed using FlexAnalysis (v.3.4) and BioTools (v.3.2), with a maximum mass tolerance of 100 ppm.
2.4. LC-TOF-MS analysis
The HSA digest samples were also analyzed with LC-TOF-MS to confirm the results from MALDI-TOF-MS analysis. The remaining sample solution was concentrated to dryness and then reconstituted in 50 μL 5% aqueous acetonitrile containing 0.1% formic acid. The LC-TOF-MS analysis was performed on an Agilent 1200 LC system coupled to an Agilent 6520A quadrupole time-of-flight mass spectrometer (LC-Q-TOF-MS) system (Agilent Technologies, Mississauga, ON, Canada) with an electrospray ionization (ESI) source. A 10 μL aliquot of sample was injected into the LC system. LC separation was performed on a Kinetex XB-C18 column (100 mm × 2.0 mm, 1.7 μm particle size) (Phenomenex Co., CA, USA) at 40 °C. The LC mobile phase consisted of A: 0.1% formic acid in water and B: 0.1% formic acid in acetonitrile. The mobile phase flow rate was 0.3 mL/min. The LC gradient started at 5% B, increasing to 30% B in 15 min, increasing to 40% in 24 min, increasing to 95% in 25 min and was held for 5 min. Thereafter, the mobile phase composition was returned to initial conditions and the column was allowed to equilibrate for 15 min prior to the next injection.
The mass spectrometer was operated in positive ion mode. The capillary voltage was 3500 V. The fragmentor and skimmer voltages were 170 V and 70 V, respectively. Nitrogen was used as drying and nebulizing gases. The gas temperature was 350 °C. Dry gas flow rate was 5 L/min. Nebulizer pressure was 20 psi. Full-scan data acquisition was performed by scanning from m/z 70 to m/z 3200. The TOF-MS was tuned and calibrated with mixture calibration solution from Agilent. The resolution of MS (TOF) was > 20,000 at m/z 622.028413. Internal mass calibration was carried out with reference masses of 121.050873 and 922.009798. Data were acquired with Mass Hunter 6.0.
3. Results and discussion
3.1. Enzymatic cleavage of human serum albumin
In the in vitro assay, pure HSA was used as a model protein to assay for the possible formation of OPE-HSA adducts. HSA has a molecular mass of 66.5 kDa and is comprised of 585 amino acids in a single peptide chain (Wa et al. 2006). Fatty acid-free HSA was used because fatty acids and the OPE might bind to the same albumin domain and therefore fatty acids could hinder the binding of OPE. The relatively high molecular weight of HSA creates a challenge to accurately measure adducts at the protein level. Therefore, in such assays, HSA and other large proteins are usually digested by a proteolytic enzyme to give characteristic peptides that can be accurately analyzed with a mass spectrometer.
Trypsin digestion is commonly used in amino acid sequence analysis among many protein digestion methods. However, trypsin digestion (cleavage occurs at arginine and lysine at the C-terminus) is known to generate a product containing Y411 that is not detectable by MALDI-TOF-MS (Li et al. 2007; John et al. 2010). In addition, some common enzymes used for protein digestion, such as pronase, are not suitable for this purpose because their products of protein digestion contain a large number of peptides at low masses (i.e., <500 Da), which cannot be identified by MALDI-TOF-MS due to overlapping signals from the matrix (Wa et al. 2006).
Therefore, in the present study Glu-C or pepsin was used to investigate HSA phosphorylation by OPEs. Pepsin (preferential cleavage occurs at F, W, or Y at the N-terminus) and Glu-C (preferential cleavage of the glutamyl bonds) are widely used peptidases (Thompson, 2010). Our experiment showed that using DTT and IAA to respectively reduce and akylate the disulfide bonds of HSA could largely improve the result repeatability and increase the responses of peptides of interest on MALDI-TOF-MS, especially for Glu-C digestion. Our result was in agreement with some earlier reports (Wa et al., 2006), though in some reported assays the process was omitted (Li et al. 2007; John et al. 2010).
3.2. Determination of HSA phosphorylation by OPE FRs and OPE plasticizers using MALDI-TOF-MS
MALDI-TOF-MS is a quick and reliable method for identification of proteins and peptides and requires no chromatographic separation (Nomura, 2015). A major advantage of MALDI is that it can give a cleaner ion profile of peptide mixtures directly because of the absence of multiple charging. This relatively simple mass spectrum makes chemical identification easy and simple (Cristadoro et al. 2008; Thompson et al. 2010; Harvey 2015).
