

Key Clinical Message
Granulomatous hypophysitis is rare pathology that mimics pituitary adenoma. Diagnosis is only confirmed by histopathology examination. Trans‐sphenoidal surgery is considered diagnostic when descent tissue specimen is obtained and therapeutic by decompressing optic pathway and the sella. Pathological findings always reveal granulomatous areas, multinucleated giant cells, plasma cells, and lymphocytes.
Keywords: diabetes insipidus, Granulomatous hypophysitis, pituitary adenoma, transsphenoidal surgery, visual field defect
Introduction
Hypophysitis is a rare condition characterized by infiltration and destruction of the pituitary gland 1. The incidence is not known, mostly from case reports in the literature. Granulomatous hypophysitis is one of five types of inflammatory hypophysitis, which are (lymphocytic, granulomatous, xanthomatous, xanthogranulomatous, and necrotizing). Granulomatous and lymphocytic hypophysitis are the most common types 1, 2, 3, 4. Hypophysitis can also be classified as primary (idiopathic) and secondary, which may develop secondary to systemic inflammatory disorders such as tuberculosis, Wegener's granulomatosis, and sarcoidosis 1, 2. The majority of inflammatory pituitary lesions occur in women related to the postpartum period. Symptoms such as headache, visual symptoms, and involvement of pituitary gland and stalk may result in endocrine dysfunction, which usually occur due to the influence of a mass lesion within the sella 3, 4.
The clinical scenario of granulomatous hypophysitis usually include headache, visual symptoms in the form of visual deterioration and double vision, constitutional symptoms; low‐grade fever, nausea and vomiting 4, 5, 6, 7 . Enhanced magnetic resonance imaging (MRI) almost always reveals sellar and pituitary stalk lesions, with iso‐ or hypointense signals on T1‐ and T2‐weighted images, a picture that resembles pituitary adenomas. Due to the presence of pituitary mass and pituitary hormonal dysfunction, trans‐sphenoidal exploration and histological examination are always offered to reach to final diagnosis. It is challenging to diagnose hypophysitis, especially when it mimics other common sellar lesions due to the lack of significant specific clinical signs. A trans‐sphenoidal biopsy with fast‐frozen pathology was able to diagnose hypophysitis likely to be granulomatous. Corticosteroid therapy may be a potential treatment for hypophysitis, as complete removal of pituitary masses may disable pituitary function 8, 9, 10.
Case History
A 37‐year‐old single female who was previously healthy, presented with 1‐week history of gradual onset of headache, which became worse 1 day prior to admission. This was associated with right eye pain and redness, blurring of vision, and diplopia on looking upwards. She felt sick and vomited twice. She denied history of trauma, polyurea, or seizures. She started her menarche at the age of 12 and used to have regular menstrual cycles; however, she missed her last period 10 days before presentation. She gave a history of benign breast lump excised 11 years ago.
Examination revealed alert, and conscious patient with low‐grade fever. Her visual acuity was OD: 20/150 and OS: 20/25. She had convergent nystagmus, limitation of lateral gaze movement of both eyes, more on the right side, upper eyelid retraction, and limitation of upward gaze with diplopia and photophobia on looking upwards in both eyes. Optic nerve cup/disk ratio was 0.5 indicating no evidence of papilloedema. The rest of neurological examination was unremarkable. Goldman visual field showed restrictive visual field in both eyes, with no specific pattern (Fig. 1).
Figure 1.
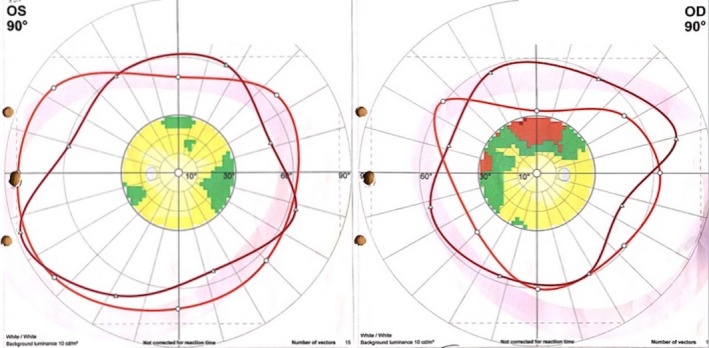
Goldman perimetry visual filed showing restrictive visual field in both eyes, with no specific pattern.
