

Abstract
Genetic variants in the triggering receptor expressed on myeloid cells 2 (TREM2) increase the risk for several neurodegenerative diseases including Alzheimer's disease and frontotemporal dementia (FTD). Homozygous TREM2 missense mutations, such as p.T66M, lead to the FTD‐like syndrome, but how they cause pathology is unknown. Using CRISPR/Cas9 genome editing, we generated a knock‐in mouse model for the disease‐associated Trem2 p.T66M mutation. Consistent with a loss‐of‐function mutation, we observe an intracellular accumulation of immature mutant Trem2 and reduced generation of soluble Trem2 similar to patients with the homozygous p.T66M mutation. Trem2 p.T66M knock‐in mice show delayed resolution of inflammation upon in vivo lipopolysaccharide stimulation and cultured macrophages display significantly reduced phagocytic activity. Immunohistochemistry together with in vivo TSPO small animal positron emission tomography (μPET) demonstrates an age‐dependent reduction in microglial activity. Surprisingly, perfusion magnetic resonance imaging and FDG‐μPET imaging reveal a significant reduction in cerebral blood flow and brain glucose metabolism. Thus, we demonstrate that a TREM2 loss‐of‐function mutation causes brain‐wide metabolic alterations pointing toward a possible function of microglia in regulating brain glucose metabolism.
Keywords: frontotemporal dementia, neurodegeneration, neuroinflammation, regulated intramembrane proteolysis, TREM2
Subject Categories: Metabolism, Molecular Biology of Disease, Neuroscience
Introduction
Genetic variations in the triggering receptor expressed on myeloid cells 2 (TREM2) have been shown to increase the risk of several neurodegenerative diseases including Alzheimer's disease (AD), frontotemporal dementia (FTD), and Parkinson's disease (PD) (Guerreiro et al, 2013a; Jonsson et al, 2013; Rayaprolu et al, 2013; Borroni et al, 2014; Cuyvers et al, 2014). Furthermore, homozygous null mutations have been linked to a rare recessive early‐onset dementia syndrome called Nasu–Hakola disease (NHD; also known as polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy) directly connecting TREM2 function with maintenance of central nervous system homeostasis (Klunemann et al, 2005). In an attempt to identify novel risk factors for FTD, homozygous TREM2 missense mutations such as the TREM2 p.T66M mutation have been identified to also cause FTD‐like syndrome (Guerreiro et al, 2013b), which has a similar clinical picture like NHD but without bone involvement. TREM2 is a type I transmembrane innate immune receptor that has been shown to be a substrate for regulated intramembrane proteolysis (Wunderlich et al, 2013; Kleinberger et al, 2014). Shedding of the TREM2 ectodomain by proteases of the ADAM (a disintegrin and metalloproteinase) family releases a soluble fragment (sTREM2) into the extracellular space that can be readily detected in biological fluids including the cerebrospinal fluid (CSF), plasma, and serum (Piccio et al, 2008, 2016; Kleinberger et al, 2014; Henjum et al, 2016; Heslegrave et al, 2016; Suarez‐Calvet et al, 2016a,b). The remaining membrane‐retained C‐terminal fragment is degraded via intramembrane proteolysis by γ‐secretase (Wunderlich et al, 2013; Glebov et al, 2016). TREM2 is expressed in all cells of the myeloid linage, but in the brain TREM2 expression is predominantly restricted to microglia (Hickman et al, 2013). Previously, we and others showed that the TREM2 mutations p.T66M and p.Y38C which are associated with early‐onset dementia (Guerreiro et al, 2013b) result in impaired cell surface trafficking of TREM2, accumulation of immature TREM2 in the endoplasmatic reticulum (Kleinberger et al, 2014; Park et al, 2015), and reduced shedding of sTREM2. In line with that patients with homozygous TREM2 p.T66M have virtually no sTREM2 in the CSF or plasma (Kleinberger et al, 2014; Henjum et al, 2016).
TREM2 has been recently reported to bind anionic lipids (Daws et al, 2003), lipoproteins (Wang et al, 2015), and apolipoproteins including ApoE and ApoJ (Atagi et al, 2015; Bailey et al, 2015; Yeh et al, 2016). TREM2 plays an important role in phagocytosis (Takahashi et al, 2005; N'Diaye et al, 2009; Kleinberger et al, 2014; Xiang et al, 2016) and facilitates microglia‐mediated clearance of fibrillar amyloid‐β peptides (Kleinberger et al, 2014; Xiang et al, 2016). Overexpressed mutant TREM2 does not stimulate phagocytosis like wild‐type TREM2 due to impaired TREM2 cell surface transport (Kleinberger et al, 2014). Besides its involvement in regulating phagocytic activity, TREM2 has also been shown to negatively regulate inflammatory responses (Ito & Hamerman, 2012). In vitro studies using Trem2 knockdown or knockout approaches in bone marrow‐derived macrophages (BMDM) revealed an exaggerated release of pro‐inflammatory cytokines upon stimulation with Toll‐like receptor ligands (Hamerman et al, 2006; Turnbull et al, 2006). Furthermore, loss of Trem2 or its signaling adaptor DNAX activation protein of 12 kDa (DAP12) has been shown to affect proliferation and survival of macrophages (Otero et al, 2012; Wu et al, 2015) and microglia (Wang et al, 2015) in a CSF1 and sTrem2 concentration‐dependent manner. Finally, we recently demonstrated that loss of Trem2 results in impaired chemotaxis and reduced responses of microglia to neuronal injury. Moreover, transcriptomic analyses revealed that microglia may be locked in a homeostatic stage in the absence of Trem2 (Mazaheri et al, 2017).
So far, all functional studies on mutant TREM2 have been performed using ectopic expression of TREM2 and animal models for the FTD‐like syndrome are currently missing. In order to establish a model that mimics the effects of the TREM2 p.T66M mutation at physiological expression conditions and also allows investigating the in vivo consequences of mutant TREM2, we generated a Trem2 knock‐in mouse using CRISPR/Cas9 genome editing (Chu et al, 2015). Using this novel mouse model, we show that immature mutant Trem2 accumulates resulting in significantly reduced levels of sTrem2 in the brain and biological fluids. Furthermore, Trem2 p.T66M knock‐in mice displayed significantly reduced activity of microglia as shown by Iba1 immunohistochemistry and longitudinal in vivo small animal positron emission tomography (μPET) for the 18‐kD translocator protein ligand (TSPO) a noninvasive method to determine microglial activity (Liu et al, 2015; Brendel et al, 2016). Reduced microglial activity correlated with reduced fluoro‐2‐deoxy‐D‐glucose (FDG) μPET signal as a surrogate marker for impaired brain glucose metabolism and function.
Results
Generation of a mouse model for a FTD‐like disease‐associated TREM2 mutation
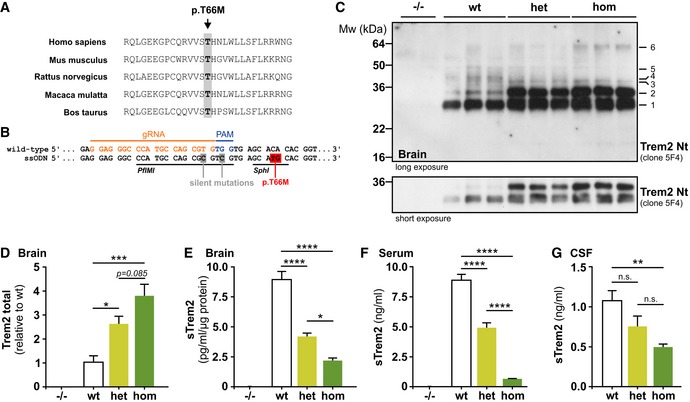
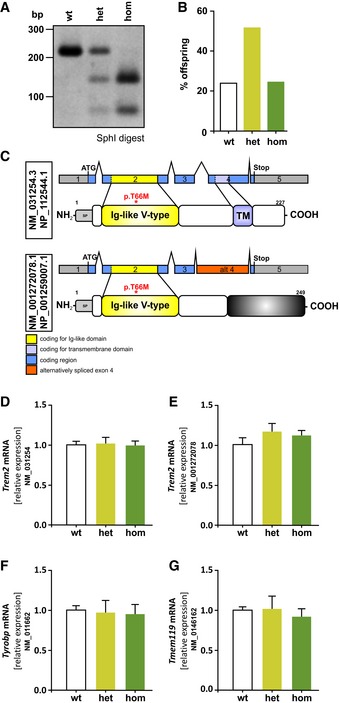
TREM2 is an evolutionary highly conserved protein. This also accounts for the threonine at position 66, which is fully conserved between various mammalian species (Fig 1A). In order to investigate the functional consequences of the disease‐associated TREM2 p.T66M mutation under physiological conditions in vivo, we generated a knock‐in mouse model using CRISPR/Cas9‐mediated genome editing by introducing a CA>TG substitution directly into the murine Trem2 gene (Fig 1B; highlighted in red). We also introduced two silent mutations (Fig 1B, highlighted in gray) allowing genotyping by restriction enzyme digestion (Fig EV1A; see also Materials and Methods). Trem2 p.T66M mice were fertile, reproduced with a normal Mendelian pattern of inheritance (Fig EV1B) and no sudden death occurred within two aging cohorts (n wt(13m) = 8; n hom(13m) = 12; n wt(19m) = 4; n hom(19m) = 8). Expression of both Trem2 mRNA transcripts (NM_031254 and NM_001272078; Fig EV1C) was not affected in brains from heterozygous (het) and homozygous (hom) Trem2 p.T66M knock‐in mice (Fig EV1D and E). Furthermore, Tyrobp (NM_011662), the signaling adaptor of Trem2, and Tmem119 (NM_0146162), a microglia‐specific gene, were also not changed in the brains of 6‐month‐old Trem2 knock‐in mice (Fig EV1F and G).
Figure 1. Trem2 p.T66M knock‐in mice recapitulate biochemical alterations observed in FTD‐like patients.
-
AAlignment of five mammalian TREM2 protein sequences demonstrates complete sequence conservation at the positions harboring the disease‐associated p.T66M mutation.
-
BStrategy to target the murine Trem2 locus indicating the protospacer region (orange), protospacer adjacent region (PAM, blue), and the introduced nucleotide changes (gray or red). Restriction sites for PfIMI and SphI are underlined.
-
CImmunoblotting with anti‐Trem2 Nt (clone 5F4) of immunoprecipitated Trem2 from wild‐type (wt), heterozygous (het), and homozygous (hom) Trem2 p.T66M knock‐in mouse brain reveals a gene dose‐dependent accumulation of Trem2 (bands 1, 2, 3, and 6) accompanied by a reduction of higher molecular weight Trem2 species (bands 4 and 5). A shorter exposure is included to highlight the gene dose‐dependent accumulation of immature Trem2 (bands 1 and 2) in Trem2 p.T66M knock‐in mice. Note that bands can potentially arise from both Trem2 isoforms as well as from sTrem2 in the total lysate. Corresponding brain homogenates from Trem2 knockout (−/−) mice (Turnbull et al, 2006) were used to demonstrate specificity of immunoblotting signal.
-
DQuantification of total Trem2 immunoreactivity (bands 1–6 in C). Data from two experiments are shown as mean ± SEM; n wt = 7, n het = 8, n hom = 8. One‐way ANOVA, Tukey's post hoc test *P < 0.05; ***P < 0.001.