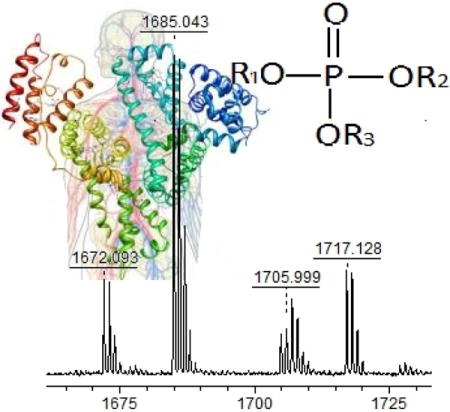
Figure 1 (a, b, c) shows the MALDI-TOF MS spectra of phosphorylated target peptides in a sample of profenofos-treated HSA, digested with pepsin (positive control sample). In comparison with the blank control sample (spectrum not shown), there were three peaks with a mass increase of 166.021 Da and each of the three corresponded to a profenofos-HSA adduction at Tyr411, with a mass tolerance of 100 ppm. Fig. 1a shows peaks corresponding to the peptide L408VRY411TKKVPQVSTPTL423 without (mono-isotopic ion at m/z 1830.222) and with (m/z 1996.259) addition of OP(OC2H5)(SC3H7) from profenofos. Similarly, Fig. 1b shows a mass spectrum of the peptide V409RY411TKKVPQVSTPTL423 without (m/z 1717.128) and with (m/z 1883.160) adduction of profenofos. Another peptide, corresponding to the non-specific cleavage of HSA, L408VRY411TKKVPQVST420, was observed in the mass spectrum without (m/z 1518.998) and with (m/z 1685.043) adduction of profenofos (Fig. 1c). This additional ion gave more information about OPE adduction to HSA and has not been reported in the literature. This additional ion most likely resulted from the relatively long digestion incubation time. These results were confirmed by MALDI-TOF/TOF MS/MS and the mass increase (166.021 Da) of the relevant fragments ions clearly showed that profenofos covalently reacted with the Tyr411 residue on HSA (Fig. 2), which was consistent with earlier studies (Li et al. 2007, John et al. 2010).
Fig. 1.
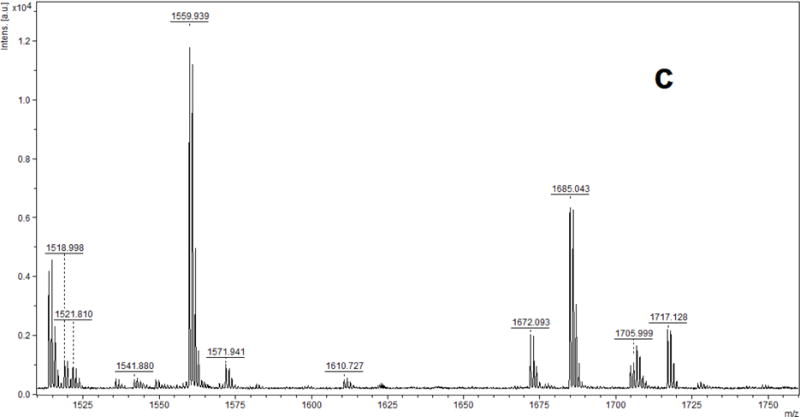
MALDI-TOF mass spectra of pepsin-digested human serum albumin incubated with profenofos. Peptide adduct ions with a Δm/z of 166 relative to the unmodified peptide were observed at (a) m/z 1996.3 originating from m/z 1830.2, (b) m/z 1883.2 originating from m/z 1717.1 and (c) m/z 1685.0 originating from m/z 1519.0.
Fig. 2.
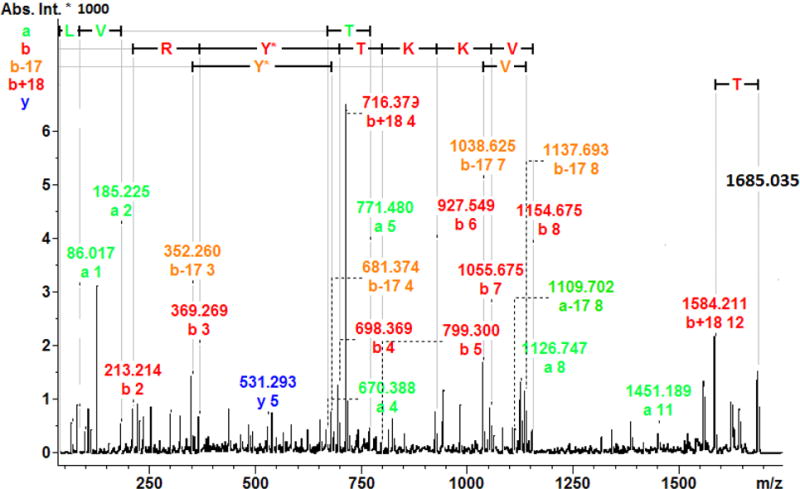
MALDI-TOF/TOF MS/MS spectrum of the human serum albumin peptide, L408VRY411TKKVPQVST420, with adduction of profenofos at Tyr411 (Y411-OP(OC2H5)(SC3H7)). (Color figure)
Eleven OPE FRs and OPE plasticizers (Table 1) were examined for OPE-HSA adduct formation along with the blank control sample and positive profenofos control sample. However, in contrast to the OP insecticide profenofos, no phosphorylated peptide products were found. Figure 3 (a, b, c) shows the MALDI-TOF-MS spectra obtained for pepsin-digested HSA after treatment with TPHP. If TPHP-HSA adducts had been formed, after pepsin digestion, their phosphorylated peptide ion would have been detected with MALDI-TOF-MS with ions of m/z 2062.11, 1949.03 and 1751.00 corresponding to the unphosphorylated peptide ions at m/z 1830.09, 1717.0 and 1519.0 (calculated m/z). For all other target OPEs, the MALDI-TOF-MS mass spectral results also showed no detectable phosphorylated peptides found for pepsin digested HSA after treatment with these compounds.