Brain CT scan showed a bulky pituitary gland in a marginally large sella with no evidence of hemorrhage or infarction (Fig. 2).
Figure 2.
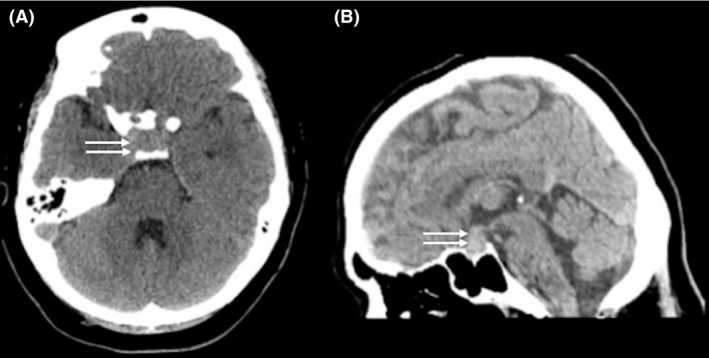
Axial CT scan (A) and sagittal reformatting (B) showing hyperdense sellar mass with suprasellar extension abutting the optic chiasm (arrows).
MRI scan revealed a large sellar mass with parasellar and suprasellar extension abutting the optic chiasm, and buckling the prechiasmatic optic nerves. The mass was isointense on T1‐ and T2‐weighted images and heterogeneously enhanced after the administration of gadolinium (Fig. 3). An incidental benign pineal body cyst, with subtle rim enhancement, was seen.
Figure 3.
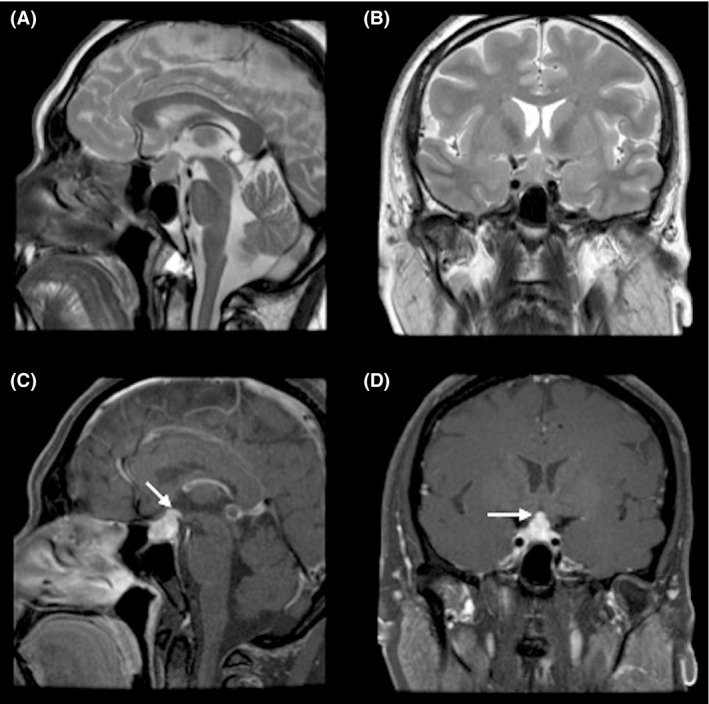
MRI scan of the brain, axial view (A) and coronal view T2WI (B) showing isointense sellar, suprasellar lesion. (C&D) after intravenous injection of gadolinium, showing enhancement of the mass lesion.
Hormonal assay of the pituitary gland showed manifestations of central hypothyroidism (low T4 free, and normal TSH), and morning cortisol level was low and other pituitary hormones were within normal level (Table 1 ).
Table 1.