-
E–GQuantifications of sTrem2 from TBS‐soluble brain extracts (E), serum (F), and cerebrospinal fluid (CSF) (G) show a gene dose‐dependent reduction of sTrem2 in Trem2 p.T66M knock‐in mice. Importantly, the Trem2 ELISA was capable of detecting both wild‐type and mutant Trem2 proteins in membrane fractions of transiently transfected HEK293 cells (Appendix Fig S1). In (E) and (F), mice of different age groups (3–13 months; brain: n wt = 12, n het = 8, n hom = 12; serum: n wt = 36, n het = 22, n hom = 35) were pooled for sTrem2 analysis and samples from homozygous Trem2 knockout mice (−/−; n = 2; (Turnbull et al, 2006)) were used to demonstrate specificity of ELISA signal. (G) n wt = 5, n het = 5, n hom = 8. Data are shown as mean ± SEM. One‐way ANOVA, Tukey's post hoc test (E and F); Kruskal–Wallis, Dunn's post hoc test (G); n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., nonsignificant.
Source data are available online for this figure.
Figure EV1. Characterization of Trem2 p.T66M knock‐in mice.
-
ARepresentative of genotyping results using SphI restriction enzyme digest resulting in bands at 215 bp (lane 1: wild‐type; wt), 215 bp + 141 bp + 74 bp (lane 2: heterozygous; het), or 141 bp + 74 bp (lane 3: homozygous; hom).
-
BMendelian pattern of inheritance in Trem2 knock‐in mice (n wt = 38, n het = 82, n hom = 39).
-
CSchematic overview of murine Trem2 transcripts and the respective proteins. Alternative splicing results in a transcript variant encoding soluble Trem2 (NM_001272078.1) lacking its transmembrane domain (NP_001259007.1).
-
D–GqRT–PCR analysis of the canonical (D) and alternative (E) Trem2 transcripts as well as the signaling adaptor Tyrobp (F) and a microglia marker Tmem119 (G) shows no significant changes due to introduction of the Trem2 p.T66M mutation in total brain of 6‐month‐old mice. Data are represented as mean ± SEM. n = 4 mice/genotype.
Source data are available online for this figure.
Reduced maturation and impaired shedding of Trem2 affects phagocytosis
Immunoprecipitation of Trem2 from total mouse brain followed by immunoblotting revealed a gene dose‐dependent accumulation of immature Trem2 (Fig 1C and D). In line with the accumulation of immature Trem2, analysis of sTrem2 by an ELISA that detects both wild‐type and mutant Trem2 protein (Appendix Fig S1) revealed a statistically significant gene dose‐dependent reduction of sTrem2 in TBS‐soluble brain extracts (Figs 1E and EV2A), serum (Figs 1F and EV2B), and CSF (Fig 1G), thereby recapitulating the findings in patients with heterozygous (Piccio et al, 2016) or homozygous TREM2 p.T66M mutation (Kleinberger et al, 2014; Henjum et al, 2016). Reduced mutant sTrem2 was observed in all age groups analyzed (Fig EV2A and B) although wild‐type (wt) sTrem2 levels in the brain significantly increased during aging. The age‐dependent increase of sTrem2 is consistent with previous studies measuring sTREM2 in human CSF (Henjum et al, 2016; Piccio et al, 2016; Suarez‐Calvet et al, 2016a,b). Together, these data confirm the strong biochemical phenotype of the Trem2 p.T66M mutation on an endogenous level and clearly rule out that the accumulation of immature Trem2 occurs only after overexpression (Kleinberger et al, 2014; Park et al, 2015).
Figure EV2. sTrem2 brain and serum analysis in different age groups of Trem2 p.T66M knock‐in mice.
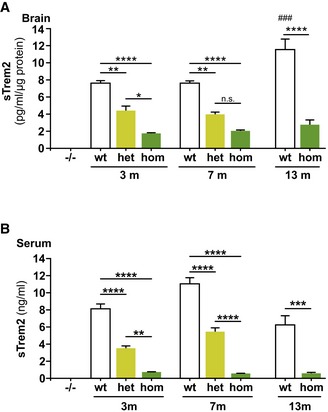
- Brain samples used in Fig 1E split up in three different age groups according to mean age of the group. Data are represented as mean ± SEM (n = 4/genotype and group). One‐way ANOVA, Tukey's post hoc test; *P < 0.05; **P < 0.01; ****P < 0.0001; ### P < 0.001; # denotes significant differences between age groups within the wt group.
- Serum samples used in Fig 1F split up in three different age groups according to mean age of the group. Data are represented as mean ± SEM (3 m: n wt = 20, n het = 8, n hom = 16; 6.5 m: n wt = 12, n het = 13, n hom = 15; 13 m: n wt = 4, n hom = 4). One‐way ANOVA, Tukey's post hoc test; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Source data are available online for this figure.
Bone marrow‐derived macrophages (BMDMs) express high levels of Trem2 (Turnbull et al, 2006) and Trem2 knockout BMDMs show an exaggerated inflammatory response upon exposure to Toll‐like receptor agonists (Turnbull et al, 2006), decreased proliferation, and survival upon CSF1 deprivation (Otero et al, 2012; Wu et al, 2015) as well as reduced phagocytic activity (Xiang et al, 2016). We therefore generated BMDMs from Trem2 p.T66M knock‐in mice by differentiating bone marrow cells with murine macrophage colony‐stimulating factor (M‐CSF). We consistently observed a significant reduction in the number of both heterozygous and homozygous Trem2 p.T66M BMDMs after 7 days of differentiation similar to Trem2 knockout (−/−) BMDMs (Fig 2A). BMDMs derived from Trem2 p.T66M knock‐in mice exhibit significantly increased caspase 3/7 activation (Fig 2B) as well as reduced proliferation as shown by reduced BrdU incorporation (Fig 2C) and decreased mRNA levels of cyclin D1 (Fig 2D). Thus, proliferation and survival of BMDMs are both affected by the Trem2 p.T66M mutation and together account for the observed reduced cell count.
Figure 2. Decreased proliferation and increased apoptosis in Trem2 p.T66M bone marrow‐derived macrophages.
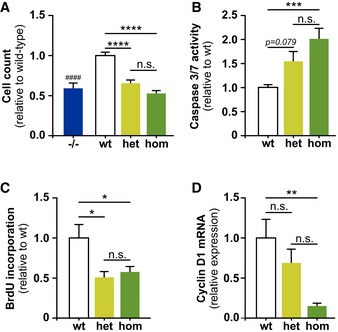
- After 7 days of differentiation with M‐CSF both heterozygous (het) and homozygous (hom), BMDMs show a significantly reduced cell number similar to the reduction seen for Trem2 knockout (−/−) BMDMs (Turnbull et al, 2006). Data are shown as mean ± SEM from at least three independent experiments (n –/– = 9, n wt = 12, n het = 14, n hom = 12). One‐way ANOVA, Tukey's post hoc test; n.s., P > 0.05; ****, P < 0.001; n.s., nonsignificant; ####, P < 0.0001; # denotes significant differences between −/− and wt.
- Caspase 3/7 activity measured using 5 × 104 cells at day 7 of differentiation by a luminescence assay. Data are shown as mean ± SEM from two independent experiments (n wt = 7, n het = 8, n hom = 8). Kruskal–Wallis, Dunn's post hoc test; n.s., P > 0.05; ***, P < 0.001.
- Bone marrow‐derived macrophages were differentiated in 12‐well plates for 6 days prior adding BrdU for the last 24 h of differentiation and BrdU incorporation was quantified by flow cytometry. Data are shown as mean ± SEM from two independent experiments (n = 5). One‐way ANOVA, Tukey's post hoc test; n.s., P > 0.05; *, P < 0.05.
- qRT–PCR analysis of cyclin D1 (Ccnd1; NM_007631) in BMDMs shows significantly reduced cyclin D1 levels in homozygous Trem2 p.T66M BMDMs. Data are shown as mean ± SEM from two independent experiments (n = 5). One‐way ANOVA, Tukey's post hoc test; n.s., P > 0.05; **, P < 0.01.
Source data are available online for this figure.
In line with data from mutant mouse brains (Fig 1C), BMDMs derived from Trem2 knock‐in mice showed a dramatic accumulation of immature Trem2 (Fig 3A, B and D) and a concomitant reduced release of sTrem2 into the supernatants as shown by immunoblotting (Fig 3A) and ELISA quantification (Fig 3C). Upon shedding of Trem2, a C‐terminal membrane‐associated fragment is retained and further degraded by γ‐secretase (Wunderlich et al, 2013). Previously, we and others could show that the TREM2 C‐terminal fragment (CTF) is dramatically reduced in cells overexpressing TREM2 p.T66M (Kleinberger et al, 2014; Park et al, 2015). γ‐Secretase‐mediated degradation of the Trem2 CTF seems to be highly efficient in BMDMs, as seen by very low constitutive levels of CTF (Fig 3A). We therefore treated cells with the γ‐secretase inhibitor DAPT (Fig 3A). This allowed the detection of reduced CTF generation in an allele‐dependent manner (Fig 3A and E).
Figure 3. Accumulation and reduced processing of mutant Trem2 in BMDMs.
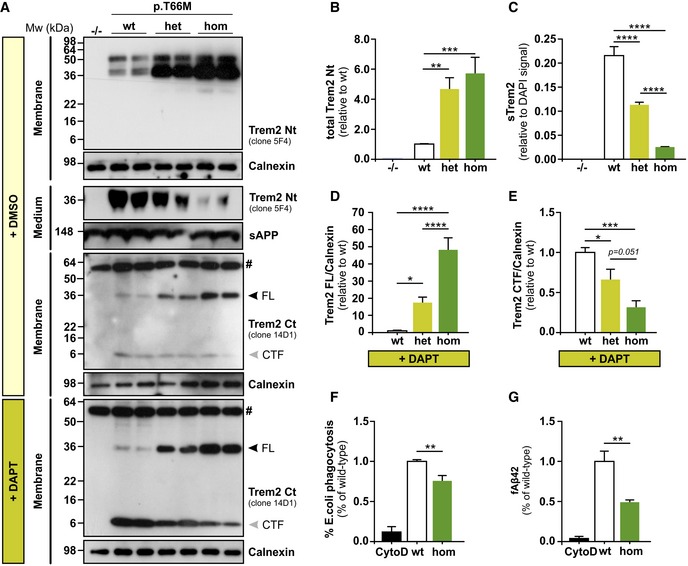
-
AImmunoblotting with anti‐Trem2 Nt (clone 5F4) and anti‐Trem2 Ct (clone 14D1) shows accumulation of mutant full‐length Trem2 in membrane fractions of heterozygous (het) and homozygous (hom) BMDMs compared to wild‐type (wt) BMDMs. BMDMs treated with DMSO show accumulation of mutant full‐length Trem2 in membrane fractions of heterozygous (het) and homozygous (hom) BMDMs compared to wild‐type (wt) BMDMs using anti‐Trem2 Ct (clone 14D1). Trem2 C‐terminal fragment (CTF) is generated in BMDMs derived from wt, heterozygous, and homozygous Trem2 p.T66M knock‐in mice, however to very moderate levels compared to DAPT‐treated BMDMs. Upon inhibition of γ‐secretase by DAPT treatment, wt BMDMs strongly accumulate a Trem2 CTF which is significantly reduced in an allele‐specific manner in Trem2 p.T66M knock‐in BMDMs. Analysis of conditioned media shows an allele‐specific reduction of sTrem2 in Trem2 p.T66M knock‐in BMDMs. Homozygous Trem2 knockout (−/−) BMDMs from a Trem2 knockout line (Turnbull et al, 2006) were used to confirm specificity of the Trem2 antibodies. Calnexin (membrane fractions) and sAPP (medium fractions) were used to confirm equal loading. FL, full‐length. # labels an unspecific band.