Fig. 3.
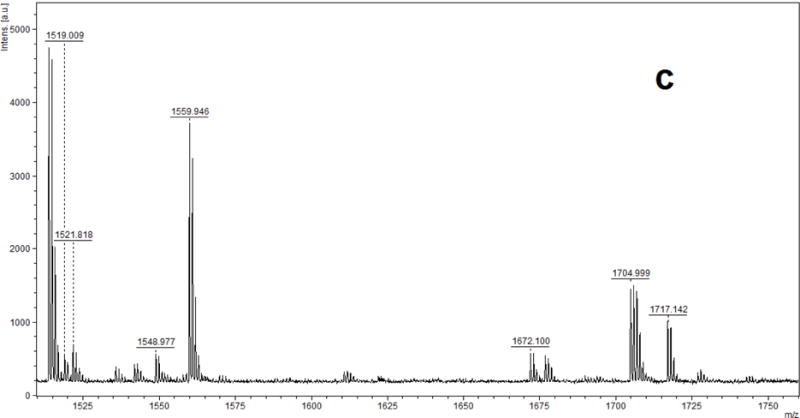
MALDI-TOF mass spectra for pepsin-digested human serum albumin incubated with triphenyl phosphate (TPHP) showing the absence of adduct peptides at (a) m/z 2062.11 (b) m/z 1949.03 and (c) m/z 1751.00.
It was reported that relative to pepsin, Glu-C appeared to be a more suitable enzyme for catalyzing albumin cleavage and allowed for the detection of more adduct sites, specifically Y148, Y150 (I142ARRHPY148FY150APE153, at m/z 1519.8), Y161 (L154LFFAKRY161KAAFTE167, at m/z 1704.9) and Y411 (Y401KFQNALLVRY411TKKVPQVSTPTLVE425, at m/z 2922.6) (John, 2010). Therefore, Glu-C digestion was also used to investigate possible phosphorylation of HSA by the target OPE FRs and OPE plasticizers under study. For the profenofos positive control sample, a mass increase (Δm/z 166.021) indicated the profenofos adducted peptides Y401KFQNALLVRY411TKKVPQVSTPTLVE425 (m/z 2922.672 and 3088.702, in Fig. 4a) and I142ARRHPY148FY150APE153 (m/z 1519.770 and 1685.799, in Fig. 4b). However, consistent with the pepsin results already described, none of the OPE FRs and OPE plasticizers tested showed such phosphorylated peptides. Fig. 5a and b show MALDI-TOF-MS spectra of HSA derived peptides treated with TPHP after Glu-C digestion. If there were TPHP-HSA adducts formed, their phosphorylated peptide ions would be detected by MALDI-TOF-MS analysis at m/z 3154.675 and 1751.810, corresponding to the unphosphorylated peptide ions at m/z 2922.646 and 1519.781 (calculated m/z).
Fig. 4.
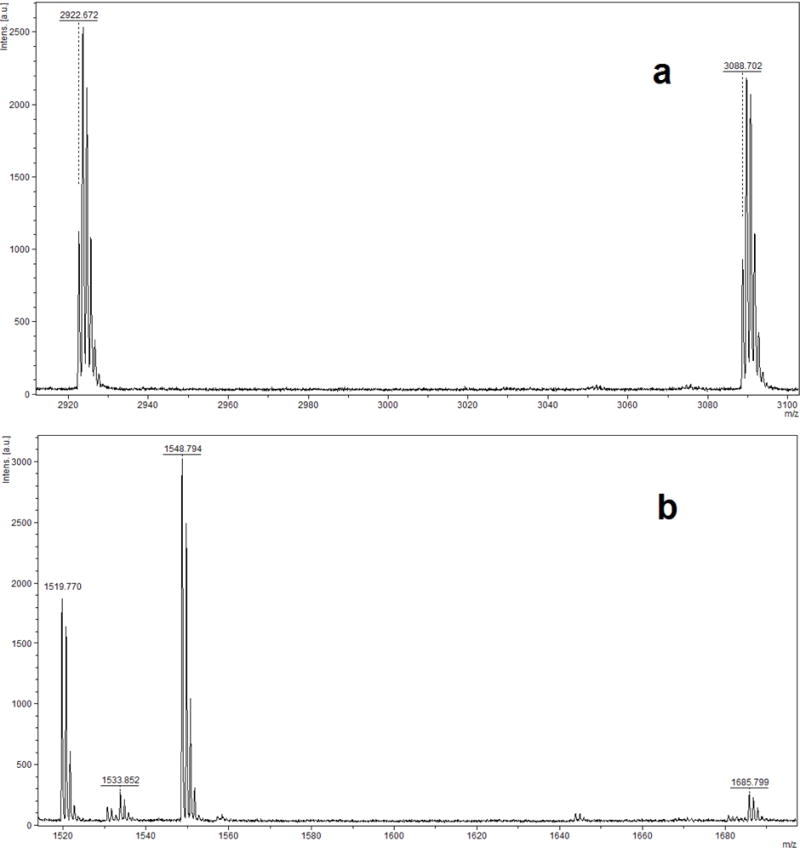
MALDI-TOF mass spectra of human serum albumin incubated with profenofos followed by digestion with Glu-C, which show the adduct peptide ions with a Δm/z of 166 at (a) m/z 3088.702 originating from m/z 2922.672 and (b) m/z 1685.788 originating from m/z 1519.770.