Pituitary function assessment
| Hormone | Level | Normal ratio |
|---|---|---|
| FSH | 4.4 mIU/mL | 3–20 |
| LH | 2.2 mIU/mL | 0.3–31.0 |
| Prolactin | 378.20 mIU/L | <440 |
| Estradiol | 18 pmol/L | 0–30 |
| Progesterone | 0.82 nmol/L | 1–3 |
| Beta hCG Quant | 0.1 IU/L | <5 |
| TSH | 0.846 mIU/L | 0.3–5.0 |
| T4 free | 9.4 pmol/L (Low) | 11.5–23.5 |
| Free T3 | 3.80 pmol/L | 3.5–6.5 |
| ACTH | 5.1 pmol/L | 0–46 |
| Cortisol (am) | 71 nmol/L (Low) | 138–635 |
FSH, Follicle‐stimulating hormone; LH, Luteinizing hormone, hCG, human chorionic gonadotrophin; TSH, thyroid‐stimulating hormone; ACTH, Adrenocorticotropic hormone.
Bold values are self descriptive, low between the brackets.
The diagnosis of pituitary macro‐adenoma and secondary hypopituitarism was made. She underwent navigation‐guided endoscopic transsphenoidal debulking of the sella and decompression of the optic chiasm. On opening the dura, greyish necrotic tissues came out under moderate pressure and with the aid of suction tip and ring curette, all abnormal tissues could be removed, till optic chiasm visualized free of compression. She tolerated the procedure well and nasal packs were removed after 48 h with no evidence of postoperative CSF leak.
She developed temporary diabetes insipidus treated by DDAVP® and spontaneously recovered within 1 month of surgery.
The patient received hydrocortisone therapy before surgery, which was tapered down after surgery to a maintenance dose. She was followed up for 6 months and remained on replacement thyroxin and hydrocortisone therapy.
Histopathology examination of the specimen showed multiple fragments of fibro‐histiocytic granulomatous lesion with multinucleated giant cell reaction. No cavitation was identified. No viral inclusion or infective pathogen was seen. One fragment showed features of pituitary tissues including nests of small round blue cell with a stippled chromatin. No eosinophils features of Langerhans’ cell histiocytosis was seen (Fig. 4).
Figure 4.
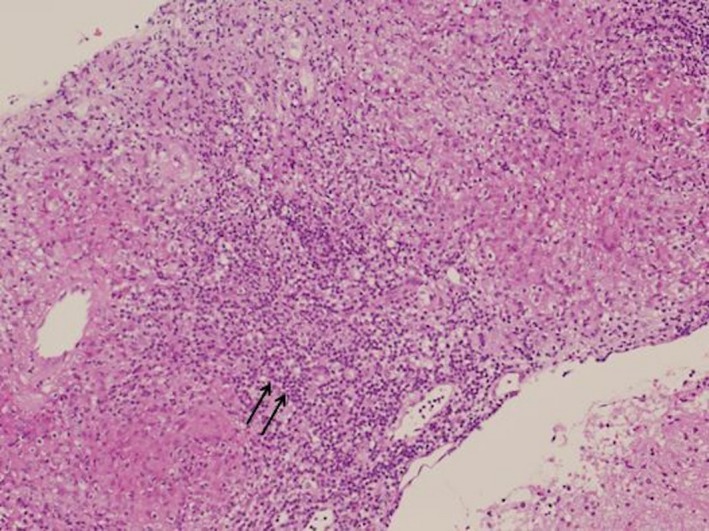
Histology section of specimen obtained by trans‐sphenoidal approach showing noncaseating granuloma with multinucleated giant sells (arrows) and surrounded by lymphocytes. (20× hematoxylin and eosin stain).
A through workup was carried out and excluded evidence of systemic granulomatous disease (e.g., tuberculosis, syphilis, sarcoidosis, brucellosis, and histiocytosis X).