-
BQuantification of total Trem2 levels from immunoblots using the anti‐Trem2 Nt (clone 5F4) antibody. Data shown relative to wt as mean ± SEM. (n –/– = 3, n wt = 8, n het = 8, n hom = 8). One‐way ANOVA, Tukey's post hoc test; **, P < 0.01; ***, P < 0.001.
-
CQuantification of sTrem2 by ELISA in supernatants from mutant BMDMs confirms an allele‐specific reduction of sTrem2 in BMDMs derived from Trem2 p.T66M knock‐in mice. Levels of sTrem2 are depicted normalized to cell number as determined by quantification of DAPI signal. Data are represented as mean ± SEM (n wt = 12, n het = 16, n hom = 16). One‐way ANOVA, Tukey's post hoc test; ****, P < 0.0001.
-
D, EQuantification of Trem2 full‐length (D) and Trem2 CTF (E) levels upon treatment with DAPT using the anti‐Trem2 Ct (clone 14D1) antibody to detect Trem2. Trem2 levels were normalized to the signal of calnexin and data shown relative to wt as mean ± SEM from three independent experiments (n wt = 9, n het = 9, n hom = 7). One‐way ANOVA, Tukey's post hoc test; *P < 0.05; ***P < 0.001; ****P < 0.0001.
-
FReduced phagocytosis of E. coli particles of BMDMs derived from homozygous Trem2 p.T66M knock‐in mice (hom) compared to wild‐type (wt). Data are represented relative to wt as mean ± SEM from three independent experiments (n CytoD = 4, n wt = 10, n hom = 9). One‐way ANOVA, Tukey's post hoc test; **P < 0.01.
-
GPhagocytosis of fAb42 by BMDMs from wild‐type (wt) and homozygous Trem2 p.T66M knock‐in mice (hom). Data are represented as relative to wt as mean ± SEM from three independent experiments (n CytoD = 3, n wt = 7, n hom = 8). One‐way ANOVA, Tukey's post hoc test; **P < 0.01.
Source data are available online for this figure.
Trem2 facilitates phagocytosis of E. coli (N'Diaye et al, 2009), apoptotic neurons (Takahashi et al, 2005; Hsieh et al, 2009), as well as fibrillar amyloid‐β peptides (Xiang et al, 2016). Similar to BMDMs derived from Trem2 knockout mice (Xiang et al, 2016), homozygous Trem2 p.T66M knock‐in BMDMs showed a significantly reduced capacity of engulfing pHrodo‐labeled E. coli particles (Fig 3F) as well as fluorescently labeled fibrillar Aβ1–42 (Fig 3G) supporting the loss‐of‐function character of the Trem2 p.T66M mutation.
Exaggerated immune response to in vivo lipopolysaccharide stimulation
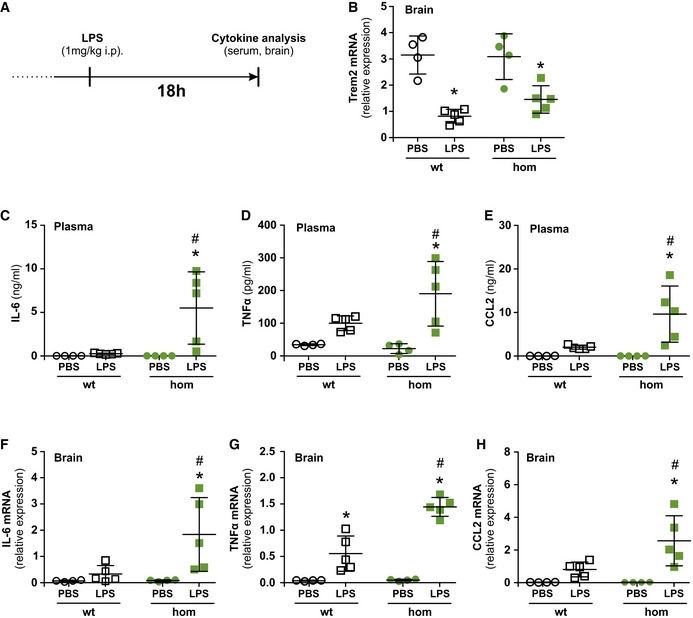
Stimulation of macrophages with lipopolysaccharide (LPS) or interferon‐γ (IFN‐γ) causes a robust downregulation of Trem2 expression (Turnbull et al, 2006) and studies using Trem2‐deficient macrophages or microglial cell lines suggested Trem2 to be involved in inhibiting cytokine production upon stimulation with LPS or IFN‐γ (Turnbull et al, 2006; Zhong et al, 2015). We therefore asked whether the Trem2 p.T66M mutation leads to an exaggerated pro‐inflammatory response in vivo during the resolution phase of the inflammatory reaction 18 h post‐intraperitoneal LPS stimulation (Fig 4A). First, we confirmed that LPS injection led to a marked reduction of Trem2 mRNA in the brain of both wild‐type and homozygous Trem2 p.T66M knock‐in mice (Fig 4B). We then compared the plasma protein (Fig 4C–E) and brain mRNA (Fig 4F–H) levels of interleukin‐6 (IL‐6), tumor necrosis factor alpha (TNF‐α), and chemokine (C‐C motif) ligand 2 (CCL2) in wild‐type and homozygous Trem2 p.T66M knock‐in mice. While only a small increase of IL‐6, TNF‐α, and CCL2 plasma and brain expression was seen 18 h after LPS injection in wild‐type mice, homozygous Trem2 p.T66M knock‐in mice showed significantly higher levels of these cytokines consistent with an exaggerated and/or prolonged inflammatory response. These data confirm the involvement of Trem2 in regulating the release of pro‐inflammatory cytokines after LPS stimulation and further support a loss of function of the Trem2 p.T66M mutation.
Figure 4. Exaggerated immune response to lipopolysaccharide infection in Trem2 p.T66M mice.
-
ASchematic representation of the experimental outline. i.p., intraperitoneal.
-
BIntraperitoneal injection of lipopolysaccharide (LPS) reduces Trem2 mRNA expression in the brain 18 h post‐injection in both wild‐type and homozygous Trem2 p.T66M knock‐in mice.
-
C–ECytometric Bead Array‐based analysis of cytokine levels in the serum 18 h post‐injection with PBS or LPS shows significant increased levels of IL‐6 (C), TNF‐α (D), and CCL2 (E) in the serum of LPS‐injected Trem2 p.T66M knock‐in mice.
-
F–HqRT–PCR‐based analysis of cytokine levels in the brain 18 h post‐injection with PBS or LPS shows significant increased levels of IL‐6 (F), TNF‐α (G), and CCL2 (H) in the brain of LPS‐injected Trem2 p.T66M knock‐in mice.
Decreased age‐dependent microglial activity
Next, we searched for a potentially age‐related microglial phenotype in Trem2 p.T66M knock‐in mice. To do so, we conducted a longitudinal study (3, 5, 8, and 12 months) using the PET tracer [18]F‐GE180 (18‐kD translocator protein ligand [TSPO]) for imaging activated microglia in vivo by μPET. In line with previous results (Liu et al, 2015; Brendel et al, 2016), we found an age‐dependent increase in TSPO μPET signal in wild‐type mice (Fig 5A and B). Visual interpretation of TSPO μPET scans indicated similar patterns of tracer uptake at 3 and 5 months. However, a globally lower retention in Trem2 p.T66M knock‐in mice at 8 months was observed which was even more pronounced at 12 months of age (Fig 5A). Quantitative cerebral TSPO μPET signals increased in parallel in 3‐ and 5‐month‐old wild‐type and Trem2 p.T66M knock‐in mice (Fig 5B). While wild‐type mice continued their increase at a flattened slope until 12 months of age, the TSPO signal in Trem2 p.T66M knock‐in mice started to decline after 8 months (Δ8%, P = 0.16), resulting in a significantly reduced TSPO μPET signal at 12 months of age (Δ23%, P < 0.01) (Fig 5B).
Figure 5. Decreased age‐dependent microglial activity in Trem2 p.T66M mice.
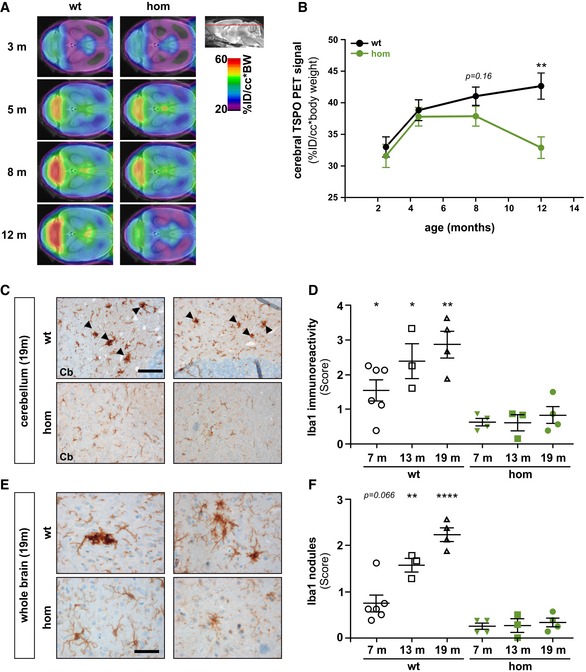
- Axial slices of TSPO μPET imaging (scaled by %ID and body weight) are projected on a T1 MRI mouse template (left: cerebellum; right: frontal pole). Averaged images (n = 7–8 mice/genotype) show a clear visual decrease of TSPO activity in Trem2 p.T66M knock‐in mice aged 8 and 12 months when compared to wt.
- Quantitative TSPO activity in wt (black) and hom (green) mice. Data are represented as mean ± SEM. n = 7–8 mice/genotype. Student's t‐test; **P < 0.01.
- Iba‐1 immunohistochemistry in brains of 19‐month‐old wild‐type (wt) or homozygous Trem2 p.T66M knock‐in mice (hom) shows a strong signal with frequent occurrence of microglia nodule‐like structures (black arrowheads) in the cerebellar white matter of wt compared to mutant mice. Scale bar, 100 μm.
- Semiquantitative analysis of Iba‐1 reactivity shows an age‐dependent increase in wt but not hom mice. Data are represented as mean ± SEM. n 6 m = 4–6 mice/genotype; n 13m = 3 mice/genotype; n 19m = 4 mice/genotype; Student's t‐test; *P < 0.05; **P < 0.01.
- Iba1‐positive (Iba1+) nodules are also frequently observed throughout the brain in wt but not in hom mice. Scale bar, 50 μm.