Fig. 5.
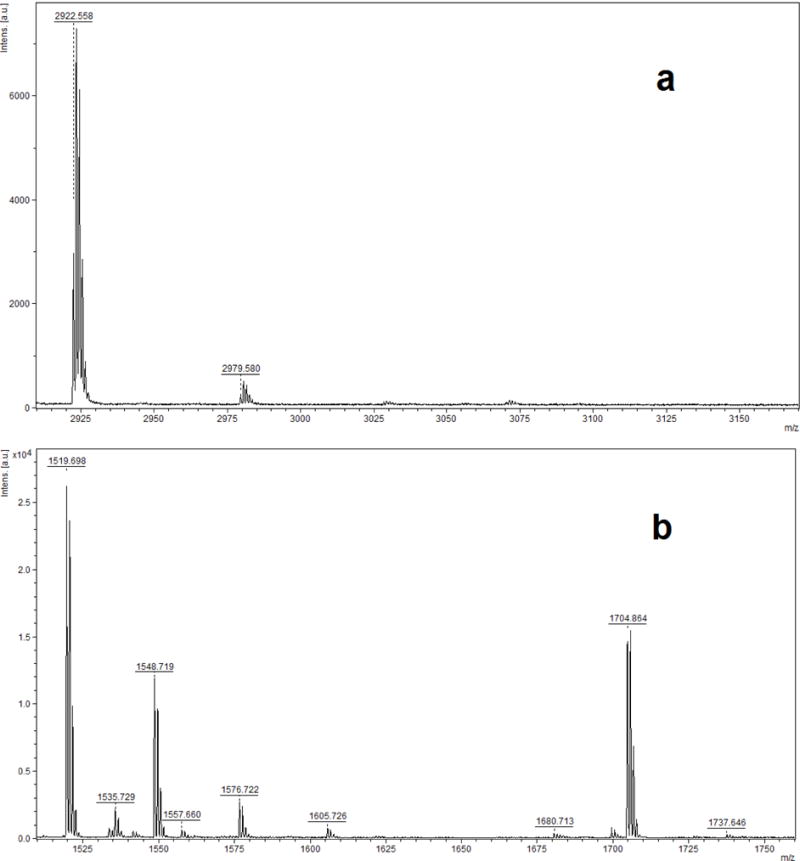
MALDI-TOF mass spectra of human serum albumin incubated with triphenyl phosphate (TPHP) followed by digestion with Glu-C, which show the absence of the adduct peptide ions (a) at m/z 3154.675 and (b) at m/z 1751.810
We suspected the high concentration of acetonitrile might have affected OPE-HSA adduct formation. In a set of OPE-HSA adduct assay experiments, individual OPE compound solutions were prepared with a concentration below its saturation limit in 0.1% aqueous acetonitrile, or at 2 mg/mL in pure water for OPEs that have high water solubility, such as TEP and TCEP, and then the adduction assay was performed in the same way as described above. The OPE-HSA adduct assay results also showed that no phosphorylated peptides were detectable by MALDI-TOF MS, whereas in digestion samples of HSA incubated with profenofos (0.1% aqueous acetonitrile) the phosphorylated HSA-derived peptide ions were detectable. Therefore, the acetonitrile concentration was not a factor that affected adduct formation in the assay.
3.3. Determination of HSA phosphorylation by OPE FRs and OPE plasticizers using LC-Q-TOF-MS
To further confirm the MALDI–TOF-MS results, all OPE-HSA adduct assay samples were reanalyzed with LC-Q-TOF-MS. LC-Q-TOF-MS can detect multiply charged peptide ions with high mass resolution, high accuracy and relative high sensitivity. It has the advantage of being able to identify large-molecular-weight peptides and provides a versatile method for the detection of protein adducts (Thompson et al. 2010; Harvey 2015).