Discussion
The authors report a rare case of primary granulomatous hypophysitis in a young lady presented with manifestations of central hypothyroidism and unusual visual symptoms associated with sellar mass with suprasellar and parasellar extension mimicking pituitary macroadenoma. The majority of sellar masses represent neoplastic lesions. However, granulomatous hypophysitis, though rare, but may also occur and include several other conditions, such as tuberculosis, sarcoidosis, Wegener's granulomatosis, syphilis, and mycotic infections 3, 4 .
Although most granulomatous hypophysitis present with diabetes insipidus (D.I.) and diffuse pituitary enlargement and pituitary stalk thickening, they can be differentiated from pituitary adenoma. In the present case, the absence of D.I. and pituitary stalk thickening mimicked pituitary macroadenomas 8, 9, 10. Headache and visual symptoms as a result of progressive chiasmal compression are the most common presenting symptoms of granulomatous hypophysitis. Patients may also present with amenorrhea–galactorrhea, hyperprolactinemia, fatigue, and aseptic meningitis 8, 9, 10, 11, 12. Granulomatous hypophysitis is difficult to differentiate from simple adenoma based on its clinical manifestations or radiological findings, and the diagnosis is usually made by histopathologic findings 8, 9, 10.
Histopathology appearance of granulomatous hypophysitis usually contains necrotizing granulomas that are mainly formed of histiocytes, multinucleated giant cells, and variable numbers of lymphocytes and plasma cells 2. The presence of noncaseating granulomatous inflammation could be compatible with tuberculosis despite negative ZN stain. Other differential diagnoses should include sarcoidosis, and other specific infections. Due to absence of significant evidences of systemic granulomatous disease, this case was labeled as a case of primary granulomatous hypophysitis 3, 9, 12, 13.
The natural history of granulomatous hypophysitis is not well understood. Although most reported cases respond to high dose of steroid therapy, its treatment is still controversial 3. Based on the benign nature of the disease and its transient course, conservative treatment with close clinical and radiological observation has been advocated as the primary therapeutic option 3. Transsphenoidal surgery is indicated to decompress the mass lesion, thereby resolving headache and visual deficits, to obtain a descent tissue specimen for histological diagnosis, and facilitates the appropriate treatment. This should be applied for all patients whose clinical and radiological findings are not typical and diagnosis is not confirmed. In fact, majority of reported cases were misdiagnosed as pituitary adenomas preoperatively, and diagnosis was confirmed only after surgery 13, 14.
The lesion described would probably explain the patient's symptomatology and ocular manifestations. Visual symptoms of the patient were confusing and not suggestive of chiasmal compression especially the presence of nonspecific visual field. Visual symptoms improved dramatically after surgical decompression of the optic pathway as usually happens after removal of pituitary adenomas 15. Morning cortisol and T4 free levels were low while the rest of pituitary hormones were within normal level indicating hypopituitarism. Consideration was given to prompt surgical intervention to decompress both optic pathway and obtain a tissue diagnosis 8, 9, 10, 15, 16.
Pituitary neurosurgeons usually preserve normal tissue to minimize the risk of permanent hypopituitarism. In our case, once frozen section result confirmed inflammatory condition excluding neoplasm and optic chiasm adequately decompressed, surgery was considered completed.
Patients present with severe endocrine defects do not improve after surgery and require lifelong hormonal replacement. This was attributed to cellular destruction associated with inflammatory hypophysitis; therefore, long‐term follow‐up is very important.
Conclusion
Granulomatous hypophysitis is rare inflammatory condition that mimics pituitary adenoma. Diagnosis is challenging and is only confirmed by histopathology examination. The outcome of surgical treatment by trans‐sphenoidal approach is favorable confirming the diagnosis and relieving symptoms related to mass effect. Pathological findings always reveal granulomatous areas, multinucleated giant cells, plasma cells, and lymphocytes.
Authorship
Mohannad E Elgamal: wrote the manuscript and edited the images. The role of other authors: is equal in obtaining the consent, reviewing and editing the manuscript, and providing advice.
Conflict of interest
There is no potential conflict of interest relevant to this article.