- Semiquantitative analysis of Iba1+ nodules shows an age‐dependent increase in wt but not hom mice. Data are represented as mean ± SEM. n 6m = 4–6 mice/genotype; n 13m = 3 mice/genotype; n 19m = 4 mice/genotype; Student's t‐test; **P < 0.01; ****P < 0.0001.
Source data are available online for this figure.
In order to relate the observed functional deficit of microglia in vivo to histological changes, we analyzed Iba1 reactivity during aging in the Trem2 p.T66M knock‐in mice. Clusters of activated microglia have been recently termed “microglial nodules” (Singh et al, 2013) and are thought to be associated with removing myelin/axonal debris (Singh et al, 2013). Strikingly, we observed an age‐dependent increase in Iba1 immunoreactivity in wild‐type but not homozygous Trem2 p.T66M knock‐in mice especially in the white matter of the cerebellum (Fig 5C) which is consistent with a strong TSPO μPET signal in the cerebellum of wild‐type but not homozygous Trem2 p.T66M knock‐in mice (Fig 5A). Furthermore, we frequently observed strongly Iba1 immunoreactive microglia nodule‐like structures in wild‐type but not homozygous Trem2 p.T66M knock‐in brains (Fig 5C, arrowheads). Iba1 reactivity increased with age in wild‐type but not in Trem2 p.T66M knock‐in mice with significant differences detected as early as 6 months of age as shown by semiquantitative image analysis (Fig 5D). An age‐dependent accumulation of nodule‐like structures of Iba1 immunoreactive microglia was also observed throughout the whole brain of wild‐type but not Trem2 p.T66M knock‐in mice (Fig 5E and F), suggesting a general reduction in microglia activity caused by loss of Trem2 function. Taken together, both Iba1 immunohistochemical and in vivo longitudinal TSPO μPET analyses indicated a significantly reduced microglia activity, which became more prominent with aging, thus indicating a probable correlate of dysfunctional microglia due to the Trem2 p.T66M mutation.
Reduced cerebral blood flow in Trem2 p.T66M knock‐in mice
In humans, homozygous TREM2 mutations lead to marked thinning of the corpus callosum, cortical atrophy, and periventricular white matter abnormalities (Guerreiro et al, 2013b). Furthermore, single positron emission computer tomography (SPECT) in a family carrying a TREM2 loss‐of‐function mutation revealed hypoperfusion of the brain in mutation carriers (Montalbetti et al, 2005). We therefore used magnetic resonance imaging (MRI) to analyze homozygous Trem2 p.T66M knock‐in mice for volumetric brain changes and performed continuous arterial spin‐labeled (CASL) MRI to detect possible abnormalities in absolute brain perfusion. Volumetric brain imaging did not reveal any global differences between homozygous Trem2 p.T66M knock‐in mice compared to wild‐type littermate controls at both 4 (P = 0.60) and 6 (P = 0.64) months of age. However, using CASL‐MRI we observed a significant difference between genotypes at 6 months of age (P = 0.029; Fig 6A and B). As cerebral blood flow has been reported to be tightly coupled to brain metabolism (Raichle, 1998), reduced brain perfusion in Trem2 p.T66M knock‐in mice at 6 months of age might indicate the first decline in brain function due to Trem2 loss of function.
Figure 6. Reduced cerebral blood flow in Trem2 p.T66M knock‐in mice.
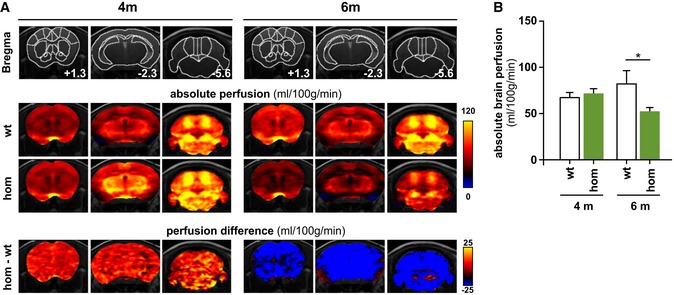
- Upper panels: mouse brain atlas superimposed on T2‐weighted anatomical images with outlined regions of interest and indicated distance to bregma. Middle panels: color‐coded MRI images indicating mean absolute brain perfusion (according to the color bar on the right) of wild‐type (wt) and homozygous Trem2 p.T66M knock‐in (hom) mice depicted in three independent coronal slices at either 4 months (n wt = 12, n hom = 15) or 6 months (n wt = 8, n hom = 8) of age. Lower panels: color‐coded MRI images indicating the differences in brain perfusion between wt and hom mice (according to the color bar on the right).
- Quantitative absolute brain perfusion showed no differences in 4‐month‐old Trem2 p.T66M knock‐in mice (n wt = 12, n hom = 15) while at 6 months of age absolute brain perfusion was significantly reduced in homozygous Trem2 p.T66M knock‐in mice compared to wild‐type controls (n wt = 8, n hom = 8). Data are represented as mean ± SEM. Two‐way mixed ANOVA; *P < 0.05.
Reduced cerebral glucose metabolism in Trem2 p.T66M knock‐in mice
Patients with homozygous TREM2 mutations develop NHD or FTD‐like syndrome manifesting with cognitive impairments that have been associated with frontal lobe hypometabolism as shown by reduced signals by FDG‐PET (Ueki et al, 2000). FDG‐PET has long been used to measure resting‐state cerebral metabolic rates of glucose, a proxy for neuronal activity (Sokoloff, 1977; Sokoloff et al, 1977). We therefore asked whether reduced microglial activity could have a functional effect on the brain using FDG‐μPET imaging of Trem2 p.T66M knock‐in mice at 12 months of age, a time point where a significant reduction in microglial activity by immunohistochemistry and TSPO‐μPET was manifested. Importantly, blood glucose levels were not significantly different between wild‐type and Trem2 p.T66M knock‐in mice (Fig 7A). First, we excluded relevant perfusion effects in μPET by initial dynamic imaging of FDG‐μPET (Fig EV3), which justified the use of common short emission recordings (see Materials and Methods). Visual interpretation of FDG‐μPET scans indicate a reduced signal throughout different areas of the brain (Fig 7B; axial and coronal slices) and quantitative analyses confirmed a significant reduction in the global cerebral FDG‐μPET signal in Trem2 p.T66M knock‐in mice compared to age‐ and sex‐matched wild‐type controls (Fig 7C; Δ23%; P < 0.01). We then performed additional TSPO‐μPET scans to study the correlation between FDG‐μPET and TSPO‐μPET signals on the level of individual mice. Correlation of dual quantitative μPET readouts showed a statistically significant positive correlation (Fig 7D; r 2 = 0.5269; P = 0.041) in Trem2 p.T66M knock‐in mice, suggesting a relationship between reduced microglial activity due to the Trem2 mutation and reduced brain function.
Figure 7. Reduced cerebral glucose metabolism in Trem2 p.T66M knock‐in mice.
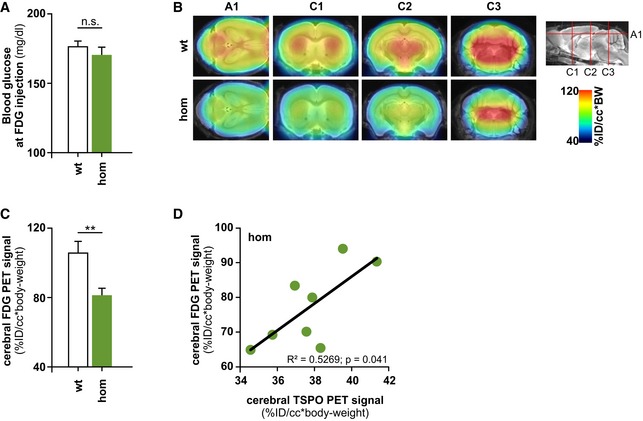
- Analysis of blood glucose levels at the time point of FDG‐μPET analysis shows no significant differences between wild‐type (wt) and homozygous Trem2 p.T66M knock‐in mice (hom) (n wt = 6; n hom = 9 mice). Data are represented as mean ± SEM. Student's t‐test; n.s., P > 0.05.
- Axial (A1) and coronal (C1–3) slices of FDG μPET imaging (scaled by %ID and body weight) and normalized to blood glucose level are projected on a T1 MRI mouse template (left: cerebellum; right frontal pole). Averaged images (n wt = 6; n hom = 9 mice) show a clear visual decrease of FDG signal in several brain areas in Trem2 p.T66M knock‐in mice at 12 months of age when compared to age‐ and sex‐matched wt controls.
- Quantitative FDG μPET signal in wt and hom mice. Data are represented as mean ± SEM. Student's t‐test; **P < 0.01 (n wt = 6; n hom = 9 mice).
- Dual quantitative μPET shows a significant positive correlation of global cerebral FDG and TSPO signals in homozygous Trem2 p.T66M knock‐in mice (hom). Pearson product–moment correlation.
Source data are available online for this figure.
Figure EV3. Dynamic FDG μPET imaging.
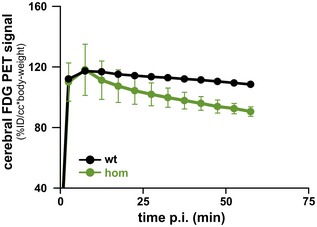
Dynamic FDG μPET imaging of two homozygous Trem2 p.T66M knock‐in mice and one wild‐type control revealed no relevant differences in the perfusion phase of μPET while in the late phase reduced FDG μPET signal was detected in homozygous (hom) Trem2 p.T66M knock‐in mice. Data are represented as mean ± SEM.Source data are available online for this figure.
Discussion
Microglia are the resident macrophages of the central nervous system accounting for 5–20% of all cells in the brain (Lawson et al, 1992) and represent the first line of defense against invading pathogens and pathological insults. Besides their function in immune surveillance, microglia also play an important role in neuronal plasticity through pruning of synapses and axon terminals (Tremblay & Majewska, 2011; Salter & Beggs, 2014). It is well known that microglia change their morphology, number, and activity during aging and pathological insults (Lourbopoulos et al, 2015). The identification of a number of microglia‐related risk variants in AD (Cuyvers & Sleegers, 2016; Villegas‐Llerena et al, 2016) and several other neurodegenerative diseases highlight an important role of microglia and neuroinflammation in the pathogenesis of neurodegenerative diseases. Genetic variants in TREM2 increase the risk of developing AD up to threefold (Guerreiro et al, 2013a; Jonsson et al, 2013) and complete loss of TREM2 leads to an early‐onset dementia known as NHD (Klunemann et al, 2005). Moreover, patients with homozygous missense mutations in TREM2 present clinically with FTD‐like syndrome (Guerreiro et al, 2013b) most likely due to loss of TREM2 function caused by misfolding (Kober et al, 2016) and reduced cell surface trafficking (Kleinberger et al, 2014). So far the effects of TREM2 variants on its biological functions have been studied mainly in tissue culture using overexpressing paradigms. In order to rule out artifacts caused by strong overexpression of TREM2, in vivo models with endogenous expression of mutant Trem2 protein are urgently needed. With the discovery and establishment of the CRISPR/Cas9 system for genome editing, it is possible to rapidly generate animal models with patient‐specific mutations (Yang et al, 2016). Using this new tool, we generated a knock‐in mouse model expressing the FTD‐like syndrome‐associated TREM2 p.T66M mutation at an endogenous level. We could demonstrate a reduced generation of sTrem2 in the brain, serum, and CSF (Fig 1E–G) due to a dramatic accumulation of mutant immature Trem2 (Figs 1C and 3A, B and D) and reduced processing (Fig 3A, C and E) confirming our previous results obtained in tissue culture (Kleinberger et al, 2014). Moreover, primary macrophages derived from bone marrow of Trem2 p.T66M knock‐in mice showed a reduced phagocytic capacity (Fig 3F and G). Furthermore, Trem2 p.T66M knock‐in mice displayed an exaggerated inflammatory response upon in vivo stimulation with LPS (Fig 4) similar to results obtained with cells derived from Trem2 knockout mice (Turnbull et al, 2006; Zhong et al, 2015). Together, this further strengthens the involvement of Trem2 in phagocytosis and regulation of inflammation and underlines the loss‐of‐function character of the Trem2 p.T66M missense mutation. However, both reduced full‐length Trem2, and consequently lowered TREM2‐dependent signaling as well as reduced sTrem2 levels may account for the observed effects on survival, phagocytosis, and regulation of an appropriate inflammatory response. While we have shown that sTrem2 is not effective in rescuing phagocytic defects (Xiang et al, 2016), a recent study linked sTrem2 to microglial survival and induction of inflammatory responses (Zhong et al, 2017).