Consistent with the MALDI-TOF-MS results, LC-Q-TOF-MS analysis revealed detection of multiply charge ions of phosphorylated peptides as a result of pepsin digestion of HSA samples incubated with the positive control profenofos. Fig. 6a shows the extracted ion chromatogram (EIC) of a phosphorylated peptide ion with multiple charges states at m/z 998.554 ([M+2H]2+), m/z 666.040 ([M+3H]3+), and m/z 499.783 [M+4H]4+). The molecular weight of the phosphorylated peptide (at retention time of 13.5 min) was calculated to be 1995.098 Da, corresponding to the profenofos adducted peptide L408VRY411TKKVPQVSTPTL423 (theoretical mass 1995.100 Da). Fig. 6b shows the EIC of a phosphorylated peptide ion at m/z 942.017 ([M+2H]2+), m/z 628.346 ([M+3H]3+), and m/z 471.511 ([M+4H]4+) in the same sample, which corresponded to the phosphorylated peptide V409RY411TKKVPQVSTPTL423, with a calculated mass of 1882.017 Da (theoretical mass 1882.016 Da). Phosphorylated peptide ions at m/z 842.966 ([M+2H]2+), m/z 562.313 ([M+3H]3+) and m/z 421.986 ([M+4H]4+) were detected in the same sample (Fig. 6c) and corresponded to the phosphorylated peptide L408VRY411TKKVPQVST420 with a calculated mass of 1683.917 Da (theoretical mass 1683.916 Da). As with the pepsin digested sample, in the Glu-C digestion sample incubated with profenofos, the LC-Q-TOF-MS spectrum showed multiply charged ions of phosphorylated peptides, which had calculated molecular weights of 3087.653 Da (theoretical mass 3087.659 Da) and 1684.801 Da (theoretical mass 1684.794 Da).
Fig. 6.
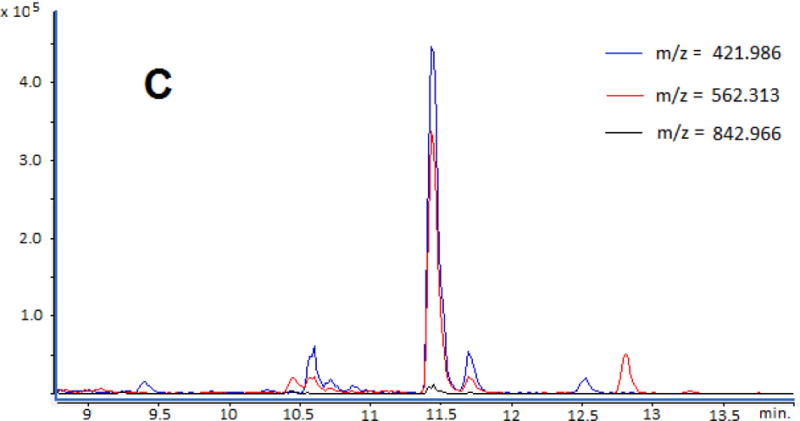
Extraction ion chromatograms of phosphorylated peptide ions (a) multiply charged at m/z 998.554 ([M+2H]2+), m/z 666.040 ([M+3H]3+) and m/z 499.783 ([M+4H]4+) corresponding a molecular weight of 1995.098 Da (1995.100 Da, theoretical value), (b) multiply charged at m/z 942.017 ([M+2H]2+), m/z 628.346 ([M+3H]3+) and m/z 471.511 ([M+4H]4+) corresponding a molecular weight of 1882.017 Da (1882.016 Da, theoretical value) and (c) multiply charged at m/z 842.966 ([M+2H]2+), m/z 562.313 ([M+3H]3+) and m/z 421.986 ([M+4H]4+) corresponding a molecular weight of 1683.917 Da (1683.916 Da, theoretical value). (Color figures)
In the present study, the signal-to-noise (S/N) ratios of analyte peaks on LC-QTOF-MS were much improved because peptides were separated by LC. For one of the profenofos-adducted peptide ions (at m/z 421.986), the S/N ratio was 294 (Fig. 6c). However, even at this high sensitivity, no peaks were detected in the corresponding EICs of HSA treated with the 11 OPE FRs and OPE plasticizers.
3.4. Explanations for no detectable HSA peptides phosphorylated by OPE FRs and OP plasticizers
Both LC-Q-TOF-MS and MALDI-TOF-MS analyses clearly showed the absence of any phosphorylation of HSA by the 11 OPE FRs and OPE plasticizers under study, despite the fact that these compounds have similar molecular structures with OP insecticides, such as profenofos and paraoxon-ethyl, which yielded detectable adducts of HSA (John et al. 2010). John et al. (2010) studied a series of toxic OP compounds including pesticides and nerve agents. Although some OP compounds have very similar molecular structures, their behaviours differed largely towards albumin phosphorylation (John et al. 2010). It is well known that HSA is phosphorylated preferentially at the nucleophilic Tyr411 located on the protein surface (John et al. 2008). Cleavage of the P-O bond is priority in the process. Our results and those described in the literature indicated that HSA phosphorylation resulted from cleavage of the aryl phosphoester bond in profenofos and attachment of OP(OC2H5)(SC3H7). If phosphorylation had instead occurred through breaking of the ethyl phosphoester bond, leading to attachment of C6H3ClBrO-PO(SC3H7), the OPE-HSA adduct should have had a Δm/z of 325.893. However, no such ion was detected by MALDI-TOF-MS and LC-ESI(+)-TOF-MS. John at el. (2010) studied albumin phosphorylation by some OP pesticides and G- and V-type nerve agents. They did not find albumin adducts resulting from cleavage of the alcohol chain from the target compounds. Comparing OP insecticides (e.g., profenofos, paraoxon-ethyl and chlorpyrifos-oxon) with the aryl OPE plasticizers TPHP and TMPP, these OP insecticides bear one aryl group with electron-withdrawing substituents at the ortho- or/and para-positions. Such good leaving-groups (more acidic conjugated acid) can result in easy cleavage of the aryl phosphoester bond to form adducts at Tyr411 in HSA. However, although the OPE plasticizers TPHP and TMPP have an aryl group, they do not have electron-withdrawing substituents, and therefore, TPHP and TMPP showed no such property.