References
- 1. Cheung, C. C. , Ezzat S., Smyth H. S., and Asa S. L.. 2006. The spectrum and significance of primary hypophysitis. J. Clin. Endocrinol. Metab. 86:1048–1053. [DOI] [PubMed] [Google Scholar]
- 2. Gutenberg, A. , Hans V., Puchner M. J. A., and Kreutzer J., Bru¨ck W., Caturegli P., Buchfelder M.. 2006. Primary hypophysitis: clinical‐pathological correlations. Eur. J. Endocrinol. 155:101–107. [DOI] [PubMed] [Google Scholar]
- 3. Padilla‐Martínez, J. J. , Alvarez‐Palazuelos L. E., Villagómez‐Méndez J. A., Chiquete E., Domínguez‐Rosales J. A., Espejo‐Plascencia I., et al. 2009. Granulomatous hypophysitis by Mycobacterium gordonae in a non HIV‐infected patient. Neurol. Int. 16:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chung, C. H. , Song M. S., Cho H. D., du Jeong S., Kim Y. J., Bae H. G., et al. 2012. A case of idiopathic granulomatous hypophysitis. Korean J. Intern. Med. 27:346–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hunn, B. H. M. , Martin W. G., Simpson S. Jr, and Mclean A. C.. 2014. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary 17:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iqbal, M. , Jamal S., Luqman M., and Mamoon N.. 2004. Granulomatous hypophysitis mimicking pituitary adenoma. J. Coll. Physicians Surg. Pak. 14:685–686. [PubMed] [Google Scholar]
- 7. Shi, J. , Zhang J.‐M., Wu Q., Chen G., Zhang H., and Bo W.‐L.. 2009. Granulomatous hypophysitis: two case reports and literature review. Univ. Sci. B 10:552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yildirim, A. E. , Divanlioglu D., Cetinalp N. E., Sahin S., and Kulacoglu S., Belen A. D.. 2014. Primary nonnecrotizing granulomatous hypophysitis mimicking pituitary adenomas. Turk. Neurosurg. 24:688–694. [DOI] [PubMed] [Google Scholar]
- 9. Unlu, E. , Puyan F. O., and Bilgi S., Kemal Hamacioglu M.. 2006. Granulomatous hypophysitis: presentation and MRI appearance. J. Clin. Neurosci. 13:1062–1066. [DOI] [PubMed] [Google Scholar]
- 10. Hunn, B. H. , Martin W. G., Simpson S. J., and Mclean C. A.. 2014. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary 17:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kong, X. , Wang R., Yang Y., Wu H., Su C., Ma W., et al. 2015. Idiopathic granulomatous hypophysitis mimicking pituitary abscess. Medicine 94:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo, S. , Wang C., Zhang J., Tian Y., and Wu Q.. 2016. Diagnosis and management of tumor‐like hypophysitis: a retrospective case series. Oncol. lett. 11:1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Manousaki, D. , Deal C., De Bruycker J. J., Ovetchkine P., Mercier C., and Alos N.. 2014. A 15‐year‐old adolescent with a rare pituitary lesion. Endocrinol. Diabetes Metab. Case Rep. 14:2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Çavuşoğlu, M. , Elverici E., Duran S., Komut E., Gürecçi S., and Sakman B.. 2015. Idiopathic Granulomatous Hypophysitis: A rare cystic lesion of the pituitary. Internal Medicine 54:1407–1410. [DOI] [PubMed] [Google Scholar]
- 15. Elgamal, E. A. , Osman E. A., El‐Watidy S. F., Jamjoom Z. B., Hazem A., Al‐Khawajah N., et al. 2007. Pituitary adenomas: patterns of visual presentation and outcome after transsphenoidal surgery‐an institutional experience. Internet J Ophthalmol Vis Sci 4.2. [Google Scholar]
- 16. Su, S. B. , Zhang D. J., Yue S. Y., and Zhang J. N.. 2011. Primary granulomatous hypophysitis: a case report and literature review. Endocr. J. 8:467–473. [DOI] [PubMed] [Google Scholar]