Patients with homozygous loss of function of TREM2 develop NHD or FTD‐like syndrome, and present with a cerebral white matter disease characterized by demyelination and occurrence of axonal spheroids (Paloneva et al, 2001). It is known that myelin‐associated lipids can trigger TREM2 signaling in vitro (Poliani et al, 2015; Wang et al, 2015) and Trem2 knockout mice are not capable of responding properly to myelin damage upon cuprizone‐induction demyelination (Cantoni et al, 2015; Poliani et al, 2015). Interestingly, in early stages of multiple sclerosis, microglia have been described to form microglia nodules even in the absence of demyelination (Singh et al, 2013) and sTREM2 levels in the CSF of patients with multiple sclerosis have been described to be increased (Piccio et al, 2008). Our findings support the idea that microglial nodule‐like structures are also formed during physiological aging and may represent a clearance mechanism of minor white matter insults. Furthermore, using Iba‐1 immunohistochemistry we have observed an age‐dependent increase in Iba‐1 immunoreactive microglia nodule‐like structures especially in the white matter of wild‐type mice that was absent in homozygous Trem2 p.T66M knock‐in mice (Fig 5C–F). This is in line with a reduced sensing of myelin‐associated lipids (Poliani et al, 2015; Wang et al, 2015) as well as a general defect of chemotaxis of Trem2‐deficient microglia (Mazaheri et al, 2017). During physiological aging, microglia become increasingly activated which is consistent with a positive correlation of sTREM2 levels in the CSF of healthy subjects with age (Henjum et al, 2016; Piccio et al, 2016; Suarez‐Calvet et al, 2016a,b).
Recently, protocols have been established to image microglial activation by in vivo PET imaging using a tracer against TSPO (Liu et al, 2014) that also has been demonstrated to be useful to monitor the microglia activation status in rodents during aging and amyloidosis (Liu et al, 2015; Brendel et al, 2016). TSPO μPET signal significantly correlates with the levels of sTrem2 in the brain of an amyloidosis mouse model (Brendel et al, 2017), suggesting that sTREM2 indirectly reflects the amount of signaling competent TREM2 on the cell surface of activated microglia. Using in vivo TSPO‐μPET imaging, we could show that the Trem2 p.T66M mutation leads to a decreased microglial activity at 12 months of age (Fig 5A and B), further supporting the loss‐of‐function hypothesis of FTD‐like syndrome‐associated TREM2 mutations. Importantly, Trem2 p.T66M knock‐in mice showed a strong reduction of TSPO‐μPET signal in the cerebellar regions supporting the results obtained by Iba‐1 immunohistochemistry.
FTD‐like syndrome and NHD clinically presents as a frontal lobe syndrome (Paloneva et al, 2001), and functional imaging of these NHD patients revealed hypoperfusion as well as alterations in glucose metabolism in bilateral frontal lobes and thalamus (Ueki et al, 2000; Klunemann et al, 2005; Takeshita et al, 2005). Functional imaging of cerebral blood flow (CBF) using either single photon emission computed tomography (SPECT) or arterial spin‐labeled MRI (ASL‐MRI) as well as measurement of cerebral metabolic rate of glucose by FDG‐PET provides images that closely reflect neuronal activity (Herholz, 2011). In line with that, we find a significantly reduced CBF in Trem2 p.T66M knock‐in mice compared to wild‐type mice at 6 months of age (Fig 6). The decline in brain function and metabolism in Trem2 p.T66M knock‐in mice was further verified with FDG‐μPET imaging at 12 months of age. Reduced FDG‐μPET was not a result of increased blood glucose levels (Fig 7A), a factor which is known to interfere with FDG‐μPET signals (Varrone et al, 2009). Neuronal activity and glucose metabolism are strongly correlated (Sokoloff, 1999), and hence, FDG‐PET imaging has been established as a routine method to measure regional brain activation. However, it is still controversially discussed, whether neurons or astrocytes are the main consumers of glucose in the brain (Mergenthaler et al, 2013). Moreover, it is not known how much microglia, a highly mobile cell population (Nimmerjahn et al, 2005), contribute to the energy consumption of the whole brain. In previous studies, it has been shown that during neurodegeneration, the pattern of CBF and metabolic deficits occur in parallel in AD and FTD patients (Verfaillie et al, 2015), and ASL‐MRI has similar diagnostic utility as FDG‐PET in both AD and FTD (Tosun et al, 2016). Therefore, our ASL‐MRI and FDG‐μPET data complementary prove an age‐dependent decline in brain perfusion and metabolism in Trem2 p.T66M knock‐in mice.
A limitation of our study is the use of separate cohorts of mice for the ASL‐MRI (Fig 6) and FDG‐μPET (Fig 7) analysis. Ideally, all parameters would have been measured in one and the same mouse at exactly the same time points resulting in a study setup that is technically highly challenging. Furthermore, analysis of cerebral blood flow at a later time point, where also reduced microglial activation and glucose metabolism was observed, would be required to strengthen the link between reduced blood flow and reduced microglial activation and glucose metabolism. As TSPO‐μPET and FDG‐μPET results showed a significant correlation at 12 months of age (Fig 7D), the findings presented in this study raise the possibility for a function of microglia in regulating brain glucose metabolism. To address this question, it will be important to study whether the observed brain‐wide hypometabolic effects are mediated by a cell autonomous mechanism (TREM2 receptor on the cell surface of microglia) or by a cell nonautonomous mechanism (e.g., via sTREM2). Our novel mouse model will provide an essential tool to investigate this important question.
In summary, we have shown the first in vivo evidence that the FTD‐like syndrome‐associated TREM2 p.T66M mutation causes a dysfunction in microglia and macrophages which is not only associated with their reduced activity, but also correlates with reduced CBF and brain energy metabolism. Moreover, homozygous Trem2 p.T66M knock‐in mice recapitulate the dramatic biochemical phenotype observed in cultured cells (Kleinberger et al, 2014; Park et al, 2015) and biological fluids of humans (Kleinberger et al, 2014; Henjum et al, 2016). This mouse model might therefore be valuable for investigating TREM2 modulating therapeutic strategies with the aim of improving microglia activity in neurodegenerative disorders.
Materials and Methods
Generation of Trem2 p.T66M knock‐in mice
Trem2 p.T66M mice were generated by nuclease‐mediated homology‐directed repair in zygotes as described previously (Wefers et al, 2013; Brandl et al, 2015). Cas9 mRNA was prepared by in vitro transcription from pCAG‐Cas9v2‐162A (linearized with XbaI) using the mMessage mMachine T7 Ultra kit (Ambion, #AM1345) and purified using the MEGAClear kit (Ambion, #AM1908). For the preparation of Trem2 exon 2‐specific sgRNAs, two annealed oligonucleotides (sgTremA 5′‐CACCTAATACGACTCACTATAGGGAGGGCCCATGCCAGCGTG‐3′, sgTremB 5′‐AAACCACGCTGGCATGGGCCCTCCCTATAGTGAGTCGTATTA‐3′) were cloned into pBS‐U6‐sgRNA, and in vitro transcribed and purified using the MEGAshortscript T7 kit (Ambion, #AM1354) and the MEGAclear kit. The single‐stranded target molecule ssODN‐Trem2T66M (5′‐ACTGGGGGAGACGCAAGGCCTGGTGTCGGCAGCTGGGTGAGGAGGGCCCATGCCAGCGCGTCGTGAGCATGCACGGTGTGTGGCTGCTGGCCTTCCTGAAGAAGCGGAATGGGAGCACAGTCATCGCAGATGACACCCTT‐3′), comprising the CA>TG substitution and two additional silent mutations for genotyping purposes, was chemically synthetized (Metabion; see also Fig 1B). For microinjection, one‐cell embryos were obtained by mating D2B6F1 males with superovulated FVB/N females (Charles River). Cas9 mRNA (5 ng/μl), sgRNA (2.5 ng/μl), and ssODN‐Trem2T66M (10 ng/μl) were injected into the male pronucleus, and injected zygotes were immediately transferred into pseudopregnant CD‐1 foster mice resulting in the strain of mice designated Trem2em1Bwef (hereafter referred to as Trem2 p.T66M knock‐in). Amplification and sequencing of eight loci with potential off‐target sites did not show any additional sequence variations (Appendix Fig S2). Mice used in all experiments were on a mixed genetic background (N1 backcross to C57BL/6J).
CSF and serum collection and tissue harvesting
Serum was harvested by collecting blood by cardiac puncture under deep anesthesia with a combination of midazolam, medetomidine, and fentanyl. Collected blood was incubated for at least 15 min at room temperature and centrifuged at 2,000 g for 10 min, and separated serum was collected and used for sTrem2 measurements. Cerebrospinal fluid (CSF) was collected from the cisterna magna according to previously published methods (Schelle et al, 2016). All mice were handled according to institutional guidelines approved by the animal welfare and use committee of the Government of Upper Bavaria and housed in standard cages in a specific pathogen‐free facility on a 12‐hr light/dark cycle with ad libitum access to food and water.
Genotyping
Genomic DNA was purified from tail biopsies or ear punches by isopropanol precipitation, and the Trem2 locus harboring the knock‐in mutation was amplified by polymerase chain reaction (PCR) using forward primer 5′‐AACACCACGGTGCTGCAGGG‐3′ and reverse primer 5′‐CCAGCAAGGGTGTCATCTGCG‐3′. Resulting PCR products of 215 base pairs (bp) were further processed using either the restriction enzyme SphI‐HF digesting only the mutant allele into 74‐ and 141‐bp fragments or the restriction enzyme PfIMI (both New England Biolabs) digesting only wild‐type allele into 86‐ and 129‐bp fragments and subsequently analyzed by 2% Tris–borate–EDTA agarose gel electrophoresis (see also Fig EV1A).