This difference between OP insecticides and OPE FRs and OPE plasticizers can also be found in their hydrolysis behavior. Most OPE FRs and OPE plasticizers are relatively stable against hydrolysis at neutral pH. In a very recent paper by Su et al. (2016), over a period of 35 days at 20 °C at pH 7, 9 out of the 16 OPEs were stable and were not significantly hydrolyzed. For the other 7 OPEs, including TPHP and TMPP, their hydrolysis half-live ranged from 23 d to 112 d at pH 7. It was reported that hydrolysis half-lives at pH 7 for TEP and TPHP are in a range of 1.2–5.5 years (Mabey and Mill 1978). However, profenofos hydrolysis half-lives were approximately 24–62 d at pH 7 and 15 d at pH 8 (Zamy et al. 2004). The results of the present study suggest that OPE molecular weight is not an important factor in HSA phosphorylation because there was no detectable phosphorylation for the low molecular weight TEP.
With little information about covalent adducts of OPs available, these explanations are far from adequately addressing this complex phosphorylation process. Covalent adduct formation might not be a simple process of cleavage and attachment. The complex albumin protein structure may play an important role in the process.
4. Conclusions
Although HSA adduct formation is a potential biomarker of exposure to OPEs, in the present study we did not detect any HSA adduct formation with any of the 11 target OPE FRs and OPE plasticizers. The results suggest that these OPEs have very different protein binding affinities in comparison to OP insecticides, or they may possess a specific form of currently unknown toxicity (Abou-Donia 2003; Casida and Quistad 2004; Schang et al. 2016). For example, ortho-TMPP (also known as tri-ortho-cresyl phosphate (ToCP)) is neurotoxic, and exposure to ToCP has been associated with the alleged Aerotoxic syndrome (Abou-Donia et al. 2013, de Ree et al. 2014). Most environmentally relevant OPE FRs and OPE plasticizers have shown low persistence and bioaccumulation potential in biota as they are rapidly metabolized (Greaves et al. 2016c). Therefore, their metabolisms and potent molecular toxicity may be very different from common persistent organic pollutants. A large amount and variety of OPE FRs and OP plasticizers have been produced and used, with more OPEs likely to be produced in future. Further studies are necessary to elucidate the behaviour of OPEs, including their toxicology and effects in exposed organisms, to facilitate assessments of risk with respect to human and environmental health.
HIGHLIGHTS.
Adduct formation between HSA and OPE flame retardants and plasticizers is explored.
Profenofos-HSA adduct preparation and analysis verify the methodology.
L408VRY411TKKVPQVST420 (m/z 1685.043) is a HSA-profenofos adduct.
No OPE-HSA adducts are observed.
Acknowledgments
This work was supported in part by grant G12 MD007601 from the National Institutes of Health Research Centers in Minority Institutions Program, NIEHS R01ES002718, and a subcontract with the Western Center for Agricultural Health and Safety (NIOSH grant 2U54OH007550).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare that there are no conflicts of interest regarding the submitted article.
References
- Abou-Donia MB. Organophosphorus ester-induced chronic neurotoxicity. Arch Environ Health. 2003;58(8):484–497. doi: 10.3200/AEOH.58.8.484-497. [DOI] [PubMed] [Google Scholar]
- Abou-Donia MB, Abou-Donia MM, ElMasry EM, Monro JA, Mulder MFA. Autoantio-bodies to nervous system specific proteins are elevated in sera of flight crew members: biomarkers for nervous system injury. J Toxicol Environ Health A. 2013;76(6):363–380. doi: 10.1080/15287394.2013.765369. [DOI] [PubMed] [Google Scholar]
- Brommer S, Harrad S. Sources and human exposure implications of concentrations of organophosphate flame retardants in dust from UK cars, classrooms, living rooms, and offices. Environ Int. 2015;83:202–207. doi: 10.1016/j.envint.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Camarasa JG, Serrabaldrich E. Allergic contact-dermatitis from triphenyl phosphate. Contact Dermatitis. 1992;26(4):264–265. doi: 10.1111/j.1600-0536.1992.tb00241.x. [DOI] [PubMed] [Google Scholar]
- Casida JE, Quistad GB. Organophosphate toxicology: Safety aspects of nonacetylcholinesterase secondary targets. Chem Res Toxicol. 2004;17(8):983–998. doi: 10.1021/tx0499259. [DOI] [PubMed] [Google Scholar]
- Cristadoro A, Rader HJ, Mullen K. Quantitative analyses of fullerene and polycyclic aromatic hydrocarbon mixtures via solvent-free matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 2008;22(16):2463–2470. [Google Scholar]
- Crow BS, Pantazides BG, Quinones-Gonzalez J, Garton JW, Carter MD, Perez JW, Watson CM, Tomcik DJ, Crenshaw MD, Brewer BN, Riches JR, Stubbs SJ, Read RW, Evans RA, Thomas JD, Blake TA, Johnson RC. Simultaneous measurement of tabun, sarin, soman, cyclosarin, VR, VX, and VM adducts to tyrosine in blood products by isotope dilution UHPLC-MS/MS. Anal Chem. 2014;86(20):10397–10405. doi: 10.1021/ac502886c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ree H, van den Berg M, Brand T, Mulder GJ, Simons R, van Zanten BV, Westerink RHS. Health risk assessment of exposure to tricresyl phosphates (TCPs) in aircraft: A commentary. Neurotoxicology. 2014;45:209–215. doi: 10.1016/j.neuro.2014.08.011. [DOI] [PubMed] [Google Scholar]
- EPA. EPA Document# 740-R1-5001. 2015 https://www.epa.gov/sites/production/files/2015-09/documents/cpe_fr_cluster_problem_formulation.pdf.
- Greaves AK, Letcher RJ, Chen D, Gauthier LT, McGoldrick D, Backus S. Retrospective trends of organophosphate flame retardants in herring gull eggs and in relation to the aquatic food web for the Laurentian Great Lakes of North America. Environ Res. 2016a;150:255–263. doi: 10.1016/j.envres.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Greaves AK, Letcher RJ. Organophosphate esters: A review on wildlife distribution, fate and toxicology. Bull Environ Contam Toxicol. 2016b doi: 10.1007/s00128-016-1898-0. in press. [DOI] [PubMed] [Google Scholar]
- Greaves AK, Su G, Letcher RJ. Environmentally relevant organophosphate triester flame retardants in herring gulls: In vitro biotransformation and kinetics and diester metabolite formation using a hepatic microsomal assay. Toxicol Appl Pharmacol. 2016;308:59–65. doi: 10.1016/j.taap.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Hallanger IG, Sagerup K, Evenset A, Kovacs KM, Leonards P, Fuglei E, Routti H, Aars J, Strom H, Lydersen C, Gabrielsen GW. Organophosphorus flame retardants in biota from Svalbard, Norway. Marine Pull Bull. 2015;101(1):442–447. doi: 10.1016/j.marpolbul.2015.09.049. [DOI] [PubMed] [Google Scholar]
- Hartmann PC, Burgi D, Giger W. Organophosphate flame retardants and plasticizers in indoor air. Chemosphere. 2004;57(8):781–787. doi: 10.1016/j.chemosphere.2004.08.051. [DOI] [PubMed] [Google Scholar]
- Harvey DJ. Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: An update for 2009–2010. Mass Spectrom Rev. 2015;34(3):268–422. doi: 10.1002/mas.21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He RW, Li YZ, Xiang P, Li C, Zhou CY, Zhang SJ, Cui XY, Ma LQ. Organophosphorus flame retardants and phthalate esters in indoor dust from different microenvironments: Bioaccessibility and risk assessment. Chemosphere. 2016;150:528–535. doi: 10.1016/j.chemosphere.2015.10.087. [DOI] [PubMed] [Google Scholar]
- John H, Worek F, Thiermann H. LC-MS-based procedures for monitoring of toxic organophosphorus compounds and verification of pesticide and nerve agent poisoning. Anal Bioanal Chem. 2008;391(1):97–116. doi: 10.1007/s00216-008-1925-z. [DOI] [PubMed] [Google Scholar]
- John H, Breyer F, Thumfart JO, Hochstetter H, Thiermann H. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) for detection and identification of albumin phosphorylation by organophosphorus pesticides and G- and V-type nerve agents. Anal Bioanal Chem. 2010;398(6):2677–2691. doi: 10.1007/s00216-010-4076-y. [DOI] [PubMed] [Google Scholar]
- Li B, Schopfer LM, Hinrichs SH, Masson P, Lockridge O. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry assay for organophosphorus toxicants bound to human albumin at Tyr411. Anal Biochem. 2007;361(2):263–272. doi: 10.1016/j.ab.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabey W, Mill T. Critical-review of hydrolysis of organic compounds in water under environmental conditions. J Phys Chem Ref Data. 1978;7(2):383–415. [Google Scholar]
- Marklund A, Andersson B, Haglund P. Organophosphorus flame retardants and plasticizers in Swedish sewage treatment plants. Environ Sci Technol. 2005;39(19):7423–7429. doi: 10.1021/es051013l. [DOI] [PubMed] [Google Scholar]
- Marklund A, Andersson B, Haglund P. Screening of organophosphorus compounds and their distribution in various indoor environments. Chemosphere. 2003;53(9):1137–1146. doi: 10.1016/S0045-6535(03)00666-0. [DOI] [PubMed] [Google Scholar]
- Marsillach J, Costa LG, Furlong CE. Protein adducts as biomarkers of exposure to organophosphorus compounds. Toxicol. 2013;307:46–54. doi: 10.1016/j.tox.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough JH, Shih TM. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci Biobehav Rev. 1997;21(5):559–57. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- McGoldrick DJ, Letcher RJ, Barresi E, Keir MJ, Small J, Clark MG, Sverko E, Backus SM. Organophosphate flame retardants and organosiloxanes in predatory freshwater fish from locations across Canada. Environ Pollut. 2014;193:254–261. doi: 10.1016/j.envpol.2014.06.024. [DOI] [PubMed] [Google Scholar]