Magnetic resonance imaging (MRI) acquisition and analysis
Animal preparation and anesthesia
For MRI perfusion measurements, primed continuous intravenous etomidate anesthesia was carried out with initial isoflurane support. Respiratory rate, rectal body temperature, and oxygen and CO2 levels in the inspired and exhaled air were continuously monitored. Body temperature was maintained at 37.3°C with a feedback‐regulated electric heating blanket. Perfusion MRI was carried out on a Bruker Biospec 94/20 MR systems (Bruker Biospin). A volume resonator was used for signal excitation, and an actively decoupled quadrature surface coil was positioned over the head of the animal for signal reception. Following localization of the most rostral extension of the corpus callosum as a landmark on scout images, 14 coronal image planes were selected between −7.70 and +4.00 mm from bregma with a 0.9‐mm interval between planes (Paxinos & Franklin, 2001). All subsequent images were acquired in these planes, with a field of view of 20 × 20 mm2 and a slice thickness of 0.6 mm. The first volume was a set of T2‐weighted anatomical RARE images (TR/TEeff = 3,150/34 ms, RARE factor 8, matrix 256 × 256). Next, a T1 image series required to quantitate perfusion was obtained using an inversion recovery snapshot FLASH sequence with eight inversion times (TR/TE = 4,000/1.6 ms, TI = 96, 263, 429, 595, 761, 927, 1,094, and 1,260 ms, matrix 64 × 64) (Haase et al, 1986). Finally, cerebral perfusion was assessed by continuous arterial spin labeling (CASL) (Williams et al, 1992) with single‐slice centered‐RARE readout (TR/TE = 3,750 ms/5.4 ms, RARE factor = 32, matrix 128 × 64, labeling pulse 3 s, post‐labeling delay 0.4 s). The total acquisition time for one volume of CASL images was 4 min, and contiguous time series of three volumes were acquired for each animal. Both acquisition and analysis of images were performed in a highly automated way as further detailed below, thus excluding user bias. CASL images were processed and analyzed using in‐house software written in IDL 6.4 (Interactive Data Language; Exelis) and MATLAB 7.14 (The MathWorks Inc.). The anatomical volume of each individual animal was co‐registered to an in‐house mouse brain template using the open‐source software SPM5 (Welcome Trust Centre for Neuroimaging). Spatial normalization comprised a 12‐parameter affine as well as a nonlinear transform, allowing for global scaling as well as for local adjustments of anatomical features. This normalization procedure was applied identically to all functional images of the same subject. The template was in alignment with an in‐house generated digital atlas delineating selected anatomical brain areas adapted from standard brain atlases (Paxinos & Watson, 1998; Paxinos & Franklin, 2001). T1 maps per animal were calculated on a voxel‐wise basis by fitting a 3‐parameter exponential to the intensities across the eight inversion times. These T1 maps were then combined with the pertinent CASL images to obtain quantitative absolute perfusion maps, as described elsewhere (Bruns et al, 2009). Mean cerebral perfusion for individual animals was determined in the entire brain, and values were averaged across the three successive CASL volumes.
Lipopolysaccharide injections and cytokine analysis
Wild‐type and Trem2 p.T66M knock‐in mice were treated i.p. with 1 mg/kg lipopolysaccharide (LPS) to investigate the impact of Trem2 deficiency on inflammatory processes. Control animals were injected with equivalent volume of vehicle (PBS). Eighteen hours after LPS injection, mice were anesthetized with pentobarbital (150 mg/kg) and blood was drawn from the heart into EDTA tubes and plasma was prepared for cytokine detection. After blood sampling, mice were transcardially perfused with ice‐cold PBS and brains collected.
For RNA extraction, brain tissue was homogenized and RNA was isolated with RNeasy Mini Kit (QIAGEN) following the manufacturer's instructions. qRT–PCR was performed using AgPath One‐Step RT–PCR kit. Gapdh was used as housekeeping gene.
Cytokine concentrations of plasma samples were measured by Cytometric Bead Array (CBA, BD Biosciences) following the manufacturer's instructions using a Guava® easyCyte 8HT flow cytometer (EMD Millipore).
μPET data acquisition and reconstruction
Eight female homozygous Trem2 p.T66M knock‐in mice and eight age‐ and sex‐matched wild‐type controls underwent μPET imaging with the TSPO ligand [18F]‐GE180 (Dickens et al, 2014; Wickstrom et al, 2014) in a longitudinal design (3, 5, 8 and 12 months). All mice were anesthetized with isoflurane (1.5%, delivered at 3.5 l/min) and placed in the aperture of the Siemens Inveon DPET, as described previously (Brendel et al, 2015). Upon tail vein injection of 13.2 ± 2.1 MBq [18F]‐GE180 (in 150 μl saline), mice were kept in anesthesia for 45 min, followed by a 15‐min transmission scan using a rotating [57Co] point source, and 60‐ to 90‐min post‐injection static emission recording (Brendel et al, 2016).
Nine homozygous Trem2 p.T66M knock‐in mice and six age‐ and sex‐matched wild‐type controls at the age of 12 months underwent dual μPET imaging with [18F]‐FDG and [18F]‐GE180. Blood glucose was measured 1 min before administration of 13.8 ± 1.5 MBq [18F]‐FDG (in 140 μl saline). After 30‐min anesthesia, a 30‐ to 60‐min post‐injection static emission recording was acquired followed by a 15‐min transmission scan using the same rotating [57Co] point source. Recordings of [18F]‐GE180 μPET were identical to the longitudinal design described above. Reconstruction was consistently performed with four OSEM3D and 32 MAP3D iterations, a zoom factor of 1.0, scatter‐, attenuation‐, and decay‐corrected, leading to a final voxel dimension of 0.78 × 0.78 × 0.80 mm.
Image co‐registration and quantitative μPET data analyses
Static 30‐ to 60‐min (FDG‐μPET) and 60‐ to 90‐min (TSPO‐μPET) datasets were co‐registered to an MRI mouse atlas (Dorr et al, 2007) by a manual rigid‐body transformation (TXrigid) using the PMOD fusion tool (V3.5, PMOD Technologies Ltd.). Tracer and age‐specific templates were generated by combining and averaging all age‐specific μPET scans from wild‐type and Trem2 p.T66M knock‐in mice. In the second step, a reader‐independent fine co‐registration to age‐specific templates was performed (Overhoff et al, 2016). Here, the initial manual μPET‐to‐MRI atlas fusion images were normalized by nonlinear brain normalization (TXBN) to the age‐specific templates using the PMOD brain normalization tool (equal modality; smoothing by 0.6 mm; nonlinear warping; 16 iterations; frequency cutoff 3; regularization 1.0; no thresholding). The concatenation of TXrigid and TXBN was then applied to μPET frames in the native space, so as to obtain optimal resampling with a minimum of interpolation. Normalization of the data was performed by dividing the data by the injected dose (%ID) and multiplicating it by the body weight. A whole‐brain volume of interest was used for calculation of the cerebral TSPO activity and glucose metabolism in individual mice as the quantitative μPET endpoints. FDG‐μPET data were corrected for blood glucose levels at the time of tracer injection.
Additionally, two homozygous Trem2 p.T66M knock‐in mice and one wild‐type mouse received dynamic FDG‐μPET imaging over 60 min (reconstructed in 5 min frames) to test for relevant effects in the perfusion phase of PET (Fig EV3).
Cell culture
Bone marrow‐derived macrophages (BMDMs) were prepared essentially as described before (Xiang et al, 2016). For γ‐secretase inhibition experiments, 1 × 106 BMDMs were seeded in 6‐well plates, allowed to attach for 6 h, and treated with 5 μM DAPT (Sigma) or DMSO (vehicle control) for 16–20 h prior to preparing membrane fractions essentially as described previously (Kleinberger et al, 2014).
For analysis of cell proliferation by bromodeoxyuridine (BrdU) incorporation or cyclin D1 expression analysis, 2 × 105 BMDMs were seeded in non‐tissue culture‐treated 12‐well plates (VWR) and differentiated for 7 days as described before (Xiang et al, 2016).
Biochemical analyses
Frozen mouse brain was crunched in liquid nitrogen and extracted in Tris‐buffered saline (TBS; 50 mM Tris pH 7.4, 150 mM NaCl). Pulverized brain tissue was homogenized in ice‐cold TBS (10 w/v) using a 26‐G needle and cleared by ultracentrifugation (186,000 × g, 1 h, 4°C). For immunoprecipitation from mouse brain, total brain lysates were prepared in immunoprecipitation lysis buffer (25 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, and 5% glycerol) and cleared by centrifugation at 16,200 g for 30 min at 4°C. Protein concentrations were measured using the bicinchoninic acid (BCA) method (Pierce).
Immunoprecipitation and immunoblotting
Trem2 was immunoprecipitated from 400 μg total brain protein using 1 μg biotinylated goat anti‐Trem2 antibody (R&D Systems; BAF1729). After overnight incubation at 4°C, Trem2 was pulled down using streptavidin sepharose (GE Healthcare) for at least 2 h at 4°C, washed three times with ice‐cold PBS, eluted with 2× Laemmli buffer supplemented with β‐mercaptoethanol, separated by standard 12% SDS–PAGE, and transferred onto polyvinylidene difluoride membranes (Hybond P; Amersham Biosciences). Bound antibodies were visualized by corresponding HRP‐conjugated secondary antibodies using enhanced chemiluminescence technique (Pierce). Images for immunoblot quantifications were obtained on a Fusion SL imager (PeqLab) and analyzed using Multi Gauge V3.0 (PLX Devices).
Antibodies
For immunoprecipitation from total mouse brain lysates, biotinylated goat anti‐mouse Trem2 (BAF1729; R&D Systems) was used. For immunoblot detection, the following antibodies were used: rat monoclonal antibody against the ectodomain of murine Trem2 (clone 5F4; Xiang et al, 2016), rat monoclonal antibody raised against a C‐terminal peptide (C‐GRQKPGTPVVRGLDCGQDAG) of murine Trem2 (clone 14D1), mouse anti‐APP (clone 22C11), and rabbit anti‐calnexin (1:3,000; Enzo Life Sciences). Secondary antibodies were HRP‐conjugated goat anti‐rabbit IgG (1:10,000; Promega), goat anti‐mouse IgG (1:10,000; Santa Cruz Biotechnology), or goat anti‐rat IgG (1:5,000–1:10,000; Santa Cruz Biotechnology).
Quantification of sTrem2
sTrem2 levels in mouse serum, mouse cerebrospinal fluid (CSF), cell culture supernatants from BMDMs and in TBS fractions from mouse brain were quantified using the Meso Scale platform with small modifications to previously described methods (Kleinberger et al, 2014). For sTrem2, streptavidin‐coated 96‐well small‐spot plates were blocked overnight at 4°C in blocking buffer [3% bovine serum albumin (BSA) and 0.05% Tween‐20 in PBS, pH 7.4]. For the detection of mouse sTrem2, plates were incubated for 1 h at RT with 0.25 μg/ml biotinylated polyclonal goat anti‐mouse TREM2 capture antibody (R&D Systems; BAF1729) diluted in sample diluent [1% bovine serum albumin (BSA) and 0.05% Tween‐20 in PBS, pH 7.4]. Plates were washed subsequently for four times with washing buffer (0.05% Tween‐20 in PBS) and incubated for 2 h at RT with samples diluted in assay buffer (serum: 1:10; CSF: 1:6; BMDMs: 1:2; brain TBS fraction: 1:2) supplemented with protease inhibitors (Sigma). A recombinant mouse Trem2 protein (Hölzel Diagnostika) was diluted in assay buffer in a twofold serial dilution and used for the standard curve (concentration range: 2,000–31.25 pg/ml). Plates were washed three times with washing buffer before incubation for 1 h at RT with 1 μg/ml rat monoclonal anti‐Trem2 antibody (R&D Systems, MAB1729) diluted in assay diluent. MAB1729 was confirmed to detect mutant Trem2 (Appendix Fig S1). After three additional washing steps, plates were incubated with 0.5 μg/ml SULFO‐TAG‐labeled anti‐rat secondary antibody (Meso Scale Discovery) and incubated for 1 h at RT. Lastly, plates were washed three times with wash buffer and developed by adding 1× Meso Scale Discovery Read buffer. The light emission at 620 nm after electrochemical stimulation was measured using the Meso Scale Discovery Sector Imager 2400 reader. Calculation of the concentration of sTrem2 was performed with the MSD Discovery Workbench v4 software (MSD). To normalize sTrem2 levels obtained from BMDMs to the cell number, BMDMs were fixed with 4% paraformaldehyde for 20 min and stained with DAPI and the DAPI signal was quantified using a Cytation 3 Imaging System (BioTek).
qRT–PCR analysis
Total RNA was isolated from BMDMs or 20 mg pulverized mouse brain using the RNeasy Plus Mini Kit (Qiagen) and reverse‐transcribed into cDNA using the SuperScript III First‐Strand Synthesis System (Thermo Fisher Scientific; BMDMs) or the RT2 First‐Strand Kit (Qiagen; brain) according to the manufacturer's recommendations. mRNA expression of cyclin D1 (NM_007631), Trem2 transcript variant 1 (NM_031254) and transcript variant 2 (NM_001272078) (see Fig EV1C), Tyrobp (NM_011662), and Tmem119 (NM_0146162) was determined using TaqMan assays (IDT) and expression normalized to the geometric mean of two (Gusb and Hsp90ab1; BMDMs) or four housekeeping genes (Gapdh, Gusb, B2M, and Hsp90ab1; brain).
Phagocytosis assays
Phagocytosis of fluorogenic E. coli particles (pHrodo™ Green, Molecular Probes) was analyzed using BMDMs derived from wild‐type or homozygous Trem2 knock‐in mice. Briefly, cells were plated in 12‐well plates at a density of 1 × 105 cells and cultured for 24 h. pHrodo E. coli bioparticles were dissolved in PBS to a concentration of 1 μg/μl, and a total of 20 μg bioparticles was added per condition and incubated for 30 min at 37 °C. As a negative control, phagocytosis was inhibited with 10 μM cytochalasin D (Sigma) added 30 min prior to addition of pHrodo E. coli bioparticles. Cells were harvested by scraping, washed two times with PBS, and analyzed by flow cytometry on a MACSQuant® VYB flow cytometer (Miltenyi Biotec). Data analysis was performed using the MACSQuantify software (Miltenyi Biotec).
Phagocytosis of aggregated FAM‐labeled amyloid beta peptide 1–42 (Aβ1–42; Anaspec) was analyzed as previously described (Xiang et al, 2016).
Bromodeoxyuridine (BrdU) incorporation assay
BMDMs were differentiated in 12‐well plates, and at day 6, BrdU was added to a final concentration of 100 μg/ml and incubated for 24 h. BMDMs were harvested and BrdU incorporation was quantified using the FITC BrdU Flow Kit (BD Pharmingen) according to the manufacturer's recommendations.
Apoptosis
The levels of active caspase 3/7 were quantified in BMDMs at day 7 of differentiation. BMDMs were harvested and counted and 5 × 104 cells incubated with Caspase‐Glo 3/7 luminescence reagent (Promega) for 1 h at RT before quantifying the luciferase signal using a MicroLumatPlus LB96V luminometer (Berthold Technologies).
Immunohistochemistry and semiquantitative image analysis
For immunohistochemistry, mice were transcardially perfused with phosphate‐buffered saline (PBS) and post‐fixed in formalin at 4°C. Immunohistochemistry was performed on 5‐ to 8‐μm‐thick sections from paraffin‐embedded tissue. Sections were deparaffinized in xylene, followed by rehydration in a series of graded ethanol, and antigen retrieval was performed by boiling in citrate buffer. Iba1 (Wako) immunohistochemistry was performed with the Ventana BenchMark XT automated staining system (Ventana) using the UltraView Universal DAB Detection Kit (Roche). Images were taken by CellD, Olympus BX50 Soft Imaging System (Olympus, Tokyo, Japan). For semiquantitative image analysis, 40 serial sections were prepared and Iba1 immunohistochemistry was performed on 8 sections/sample. Iba1 immunoreactivity was scored on 8 sections/sample as outlined:
| Score | Cerebellum | Cerebrum |
|---|---|---|
| 0 | Weak Iba1 reactivity | No Iba1 IR nodules |
| 1 | Moderate Iba1 reactivity | < 3 Iba1 IR nodules |
| 2 | Strong Iba1 reactivity | 3–10 Iba1 IR nodules |
| 3 | Moderate Iba1 reactivity with frequent nodules | > 10 Iba1 IR nodules |
| 4 | Strong Iba1 reactivity with frequent nodules |
Statistical analysis
Statistical significance was either calculated by one‐way ANOVA with Tukey's post hoc test (Figs 1D–F, 2A, C and D, and 3B–G), Kruskal–Wallis test followed by Dunn's post hoc test (Figs 1G and 2B), two‐way ANOVA with Tukey's post hoc test (Fig 4B–H), or Student's t‐test (Figs 5 and 7A and C). Statistical analysis of the CASL‐MRI perfusion values was performed using a two‐way mixed ANOVA with genotype as between‐subject factor and age as within‐subject factor. Comparisons of interest were implemented as post hoc contrasts within the framework of this model (Fig 6B). Correlation between TSPO and FDG μPET signals was calculated using Pearson product–moment correlation coefficient. Statistical significance was set to 5% (α = 0.05). All tests were two‐sided, data were analyzed using GraphPad Prism 7, and figures were build using CorelDRAW X5.
Author contributions
CH and GK conceived the study and analyzed the results. CH and GK wrote the manuscript with input from all co‐authors. BW and WW generated the Trem2 knock‐in mouse line. GK, XX, and FM performed phagocytosis assays. GK and NP performed RNA analysis and immunoblotting. EM and LG performed and analyzed the in vivo LPS experiments. MS helped with ELISA measurements, statistical analysis, and interpretation of the results. SP collected CSF samples and performed ELISA measurements. RF generated monoclonal antibodies. TA and GK performed and analyzed immunohistochemistry. MB, PB, AR, CF, and MD conducted the μPET studies. EM, TM, and IK conducted volumetric brain imaging and CASL‐MRI measurements and interpretation of the results.
Conflict of interest
C.H. is an advisor of F. Hoffmann‐La Roche. C.H. E.M, L.G., and I.K. are full‐time employees of Hoffmann‐La Roche. All other authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy) and FOR2290, the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013)/ERC Grant Agreement No. 321366‐Amyloid, the general legacy of Mrs. Ammer, the MetLife award, and the Cure Alzheimer's Fund. W.W was further supported by SFB870. We would like to thank Dr. Marco Colonna (Washington University, School of Medicine) for kindly providing the Trem2 knockout mouse line and Mrs. Brigitte Kraft for technical assistance with immunohistochemistry. We thank Dr. Mikael Simons for critically reading the manuscript.
The EMBO Journal (2017) 36: 1837–1853
See also: L Bonanno & T Wyss‐Coray (July 2017)
References
- Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, Younkin S, Das P, Fryer JD, Bu G (2015) Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 290: 26043–26050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CC, DeVaux LB, Farzan M (2015) The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem 290: 26033–26042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, Fenoglio C, Piaceri I, Archetti S, Bonvicini C, Gennarelli M, Turla M, Scarpini E, Sorbi S, Padovani A (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35: 934 e937–910 [DOI] [PubMed] [Google Scholar]
- Brandl C, Ortiz O, Rottig B, Wefers B, Wurst W, Kuhn R (2015) Creation of targeted genomic deletions using TALEN or CRISPR/Cas nuclease pairs in one‐cell mouse embryos. FEBS Open Bio 5: 26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, Jaworska A, Herms J, Trambauer J, Rotzer C, Gildehaus FJ, Carlsen J, Cumming P, Bylund J, Luebbers T, Bartenstein P, Steiner H, Haass C, Baumann K, Rominger A (2015) Amyloid‐PET predicts inhibition of de novo plaque formation upon chronic gamma‐secretase modulator treatment. Mol Psychiatry 20: 1179–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, Probst F, Jaworska A, Overhoff F, Korzhova V, Albert NL, Beck R, Lindner S, Gildehaus FJ, Baumann K, Bartenstein P, Kleinberger G, Haass C, Herms J, Rominger A (2016) Glial activation and glucose metabolism in a transgenic amyloid mouse model: a triple‐tracer PET study. J Nucl Med 57: 954–960 [DOI] [PubMed] [Google Scholar]
- Brendel M, Kleinberger G, Probst F, Jaworska A, Overhoff F, Blume T, Albert NL, Carlsen J, Lindner S, Gildehaus FJ, Ozmen L, Suarez‐Calvet M, Bartenstein P, Baumann K, Ewers M, Herms J, Haass C, Rominger A (2017) Increase of TREM2 during aging of an Alzheimer's disease mouse model is paralleled by microglial activation and amyloidosis. Front Aging Neurosci 9: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns A, Kunnecke B, Risterucci C, Moreau JL, von Kienlin M (2009) Validation of cerebral blood perfusion imaging as a modality for quantitative pharmacological MRI in rats. Magn Reson Med 61: 1451–1458 [DOI] [PubMed] [Google Scholar]
- Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS, Cross AH, Otero K, Piccio L (2015) TREM2 regulates microglial cell activation in response to demyelination in vivo . Acta Neuropathol 129: 429–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R (2015) Increasing the efficiency of homology‐directed repair for CRISPR‐Cas9‐induced precise gene editing in mammalian cells. Nat Biotechnol 33: 543–548 [DOI] [PubMed] [Google Scholar]
- Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, Engelborghs S, Vandenbulcke M, Van Dongen J, Geerts N, Maes G, Mattheijssens M, Peeters K, Cras P, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Cruts M, Sleegers K, Consortium B (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging 35: 726.e11–726.e19 [DOI] [PubMed] [Google Scholar]
- Cuyvers E, Sleegers K (2016) Genetic variations underlying Alzheimer's disease: evidence from genome‐wide association studies and beyond. Lancet Neurol 15: 857–868 [DOI] [PubMed] [Google Scholar]
- Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE (2003) Pattern recognition by TREM‐2: binding of anionic ligands. J Immunol 171: 594–599 [DOI] [PubMed] [Google Scholar]
- Dickens AM, Vainio S, Marjamaki P, Johansson J, Lehtiniemi P, Rokka J, Rinne J, Solin O, Haaparanta‐Solin M, Jones PA, Trigg W, Anthony DC, Airas L (2014) Detection of microglial activation in an acute model of neuroinflammation using PET and radiotracers 11C‐(R)‐PK11195 and 18F‐GE‐180. J Nucl Med 55: 466–472 [DOI] [PubMed] [Google Scholar]
- Dorr A, Sled JG, Kabani N (2007) Three‐dimensional cerebral vasculature of the CBA mouse brain: a magnetic resonance imaging and micro computed tomography study. NeuroImage 35: 1409–1423 [DOI] [PubMed] [Google Scholar]