- Meeker JD, Stapleton HM. House dust concentrations of organophosphate flame retardants in relation to hormone levels and semen quality parameters. Environ Health Persp. 2010;118(3):318–323. doi: 10.1289/ehp.0901332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier F, Glorennec P, Thomas O, Le Bot B. Organic contamination of settled house dust, A review for exposure assessment purposes. Environ Sci Technol. 2011;45(16):6716–6727. doi: 10.1021/es200925h. [DOI] [PubMed] [Google Scholar]
- Morgan AB, Gilman JW. An Overview of flame retardancy of polymeric materials: application, technology, and future directions. Fire Mater. 2013;37(4):259–279. [Google Scholar]
- Nomura F. Proteome-based bacterial identification using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS): A revolutionary shift in clinical diagnostic microbiology. Biochim Biophys Acta. 2015;1854(6):528–537. doi: 10.1016/j.bbapap.2014.10.022. [DOI] [PubMed] [Google Scholar]
- Peeples ES, Schopfer LM, Duysen EG, Spaulding R, Voelker T, Thompson CM, Lockridge O. Albumin, a new biomarker of organophosphorus toxicant exposure, identified by mass spectrometry. Toxicol Sci. 2005;83(2):303–312. doi: 10.1093/toxsci/kfi023. [DOI] [PubMed] [Google Scholar]
- Reemtsma T, Quintana JB, Rodil R, Garcia-Lopez M, Rodriguez I. Organophosphorus flame retardants and plasticizers in water and air I. Occurrence and fate. Trends Anal Chem. 2008;27(9):727–737. [Google Scholar]
- Schang G, Robaire B, Hales BF. Organophosphate flame retardants act as endocrine-disrupting chemicals in MA-10 mouse tumor Leydig cells. Toxicol Sci. 2016;150(2):499–509. doi: 10.1093/toxsci/kfw012. [DOI] [PubMed] [Google Scholar]
- Schopfer LM, Furlong CE, Lockridge O. Development of diagnostics in the search for an explanation of aerotoxic syndrome. Anal Biochem. 2010;404(1):64–74. doi: 10.1016/j.ab.2010.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreder ED, Uding N, La Guardia MJ. Inhalation a significant exposure route for chlorinated organophosphate flame retardants. Chemosphere. 2016;150:499–504. doi: 10.1016/j.chemosphere.2015.11.084. [DOI] [PubMed] [Google Scholar]
- Su G, Letcher RJ, Yu H. Organophosphate flame retardant and plasticizer chemicals in aqueous solutions: Hydrolytic pH stability, kinetics and mechanisms. Environ Sci Technol. 2016;50(15):8103–8111. doi: 10.1021/acs.est.6b02187. [DOI] [PubMed] [Google Scholar]
- Thompson CM, Prins JM, George KM. Mass spectrometric analyses of organophosphate insecticide oxon protein adducts. Environ Health Perspect. 2010;118(1):11–19. doi: 10.1289/ehp.0900824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen I, de Boer J. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere. 2012;88(10):1119–1153. doi: 10.1016/j.chemosphere.2012.03.067. [DOI] [PubMed] [Google Scholar]
- Wa CL, Cerny R, Hage DS. Obtaining high sequence coverage in matrix-assisted laser desorption time-of-flight mass spectrometry for studies of protein modification: Analysis of human serum albumin as a model. Anal Biochem. 2006;349(2):229–241. doi: 10.1016/j.ab.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu J, Yin Y. The pollution status and research progress on organophosphate ester flame retardants. Prog Chem. 2010;22(10):1983–1992. [Google Scholar]
- Wei GL, Li DQ, Zhuo MN, Liao YS, Xie ZY, Guo TL, Li JJ, Zhang SY, Liang ZQ. Organophosphorus flame retardants and plasticizers: Sources, occurrence, toxicity and human exposure. Environ Poll. 2015;196:29–46. doi: 10.1016/j.envpol.2014.09.012. [DOI] [PubMed] [Google Scholar]
- Williams NH, Harrison JM, Read RW, Black RM. Phosphorylated tyrosine in albumin as a biomarker of exposure to organophosphorus nerve agents. Arch Toxicol. 2007;81:627–639. doi: 10.1007/s00204-007-0191-8. [DOI] [PubMed] [Google Scholar]
- Zamy C, Mazellier P, Legube B. Analytical and kinetic study of the aqueous hydrolysis of four organophosphorus and two carbamate pesticides. In J Environ Anal Chem. 2004;84:1059–1068. [Google Scholar]