- Glebov K, Wunderlich P, Karaca I, Walter J (2016) Functional involvement of gamma‐secretase in signaling of the triggering receptor expressed on myeloid cells‐2 (TREM2). J Neuroinflammation 13: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K et al (2013a) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H, Emre M, Singleton A, Hardy J (2013b) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia‐like syndrome without bone involvement. JAMA Neurol 70: 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A, Frahm J, Matthaei D, Hanicke W, Bomsdorf H, Kunz D, Tischler R (1986) MR imaging using stimulated echoes (STEAM). Radiology 160: 787–790 [DOI] [PubMed] [Google Scholar]
- Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)‐2 and DAP12. J Immunol 177: 2051–2055 [DOI] [PubMed] [Google Scholar]
- Henjum K, Almdahl IS, Arskog V, Minthon L, Hansson O, Fladby T, Nilsson LN (2016) Cerebrospinal fluid soluble TREM2 in aging and Alzheimer's disease. Alzheimer's Res Ther 8: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herholz K (2011) Perfusion SPECT and FDG‐PET. Int Psychogeriatr 23(Suppl. 2): S25–S31 [DOI] [PubMed] [Google Scholar]
- Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Ohrfelt A, Blennow K, Hardy J, Schott J, Mills K, Zetterberg H (2016) Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener 11: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109: 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Hamerman JA (2012) TREM‐2, triggering receptor expressed on myeloid cell‐2, negatively regulates TLR responses in dendritic cells. Eur J Immunol 42: 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim‐Verbaas C, Hofman A, Ikram MA et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez‐Valle R, Antonell A, Ramirez A et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6: 243ra286 [DOI] [PubMed] [Google Scholar]
- Klunemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64: 1502–1507 [DOI] [PubMed] [Google Scholar]
- Kober DL, Alexander‐Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, Brett TJ (2016) Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss‐of‐function mechanisms. eLife 5: e20391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48: 405–415 [DOI] [PubMed] [Google Scholar]
- Liu GJ, Middleton RJ, Hatty CR, Kam WW, Chan R, Pham T, Harrison‐Brown M, Dodson E, Veale K, Banati RB (2014) The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol 24: 631–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Le KX, Park MA, Wang S, Belanger AP, Dubey S, Frost JL, Holton P, Reiser V, Jones PA, Trigg W, Di Carli MF, Lemere CA (2015) In vivo detection of age‐ and disease‐related increases in neuroinflammation by 18F‐GE180 TSPO microPET imaging in wild‐type and Alzheimer's transgenic mice. J Neurosci 35: 15716–15730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourbopoulos A, Erturk A, Hellal F (2015) Microglia in action: how aging and injury can change the brain's guardians. Front Cell Neurosci 9: 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trümbach D, Wurst W, Brunner B, Bultmann S, Tahirovic S, Kerschensteiner M, Misgeld T, Butovsky O, Haass C (2017) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18: 1186–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36: 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbetti L, Ratti MT, Greco B, Aprile C, Moglia A, Soragna D (2005) Neuropsychological tests and functional nuclear neuroimaging provide evidence of subclinical impairment in Nasu‐Hakola disease heterozygotes. Funct Neurol 20: 71–75 [PubMed] [Google Scholar]
- N'Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, Hamerman JA, Seaman WE (2009) TREM‐2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol 184: 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science 308: 1314–1318 [DOI] [PubMed] [Google Scholar]
- Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, Faccio R, Ross FP, Teitelbaum SL, Takayanagi H, Colonna M (2012) TREM2 and beta‐catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol 188: 2612–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overhoff F, Brendel M, Jaworska A, Korzhova V, Delker A, Probst F, Focke C, Gildehaus FJ, Carlsen J, Baumann K, Haass C, Bartenstein P, Herms J, Rominger A (2016) Automated spatial brain normalization and hindbrain white matter reference tissue give improved [(18)F]‐Florbetaben PET quantitation in Alzheimer's model mice. Front Neurosci 10: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M (2001) CNS manifestations of Nasu‐Hakola disease: a frontal dementia with bone cysts. Neurology 56: 1552–1558 [DOI] [PubMed] [Google Scholar]
- Park JS, Ji IJ, An HJ, Kang MJ, Kang SW, Kim DH, Yoon SY (2015) Disease‐associated mutations of TREM2 alter the processing of N‐linked oligosaccharides in the golgi apparatus. Traffic 16: 510–518 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates, 4th edn San Diego, CA: Academic Press; [Google Scholar]
- Paxinos G, Franklin K (2001) The mouse brain in stereotaxic coordinates. New York: Academic Press; [Google Scholar]
- Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J II, Naismith RT, Panina‐Bordignon P, Passini N, Galimberti D, Scarpini E, Colonna M, Cross AH (2008) Identification of soluble TREM‐2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131: 3081–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Deming Y, Del‐Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C (2016) Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol 131: 925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M (2015) TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest 125: 2161–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME (1998) Behind the scenes of functional brain imaging: a historical and physiological perspective. Proc Natl Acad Sci USA 95: 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen‐Hoerth C, Kertesz A, Bigio EH, Lippa C, Josephs KA, Knopman DS, White CL III, Caselli R, Mackenzie IR, Miller BL, Boczarska‐Jedynak M, Opala G, Krygowska‐Wajs A et al (2013) TREM2 in neurodegeneration: evidence for association of the p. R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Beggs S (2014) Sublime microglia: expanding roles for the guardians of the CNS. Cell 158: 15–24 [DOI] [PubMed] [Google Scholar]
- Schelle J, Hasler L, Gopfert JC, Joos TO, Vanderstichele H, Stoops E, Mandelkow EM, Neumann U, Shimshek DR, Staufenbiel M, Jucker M, Kaeser SA (2016) Prevention of tau increase in cerebrospinal fluid of APP transgenic mice suggests downstream effect of BACE1 inhibition. Alzheimer's Dement 13: 701–709 [DOI] [PubMed] [Google Scholar]
- Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W (2013) Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 125: 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L (1977) Relation between physiological function and energy metabolism in the central nervous system. J Neurochem 29: 13–26 [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28: 897–916 [DOI] [PubMed] [Google Scholar]
- Sokoloff L (1999) Energetics of functional activation in neural tissues. Neurochem Res 24: 321–329 [DOI] [PubMed] [Google Scholar]
- Suarez‐Calvet M, Caballero M, Kleinberger G, Bateman R, Fagan AM, Morris J, Levin J, Danek A, Ewers M, Haass C, Network ftDIA (2016a) Early changes of CSF sTREM2 in dominantly inherited Alzheimer's disease follow markers of amyloid deposition and neuronal injury. Sci Transl Med 8: 369ra178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez‐Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD, Sanchez‐Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschutz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K et al (2016b) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early‐stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med 8: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells‐2. J Exp Med 201: 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita T, Kaminaga T, Tatsumi T, Hatanaka Y, Furui S (2005) Regional cerebral blood flow in a patient with Nasu‐Hakola disease. Ann Nucl Med 19: 309–312 [DOI] [PubMed] [Google Scholar]
- Tosun D, Schuff N, Rabinovici GD, Ayakta N, Miller BL, Jagust W, Kramer J, Weiner MM, Rosen HJ (2016) Diagnostic utility of ASL‐MRI and FDG‐PET in the behavioral variant of FTD and AD. Ann Clin Transl Neurol 3: 740–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Majewska AK (2011) A role for microglia in synaptic plasticity? Commun Integr Biol 4: 220–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM‐2 attenuates macrophage activation. J Immunol 177: 3520–3524 [DOI] [PubMed] [Google Scholar]
- Ueki Y, Kohara N, Oga T, Fukuyama H, Akiguchi I, Kimura J, Shibasaki H (2000) Membranous lipodystrophy presenting with palilalia: a PET study of cerebral glucose metabolism. Acta Neurol Scand 102: 60–64 [DOI] [PubMed] [Google Scholar]
- Varrone A, Asenbaum S, Vander Borght T, Booij J, Nobili F, Nagren K, Darcourt J, Kapucu OL, Tatsch K, Bartenstein P, Van Laere K, European Association of Nuclear Medicine Neuroimaging C (2009) EANM procedure guidelines for PET brain imaging using [18F]FDG, version 2. Eur J Nucl Med Mol Imaging 36: 2103–2110 [DOI] [PubMed] [Google Scholar]
- Verfaillie SC, Adriaanse SM, Binnewijzend MA, Benedictus MR, Ossenkoppele R, Wattjes MP, Pijnenburg YA, van der Flier WM, Lammertsma AA, Kuijer JP, Boellaard R, Scheltens P, van Berckel BN, Barkhof F (2015) Cerebral perfusion and glucose metabolism in Alzheimer's disease and frontotemporal dementia: two sides of the same coin? Eur Radiol 25: 3050–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas‐Llerena C, Phillips A, Garcia‐Reitboeck P, Hardy J, Pocock JM (2016) Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol 36: 74–81 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefers B, Panda SK, Ortiz O, Brandl C, Hensler S, Hansen J, Wurst W, Kuhn R (2013) Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nat Protoc 8: 2355–2379 [DOI] [PubMed] [Google Scholar]
- Wickstrom T, Clarke A, Gausemel I, Horn E, Jorgensen K, Khan I, Mantzilas D, Rajanayagam T, in‘t Veld DJ, Trigg W (2014) The development of an automated and GMP compliant FASTlab Synthesis of [(18) F]GE‐180; a radiotracer for imaging translocator protein (TSPO). J Labelled Compd Radiopharm 57: 42–48 [DOI] [PubMed] [Google Scholar]
- Williams DS, Detre JA, Leigh JS, Koretsky AP (1992) Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proc Natl Acad Sci USA 89: 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Byers DE, Jin X, Agapov E, Alexander‐Brett J, Patel AC, Cella M, Gilfilan S, Colonna M, Kober DL, Brett TJ, Holtzman MJ (2015) TREM‐2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med 212: 681–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J (2013) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells‐2 (TREM2) protein by ectodomain shedding and gamma‐secretase‐dependent intramembranous cleavage. J Biol Chem 288: 33027–33036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, Feederle R, Knuesel I, Kleinberger G, Haass C (2016) TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med 8: 992–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Tu Z, Sun Q, Li XJ (2016) CRISPR/Cas9: implications for modeling and therapy of neurodegenerative diseases. Front Mol Neurosci 9: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid‐beta by microglia. Neuron 91: 328–340 [DOI] [PubMed] [Google Scholar]
- Zhong L, Chen XF, Zhang ZL, Wang Z, Shi XZ, Xu K, Zhang YW, Xu H, Bu G (2015) DAP12 stabilizes the C‐terminal fragment of the triggering receptor expressed on myeloid cells‐2 (TREM2) and protects against LPS‐induced pro‐inflammatory response. J Biol Chem 290: 15866–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, Huang R, Wang D, Li X, Wu L, Jia L, Zheng H, Painter M, Atagi Y, Liu CC, Zhang YW, Fryer JD, Xu H, Bu G (2017) Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med 214: 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7